

Abstract
The human immunodeficiency virus 1 (HIV-1) viral protein R (Vpr) is an accessory protein that has been shown to have multiple roles in HIV-1 pathogenesis. By screening chemical libraries in the RIKEN Natural Products Depository, we identified a 3-phenyl coumarin-based compound that inhibited the cell cycle arrest activity of Vpr in yeast and Vpr-dependent viral infection of human macrophages. We determined its minimal pharmacophore through a structure-activity relationship study and produced more potent derivatives. We detected direct binding, and by assaying a panel of Vpr mutants, we found the hydrophobic region about residues Glu-25 and Gln-65 to be potentially involved in the binding of the inhibitor. Our findings exposed a targeting site on Vpr and delineated a convenient approach to explore other targeting sites on the protein using small molecule inhibitors as bioprobes.
Keywords: AIDS, Protein Drug Interactions, Protein Structure, Viral Protein, Yeast, Small Molecule Compounds
Introduction
The HIV-1 Vpr4 is a 14-kDa accessory protein that is implicated in viral pathogenesis, and the mechanisms underlying the cytopathic effects of this protein have been reviewed extensively (1–3). The Vpr gene is highly conserved in all HIV and Simian immunodeficiency virus members of the lentivirus family (4), denoting the importance of this gene in viral assault. The modest structure of the Vpr protein, three α-helices flanked by two flexible regions, belies its numerous roles in the virus life cycle and its effects on infected host cells (5–7). Vpr has been found to hijack various cellular pathways thought to be independent of one another, such as G2 cell cycle arrest, apoptosis, nuclear import, retrotranscription, the integration of the provirus in the nucleus, and transactivation of HIV-1 long terminal repeats (1, 3). Vpr has been detected in the sera and cerebrospinal fluids of AIDS patients (8), and a recent study showed that extracellular Vpr can reactivate viral production from latently infected cells (9). Vpr can form functional dimers and oligomers (10, 11), possibly explaining its breadth of biological abilities and its many protein-protein interactions with various host cell factors (12–14).
In the case of Vpr-caused cell cycle arrest, different mechanisms have been proposed for its mode of action, yet to date the exact mechanisms are still unknown. For example, the Vpr-DCAF1-DDB1 (DCAF1 is also termed VprBP, binding protein) ternary complex was said to trigger G2 cell cycle arrest (15). It was recently demonstrated that Vpr is able to form a mobile chromatin-associated nuclear focus containing VprBP, suggesting that Vpr recruits the VprBP-DDB1 complex within mobile nuclear structures to target a chromatin-bound substrate whose ubiquitination and proteolysis activate the ataxia telangiectasia-mutated and Rad3-related-mediated G2/M checkpoint and induce G2 arrest (16, 17). However, another report showed that Vpr triggers activation of Chk1 (a cell cycle checkpoint control kinase) through Ser345 phosphorylation in an S phase-dependent manner, subsequently leading to G2 arrest (18). These two examples, along with others (6, 7, 19), illustrate more than one potential cell cycle arrest mechanism for Vpr. To complicate matters, Vpr has also been shown to bind to nucleic acids, implying its regulatory role in viral and cellular transcription machineries (20, 21).
Despite extensive research into the biology of Vpr, to date only two small molecule inhibitors of Vpr derived from natural compounds have been reported. Our group first reported that fumagillin (1) (22), isolated from fungal metabolites, was able to inhibit cell cycle arrest activity of Vpr in both yeast and mammalian cells, and in the same year, another group reported that damnacanthal (2) (23), a component of noni, is a specific inhibitor of Vpr-associated cell death with no effect on cell cycle arrest.
Although extensive efforts have been channeled toward the understanding of the molecular and structural biology of the HIV-1 Vpr, the lack of a crystal structure for this protein frustrates our attempts to further understand it. Increasing evidence has suggested that Vpr and other HIV accessory proteins are promising drug targets for a comprehensive AIDS therapy (24). Thus, small-molecule inhibitors can act as potential lead compounds for the development of effective inhibitors and also as bioprobes (also called chemical probes) to study Vpr.
In view of this, our aims were to first develop a screening system to interrogate chemical libraries in the RIKEN NPDepo (25), an annotated library of microbial secondary metabolites, for Vpr inhibitors and then use the identified small molecule inhibitors to investigate Vpr. Through this, we identified an inhibitor with a 3-phenyl coumarin scaffold, and noting the relatively weak potency and solubility of the hit molecule, employed further chemistry for structural optimization as well as structure-activity relationship (SAR) studies. The hit and derivatives were then used on a panel of single point alanine mutants of Vpr to identify residues with which it may interact. We also prepared small molecule-immobilized beads for use in pull-down assays to confirm Vpr-inhibitor binding. To our knowledge, this is the first report to show Vpr-inhibitor interaction on the amino acid level.
EXPERIMENTAL PROCEDURES
Yeast Strain, Plasmids, and General Methods
Construction of the wild-type and mutant Vpr expression plasmids, the yeast strain (MLC30), and culture conditions for the expression of Vpr used in this study were as described previously (22). All proteins were FLAG-tagged at the N-terminal for immunodetection in a Western blot analysis. Oligonucleotide sequences to induce alanine mutations in Vpr are available upon request. Standard molecular biology methods were used.
