

Abstract
Tau is a multiply phosphorylated protein that is essential for the development and maintenance of the nervous system. Errors in Tau action are associated with Alzheimer disease and related dementias. A huge literature has led to the widely held notion that aberrant Tau hyperphosphorylation is central to these disorders. Unfortunately, our mechanistic understanding of the functional effects of combinatorial Tau phosphorylation remains minimal. Here, we generated four singly pseudophosphorylated Tau proteins (at Thr231, Ser262, Ser396, and Ser404) and four doubly pseudophosphorylated Tau proteins using the same sites. Each Tau preparation was assayed for its abilities to promote microtubule assembly and to regulate microtubule dynamic instability in vitro. All four singly pseudophosphorylated Tau proteins exhibited loss-of-function effects. In marked contrast to the expectation that doubly pseudophosphorylated Tau would be less functional than either of its corresponding singly pseudophosphorylated forms, all of the doubly pseudophosphorylated Tau proteins possessed enhanced microtubule assembly activity and were more potent at regulating dynamic instability than their compromised singly pseudophosphorylated counterparts. Thus, the effects of multiple pseudophosphorylations were not simply the sum of the effects of the constituent single pseudophosphorylations; rather, they were generally opposite to the effects of singly pseudophosphorylated Tau. Further, despite being pseudophosphorylated at different sites, the four singly pseduophosphorylated Tau proteins often functioned similarly, as did the four doubly pseudophosphorylated proteins. These data lead us to reassess the conventional view of combinatorial phosphorylation in normal and pathological Tau action. They may also be relevant to the issue of combinatorial phosphorylation as a general regulatory mechanism.
Keywords: Alzheimer Disease, Cytoskeleton, Microtubules, Neurobiology, Tau, Microtubule Dynamics, Phosphorylation
Introduction
The neural microtubule-associated protein Tau is critical for promoting neuronal cell polarity and axonal outgrowth during development as well as maintaining axonal morphology and function in mature cells (1–4). On the other hand, abnormal Tau behavior has long been associated with Alzheimer disease and related dementias (5, 6).
Mechanistically, Tau binds directly to microtubules, promotes microtubule assembly, and regulates microtubule dynamic instability (7–15). Indeed, proper regulation of the growing and shortening dynamics of microtubules is essential for many cellular functions, including neuronal axonal transport (16–20). Further, misregulation of microtubule dynamic instability can lead to cell cycle arrest followed by cell death in dividing cells (21, 22).
Given the importance of properly regulating microtubule dynamic instability, it is not surprising that a regulator of microtubule dynamic instability such as Tau is itself tightly regulated. Indeed, Tau action is regulated by both alternative RNA splicing (23, 24) and phosphorylation (25). Alternative splicing generates six CNS Tau isoforms, each possessing either three or four imperfect repeats in the C terminus, separated from one another by shorter interrepeats (Fig. 1). This region of repeats and interrepeats harbors inherent microtubule binding activity (12–14, 26, 27) and is capable of regulating microtubule dynamic instability (9, 11, 14, 27–30). Importantly, whereas fetal human brain expresses only three-repeat (3R)2 Tau, adult human brain expresses approximately equal amounts of four-repeat (4R) and 3R Tau (28, 31). With respect to phosphorylation, normal adult Tau possesses an average of ∼2–3 phosphates/molecule, whereas fetal Tau possesses an average of ∼6–7 phosphates/molecule (32–34). Remarkably, phosphorylation can occur at ∼30 different amino acid positions in Tau (35–38). It follows that the potential structural and functional complexity of Tau is enormous. Unfortunately, our mechanistic and functional understanding of combinatorial Tau phosphorylation remains extremely limited.
FIGURE 1.

Construction of pseudophosphorylated 4-repeat Tau. Schematic of four-repeat Tau, with positions of pseudophosphorylated Tau marked with arrows. Boxes labeled R1, R2, R3, and R4 correspond to the 18-amino acid imperfect repeats. Boxes labeled IR correspond to the three interrepeats. Exons 2, 3, and 10 are regulated by alternative splicing. All three exons are present in the Tau constructs used in this study.
Among the most prominent pathologies of Alzheimer disease and related dementias are neurofibrillary tangles, abnormal intracellular aggregates composed primarily of hyperphosphorylated Tau (39–41). Indeed, Tau phosphorylation is increased ∼3–4-fold in Alzheimer disease brain compared with normal adult human brain (33, 34, 42). These observations have led to a long-standing and widely accepted hypothesis that abnormal Tau hyperphosphorylation is a key step in the Alzheimer disease pathway (25, 43). This notion has been supported by numerous cell culture and transgenic Tau models (44–51).
