

Abstract
Insulin-degrading enzyme (IDE) exists primarily as a dimer being unique among the zinc metalloproteases in that it exhibits allosteric kinetics with small synthetic peptide substrates. In addition the IDE reaction rate is increased by small peptides that bind to a distal site within the substrate binding site. We have generated mixed dimers of IDE in which one or both subunits contain mutations that affect activity. The mutation Y609F in the distal part of the substrate binding site of the active subunit blocks allosteric activation regardless of the activity of the other subunit. This effect shows that substrate or small peptide activation occurs through a cis effect. A mixed dimer composed of one wild-type subunit and the other subunit containing a mutation that neither permits substrate binding nor catalysis (H112Q) exhibits the same turnover number per active subunit as wild-type IDE. In contrast, a mixed dimer in which one subunit contains the wild-type sequence and the other contains a mutation that permits substrate binding, but not catalysis (E111F), exhibits a decrease in turnover number. This indicates a negative trans effect of substrate binding at the active site. On the other hand, activation in trans is observed with extended substrates that occupy both the active and distal sites. Comparison of the binding of an amyloid β peptide analog to wild-type IDE and to the Y609F mutant showed no difference in affinity, indicating that Y609 does not play a significant role in substrate binding at the distal site.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, Metalloprotease, Peptidases
Introduction
Insulysin (insulin-degrading enzyme, IDE,2 EC 3.4.14.56) is one of the major enzymes involved in Aβ clearance as evidenced by genetic studies with IDE-deficient mice (1) as well as by over expression of IDE in transgenic mouse brain (2). IDE is also the major enzyme involved in cellular insulin degradation (3–5) and mutations in IDE in the GK rat have been linked to type 2 diabetes (6). IDE is made up of four structurally similar domains arranged into a U-like conformation, with only the N-terminal most domain retaining a functional active site (9). The enzyme is primarily dimeric (7–10) and unique among the zinc metalloproteases in that it exhibits non-classical kinetics with small synthetic peptide substrates (8). This property appears dependent on association to form a homodimer (8). With small synthetic peptide substrates IDE exhibits a sigmoidal rate versus substrate response indicative of allosteric enzymes showing homotropic activation or activation by substrate. In addition IDE is activated by small physiological peptides such as bradykinin and dynorphin (8) by what can be termed “heterotrophic activation” or activation by molecules other than the substrate. We recently obtained evidence that peptide activators bind to a site on the enzyme we refer to as the distal site.3 Both this distal site as well as the active site was found to contain a peptide derived from TEV protease release of the polyHis and linker region from an inactive IDE fusion protein. As might be expected with active IDE neither the active site nor the distal site is occupied by the same TEV protease treatment, presumably due to degradation of the released polyHis-containing peptide. This distal site makes extended binding contacts with larger peptide substrates such as Aβ and insulin (11) and has been reported to bind a small substrate in a non-catalytic mode (12).
In addition to activation by small peptides, IDE is activated by polyphosphates such as ATP and triphosphate (13, 14). The distal site where small peptides bind and the site where ATP binds have been shown to be distinct (13).4
The finding that IDE can be activated by peptides and polyphosphates makes it an excellent target for the development of small molecules that could be used in the treatment of Alzheimer disease (15). A better understanding of the mechanism of activation of IDE should facilitate such efforts. To this end we have made use of mutations that affect catalytic activity, substrate binding at the active site, and activation via the distal site to further study the mechanism of activation of IDE in the context of the IDE dimer.
EXPERIMENTAL PROCEDURES
Preparation of Mixed Oligomers of IDE
Mixed oligomers, of IDE were generated by using baculovirus to co-express wild type and IDE mutant forms in Sf9 insect cells (8). Mutations introduced into IDE included E111F, which removes the catalytic glutamate that serves to facilitate attack of water on the scissile bond, H112Q, which removes this zinc binding residue, and Y609F, which reduces allosteric activation produced at a distal substrate binding site. None of these mutations affect the overall structure of the enzyme as shown by circular dichroism measurements, Fig. 1. In each case we added to Sf9 cells baculovirus coding for an inactive form of IDE containing a hexahistidine affinity tag attached through a linker containing a TEV protease site mixed with baculovirus expressing an active form of IDE devoid of an affinity tag. Preliminary experiments were conducted in which each baculovirus culture was titered with respect to expression of its respective IDE form. Different viruses were then mixed to optimize equal co-expression of each IDE form. As noted under “Results,” we usually obtained lower yields of mixed dimer than anticipated. Although not investigated it is possible that the inactive enzyme forms tend to generate homodimers rather than heterodimers. Oligomeric forms were generated in 200-ml cultures expressed for 72 h and isolated by Ni-affinity chromatography using a 2-ml HIS-select Ni-NTA-agarose column as previously described (16, 17). This method yielded catalytically active oligomers composed of the His-tagged inactive subunit and non-tagged catalytically active subunits. Oligomers containing both inactive subunits copurified using this approach, but did not contribute to activity measurements.
FIGURE 1.