Screening for Vpr Inhibitors
The 96-well plate format HTS assays were performed using a modification of the described protocols. Briefly, 99 μl of yeast cells (A620 = 0.05) in inducing medium (synthetic dextrose growth medium without leucine and uracil, with 0.5 mm copper sulfate) were pipetted into each well, along with 1 μl of compounds from NPDepo (each at final concentrations of 33, 11, 3.7 and 1.2 μg/ml), fumagillin (22 μg/ml), or DMSO. Duplicate plates were incubated at 30 °C, and spectrophotometric readings were recorded with a multilabel counter (PerkinElmer Life Sciences) at 0, 12, 24, 36, and 48 h. The automated work station Biomek 2000 was used for all liquid handling. For the HTS parameters, see the supplemental “Experimental Procedures”. Secondary confirmatory assays were also performed on 96-well plates and included visual inspection under a low-power microscope (Olympus) at 0 and 48 h to identify optical interfering compounds. For the tertiary screen, cells were grown in inducing medium with compounds or DMSO at 30 °C with shaking (150 rpm) in a 50-ml conical tube (Iwaki). Cells were harvested at 36 h for SDS-PAGE and Western blot analyses.
SAR Study
The synthesis and characterization of the 3-phenyl coumarin compounds (3-10) are described in the supplemental “Experimental Procedures”.
Determination of Hit and Analog Potencies
Similar to the primary HTS screen, 1 μl of compounds (3-10) (final concentration, 5–100 μm) or DMSO was added to 99 μl of yeast cells in each well of a 96-well plate in duplicate. Spectrophotometric readings were taken at 0, 12, 24, and 36 h. Paper disc assays were performed as described previously (22).
Inhibition of Vpr Mutants
Cells were prepared similar to the HTS screen. Wild-type and mutant Vpr-expressing yeast strains were grown, washed, and recalibrated in inducing media before being dispensed into 96-well plates. 1 μl of compound at the optimum inhibitory concentration was added to each well containing 99 μl of yeast culture (final concentrations are 3, 100 μm; 5, 25 μm; and 9, 10 μm). Spectrophotometric readings were taken at 0, 12, 24, and 36 h.
Vpr Binding and Competition Assay
Beads containing immobilized 3 and vipirinin were prepared as described (26). Yeast cells expressing Vpr were grown overnight in 50 ml inducing medium (30 °C, 150 rpm) to an A600 of 1.5. Cells were harvested and resuspended in 1 ml of ice-cold lysis/binding buffer (200 mm sorbitol, 50 mm potassium acetate, 20 mm HEPES (pH 7.2), 2 mm EDTA, 0.05% Tween 20) with protease inhibitors (Roche). An equal volume of acid-washed glass beads (Sigma) was added to each tube. The tubes were vortexed at top speed for 30 s and cooled on ice for 30 s. This cycle was repeated for a total of 10 min. Insoluble material was removed by centrifugation twice at top speed for 15 min (4 °C). The total cell lysate was assayed (Pierce BCA protein assay kit), and 2 μg (in 500 μl of binding buffer) of Vpr-containing lysate was incubated with 10 μl of beads at room temperature for 8 h. The beads were washed twice with 500 μl of washing buffer (binding buffer containing 0.1% Tween 20), and the bound proteins were eluted by boiling in SDS-PAGE sample buffer followed by electrophoresis in 12% denaturing gels and detection by Western blotting.
Infectivity Assays
Human cell culture, the preparation of virus, and immunological techniques for the infectivity assays used in this study were used as described previously (22).
RESULTS
HTS of RIKEN NPDepo
We described previously a yeast-based screening system for Vpr inhibitors using paper discs containing fungal metabolites on agar plates and have identified 1 as a potent inhibitor of Vpr. We found that compound 1 was able to reverse the cell cycle arrest of Vpr in yeast and human cells and suppress HIV-1 replication in human macrophages (22).
Here, we developed a faster, quantifiable primary high-throughput screening (HTS) system to facilitate the screening of a library of compounds in the RIKEN NPDepo for Vpr inhibitors. For expediency, we screened compounds on 96-well plates using optical density as an index of growth recovery, as described for yeasts (27). Growth of Vpr-expressing yeast in the presence or absence of small molecules was measured spectrophotometrically and plotted as percentage growth (with the growth recovery of yeast in the presence of 1, our positive control in the screen, expressed as 100%). The screening library was provided in 96-well plates at the stock concentration of 10 mg/ml (in DMSO), and it was serially diluted to 3.3, 1.1, 0.37, and 0.12 mg/ml. We screened 6664 small molecules at these four concentrations in duplicate plates by adding 1 μl of each to 99 μl of Vpr-expressing yeast (to final concentrations of compounds of 33, 11, 3.7, and 1.2 μg/ml, respectively). The screening process is summarized in Fig. 1A.
FIGURE 1.

HTS of Vpr inhibitors. A, schematic diagram of the screening process. B, for simplicity, only the results for the plate containing hit inhibitor 3 (arrow) are shown. The hit zone is denoted by the area above 100% (growth in the presence of control 1). Symbols represent average of readings from two plates at 48 h. C, growth curves of hit compound 3, compound 1, and DMSO. D, Western blotting assays of Vpr expression from plasmid in the presence of DMSO (D), compound 1 (5.5 μm) (1), hit compound 3 (87 μm) (3), leucine (360 μg/ml) (L), or DAPI (120 μm) (Dp). Leucine reduced the gene expression in this expression system. E, structure of the hit compound Natural Product Derivative (NPD) 4456 (compound 3).