Genetic analyses in 1998 (52–55) demonstrated that mutations in the Tau gene were linked to frontal-temporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) as well as progressive supranuclear palsy and cortico-basal degeneration. More specifically, molecular analyses demonstrated that errors in Tau action, caused by either structural alterations of Tau or misregulation of Tau RNA splicing can cause neuronal cell death and dementia. Because phosphorylation is a major mechanism regulating Tau action, it follows that aberrant Tau phosphorylation could also have catastrophic consequences. Unfortunately, the precise molecular mechanisms by which Tau mediates neuronal cell death remain elusive. A widely held “gain of toxic function” model suggests that abnormal Tau phosphorylation increases its propensity to form abnormal Tau fibers, which are proposed to be cytotoxic. However, it has been shown that Tau-dependent neurodegeneration can occur in the absence of Tau aggregates in some model systems and in culture (48, 51, 56–58), suggesting that Tau aggregates may not be necessary for Tau-mediated cell death. A more recent gain-of-function model suggests that small Tau oligomers might be toxic (59). Alternatively, a loss-of-function “misregulation of microtubule dynamics” model suggests that mutation and/or abnormal phosphorylation of Tau might compromise its ability to regulate microtubule dynamic instability, which in turn causes cell death and subsequent dementia (10, 11, 60–62). This model is based on extrapolation from pharmacological and somatic cell genetics studies in dividing cells (21, 22).
Despite the paucity of work assessing the functional and mechanistic effects of combinatorial Tau phosphorylation (which is the state of both normal and pathological Tau), it has nonetheless become widely held that the combinatorial effects of multiple phosphorylation events within individual Tau molecules are simply the sum of the effects of the constituent single phosphorylation events, which have generally been observed to diminish Tau activities. However, basic protein biochemistry raises the possibility that this may be too simple a point of view. In an attempt to better understand the regulatory and mechanistic effects of multiple versus single phosphorylation events upon protein action, focusing on Tau, we have methodically examined the effects of pseudophosphorylation of Tau at four key phosphorylation sites (Thr231, Ser262, Ser396, and Ser404), individually and combinatorially, upon the abilities of Tau (i) to bind and assemble microtubules and (ii) to regulate microtubule dynamic instability. These analyses provide novel insights into phosphorylation-mediated regulation of Tau action upon microtubules, functional relationships between singly and multiply phosphorylated proteins, and the plausibility of integrating aberrant phosphorylation into the misregulation of microtubule dynamic instability model for Tau-mediated neuronal cell death.
EXPERIMENTAL PROCEDURES
Tau Protein Purification
A human cDNA encoding the longest four-repeat Tau isoform (441 amino acids) was the starting wild-type Tau sequence used in these studies. Specific serine/threonine to aspartic acid amino acid substitutions to pseudophosphorylate Tau were introduced into the four-repeat Tau cDNA using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). All sequences were verified by sequence analysis prior to use (Iowa State DNA Sequencing Facility). Recombinant Tau proteins were expressed and purified as described previously (10). Briefly, Tau expression was induced in BL21 (DE3) cells (Novagen, Madison, WI). Bacteria were lysed by sonication, and the lysate was clarified by centrifugation (12,000 × g, 15 min, 4 °C). Supernatants were boiled to precipitate non-heat-stable proteins and recentrifuged. The heat-stable proteins were adsorbed to a phosphocellulose column and eluted with a 0.2–1.0 m NaCl gradient. Fractions containing Tau were identified by SDS-PAGE followed by Coomassie Blue staining. Tau-containing fractions were pooled and further purified by reverse-phase HPLC (DeltaPak-C18, Millipore (Billerica, MA)). Tau-containing HPLC fractions were pooled, lyophilized, and resuspended in BRB-80 buffer (80 mm Pipes, pH 6.8, 1 mm EGTA, 1 mm MgSO4) with 0.1% β-mercaptoethanol. Previous work has demonstrated that Tau synthesized and isolated from bacteria is unphosphorylated (63). Tau concentrations were determined by a quantitative SDS-polyacrylamide gel comparison against a known “4R Tau mass standard,” the concentration of which was established by amino acid analysis (8). Bovine microtubule-associated protein-free brain tubulin was prepared, and the concentrations were determined as described previously (64, 65).
Tau-Microtubule Co-sedimentation Assays
15 μm microtubule-associated protein-free tubulin dimer was mixed with pseudophosphorylated 4R Tau constructs at 0.25 μm (1:60 Tau/tubulin), 0.375 μm (1:40 Tau/tubulin), 0.75 μm (1:20 Tau/tubulin), and 1.5 μm (1:10 Tau/tubulin) in PEM buffer (100 mm Pipes, pH 6.8, 1 mm MgCl2, and 1 mm EGTA) with 1 mm GTP. Microtubules were assembled at 35 °C until steady state was achieved (1.5 h), layered over an 80-μl sucrose cushion (50% sucrose in PEM, 2 mm GTP) in 5 × 20-mm ultraclear centrifuge tubes (Beckman Instruments, Palo Alto, CA), and centrifuged in a Sorvall RC70 swinging bucket rotor for 12 min at 35,000 rpm (150,000 × g) at 35 °C. Supernatants and pellets were harvested and solubilized in SDS-PAGE sample buffer. The quantity of tubulin in the supernatants and pellets were determined by SDS-PAGE. The quantity of Tau in the supernatants and pellets were determined by using Western blot analysis with the non-phosphospecific monoclonal antibody Tau-1 (60). Values presented in the figures and tables were corrected for a small amount of non-Tau-dependent tubulin that pellets in control reactions, generally in the range of 10–15% of total tubulin in the reaction. The values represent the mean ± S.E. from at least three independent experiments.