Comparison of the structure of IDE mutants by circular dichroism. Circular dichroism expressed as mean molar ellipticity per residue is plotted as a function of wavelength for IDE, IDEE111F, IDEH112Q, and IDEY609F. Protein was present at a concentration around 0.5 mg/ml. Spectra were collected at 20 °C in 50 mm Tris-HCl buffer, pH 7.4, and normalized based on values at 214 nm. Only every fourth experimental point is shown for clarity.
To determine the concentration of active (non-affinity tagged) IDE the isolated oligomers were subjected to SDS-PAGE on duplicate gels along with IDE-His6 as a standard. Following transfer to PDVF membranes one membrane was treated with a rabbit anti-IDE antibody (1) at 1/1,000 while the other was treated with mouse anti-His6 antibody (GE, Healthcare Life, Sciences) at 1/1,000. Following incubation for 1 h with HRP-conjugated goat anti-rabbit IgG (1/20,000) (Zymed Laboratories Inc., Inc.) or HRP-conjugated goat anti-mouse IgG-HRP antibody (1/5,000) (Zymed Laboratories Inc., Inc.) the immunoreactive bands were visualized by ECL using Western blotting Detection Reagents from GE Healthcare. The ratio of the intensity of anti-His antibody staining (inactive IDE) and the intensity of staining obtained using an anti-IDE antibody (total IDE) was calibrated on theWestern blot using fully His-tagged IDE. The ratio of anti-His/anti-total IDE was assigned a value, N. The amount of active IDE subunit was calculated as follows: The observed anti-His staining/N yields the expected anti-IDE staining for the fraction of His-tagged IDE present in the mixture and is designated as X.
Total anti-IDE immunostaining is equal to the anti-IDE staining for the His-tagged IDE (X) plus the amount of non-His-tagged IDE present. Thus the amount of non-His-tagged IDE is equal to the total staining minus X and is designated Y. The fraction of non-His-tagged IDE present is calculated as Equation 1.
We found more reproducible results were obtained with the His tag removed by treatment with TEV protease, so the above analysis was conducted prior to His tag removal.
Enzyme Activity Assays
The fluorogenic peptide Abz-GGFLRKHGQ-EDDnp was routinely used for measuring IDE activity. This internally quenched FRET peptide is cleaved at the RK bond resulting in an increase in fluorescence as previously described (16). Reaction mixtures contained 50 mm Tris-HCl, pH 7.4, IDE, 10 μm Abz-GGFLRKHGQ-EDDnp, and the indicated concentration of peptides when added in a total volume of 200 μl. The reaction was continuously monitored at 37 °C for 30 min. using a SpectraMax Gemini XS fluorescence plate reader. Kinetic data were fit to either a hyperbolic substrate versus velocity response curve (Michaelis-Menten equation) or to a sigmoidal response curve using Graphpad software. Experiments were repeated two or three times with different enzyme preparations. For monitoring the activity of fractions eluted from a Amersham Biosciences Superdex S200 column, the reaction contained 195 μl of the column fraction prewarmed to 37 °C, with the reaction initiated by adding 5 μl of 400 mm Abz-GGFLRKHGQ-EDDnp. Assay times varied from 30 min to 240 min depending on the activity of the eluted enzyme.
The reaction of IDE with amyloid β peptide (Aβ1–40) was measured by two different methods. In one method we used a biotinylated/fluorescently labeled Aβ analog Aβ1–40-Lys(Biotin)FAM-labeled (AnaSpec, Inc.). Reactions (200 μl) containing 0.5 μm Aβ1–40-Lys(Biotin)FAM-labeled in 50 mm Tris-HCl buffer, pH 7.4, plus 0.05% Tween-20 were incubated with 50–400 ng of IDE for 30 min. at 37 °C. The reaction was stopped by adding 1 μl of 200 mm 1–10-phenanthroline to produce a final concentration of 1 mm and then 15 μl of agarose bound avidin D (Vector Laboratories, Inc.) was added to bind any unreacted Aβ1–40-Lys(Biotin)FAM-labeled and C-terminally biotin labeled reaction products. After centrifugation for 5 min, the supernatant containing released FAM-labeled peptide products was measured on a Spectramax plate reader at an excitation of 485 nm and an emission of 528 nm. Total fluorescence released from Aβ1–40-Lys(Biotin)FAM-labeled was determined by the same procedure using an excess of IDE and used to calculate fractional release in the various incubations.