Hits from each plate were identified by comparing the growth of yeasts at 48 h to that of yeasts incubated with compound 1, our positive control, which was set at 100% (Fig. 1B and supplemental Fig. S1). All 212 compounds from the primary HTS that fell within the “hit zone” from the primary HTS were cherry-picked for a confirmatory secondary screening. This screening included visual inspections under a low-power microscope to detect false positives caused by optical interference (because of insoluble compounds or aggregated cells). Thirty-two positive hits from the secondary screen were then tested for their reproducibility in a 5 ml of shaking culture and analyzed by Western blotting for their effects on Vpr expression from the expression plasmid. From these tests, we selected compounds that inhibited the growth-suppressing effect of Vpr in yeast without any noticeable reduction of Vpr expression. As an example of a false-positive hit, DAPI induced yeast growth recovery in the primary and secondary screenings; however, this compound is a DNA intercalating agent that has been reported to inhibit plasmid expression in Escherichia coli (28). Indeed, we found that DAPI inhibited the expression of Vpr, resulting in false-positive growth of yeast cells (Fig. 1D). Following this rigorous tri-level screen, we identified a 3-phenyl coumarin Vpr inhibitor, Natural Product Derivative 4456 (3) (Fig. 1E).
Structure-Activity Relationship of Hit Compound 3
To improve the potency of hit compound 3, we synthesized a series of structural derivatives (4–10) (Fig. 2), maintaining the 3-phenyl coumarin scaffold both for structural optimization and to explore its SAR. Hit compound 3 was also resynthesized and purified by HPLC to validate the source compound from the RIKEN NPDepo. The derivatives were tested using a paper disc assay (supplemental Fig. S2), and the potency of each active derivative was accurately determined in a 96-well plate dose-response format.
FIGURE 2.

Structures of coumarin-based derivatives for structure-activity relationship study. The morpholine derivative vipirinin (9) was the most potent, whereas 5 represents the minimal pharmacophore. The relative inhibitory activities of the derivatives are indicated by the − or + signs in parentheses.
Initially, we found that removal of the 5-methoxy moiety from the 3-phenyl ring of 3, which resulted in 4, completely abolished its Vpr inhibitory activity. In contrast, elimination of the methoxy group from position 2 (5) enhanced Vpr inhibitory potency 4-fold (Fig. 3). Henceforth, subsequent derivatives were without the 2-methoxy moiety. Although we thought that fluorination of the methoxy moiety of 5 might improve its bioavailability (29), the substitution was not tolerated, and this resulted in a null derivative (6).
FIGURE 3.

Potency of the hit compound and its derivatives (3–10). Growth of Vpr-expressing yeasts at 36 h in the presence or absence of compounds. Vipirinin (9) was the most potent at lower concentrations. The higher readings seen on the curve for compound 8 were due to precipitation at higher concentrations. Open symbols indicate inactive derivatives. Derivatives 6 and 7 were toxic at higher concentrations. All points are averages of duplicate readings.
To determine the functionality of the amino moiety of 3 and to assess whether the carbamate moiety is also necessary for activity, we substituted the amide nitrogen with a carbon, resulting in a dimethylpropanoate group (7). This resulted in the complete loss of Vpr inhibitory activity, clearly demonstrating the importance of the carbamoyl moiety. We therefore replaced this group with the cyclic carbamates derived from piperidine and morpholine (8 and 9), and found that the potency of these derivatives was 10-fold higher than that of 3 (Fig. 3). However, a methylpiperazine surrogate (10) resulted in a potency that is similar to that of 3. As 9 was more potent than 8 at the lowest concentration and did not aggregate in solution at higher concentrations, compound 9, which we named vipirinin, was selected for use in subsequent analyses.
Inhibitory Assay of Vpr Mutants
We previously found that the Vpr mutant E25K was resistant toward 1 on a paper disc assay (22). Because this mutant was also resistant to the inhibitory activity of 3 by maintaining the growth arrest (supplemental Fig. S3), we hypothesized that residue Glu-25 may be important for the growth arrest activity of Vpr and that both compounds 1 and 3 interact with Vpr within the vicinity of residue Glu-25. Therefore, we constructed a series of single point alanine mutants (amino acids 21ELLEELKS28 on α-helix 1) of Vpr as well as a second series of mutants (amino acids 61IRILQQLL68 on α-helix 3) in a leucine-rich domain of Vpr, a domain found to be important for binding to VprBP, a precursor in its G2 arrest activity (30). These two stretches of amino acids (21–28 and 61–68) are adjacent to each other, and a hydrophobic region is present between them (7). The hit compound 3 and active derivatives were then used to assay the Vpr mutants on a 96-well plate format akin to the HTS process described earlier.
We found that the effects of these mutations on the cell cycle arrest activity of Vpr were similar to previous findings (6, 7), as reflected in the overall growth profile of the two series of mutants (supplemental Fig. S3), which also revealed a profile of the inhibitors tested. As the weakened activity of certain Vpr mutants may obscure the inhibitory effect of the inhibitors, we utilized only selected Vpr mutants that maintained significant cell cycle arrest activity in further assays with the derivatives. In this analysis, the hit and derivatives were used at their optimum concentrations. We found that the Vpr mutants L22A and E25A (Fig. 4), which were responsive to 1, were resistant to 3. Furthermore, alanine mutations at residues Arg-62, Ile-63, and Gln-65, which resulted in poor responses to the inhibitory action of 3, were much more responsive toward 5 (Fig. 4). Mutant S28A, however, appeared resistant to the effects of all three compounds. Interestingly, vipirinin was more potent than 5 in inhibiting the activity of Vpr mutants L22A, E25A, and I63A, suggesting a possible effect of the morpholine moiety on these residues. The contrasting profile of the inhibitors 3 and 5 on the Vpr mutants shows a clear structure-activity relationship between the 2-methoxy moiety on the 3-phenyl ring of hit compound 3 and its Vpr inhibitory activity. This inhibitory assay also identified several key residues on the hydrophobic region of Vpr with which these inhibitors interacted.