Analysis of Dynamic Instability of Individual Microtubules by Video Microscopy
Purified tubulin (11.7 μm tubulin dimer) was polymerized at the ends of sea urchin (Strongylocentrotus purpuratus) axonemal seeds at 35 °C in the presence or absence of 0.29 μm purified pseudophosphorylated Tau (1:40 Tau/tubulin molar ratio) in PMME buffer (87 mm Pipes, 36 mm MES, 1.4 mm MgCl2, 1 mm EDTA, pH 6.8) containing 2 mm GTP and incubated at 35 °C for 30 min to achieve steady state. Time lapse images of individual microtubules were obtained at 35 °C by video-enhanced differential interference contrast microscopy, as described previously (8). The number concentration of axoneme seeds was adjusted to 4–6 axonemes/field of view. Dynamic instability of plus ends was analyzed once the reaction reached steady state. Plus ends were distinguished from minus ends on the basis of their fast growth rates, the number of microtubules that grew at the ends, and the relative lengths of the microtubules (17, 30, 66). Life histories of individual microtubules were collected as described (8) with modifications. Images were collected at 1–3-s intervals. We defined microtubules to be growing if they increased in length >0.3 μm at a rate of >0.3 μm/min. Shortening events were defined by a >1-μm length change at a rate of >2 μm/min. Microtubules that changed <0.3 μm/min over a duration of four images were considered to be in an attenuated state. Segmentation of whole life histories into shorter defined events was performed manually using a custom built Matlab (The MathWorks, Inc.)-based program that determines averages, S.D. values, and S.E. values for each parameter/condition. We next used these values to determine the statistical confidence levels using the GraphPad program (GraphPad Software, Inc., La Jolla, CA). Similarly, for the detailed statistical analysis shown in Table 4 (A–G), we used a custom built GNU Octave software (freely available on the World Wide Web)-based program to determine the percent confidence levels between conditions for each microtubule dynamicity parameter. Length sequence for each event was calculated by fitting a first order polynomial (i.e. a line). The slope of each line gives the rate of the event. The length change observed within each event was calculated with the rate multiplied by the duration of the event. A catastrophe was defined as a transition from either growth or attenuation to shortening. The catastrophe frequency was calculated as the total number of catastrophes divided by the total time spent growing and attenuated. A rescue was defined as a transition from shortening to either growth or attenuation. Frequencies of rescue were calculated as the total number of rescues divided by the total amount of time spent shortening. Average growth and shortening lengths represent the average of individual growth and shortening events. The reported average growth and shortening rates were calculated as the total growth or shortening length change for a particular condition divided by the total time spent for growth or shortening events in the condition. The percentages of time the population of microtubules spent growing, shortening, and attenuated were determined as described previously (67). Dynamicity is a calculated measure of overall dynamic activity (i.e. total length grown plus total length shortened divided by total time observed). Between 30 and 40 microtubules (with a minimum of 230 min of tracking time for each condition) were analyzed for each experimental condition. Each condition was imaged over multiple days using 3–5 different Tau/tubulin/GTP mixes (4–5 slides each).
TABLE 4.
Percent statistical confidence levels between conditions for each microtubule dynamicity parameter


RESULTS
Single and Double Pseudophosphorylation Confer Opposite Effects upon the Ability of Four-repeat Tau to Promote Microtubule Assembly
We first sought to determine whether or not pseudophosphorylation of Tau at either one or two key known phosphorylation sites per molecule affects its ability to promote microtubule assembly. As a prelude, we first established the quantitative validity of our microtubule assembly assay. Briefly, we co-incubated 15 μm tubulin dimers with varying amounts of Tau (Tau/tubulin molar ratios of 1:60, 1:40, 1:20, and 1:10), allowed microtubule assembly to proceed to completion, and then centrifuged the assembled microtubules through a sucrose cushion. These concentrations of tubulin and Tau approximate in vivo conditions (i.e. the intracellular tubulin concentration is based on the work of Hiller and Weber (68) and Zhai and Borisy (69) and the range of Tau/tubulin ratios is based upon estimates believed to be present in neuronal cells (70). Microtubules and microtubule-bound Tau pelleted, whereas non-polymerized tubulin and non-microtubule-bound Tau remained in the supernatant on top of the cushion. Negligible amounts of tubulin or Tau were present in the sucrose cushion itself. The percentages of Tau and tubulin in each fraction were determined by SDS-PAGE/immunoblotting analysis of the pellet and supernatant. As seen in Fig. 2, increased amounts of Tau in the reaction resulted in increased microtubule assembly. The specificity of the reaction is demonstrated by the lack of any increase in microtubule assembly when Tau was replaced by the nonspecific protein BSA.
FIGURE 2.