The second method employed was a sandwich ELISA assay for Aβ. Reaction mixtures containing 50 mm Tris-HCl buffer, pH 7.4, 100 nm Aβ1–40 and enzyme in a total volume of 20 μl were incubated at room temperature for 1 h. The reaction was stopped by diluting >1,000-fold into PBS-T (10 mm sodium phosphate buffer, pH 7.5 containing 150 mm NaCl and 0.05% Tween-20) to yield ∼100 pm Aβ. A 100-μl aliquot was transferred to a Maxisorp Nunc plate precoated with anti-Aβ antibody 6E10 (anti-Aβ1–6) (Covance Signet) diluted 1:500. After incubating for 2–3 h at room temperature and then at 4 °C overnight, the plate was washed and 100 μl of biotinylated anti-Aβ antibody 4G8 (anti-17–24) (Covance Signet) at a 1/2,000 dilution was added. After incubating for 2 h at room temperature, the plate was washed three times with TBST, and 100 μl of streptavidin-HRP (1:10,000) (KPL) was added followed by a 1.5 h incubation at room temperature. After removal of the streptavidin-HRP, 100 μl of a 1:1 mix of TMB Microwell Peroxidase Substrate (Kirkegaard & Perry Laboratories, Inc. Gaithersburg, MD) was added, and the reaction allowed to proceed for ∼30 min. The reaction was stopped by adding 100 μl of 1% H2SO4, and samples were read on a plate reader at 450 nm. A standard curve was constructed with varying amounts of Aβ1–40.
Amyloid Peptide Binding to IDE
The binding of the fluorescent Aβ1–40 analog Aβ1–40 FAM-labeled (AnaSpec, Inc.) to IDE was measured by following the change in fluorescence anisotropy when increasing amounts of IDE (0–3 μm) was added to 1 μm FAM-labeled Aβ1–40 in 50 mm Tris-HCl, pH 7.4 buffer in a total volume of 200 μl. Fluorescence anisotropy was measured at 23 °C using a SpectraMax M5 fluorescence plate reader at an excitation wavelength of 494 nm and an emission wavelength of 521 nm. Dissociation constants (Kd) were calculated by fitting the data using Prism software to Equation 2 (18),
where r is the observed anisotropy, ro is the initial anisotropy, and ra is the difference in anisotropy between bound and free fluorescent Aβ1–40.
Molecular Weight Determination
The oligomerization state of the various IDE preparations was determined by gel filtration using a Amersham Biosciences Superdex S200 column equilibrated with 50 mm Tris-HCl buffer, pH 7.4, and run at a flow rate of 0.4 ml/min at 4 °C. The column was standardized with a gel filtration calibration kit (GE, Healthcare Life Sciences). A 0.5-ml IDE sample was loaded at 1 mg/ml, with the column run at 0.4 ml/min with 1-ml fractions collected. Using this concentration of protein, the IDE activity in the eluted fractions was assayed without further dilution so as to approximate the same active enzyme concentrations used in kinetic experiments.
Circular Dichroism Measurements
All samples were diluted to ∼0.5 mg/ml in 50 mm Tris buffer, pH 7.4, and placed in a 0.1-cm path length cuvette prior to measuring circular dichroism spectra. Spectra were taken over a range of 200–250 nm in a Jasco J-810 spectropolarimeter (Easton, MD), measuring values every 0.2 nm. Four spectra were averaged for each IDE construct.
RESULTS
It has been established that the distal part of the extended substrate binding site of IDE can bind peptides and reaction products (11).3 We have found that mutation of a tyrosine within this distal binding site, Tyr-609, disrupts allosteric activation, and thus we have proposed that this distal site is an allosteric binding site.3 Because IDE functions primarily as a dimer we set out to determine whether binding at the substrate binding site and binding at the distal binding site can produce allosteric activation by a cis or trans mechanism. To accomplish this we generated a series of oligomeric IDE molecules in which mutations were introduced into the active site, the distal allosteric site, or both. These oligomeric IDE variants were expressed with one subunit, generally an inactive subunit, containing a hexahistidine affinity tag, and the active subunit having no affinity tag. Isolation of the oligomers on a nickel resin produced enzyme that contains at least one inactive subunit.
We first compared the kinetics of IDE derived by expressing the enzyme with both subunits containing a hexahistidine affinity tag (His6IDE)2 to IDE in which one subunit contained a His affinity tag and the other did not (His6IDE-IDE). Kinetic analysis showed that expressing IDE where both subunits contained a His tag or where only one subunit contained a His tag gave the same kinetic constants with the synthetic substrate Abz-GGFLRKHGQ-EDDnp and that removal of the His tag did not affect kinetic properties. However, we did note that the enzyme containing a His affinity tag on both subunits seemed to be activated to a slightly greater extent (5–6-fold versus ∼3–4-fold) by small peptides. To avoid any potential affect of the His affinity tag we routinely removed it prior to analysis.