FIGURE 4.

Inhibitory assay of Vpr mutants with hit and potent derivatives. Growth of wild-type and mutant Vpr-expressing yeasts at 36 h in the presence or absence of compounds. Wild-type and L68A were representative of standard and weakened Vpr cell cycle arrest activity, respectively. Hit compound 3 was no longer potent against most of the mutants shown here, but their resistance was overcome by 5 and vipirinin. Notably, S28A was resistant to all inhibitors, whereas Q65A had good growth arrest activity in yeast, although reported otherwise in mammalian cells. In this assay, the hit compound and derivatives were used at their optimum concentrations (final concentrations are 3, 100 μm; 5, 25 μm; vipirinin, 10 μm). H, hydrophobic residues. The error bars denote S.D.
Vpr binds to Hit Compound 3 and Vipirinin
To confirm that 3 and vipirinin are directly interacting with Vpr, we prepared 3- and vipirinin-immobilized-agarose beads using an established photoimmobilization method that allows the introduction of a variety of small molecules onto solid support in a functional group-independent manner (26). We had successfully used this method to identify the target of an osteoclastogenesis inhibitor (31). Using these beads, we were able to detect the direct binding of Vpr in yeast total cell lysate to 3 by Western blot analysis (supplemental Fig. S4). However, we were unable to perform a competition assay using free 3, which may have been due to the poor water solubility of coumarins. We observed a precipitate after the addition of excess 3 to the binding buffer, with the soluble portion unable, or containing insufficient amount of inhibitors, to bind with Vpr in the solution (data not shown).Because morpholine has better miscibility (32), we predicted that vipirinin, with a morpholine moiety, may perform better in the competition assay. Although there was slight precipitation when excess vipirinin was added to the binding buffer, it was less than observed with 3, and in this instance, the amount of vipirinin in solution was sufficient to compete for Vpr on both 3- and vipirinin-immobilized beads (Fig. 5A). These results indicate that 3 and vipirinin most likely inhibit Vpr by directly binding to it. Pull-down assays with selected Vpr mutants using 3- and vipirinin immobilized beads showed that mutant S28A did not bind to either (Fig. 5B), in agreement with findings of the Vpr mutants inhibitory assay that showed Vpr mutant S28A responding poorly to compound 3 and vipirinin.
FIGURE 5.

Vpr binds to hit compound 3 and vipirinin in a competitive manner. All bands are FLAG-tagged Vpr proteins bound to compound-immobilized beads. A, Vpr in yeast total cell lysates (2 μg in 500 μl of binding buffer) was preincubated in binding buffer containing vipirinin (+) or DMSO (−). Precipitants were allowed to settle, and soluble fractions were carefully removed and incubated with 10 μl of 3- or vipirinin-immobilized beads. Competition was observed when increasing amounts of vipirinin were added. B, Vpr mutant S28A does not bind to either 3- or vipirinin-immobilized beads. No chemiluminescent bands were detected despite prolonged exposure times. All bound proteins were detected by Western blotting after SDS-PAGE. V, vipirinin.
Vipirinin Inhibits Vpr-dependent Viral Gene Expression
Vpr is necessary for HIV-1 infection of nondividing cells such as macrophages (33). As compound 1 was found to be able to inhibit both Vpr-caused growth arrest and Vpr-dependent viral gene expression in our previous study (22), we wanted to see if the coumarin-based inhibitors are also able to act in the same way. To determine the effect of vipirinin on proviral transcription after infection, we tested the hit compound 3 and vipirinin in a luciferase gene reporter system (33). Briefly, luciferase activity, corresponding to proviral transcription, was measured 6 days after infection in cells that had been treated with the inhibitors at the time of infection. Treatment with 1, 3, or vipirinin inhibited luciferase expression from the virus in a dose dependent manner (Fig. 6) without any signs of toxicity toward the macrophages (supplemental Fig. S5A). In addition, these compounds at these concentrations did not significantly affect the growth of human T cells and osteosarcoma cells (supplemental Fig. S5B and data not shown). Infection by Vpr-deficient virus (used as negative control) was about 10% of that of virus carrying wild-type Vpr and was not significantly affected by the compounds (Fig. 6). These findings show that, like 1, the hit compound 3 and vipirinin can inhibit Vpr-dependent viral infection of human macrophages.
FIGURE 6.