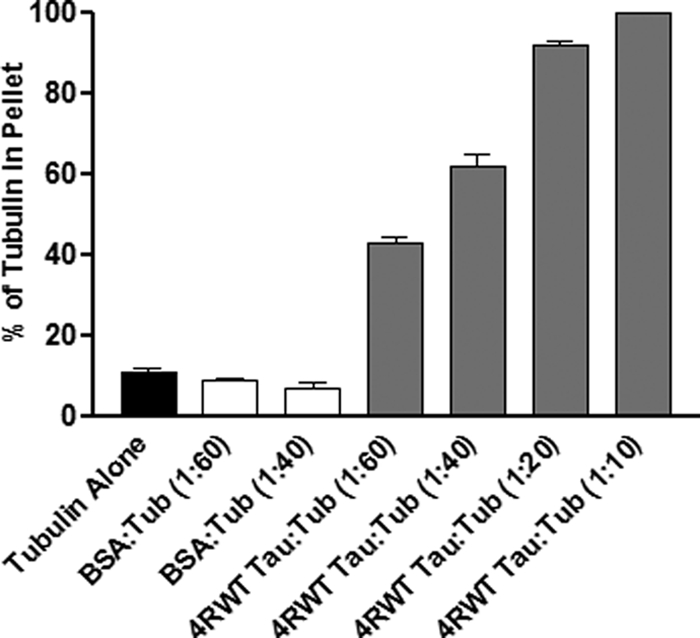
Tau-microtubule co-sedimentation assay. 15 μm tubulin dimers were incubated in the presence or absence of varying amounts of four-repeat Tau or the nonspecific protein BSA at 35 °C until steady state was achieved. The reaction was then centrifuged through a sucrose cushion. Microtubules and microtubule-bound Tau pellet, whereas non-polymerized tubulin and non-microtubule-bound Tau remain in the supernatant. SDS-PAGE analysis of the supernatant and pellet defines the percentages of tubulin in each fraction. The percentage of polymerized tubulin values are presented as mean ± S.E. (error bars).
We next sought to determine if pseudophosphorylation of Tau at one or two key sites affects its ability to promote microtubule assembly. First, we assayed Tau pseudophosphorylated at only one site per molecule (i.e. either T231D, S262D, S396D, or S404D). These are all believed to be important sites of Tau phosphorylation (for more details, see “Discussion”). In all four cases, singly pseudophosphorylated Tau promoted less microtubule assembly than control, non-pseudophosphorylated Tau (referred to as WT in the figures), most significantly in the cases of S262D and S396D (Fig. 3A and Table 1). Additionally, as expected, the quantity of microtubule assembly increased with increasing Tau concentration in all cases.
FIGURE 3.

Effects of single and double pseudophosphorylation upon the ability of Tau to promote microtubule assembly. A, 15 μm tubulin dimers were incubated in the presence or absence of varying amounts of four-repeat Tau at 35 °C until steady state was achieved. The reaction was then centrifuged through a sucrose cushion. Microtubules and microtubule-bound Tau pellet, whereas non-microtubule bound Tau and non-polymerized tubulin remain in the supernatant. SDS-PAGE analysis of the supernatant and pellet defines the percentages of tubulin in each fraction. Statistical significance of the data based on Student's t test is presented in Table 1. B, effects of Tau pseudophosphorylation on the tubulin/Tau “packing ratio” in assembled microtubules. Data for tubulin and Tau content in assembled microtubules are from Fig. 3A and Tables 1 and 2. The values are given as mean ± S.E. (error bars) from at least three independent experiments.
TABLE 1.
Dose-response analysis of pseudophosphorylated Tau-mediated microtubule assembly
Percentage of polymerized tubulin values are given as mean ± S.E.
| Constructs | Percentage of tubulin in pellet |
|||
|---|---|---|---|---|
| 1:60 Tau/tubulin ratio (0.25 μm Tau) | 1:40 Tau/tubulin ratio (0.375 μm Tau) | 1:20 Tau/tubulin ratio (0.75 μm Tau) | 1:10 Tau/tubulin ratio (1.5 μm Tau) | |
| % | % | % | % | |
| 4R WT | 43 ± 1.31 | 62 ± 2.89 | 92 ± 1.23 | 100 ± 0.07 |
| 4R T231D | 28 ± 5.91a | 50 ± 6.80 | 68 ± 2.85b | 93 ± 0.75c |
| 4R S262D | 14 ± 1.14a | 34 ± 3.37d | 53 ± 6.85b | 71 ± 5.18c |
| 4R S396D | 17 ± 2.12a | 40 ± 1.05d | 70 ± 4.02d | 87 ± 0.40c |
| 4R S404D | 25 ± 5.68a | 45 ± 3.02d | 67 ± 2.92 | 83 ± 2.80c |
| 4R T231D/S262D | 57 ± 3.21a | 72 ± 1.93 | 92 ± 2.12 | 100 ± 0.0 |
| 4R S262D/S396D | 63 ± 2.50a | 81 ± 1.68d | 100 ± 0.0 | 100 ± 0.0 |
| 4R S262D/S404D | 68 ± 4.23a | 76 ± 1.66e | 96 ± 0.47 | 100 ± 0.0 |
| 4R S396D/S404D | 53 ± 1.0a | 66 ± 2.0 | 80 ± 0.58 | 89 ± 0.88c |
a Values significant at 99.9% confidence level compared with WT control at a 1:60 Tau/tubulin dimer ratio.
b Values significant at 99.9% confidence level compared with WT control at a 1:20 Tau/tubulin dimer ratio.
c Values significant at 99.9% confidence level compared with WT control at a 1:10 Tau/tubulin dimer ratio.
d Values significant at 99.9% confidence level compared with WT control at a 1:40 Tau/tubulin dimer ratio.
e Value significant at 95% confidence level compared with WT control at a 1:40 Tau/tubulin dimer ratio.