We next compared the kinetic properties of mixed oligomers of IDE in which one subunit contained the wild-type sequence and the other contained either the E111F mutation or the H112Q mutation. Both of these mutations decrease enzyme activity to less than 1% of the wild-type enzyme (17) however the E111F mutation does not affect substrate binding. In contrast the H112Q mutation increases Km 5–10-fold (17). As shown in Table 1 when IDE contains one active subunit and an inactive subunit that binds substrate poorly (IDE:IDEH112Q) there is little or no effect on kcat (expressed as turnover per active subunit), Km, or the extent of cooperativity as measured by the Hill coefficient with Abz-GGFLRKHGQ-EDDnp as substrate. This indicates that substrate turnover by IDE can occur through one subunit independent of the other. However, expressing IDE containing one active subunit and an inactive subunit that retains substrate binding (IDE:IDEE111F) lowers the observed kcat more than 10-fold, but does not affect Km or the Hill coefficient. This finding suggests that once bound, substrate induces a conformational change in the adjacent subunit that lowers its activity. This effect could result from intervals of activity alternating between subunits, although the observed similar kcat values for the wild-type dimer (IDE:IDE) and the dimer with one non-binding subunit (IDE:IDEH112Q) indicates that the alternating between subunits in itself is not rate-limiting. This follows since if alternating between subunits were slow relative to catalysis, then on a per subunit basis the dimeric enzyme with two active subunits would exhibit a lower kcat than a dimeric enzyme with only one non-interacting active subunit. Comparing IDE:IDE and IDE:IDEH112Q, which as noted exhibit the same kcat, indicates that this is not the case. The effect is marked in IDE:IDEE111F presumably because substrate bound to the catalytically compromised subunit has a long residence time when it is not cleaved. Even though binding to the inactive subunit in IDE:IDEE111F decreases activity, cooperativity is still observed. This observation plus the finding that IDE:IDEH112Q, which binds substrate poorly, also exhibits full cooperativity shows that activation by substrate (homotropic allosteric activation) is independent of whether or not substrate is bound to the active site of the adjacent subunit. Thus homotropic allosteric affects can be attributed to binding at a site other than the active site. This is consistent with our recent finding that peptide-dependent activation occurs though binding to what we have termed the “distal site” on IDE.3
TABLE 1.
Kinetic properties of mixed oligomers of IDE
IDE forms were co-expressed in Sf9 cells with one subunit (when applicable the inactive subunit) containing a hexahistidine affinity tag. The mixed oligomers were purified on a Ni affinity resin and the fraction of total IDE protein as active IDE determined as described under “Experimental Procedures.” The kcat values are expressed per active subunit. Reaction rates were measured as described under “Experimental Procedures” at 37 °C in 50 mm Tris-HCl buffer, pH 7.4, with variable concentration of the fluorogenic substrate Abz-GGFLRKHGQ-EDDnp.
| IDE oligomer | kcata | Km | Hill coefficient | Fraction of total IDE as active IDE |
|---|---|---|---|---|
| min−1 | μm | |||
| IDE:IDE | 8.3 ± 1.0 | 31.5 ± 5.9 | 1.8 ± 0.2 | 1.0 |
| IDE:IDEH112Q | 7.1 ± 0.6 | 23.2 ± 2.0 | 1.7 + 0.3 | 0.17 ± 0.03 |
| IDE:IDEE111F | 0.21 ± 0.01 | 21.7 ± 1.8 | 1.8 + 0.3 | 0.41 ± 0.05 |
| Y609FIDE:IDEY609F | 1.7 ± 0.1 | 26.8 ± 9.8 | 1.1 ± 0.07 | 1.0 |
| IDE:IDEH112Q,Y609F | 1.7 ± 0.09 | 13.5 ± 1.1 | 1.9 + 0.3 | 0.14 ± 0.02 |
| IDE:IDEE111F,Y609F | 0.6 ± 0.06 | 9.1 ± 1.5 | 1.8 + 0.1 | 0.24 ± 0.01 |
| Y609FIDE:IDEH112Q | 2.8 ± 0.22 | 11.3 ± 2.5 | 1.2 ± 0.04 | 0.29 ± 0.04 |
| Y609FIDE:IDEE111F | 0.3 ± 0.02 | 10.9 ± 1.2 | 1.2 ± 0.04 | 0.25 ± 0.08 |
| Y609FIDE:IDEH112Q,Y609F | 3.7 ± 0.35 | 19.2 ± 2.6 | 1.3 ± 0.18 | 0.18 ± 0.01 |
| Y609FIDE:IDEE111F,Y609F | 0.20 ± .06 | 14.8 ± 1.8 | 1.1 ± 0.10 | 0.24 ± 0.04 |
a kcat expressed as μmol/min/μmol active subunit.
Gel filtration analysis showed that all of the oligomers regardless of their composition or enzymatic activity were predominantly dimers but contained some tetramer and to a much lesser extent higher molecular weight oligomers. Thus the presence of a catalytically inactive subunit does not produce a significant affect on the oligomerization state of IDE. This is shown in Fig. 2. Molecular weight determinations were performed by gel filtration loading IDE at 1 mg/ml such that the eluted enzyme concentration approximated the IDE concentration in the assaysused for kinetic analysis and thus reflects the oligomeric state of the various IDE forms during activity assays.
FIGURE 2.

IDE mutations do not alter the oligomeric state of the enzyme. The indicated dimeric form of IDE was chromatographed on a Superdex S200 column at 4 °C equilibrated with 50 mm Tris-HCl buffer, pH 7.4 and run at a flow rate of 0.4 ml/min column. Proteins were applied in a 0.5 ml volume at 1 mg/ml. Fractions of 1 ml were collected and assayed for IDE activity without further dilution using 10 μm Abz-GGFLRKHGQ-EDDnp as substrate as described under “Experimental Procedures.”