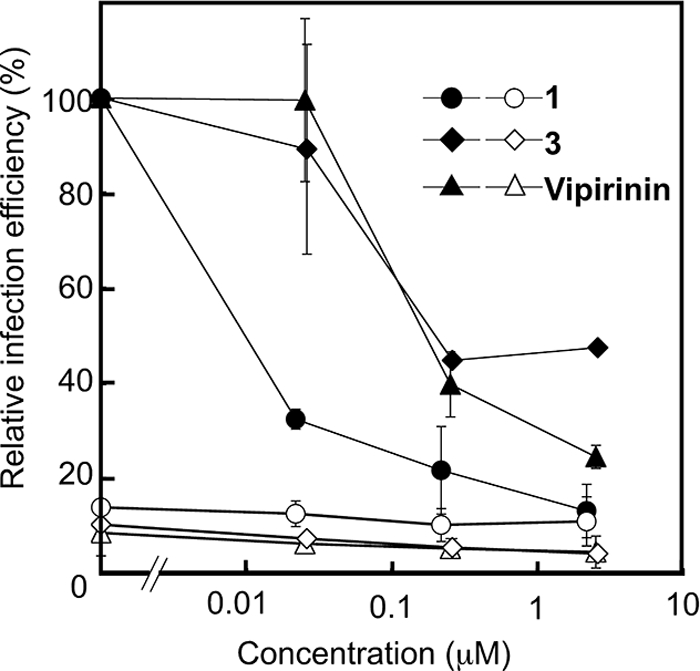
Vipirinin inhibits HIV-1 infection of human macrophages. Relative infection efficiency was estimated from the activity of the luciferase encoded in the viral genome. Activities were measured 6 days after infection of HIV-1 reporter viruses with wild-type (closed symbols) or truncated (filled symbols) Vpr. Activities from cells treated with each compound are shown in relative values to that of cells infected with wild-type Vpr containing viruses without compounds.
Because Vpr has several functions, including pre-integration complex transport and transactivation of proviral LTR, it is interesting to determine which of these steps were inhibited by vipirinin and compounds 1 and 3. We examined the effects of these three compounds on PIC transport by semi-quantitative estimation of viral DNA synthesis upon the infection of macrophages. We found that vipirinin and compounds 1 and 3 did not inhibit PIC transport at the early infection, indicating that these compounds inhibited viral infection by perturbing the Vpr-dependent transactivation of proviral LTR (supplemental Fig. S6).
Previously, we have shown that compound 1 abrogated the activity of G2 arrest by Vpr in mammalian cells (18). Interestingly, however, we found that vipirinin did not have a significant effect on this activity (data not shown). Consistent with the effects of these compounds on mutated Vpr, these results suggest a different mechanism of action between vipirinin and compound 1.
DISCUSSION
Anti-HIV agents have been largely successful in reducing viral load in HIV-positive individuals, thus enhancing their quality of life, but the long-term side effects of these drugs, coupled with increased drug resistance, complicate treatment. Current combination therapy, known as highly active antiretroviral therapy, only targets the structural proteins of HIV, but accumulative knowledge now shows that the targeting of accessory proteins is imperative in the strategy to combat HIV (24). The identification of small molecule inhibitors is the first step toward this direction, and given the still ongoing efforts to understand Vpr, these inhibitors can act as bioprobes and expand the molecular toolbox to glean further insights into it.
The initial motivation of this research was to develop an HTS system to screen chemical compounds in the RIKEN NPDepo, an annotated repository containing more than 24,000 natural products and their derivatives (25). We therefore built on our previous screening experience and capitalized on the measurable growth of yeasts in liquid culture to this end. The simple induction of yeast growth arrest by Vpr (34) and the virtues of yeast as a good model for drug discovery have been well documented (35–37). Suffice it to say, in this context it offered the advantages of detecting and precluding impermeable or toxic compounds at the early stages of screening and affords further experiments in the same biological system.
Our SAR studies showed that the 5-methoxy moiety of the phenyl ring of 3 is essential for activity, whereas the absence of the 2-methoxy in 5 enhanced activity 4-fold. The 2-methoxy moiety may have sterically hindered binding to the target region, and its removal may have produced a more favorable stereochemistry by allowing freer rotation of the methoxyphenyl group. It was unclear, however, why fluorination of the methyl group of the methoxy moiety resulted in a loss of activity. This fluorination yielded the more hydrophobic and lipophilic trifluoromethyl moiety, which has been shown to enhance drug effectiveness (29). Next, we checked the functionality of the amide and found it to be critical for activity. This clearly indicated that 5 is the minimal functional pharmacophore for inhibitory activity. The additional introductions of the polar cyclohexyls piperidine and morpholine were well tolerated and further enhanced potency (10-fold compared with 3). The potency of the methylpiperazine substitute (10), however, was similar to that of 3, most likely because of the low solubility of this compound (32). The higher potency of vipirinin, the morpholine analog, may be due, at least in part, to the aqueous miscibility associated with its morpholine moiety and to its higher surface activity compared with piperidine (38). Surface activity, although not solely responsible, corresponds to biological activity in many drugs (39).
The identification of this 3-phenyl coumarin inhibitor was not surprising, as coumarins have been found to have various pharmacological properties, with a number of its derivatives reported to have anti-HIV activity, inhibiting viral reverse transcriptase, integrase, and protease (40, 41). This correlation is fascinating and is an apposite for the development of a multipotent coumarin-based anti-HIV agent.