We next compared the microtubule assembly activities of doubly pseudophosphorylated four-repeat Tau with those of their singly pseudophosphorylated counterparts and control, non-pseudophosphorylated Tau. Based on the observation that all singly pseudophosphorylated Tau molecules exhibited reduced microtubule assembly activities relative to control, non-pseudophosphorylated Tau, the simplest prediction is that doubly pseudophosphorylated Tau should be even further compromised. In marked contrast, three of the four tested doubly pseudophosphorylated Tau molecules (T231D/S262D, S262D/S396D, and S262D/S404D) exhibited greater microtubule assembly-promoting activity than control, non-pseudophosphorylated Tau (Fig. 3A and Table 1). The fourth doubly pseudophosphorylated construct, S396D/S404D, exhibited complex microtubule assembly activity, possessing increased potency at lower concentrations and reduced potency at higher concentrations. Taken together, the simple prediction that multiple pseudophosphorylation events would be additive and further reduce microtubule assembly activity below that of the individual pseudophosphorylation events was not fulfilled. Rather, the data demonstrate that the combinatorial effect of two pseudophosphorylation events need not be the additive effects of the two individual events. Indeed, double pseudophosphorylation events generally enhanced microtubule assembly activity even beyond the level of control, non-pseudophosphorylated Tau, whereas single pseudophosphorylation at each of the sites tested suppressed Tau-mediated microtubule assembly activity.
In order to better understand mechanisms underlying these observations, we next examined the quantity of Tau bound to the pelleted microtubules in each assembly reaction. As seen in Table 2, essentially all of the control, non-pseudophosphorylated Tau was bound to the microtubules at all concentrations tested. In contrast, all of the singly pseudophosphorylated Tau molecules were, to varying extents, less effective at interacting with microtubules. Interestingly, as the concentration of Tau in the S262D and S404D reactions increased, the percentage of bound Tau decreased, perhaps suggesting a saturation effect that was not observed over the same concentration range for control, non-pseudophosphorylated Tau. Alternatively, it is possible that S262D and S404D could be aggregating. However, we believe that this is unlikely because (i) recombinant Tau, even mutants that are strong aggregators, does not aggregate under physiological in vitro conditions without the addition of an inducer, such as arachidonic acid (71, 72), and (ii) we have performed direct aggregation studies (using dynamic light scattering) on all of the pseudophosphorylated Tau proteins used in this study, and none aggregate in the absence of an inducer.3
TABLE 2.
Effects of Tau pseudophosphorylation on Tau-microtubule interaction
Percentage of bound Tau values are given as mean ± S.E.
| Constructs | Percentage of Tau in pellet |
|||
|---|---|---|---|---|
| 1:60 Tau/tubulin ratio (0.25 μm Tau) | 1:40 Tau/tubulin ratio (0.375 μm Tau) | 1:20 Tau/tubulin ratio (0.75 μm Tau) | 1:10 Tau/tubulin ratio (1.5 μm Tau) | |
| % | % | % | % | |
| 4R WT | 93 ± 1.49 | 95 ± 1.19 | 98 ± 0.90 | 97 ± 1.60 |
| 4R T231D | 83 ± 9.29 | 81 ± 9.24 | 83 ± 1.76 | 87 ± 3.84 |
| 4R S262D | 89 ± 2.76 | 83 ± 2.77 | 79 ± 1.97 | 72 ± 3.61 |
| 4R S396D | 94 ± 1.86 | 91 ± 1.76 | 86 ± 5.40 | 88 ± 5.19 |
| 4R S404D | 92 ± 5.10 | 85 ± 4.50 | 76 ± 4.67 | 73 ± 8.11 |
| 4R T231D/S262D | 93 ± 3.93 | 87 ± 4.04 | 90 ± 5.20 | 89 ± 4.00 |
| 4R S262D/S396D | 93 ± 1.15 | 98 ± 1.67 | 99 ± 0.67 | 100 ± 0.0 |
| 4R S262D/S404D | 93 ± 2.85 | 97 ± 1.53 | 100 ± 0.33 | 100 ± 0.0 |
| 4R S396D/S404D | 89 ± 2.33 | 87 ± 3.18 | 81 ± 3.18 | 81 ± 0.0 |
Similar analysis of Tau levels present in the microtubules assembled by the doubly pseudophosphorylated Tau molecules revealed a different situation (Table 2). Two of the molecules, S262D/S396D and S262D/S404D, showed no difference at all relative to control, non-pseudophosphorylated Tau despite the fact that the microtubule assembly activities of singly pseudophosphorylated S262D and S404D were strongly compromised. The remaining two doubly pseudophosphorylated molecules, T231D/S262D and S396D/S404D, bound only slightly less well than control, non-pseudophosphorylated Tau. Taken together, and as was true for the tubulin analysis shown in Fig. 3A, the combined effects of two phosphorylation events within a single Tau molecule can be very different from the sum of the effects of the two constituent events when present in individual molecules.
As a final measure of the ability of different Tau constructs to promote microtubule assembly, we used the data in Tables 1 and 2 to calculate the tubulin/Tau “packing” ratio in the assembled microtubules for each reaction (Fig. 3B). At the lowest tested Tau/tubulin ratio used in the initial reaction mixtures (1:60; 0.25 μm Tau), control, non-pseudophosphorylated Tau assembles microtubules composed of ∼28 tubulin dimers/Tau molecule. Under similar conditions, all four of the singly pseudophosphorylated Tau constructs were less effective, assembling between 8 and 20 tubulin dimers/Tau. In marked contrast, all four of the doubly pseudophosphorylated Tau constructs were much more effective than even control, non-pseudophosphorylated Tau, assembling microtubules with between 36 and 45 tubulin dimers/Tau, consistent with their increased ability to promote microtubule assembly (Table 1 and Fig. 3A). As the concentration of Tau increased in the reactions, the differences between the different Tau constructs decreased. At the highest concentration of Tau (1:10 Tau/tubulin ratio), all of the reactions converged on a tubulin/Tau ratio in assembled microtubules of ∼12.