IDE has been shown to be subject to heterotrophic activation by small peptide substrates such as bradykinin and dynorphin (8). These peptides appear to bind to the distal site (11).3 Therefore we looked at the effect of the catalytically inactive subunit on the ability of peptide activators to increase the activity of the mixed dimers. Peptide activation did not seem to be affected to any significant extent by either type of mutation in the active site of one subunit; compare IDE:IDE, IDE:IDEH112Q, and IDE:IDEE111F in Table 2.
TABLE 2.
Activation of IDE dimeric forms by dynorphin B9 and bradykinin
Reaction mixtures contained 10 μm Abz-GGFLRKHGQ-EDDnp, 50 mm Tris-HCl, pH 7.4, and 100 μm peptide when added. Activity was measured at 37 °C as described under “Experimental Procedures.”
| IDE oligomer | Fold activation by dynorphin B9 | Fold activation by bradykinin |
|---|---|---|
| IDE:IDE | 2.3 | 2.8 |
| IDE:IDEH112Q | 2.5 | 3.0 |
| IDE:IDEE111F | 3.2 | 3.7 |
| IDE:IDEH112Q,Y609F | 2.3 | 2.7 |
| IDE:IDEE111F,Y609F | 3.2 | 3.3 |
| Y609FIDE:IDEY609F | 1.4 | 1.9 |
| Y609FIDE:IDEH112Q | 1.7 | 1.8 |
| Y609FIDE:IDEE111F | 1.2 | 1.4 |
| Y609FIDE:IDEH112Q,Y609F | 1.2 | 1.7 |
| Y609FIDE:IDEE111F,Y609F | 0. 8 | 1.2 |
As alluded to above, we previously identified a distal site that is comprised of a portion of the extended substrate-binding site. We previously showed that binding to the distal site is what actually produces the observed allosteric activation.3 Larger IDE peptide substrates such as insulin or amyloid β-peptide bind to the active site and are large enough to make extended binding interactions at the distal site and thus do not exhibit homotropic cooperativity. The substrate Abz-GGFLRKHGQ-EDDnp is not large enough to extend binding interactions to the allosteric distal site, however either its hydrolytic product or a second molecule of substrate can bind to this distal site. We thus asked the question as to whether activation through the distal site can occur on the same subunit, the adjacent subunit, or on either subunit. To answer this question we introduced a mutation into the distal site, Y609F, which we have shown eliminates allosteric kinetics.3
As shown in Table 1 when the Y609F mutation is introduced into both active subunits (Y609FIDE:IDEY609F) there is a 4–5-fold decrease in catalytic activity accompanied by a change in the Hill coefficient from ∼2 for wild-type IDE to ∼1 for the Y609F mutant. Mutation of the distal binding site on only the inactive subunit (IDE:IDEH112Q,Y609F or IDE:IDEE111F,Y609F) does not affect the allosteric nature of the reaction as judged by the absence of a change in the Hill coefficient. This strongly supports the concept of cis activation and suggests that allosteric activation by substrate, reaction products, or other peptides occurs within the same catalytically active subunit. However, a lower kcat value is observed when the distal site is mutated in the catalytically incompetent non-binding subunit; compare IDE:IDEH112Q,Y609F to IDE:IDEH112Q. This further suggests that although peptide binding to the distal site of the catalytic subunit produces cis activation, disruption of the allosteric site of the second subunit in the dimer can affect the activity of the adjacent subunit.
We next examined the effect of mutating the distal site on the catalytic subunit, but having this site available on the inactive subunit, Table 1. The absence of the distal site on the catalytic subunit eliminates cooperativity, regardless of whether or not the inactive subunit has an intact distal binding site as seen in the Hill coefficients for Y609FIDE:IDEH112Q, Y609FIDE:IDEE111F, Y609FIDE:IDEH112Q,Y609F, or Y609FIDE:IDEE111F,Y609F. This finding further confirms homotropic cooperativity as resulting from cis binding to the distal site on the catalytically active subunit. With the IDEH112Q mutant, the Y609F mutation on the active subunit decreases kcat as expected (compare IDE:IDEH112Q to Y609FIDE:IDEH112Q). Interestingly, however a different situation occurs when the inactive subunit can bind substrate. In this case introducing the Y609F mutation into the active subunit has no additional effect on kcat (compare IDE:IDEE111F to Y609FIDE:IDEE111F). Here the effect of maintaining bound peptide at the active site of the catalytically compromised subunit must dominate the functioning of the active subunit.