In this study, we focused on the hydrophobic region present on the first and third α-helices (Fig. 7A), as we found 3 to be inactive in mutant E25K, and it was shown that residue Gln-65 is important for the cell cycle arrest activity of Vpr (13). To investigate the role of this region in the recognition of small molecule inhibitors, we introduced single point alanine substitutions in the amino acids 21ELLEELKS28 and 61IRILQQLL68. In those two regions, the hydrophobic side face is formed by residues Leu-22, Leu-23, Leu-26, Ile-61, Leu-63, Leu-64, and Leu-68, respectively (6). When tested with the hit 3 and derivatives, 5 and vipirinin, we found that substitution with the smaller hydrophobic alanine at residues Leu-22, Glu-25, and Ser-28 on α1, and Arg-62, Ile-63, and Gln-65A on α3 (Figs. 4 and 7A), conferred resistance to 3 or responded poorly to this compound. These mutants, however, were not resistant to 5, suggesting that the methoxyphenyl moiety in the latter is important for its interaction with residues near the exposed hydrophobic face (Fig. 7B). Potentially, the hydrophobic morpholine moiety may also have enhanced its binding affinity, as observed in mutants L22A and I63A (Fig. 4). Although the Q65A mutant was reported to have reduced cell cycle arrest activity compared with wild-type Vpr in mammalian cells (42), in this study with yeast, its growth arrest activity remained unaffected and appeared slightly stronger compared with the wild-type, implying a different binding model to its yeast cellular partner(s).
FIGURE 7.

Inhibitory assay reveals key residues for inhibitor interaction on a hydrophobic region of Vpr. A, ribbon diagram showing two regions (amino acids 21–28 and 61–68) that contained single point substitutions of alanine (blue). The three α–helices are also indicated. B, hybrid diagram showing the molecular surface and sticks diagram of the hydrophobic region between α1 and α3. Hydrophobic residues are in blue, except Leu-22 and Ile-63. Residues conferring resistance to 3 after substitution with alanine are colored yellow. These resistances were overcome by 5 and vipirinin. PyMOL molecular visualizations (49) are derived from the reported NMR structure of Vpr (5).
The contrasting inhibitory activities of 3 and 5 could be used to identify amino acid residues that are most likely important for the cell cycle arrest activity of Vpr. Of note, the Leu-26 residue, which is located next to Gln-65, may also be a key residue for the cell cycle arrest activity of Vpr, as its substitution by the less hydrophobic alanine resulted in a loss of growth arrest activity (supplemental Fig. S3). The reported NMR structure of this protein shows that Leu-26 together with residues Leu-23, Ile-61, and Leu-64 most likely form a hydrophobic pocket flapped by residue Gln-65 (5, 7).
Direct binding analysis using 3 and vipirinin-immobilized beads in pull-down and competition assays confirmed the interaction between Vpr and its inhibitors (Fig. 5). Although inhibitor binding does not necessarily correspond with functional activity, our Vpr mutants analysis strongly suggests that when an inhibitor is bound to the leucine-rich, hydrophobic region, it prevents Vpr from binding to other cellular partners that ultimately lead to cell cycle arrest. The initial hit 3 most likely binds to this hydrophobic region, as the Q65A mutation was less responsive toward 3 but was more responsive to 5 and vipirinin (Fig. 4). As the mutated residues are located on two different stretches on the surface of the protein, it is likely that mutations on both sides of this hydrophobic region (Fig. 7B) alter the structure of Vpr and thus affect the shape of the hydrophobic region, resulting in resistance to 3. Unlike residue Leu-26, alanine substitution on Leu-61 did not abolish Vpr activity but also did not confer resistance toward 3, indicating a tolerable change that does not disrupt inhibitor binding. Moreover, in agreement with previous findings (43), we found that substitution at the other hydrophobic residue, L68A, weakened cell cycle arrest activity.
Although both the hit compound 3 and vipirinin could inhibit proviral transcription and suppress HIV-1 replication in macrophages, their potency was less than that of 1, with vipirinin slightly more potent than 3. Despite the higher potencies of 3 and vipirinin than 1 in the yeast assays, it was not translated in the infectivity assay in the inhibition of viral-dependent gene expression and G2 arrest in mammalian cells. These suggest that, unlike 1, the coumarin-based Vpr inhibitors are much more specific in targeting Vpr in the context of its cell cycle arrest activity. It was reported that the transactivation function of Vpr is independent of its cell cycle arrest activity (44, 45). Additionally, we could not determine whether the poor solubility of the coumarin-based inhibitors is a factor here.
Vpr was shown to be nonessential for viral replication in T cell lines in vitro but is important for efficient infection of nondividing cells and differentiated macrophages (33, 46). As our previous work found that 1 was able to inhibit both Vpr-caused growth arrest and Vpr-dependent viral gene expression (22), we speculated that the structural domain of the Vpr protein that is responsible for its cell cycle arrest activity may also play a role in its promotion of viral gene expression in macrophages or that the domains for both those functions may be overlapping. Based on the resistance of the E25K Vpr mutant toward 1 (Fig. supplemental Fig. S3), it was thought that the inhibitor 1 interacts with Vpr at residues surrounding Glu-25, and this speculation was further supported by the resistance of Vpr mutant S28A toward 1 (supplemental Fig. S3). Both Ser-28 and Glu-25 are located near each other on the exposed surface of α1 (Fig. 7).
In the inhibitory assay of Vpr mutants, we found compound 3 to be ineffective on Vpr mutants E25K, E25A, and S28A (supplemental Fig. S3). Hence, we predicted that 3, like 1, would also show good activity in the viral gene expression assay. However, this did not turn out to be true, as compounds 3 and vipirinin were weaker than 1 at all concentrations tested in the viral gene expression assay (Fig. 6).
An earlier study, through the use of Vpr mutants R62P and R80A that were defective in nuclear localization and G2 arrest, respectively, showed that the G2 arrest ability is essential to induce productive infection if efficient proviral nuclear targeting can be achieved (47). The authors proposed that Vpr-induced biochemical changes in macrophages may lead to cell cycle arrest in the context of dividing cells, and suggested that access to the nucleus by itself is not sufficient to ensure productive infection. In contrary, a more recent study on the localization of Vpr to the nuclear envelope found that Vpr mutants L23F and K27M, which failed to localize on the nuclear envelope, not only impaired G2 arrest activity but also affected the HIV-1 replication in macrophages (48).