Taken together, the data are consistent with the conclusion that the effects of double pseudophosphorylation of Tau at positions Thr231, Ser262, Ser396, and/or Ser404 upon MT assembly activity are not simply the sum of the parts. Indeed, in almost all cases, the effects of doubly pseudophosphorylated Tau molecules are opposite to the effects of the same pseudophosphorylation events in a singly pseudophosphorylated context. It is also interesting to note the general clustering of activities of the four singly pseudophosphorylated Tau proteins and the four doubly pseudophosphorylated Tau proteins despite the different locations of their pseudophosphorylation mutations.
Doubly Pseudophosphorylated Tau Proteins Regulate Microtubule Dynamic Instability Much Differently than the Sum of the Effects of the Two Constituent Singly Pseudophosphorylated Tau Proteins
“Dynamic instability” is the term used to describe the dynamic behavior of microtubules in which microtubule ends switch between phases of relatively slow growth and rapid shortening, interspersed with periods of pause or “attenuation.” As noted above, proper regulation of microtubule dynamic instability is critical for proper cell function and viability (16, 21, 22).
As another assessment of the effects of single and double pseudophosphorylation upon Tau action, we examined the abilities of our pseudophosphorylated Tau proteins to regulate the dynamic instability behavior at the plus end of individual microtubules in vitro at steady state. Examples of the life history plots of changes in length of individual microtubules versus time are shown in Fig. 4. Because our studies were performed at steady state (i.e. when microtubule mass and soluble tubulin concentrations were not changing), we could observe all three phases of dynamic instability (growth, shortening, and attenuation). The following parameters were determined: (i) average shortening rate; (ii) average length shortened per shortening event; (iii) average growth rate; (iv) average length grown per growth event; (v) percentage of time spent growing, shortening, and attenuated; (vi) rescue frequency; (vii) catastrophe frequency; and (viii) overall dynamicity. The data (described below) are presented graphically in Figs. 5–8 and are summarized quantitatively in Table 3. Comparative statistical analyses of the data are presented in Table 4 (A–G).
FIGURE 4.

Examples of microtubule life history plots. Changes in the length of the microtubules grown in the absence of Tau (top), in the presence of 0.293 μm 4R WT Tau (1:40 Tau/tubulin dimer ratio; middle), or in the presence of 0.293 μm 4R S262D pseudophosphorylated Tau (1:40 Tau/tubulin dimer ratio; bottom) were plotted as a function of time. From these and additional life history plots, individual growth, shortening, and attenuation events were identified, and rates and lengths of each event were determined, as described under “Experimental Procedures.”
FIGURE 5.

Effects of various single and double pseudophosphorylation upon microtubule shortening (A) and growth (B) events. The positions of the ends of individual microtubules were tracked in the presence or absence of WT or pseudophosphorylated Tau at a 1:40 molar ratio of Tau/tubulin dimers, and life history plots were generated. From these data, individual growth, shortening, and attenuation events were identified. A, the average growth and shortening rates were calculated as the total growth or shortening length change for a particular reaction condition divided by the total time spent for growth or shortening events in that condition. Average growth and shortening lengths represent the average of independent growth and shortening events. Values are given as mean ± S.E. (error bars). n values for each condition are presented in Table 3. *, value significant at 95% confidence level compared with 4R WT Tau control conditions. **, value significant at 99.9% confidence level compared with 4R WT Tau control conditions.
FIGURE 6.

Effects of various single and double pseudophosphorylation upon catastrophe (A) and rescue (B) frequencies. A catastrophe is defined as conversion of a growing or attenuation event to a shortening event. Catastrophe frequencies were calculated as the total number of catastrophes divided by the total time spent growing and attenuated. A rescue is defined as a transition from shortening to either growth or attenuation. Rescue frequencies were calculated as the total number of rescues divided by the total amount of time spent shortening. Based upon the life history plots described in Fig. 4, catastrophe and rescue frequencies were calculated. Values are given as mean ± S.E. (error bars). *, value significant at 95% confidence level compared with 4R WT Tau control conditions. **, values significant at 99.9% confidence level compared with 4R WT Tau control conditions.
FIGURE 7.