That the distal site can work in cis with the catalytic site is further supported by the data obtained when measuring the ability of the peptides dynorphin B9 and bradykinin to activate hydrolysis of Abz-GGFLRKHGQ-EDDnp as substrate. As illustrated in Table 2 all of the enzyme forms that contain an intact distal site on the catalytic subunit, regardless of the presence (IDE:IDE, IDE:IDEH112Q, IDE:IDEE111F) or absence (IDE:IDEH112Q,Y609F, IDE:IDEE111F,Y609F) of an intact distal site on the non catalytic subunit exhibit the same 2–3-fold activation. In contrast, when the distal site is mutated on the catalytic subunit in the absence (Y609FIDE:IDEY609F, Y609FIDE:IDEH112Q, Y609FIDE:IDEE111F) or presence (Y609FIDE:IDEH112Q,Y609F, Y609FIDE:IDEE111F,Y609F) of a second mutation on the inactive subunit, activation is diminished.
We also examined the effect of mutations in the distal site on the hydrolysis of a substrate that spans the active site and binds at the distal site, amyloid β peptide 1–40 (Aβ1–40). We employed two different assays; for one assay we used a biotinylated, N-terminal carboxyl fluorescein (FAM) labeled Aβ1–40 and in the other we used an Aβ sandwich ELISA. As shown in Table 3 there was generally good agreement between the two methods. The mixed dimer in which the inactive subunit does not bind substrate (IDE:IDEH112Q) exhibited a slightly higher activity than wild-type IDE as confirmed in both assays; however, there is a decrease in the rate when the inactive subunit can bind substrate (IDE:IDEE111F). This is the same effect observed with Abz-GGFLRKHGQ-EDDnp as substrate as noted in Table 1. When the distal site is mutated on both subunits (Y609FIDE:IDEY609F) there is an even greater decrease in the reaction rate as would be expected if the Y609F mutation interfered with substrate activation through the distal site.
TABLE 3.
Reaction of dimeric IDE with amyloid β peptide
| IDE oligomer | Fluorescent Aba | ELISAb |
|---|---|---|
| Relative activity | Relative activity | |
| IDE:IDE | 1.0 (79)c | 1.0 (135)c |
| IDE:IDEH112Q | 1.19 (94) | 1.33 (180) |
| IDE:IDEE111F | 0.37 (29) | 0.46 (62) |
| Y609FIDE:IDEY609F | 0.13 (43) | 0.19 (26) |
| IDE:IDEH112Q,Y609F | 0.11 (9) | 0.22 (30) |
| IDE:IDEE111F,Y609F | 0.20 (16) | 0.15 (20) |
| Y609FIDE:IDEH112Q | 2.02 (160) | 2.60 (351) |
| Y609FIDE:IDEE111F | 0.24 (19) | 0.11 (15) |
| Y609FIDE:IDEH112Q,Y609F | 0.55 (45) | 0.41 (55) |
| Y609FIDE:IDEE111F,Y609F | 0.08 (6) | <1 |
a Reaction mixtures contained 0.5 μm Aβ1–40-Lys(Biotin)FAM-labeled and 50–400 ng of IDE in 50 mm Tris-HCl buffer, pH 7.4. Following a 30-min incubation at 37 °C, the reaction was terminated by adding 1–10-phenanthroline followed agarose-bound avidin D. Following centrifugation the released FAM-labeled peptide products was measured on a Spectramax plate reader.
b Reaction mixtures (20 μl) contained 50 mm Tris-HCl buffer, pH7.4, 100 nM Aβ1–40 and enzyme in a total volume of 20. Following incubation at room temperature for 1 h, the reaction was terminated by diluting >1,000-fold, and the remaining Aβ was determined by sandwich ELISA assay as described under “Experimental Procedures.”
c Values in parenthesis are expressed as nmols/h/mg active subunit.
When the distal site is mutated on the inactive subunit regardless of whether or not the inactive subunit can bind substrate (IDE:IDEH112Q,Y609F and IDE:IDEE111F,Y609F) there is a decrease in the reaction rate, a result in contrast to our findings with smaller peptides. This is most evident for IDE:IDEH112Q,Y609F and suggests that Aβ1–40 can interact at the distal site on the non-catalytic subunit and that this interaction enhances catalysis. This seems to occur whether or not binding at the active site is impaired. That bound Aβ1–40 can activate in a trans manner is further supported by the relatively high reaction rates of the Y609FIDE:IDEH112Q and IDE:IDEH112Q mutants. The low reaction rate of Y609FIDE:IDEE111F and particularlyY609FIDE:IDEE111F, Y609F may reflect, as seen with Abz-GGFLRKHGQ-EDDnp as substrate, high occupancy binding to the noncatalytic subunit that inhibits the active subunit.
We next used the fluorescent N-terminal FAM-labeled Aβ1–40 to directly measure binding to IDE by following the increase in anisotropy that occurs when the fluorescent Aβ is bound to the enzyme. No binding was observed with the homodimeric (IDEH112Q)2 or (IDEH112Q,Y609F)2 confirming our previous report that substrate binding is diminished by the H112Q mutation. IDE forms containing the E111F mutation showed a stable anisotropy reading over at least 60 min. We thus measured the binding of fluorescent Aβ to (IDEE111F,Y609F)2 and (IDEE111F)2 (Fig. 3). The binding of the fluorescent Aβ to (IDEE111F)2, exhibited a slightly higher Kd (0.63 ± 0.03 μm) compared with (IDEE111F,Y609F)2 which exhibited a Kd of 0.25 ± 0.05 μm). The same Kd values were obtained using two different enzyme preparations. Thus the Y609F mutation does not in itself prevent binding, but is involved in allosteric activation.