From the findings above, taken together with our observations pertaining to the effects of the inhibitors (1 and 3) on Vpr mutants E25K, S28A, and R62A (supplemental Fig. S3), it can be deduced that the structural domain of Vpr that is responsible for its cell cycle arrest activity possibly overlaps with the domain that promotes viral gene expression. However, because of the confines of this report, this observation was not further explored. More analyses are necessary to determine the exact functional link between the different functions of Vpr and the structural domains of the protein that are involved. Compounds 3 and vipirinin clearly interacted with Vpr in different manners compared with 1, and this is reflected in their contrasting potencies in the Vpr mutants inhibitory assay and Vpr-dependent viral gene expression (Fig. 6 and supplemental Fig. S3).
From this vantage point, although our approach delineated a stable platform for the screening of NPDepo for Vpr inhibitors, we also demonstrated the utility of small molecule inhibitors as bioprobes in identifying key residues for cell cycle arrest on a hydrophobic region of Vpr. As noted earlier, further understanding of the protein-inhibitor binding tendencies will guide drug design in addition to the identification of crucial targeting sites. This approach can also be adopted to complement established methods in revealing host cell partners of Vpr and in dissecting its other functions.
Acknowledgments
We thank the members of the RIKEN Chemical Biology Core Facility for discussions and technical support.
This study was supported in part by the Chemical Biology Project of RIKEN and grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. E. B. O. is pursuing this work as a RIKEN Asia Program Associate.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and experimental procedures.
- Vpr
- viral protein R
- BP
- binding protein
- NPDepo
- Natural Products Depository
- SAR
- structure-activity relationship
- DMSO
- dimethyl sulfoxide
- HTS
- high-throughput screening.
REFERENCES
- 1. Andersen J. L., Planelles V. (2005) Curr. HIV Res. 3, 43–51 [DOI] [PubMed] [Google Scholar]
- 2. Henklein P., Bruns K., Sherman M. P., Tessmer U., Licha K., Kopp J., de Noronha C. M., Greene W. C., Wray V., Schubert U. (2000) J. Biol. Chem. 275, 32016–32026 [DOI] [PubMed] [Google Scholar]
- 3. Le Rouzic E., Benichou S. (2005) Retrovirology 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tristem M., Purvis A., Quicke D. L. (1998) Virology 240, 232–237 [DOI] [PubMed] [Google Scholar]
- 5. Morellet N., Bouaziz S., Petitjean P., Roques B. P. (2003) J. Mol. Biol. 327, 215–227 [DOI] [PubMed] [Google Scholar]
- 6. Morellet N., Roques B. P., Bouaziz S. (2009) Curr. HIV. Res. 7, 184–210 [DOI] [PubMed] [Google Scholar]
- 7. Pandey R. C., Datta D., Mukerjee R., Srinivasan A., Mahalingam S., Sawaya B. E. (2009) Curr. HIV Res. 7, 114–128 [DOI] [PubMed] [Google Scholar]
- 8. Xiao Y., Chen G., Richard J., Rougeau N., Li H., Seidah N. G., Cohen E. A. (2008) Virology 372, 384–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoshino S., Konishi M., Mori M., Shimura M., Nishitani C., Kuroki Y., Koyanagi Y., Kano S., Itabe H., Ishizaka Y. (2010) J. Leukocyte Biol. 87, 1133–1143 [DOI] [PubMed] [Google Scholar]
- 10. Bourbigot S., Beltz H., Denis J., Morellet N., Roques B. P., Mély Y., Bouaziz S. (2005) Biochem. J. 387, 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fritz J. V., Didier P., Clamme J. P., Schaub E., Muriaux D., Cabanne C., Morellet N., Bouaziz S., Darlix J. L., Mély Y., de Rocquigny H. (2008) Retrovirology 5, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kino T., Gragerov A., Slobodskaya O., Tsopanomichalou M., Chrousos G. P., Pavlakis G. N. (2002) J. Virol. 76, 9724–9734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Le Rouzic E., Morel M., Ayinde D., Belaïdouni N., Letienne J., Transy C., Margottin-Goguet F. (2008) J. Biol. Chem. 283, 21686–21692 [DOI] [PubMed] [Google Scholar]
- 14. Zhao Y., Elder R. T. (2000) Front Biosci. 5, D905–916 [DOI] [PubMed] [Google Scholar]
- 15. Belzile J. P., Duisit G., Rougeau N., Mercier J., Finzi A., Cohen E. A. (2007) PLoS Pathog. 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belzile J. P., Abrahamyan L. G., Gerard F. C. A., Rougeau N., Cohen E. A. (2010) PLoS Pathog. 6, - [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belzile J. P., Richard J., Rougeau N., Xiao Y., Cohen E. A. (2010) J. Virol. 84, 3320–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li G., Park H. U., Liang D., Zhao R. Y. (2010) Retrovirology 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao R. Y., Li G., Bukrinsky M. I. (2011) J. Neuroimmune Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amini S., Saunders M., Kelley K., Khalili K., Sawaya B. E. (2004) J. Biol. Chem. 279, 46046–46056 [DOI] [PubMed] [Google Scholar]