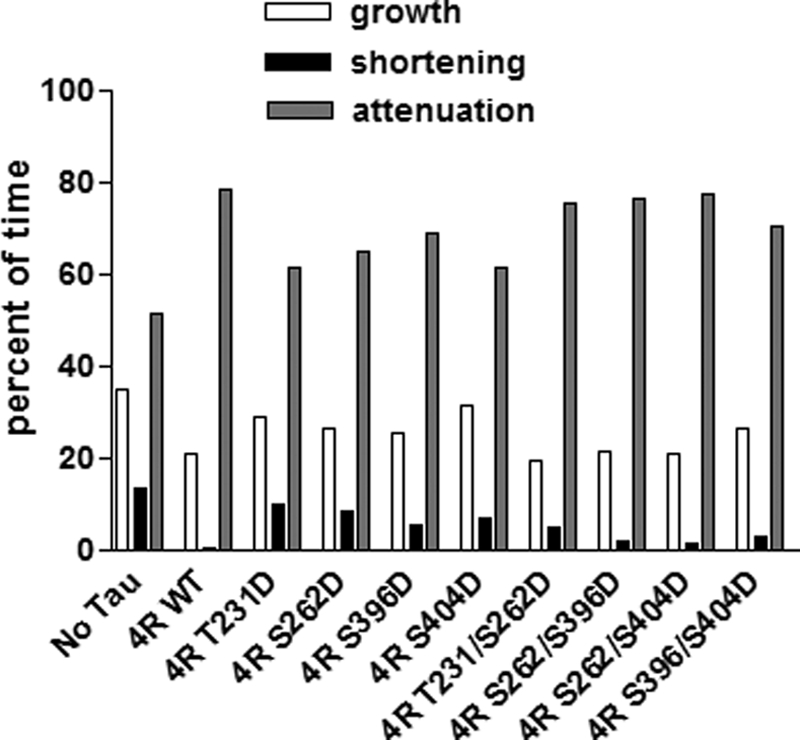
Effects of various single and double pseudophosphorylation upon total time spent for attenuation, growth, or shortening. Based upon the life history plots described in Fig. 4, the percentage of time spent in each phase was calculated.
FIGURE 8.
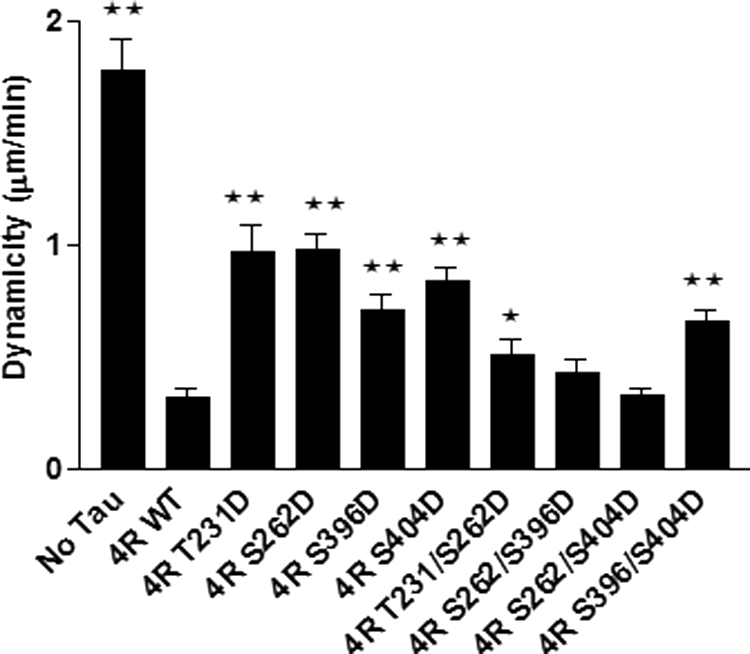
Effects of various single and double pseudophosphorylation upon overall microtubule dynamics at steady state in vitro. Dynamicity is a calculated measure of overall dynamic activity (i.e. total length grown plus total length shortened divided by total time observed). Based upon the life history plots described in Fig. 4, dynamicity was calculated for all conditions. Values are given as mean ± S.E. (error bars). *, value significant at 95% confidence level compared with 4R WT Tau control conditions. **, values significant at 99.9% confidence level compared with 4R WT Tau control conditions.
TABLE 3.
Effects of the pseudophosphorylation events upon the microtubule dynamicity parameters at steady state in vitro

Shortening Events
The ability of 4R Tau to suppress the average shortening rate was markedly reduced for two singly pseudophosphorylated Tau constructs, S262D and S396D. On the other hand, relative to control, non-pseudophosphorylated Tau, the shortening rate for T231D was unaffected, and it was slightly increased for Ser404 (Fig. 5A and Table 3). In contrast, all four doubly pseudophosphorylated Tau constructs exhibited either control, non-pseudophosphorylated Tau levels of activity (T231D/S262D and S262D/S396D) or significantly enhanced activity (S262D/S404D and S396D/S404D) relative to control Tau. A particularly glaring example of the lack of simple additivity of single and double pseudophosphorylation is to compare S262D and S396D as singly pseudophosphorylated constructs (both severely compromised relative to control Tau) and as a doubly pseudophosphorylated construct (S262/S396D; no loss-of-function at all relative to control, non-pseudophosphorylated Tau).
The ability of control Tau to reduce the average lengths of shortening events was reduced strongly for three of the singly pseudophosphorylated Tau constructs (T231D, S262D, and S396D; Fig. 5A and Table 3). S404D also exhibited reduced ability to regulate the average length of shortening events, but the effect was weaker than for the other singly pseudophosphorylated Tau proteins. Similarly, all four doubly pseudophosphorylated Tau constructs exhibited compromised Tau-mediated reduction in the average length of shortening events; however, the magnitude of the effects was less than was the case for the singly pseudophosphorylated Tau constructs. Again, the effects of single pseudophosphorylation events are not additive when the constituent pseudophosphorylation events are combined in doubly pseudophosphorylated Tau molecules.