FIGURE 3.
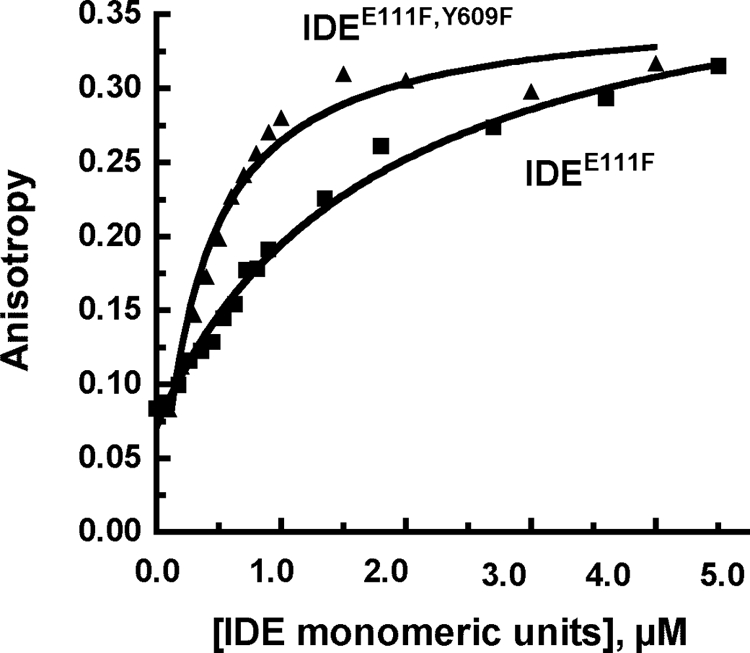
Binding of Aβ to IDEE111F and to IDEE111F, Y609F. Binding of the Aβ1–40 FAM-labeled to IDE was measured by following the change in fluorescence anisotropy at the indicated concentrations of IDE added to 1 μm FAM-labeled Aβ1–40 in 50 mm Tris-HCl, pH 7.4 buffer in a total volume of 200 μl. The stock concentration for IDE monomeric units was 10 μm (∼1 mg/ml)). Fluorescence anisotropy was measured at 23 °C using a SpectraMax 5 plate reader at an excitation wavelength of 494 nm and an emission wavelength of 521 nm.
DISCUSSION
By utilizing combinations of point mutations that affect catalysis, substrate binding, and allosteric activation we have further explored the IDE reaction mechanism and uncovered new insights into the complexity of the reaction. For example when only one subunit in the dimeric enzyme binds substrate, the single active site can function as well as the dimeric enzyme in terms of kcat, Km, and Hill coefficient. This means that under some circumstances a subunit within the IDE dimer can function independently. However, when the adjacent catalytically compromised subunit is capable of binding substrate the reaction rate at the other subunit is substantially reduced. Therefore an intact substrate bound in the active site appears to transmit a conformational change to the other subunit that affects catalysis. Because there is not a significant difference per active subunit between the kcat values for a dimer with two fully active subunits compared with a dimer with one subunit inactive in terms of both substrate binding and turnover (IDE:IDE versus IDE:IDEH112Q), the resident time of the intact substrate at a functioning active site must be short relative to the overall catalytic cycle and contribute little to reducing kcat.
We have also found that the observed cooperativity in the IDE reaction with Abz-GGFLRKHGQ-EDDnp as substrate can be attributed to cis binding to the distal part of the extended substrate binding site. That is, binding to the active site and to the distal site on the same subunit can produce cooperativity as evidenced by a Hill coefficient of ∼2 with all dimeric enzyme forms having an intact cis distal site, and a Hill coefficient of ∼1 with all dimeric enzyme forms containing a distal site Y609F mutation on the catalytic subunit. Interestingly, with both the H112Q and E111F mutants and Aβ1–40 as substrate there appears to be a decrease in catalysis when the distal site is mutated on the non-catalytic subunit, indicating that under some circumstances there also can be communication between subunits via the distal site.
A cis activation mechanism is further supported by the finding of the increase in the rate of hydrolysis of Abz-GGFLRKHGQ-EDDnp by added dynorphin B9 and bradykinin. This heterotropic activation is observed when the distal site on the catalytic subunit is intact regardless of whether the active site is mutated on the adjacent catalytically compromised subunit (IDE:IDEH112Q and IDE:IDEE111F) or when both the active site and distal site on the adjacent catalytically compromised subunit are mutated (IDE:IDEH112Q,Y609F and IDE:IDEE111F,Y609F). In contrast when the distal site on the catalytic subunit is mutated, activation by dynorphin B9 and bradykinin is diminished regardless of the mutational status of the non-catalytic subunit.