- 21. Zhang S., Pointer D., Singer G., Feng Y., Park K., Zhao L. J. (1998) Gene 212, 157–166 [DOI] [PubMed] [Google Scholar]
- 22. Watanabe N., Nishihara Y., Yamaguchi T., Koito A., Miyoshi H., Kakeya H., Osada H. (2006) FEBS Lett. 580, 2598–2602 [DOI] [PubMed] [Google Scholar]
- 23. Kamata M., Wu R. P., An D. S., Saxe J. P., Damoiseaux R., Phelps M. E., Huang J., Chen I. S. (2006) Biochem. Biophys. Res. Commun. 348, 1101–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richter S. N., Frasson I., Palù G. (2009) Curr. Med. Chem. 16, 267–286 [DOI] [PubMed] [Google Scholar]
- 25. Tomiki T., Saito T., Ueki M., Konno H., Asaoka T., Suzuki R., Uramoto M., Kakeya H., Osada H. (2006) J. Comp. Aid. Chem. 7, 157–162 [Google Scholar]
- 26. Kanoh N., Honda K., Simizu S., Muroi M., Osada H. (2005) Angew. Chem. Int. Ed. Engl. 44, 3559–3562 [DOI] [PubMed] [Google Scholar]
- 27. Toussaint M., Conconi A. (2006) Nat. Protoc. 1, 1922–1928 [DOI] [PubMed] [Google Scholar]
- 28. Parolin C., Montecucco A., Ciarrocchi G., Pedrali-Noy G., Valisena S., Palumbo M., Palu G. (1990) FEMS Microbiol. Lett. 56, 341–346 [DOI] [PubMed] [Google Scholar]
- 29. Leroux F. R., Manteau B., Vors J. P., Pazenok S. (2008) Beilstein J. Org. Chem. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeHart J. L., Zimmerman E. S., Ardon O., Monteiro-Filho C. M., Argañaraz E. R., Planelles V. (2007) Virol. J. 4, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawatani M., Okumura H., Honda K., Kanoh N., Muroi M., Dohmae N., Takami M., Kitagawa M., Futamura Y., Imoto M., Osada H. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11691–11696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Budavari S., O'Neil M. J. (2001) The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 13th ed., Merck Research Laboratories, Division of Merck & Co., Inc., Whitehouse Station, NJ [Google Scholar]
- 33. Connor R. I., Chen B. K., Choe S., Landau N. R. (1995) Virology 206, 935–944 [DOI] [PubMed] [Google Scholar]
- 34. Macreadie I. G., Castelli L. A., Hewish D. R., Kirkpatrick A., Ward A. C., Azad A. A. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 2770–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baetz K., McHardy L., Gable K., Tarling T., Rebérioux D., Bryan J., Andersen R. J., Dunn T., Hieter P., Roberge M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 4525–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barberis A., Gunde T., Berset C., Audetat S., Lüthi U. (2005) Drug Discov. Today 2, 187–192 [DOI] [PubMed] [Google Scholar]
- 37. Toussaint M., Levasseur G., Gervais-Bird J., Wellinger R. J., Elela S. A., Conconi A. (2006) Mutat. Res. 606, 92–105 [DOI] [PubMed] [Google Scholar]
- 38. Fujino Y., Yokoyama S. (2000) Chem. Pharm. Bull. 48, 298–300 [DOI] [PubMed] [Google Scholar]
- 39. Felmeister A. (1972) J. Pharm. Sci. 61, 151–164 [DOI] [PubMed] [Google Scholar]
- 40. Kostova I., Raleva S., Genova P., Argirova R. (2006) Bioinorg. Chem. Appl. 2006, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao H., Neamati N., Hong H., Mazumder A., Wang S., Sunder S., Milne G. W., Pommier Y., Burke T. R., Jr. (1997) J. Med. Chem. 40, 242–249 [DOI] [PubMed] [Google Scholar]
- 42. Le Rouzic E., Belaïdouni N., Estrabaud E., Morel M., Rain J. C., Transy C., Margottin-Goguet F. (2007) Cell Cycle 6, 182–188 [DOI] [PubMed] [Google Scholar]
- 43. Thotala D., Schafer E. A., Tungaturthi P. K., Majumder B., Janket M. L., Wagner M., Srinivasan A., Watkins S., Ayyavoo V. (2004) Virology 328, 89–100 [DOI] [PubMed] [Google Scholar]
- 44. Vanitharani R., Mahalingam S., Rafaeli Y., Singh S. P., Srinivasan A., Weiner D. B., Ayyavoo V. (2001) Virology 289, 334–342 [DOI] [PubMed] [Google Scholar]
- 45. Wang L., Mukherjee S., Jia F., Narayan O., Zhao L. J. (1995) J. Biol. Chem. 270, 25564–25569 [DOI] [PubMed] [Google Scholar]
- 46. Eckstein D. A., Sherman M. P., Penn M. L., Chin P. S., De Noronha C. M., Greene W. C., Goldsmith M. A. (2001) J. Exp. Med. 194, 1407–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Subbramanian R. A., Kessous-Elbaz A., Lodge R., Forget J., Yao X. J., Bergeron D., Cohen E. A. (1998) J. Exp. Med. 187, 1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jacquot G., Le Rouzic E., David A., Mazzolini J., Bouchet J., Bouaziz S., Niedergang F., Pancino G., Benichou S. (2007) Retrovirology 4, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 0.99rc6, DeLano Scientific, Palo Alto, CA [Google Scholar]