When measuring the direct binding of Aβ to IDE we were surprised to find that the Y609F mutation did not decrease binding affinity, and if anything increased the affinity of the enzyme for Aβ. The simplest interpretation is that the Y609F mutation still supports binding of peptide to the distal site, but prevents the bound peptide from triggering the conformational changes that activate the enzyme. That is Tyr609 does not contribute to Aβ binding per se even though its appears well positioned to do so based on crystal structures with peptide bound at the distal site (11).3 Instead, we suggest that Tyr609 facilitates a conformational change leading to IDE activation. As we have recently shown this conformational change is likely to involve a small translation of the two haves of the IDE molecule relative to each other,3 which likely alters the partitioning between the open and closed forms of the clamshell-like enzyme.
In summary we have shown that the individual subunits in the IDE oligomer have the capacity to function independently, but there is also communication between the subunits. Activation by binding to the distal region of the substrate-binding site occurs primarily through a cis mechanism, but the distal site of the adjacent subunit can affect activity in trans. The hydrolysis rate of large substrates that interact with both the active and distal sites is particularly dependent on the interaction at the distal site.
This work was supported, in whole or in part, by National Institutes of Health Grants DA02243, NS38041, and P20 RR020171.
N. Noinaj, S. K. Bhasin, E. S. Song, K. Scoggin, M. A. Juliano, L. Juliano, L. B. Hersh, and D. W. Rodgers, submitted manuscript.
N. Noinaj, E. S. Song, S. Bhasin, B. J. Alper, W. K. Schmidt, L. B. Hersh, and D. W. Rodgers, submitted manuscript.
- IDE
- insulin-degrading enzyme
- Aβ
- amyloid β.
REFERENCES
- 1. Miller B. C., Eckman E. A., Sambamurti K., Dobbs N., Chow K. M., Eckman C. B., Hersh L. B., Thiele D. L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6221–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leissring M. A., Farris W., Chang A. Y., Walsh D. M., Wu X., Sun X., Frosch M. P., Selkoe D. J. (2003) Neuron 40, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 3. Shii K., Roth R. A. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 4147–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hari J., Shii K., Roth R. A. (1987) Endocrinology 120, 829–831 [DOI] [PubMed] [Google Scholar]
- 5. Leissring M. A., Malito E., Hedouin S., Reinstatler L., Sahara T., Abdul-Hay S. O., Choudhry S., Maharvi G. M., Fauq A. H., Huzarska M., May P. S., Choi S., Logan T. P., Turk B. E., Cantley L. C., Manolopoulou M., Tang W. J., Stein R. L., Cuny G. D., Selkoe D. J. (2010) PLoS One 5, e10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fakhrai-Rad H., Nikoshkov A., Kamel A., Fernström M., Zierath J. R., Norgren S., Luthman H., Galli J. (2000) Hum. Mol. Genet. 9, 2149–2158 [DOI] [PubMed] [Google Scholar]
- 7. Safavi A., Miller B. C., Cottam L., Hersh L. B. (1996) Biochemistry 35, 14318–14325 [DOI] [PubMed] [Google Scholar]
- 8. Song E. S., Juliano M. A., Juliano L., Hersh L. B. (2003) J. Biol. Chem. 278, 49789–49794 [DOI] [PubMed] [Google Scholar]
- 9. Shen Y., Joachimiak A., Rosner M. R., Tang W. J. (2006) Nature 443, 870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song E. S., Rodgers D. W., Hersh L. B. (2010) PLoS One 5, e9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Manolopoulou M., Guo Q., Malito E., Schilling A. B., Tang W. J. (2009) J. Biol. Chem. 284, 14177–14188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Malito E., Ralat L. A., Manolopoulou M., Tsay J. L., Wadlington N. L., Tang W. J. (2008) Biochemistry 47, 12822–12834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song E. S., Juliano M. A., Juliano L., Fried M. G., Wagner S. L., Hersh L. B. (2004) J. Biol. Chem. 279, 54216–54220 [DOI] [PubMed] [Google Scholar]
- 14. Yao H., Hersh L. B. (2006) Arch. Biochem. Biophys. 451, 175–181 [DOI] [PubMed] [Google Scholar]
- 15. Cabrol C., Huzarska M. A., Dinolfo C., Rodriguez M. C., Reinstatler L., Ni J., Yeh L. A., Cuny G. D., Stein R. L., Selkoe D. J., Leissring M. A. (2009) PLoS One 4, e5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song E. S., Mukherjee A., Juliano M. A., Pyrek J. S., Goodman J. P., Jr., Juliano L., Hersh L. B. (2001) J. Biol. Chem. 276, 1152–1155 [DOI] [PubMed] [Google Scholar]
- 17. Song E. S., Daily A., Fried M. G., Juliano M. A., Juliano L., Hersh L. B. (2005) J. Biol. Chem. 280, 17701–17706 [DOI] [PubMed] [Google Scholar]
- 18. Parker M. W., Hellman L. M., Xu P., Fried M. G., Vander Kooi C. W. (2010) Biochemistry 49, 4068–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]