Growth Events
The ability of three of the four singly pseudophosphorylated Tau constructs to suppress the average growth rate was somewhat compromised relative to control Tau (T231D, S262D, and S404D), whereas S396D was not affected significantly (Fig. 5B and Table 3). In contrast, although three of the four doubly pseudophosphorylated Tau constructs were unaffected relative to control Tau, S396D/S404D was very significantly compromised in its ability to regulate the rate of growth events.
Normal Tau exerts only a relatively modest effect on the length of MT growth events. Therefore, it was not surprising that the average growth length during individual growth events for singly and doubly pseudophosphorylated Tau constructs was not affected much (Fig. 5B and Table 3), with the exception of S262D/R404D, which suppressed the average length of growth events slightly more potently than did control Tau.
Catastrophe and Rescue Frequencies
A catastrophe is defined as the conversion of a growth or attenuation event to a shortening event. Control, non-pseudophosphorylated Tau greatly decreases the catastrophe frequency (Fig. 6A and Table 3). All four singly pseudophosphorylated Tau constructs, especially T231D and S262D, exhibit much less ability to prevent catastrophes than does control, non-pseudophosphorylated Tau. In contrast, all four doubly pseudophosphorylated Tau proteins exhibit greater catastrophe-preventing activity than their singly pseudophosphorylated counterparts, with two of the doubly pseudophosphorylated Tau proteins (S262D/S396D and S262D/S404D) exhibiting near control Tau levels of activity. Again, doubly pseudophosphorylated Tau molecules exhibit less, not more, loss of function than do their singly pseudophosphorylated counterparts. Additionally, it is notable that all four singly pseudophosphorylated Tau proteins exhibit quite similar levels of activity with one another, as do the four doubly pseudophosphorylated Tau proteins.
A rescue event is defined as conversion of a shortening event to either attenuation or growth. Control, non-pseudophosphorylated Tau significantly increases the rescue frequency (Fig. 6B and Table 3). In contrast, all of the singly and doubly pseudophosphorylated Tau constructs are defective in promoting rescue events.
Percentage of Time in Each Phase
As seen in Fig. 7 and Table 3, in the absence of Tau, the microtubules used in this work spend 51% of their time attenuated, 35% of their time growing, and 14% of their time shortening. The presence of control, non-pseudophosphorylated Tau increased the percentage of time attenuated, reduced the percentage of time growing, and almost eliminated time shortening. All four of the singly pseudophosphorylated Tau molecules were compromised in their abilities to influence the percentage of time microtubules are in growth, shortening, or attenuated phases. In contrast, all four doubly pseudophosphorylated Tau constructs exhibit near control Tau percentages. This provides yet another example of the doubly pseudophosphorylated constructs not exhibiting the predicted effects of a simple additive model for combinatorial pseudophosphorylation.
Dynamicity
Dynamicity is a calculated measure of overall dynamic activity (i.e. total length grown plus total length shortened divided by total time observed). Control Tau reduces microtubule dynamicity by over 5-fold (Fig. 8 and Table 3). All four singly pseudophosphorylated Tau constructs suppressed dynamicity much less effectively than did control, non-pseudophosphorylated Tau. In contrast, one of the four doubly pseudophosphorylated Tau proteins exhibited dynamicity-suppressing activity that was very similar to that of control Tau, whereas the other three remained compromised but to a lesser extent than their singly pseudophosphorylated counterparts.
DISCUSSSION
Among the primary goals of the work presented here was to better understand molecular mechanisms by which phosphorylation regulates Tau-mediated action, focusing on microtubule assembly and the regulation of microtubule dynamic instability. Importantly, we sought to begin investigating the mechanistic effects of combinatorial Tau phosphorylation, which is of certain in vivo relevance yet remains an almost completely unstudied question. Further, understanding basic principles of combinatorial phosphorylation as a regulatory mechanism is a critically important general question far beyond just Tau and microtubules.
In the simplest case, one could imagine that multiple phosphorylation events located in functionally and physically distinct regions of a protein could act completely independently of one another. For example, one phosphorylation event might regulate an SH2 domain interaction, and another event located elsewhere in the protein might regulate some local conformational change. There are many such examples (e.g. the Src kinase) (73). On the other hand, one could also imagine that multiple phosphorylation events on a single protein molecule could cause integrated structural effects such that the overall mechanistic effect is different from the sum of the parts.
The first and most important outcome of this work was that the activities exhibited by Tau molecules harboring pseudophosphorylation at two sites per molecule were not simply the sum of the activities of the constituent two Tau molecules harboring the corresponding individual pseudophosphorylations. In fact, no clear cases of simple additivity were observed (i.e. there were no cases in which both singly pseudophosphorylated Tau proteins exhibited loss-of-function effects and the doubly pseudophosphorylated protein exhibited a larger loss-of-function effect). Rather, we observed (i) several cases in which the doubly pseudophosphorylated Tau protein exhibited an opposite effect from that exhibited by its singly pseudophosphorylated counterparts (such as T231D/S262D (microtubule assembly reactions, shortening length, and catastrophe frequency), S262D/S396D (microtubule assembly reactions, shortening length, and catastrophe frequency), and S262D/S404D (microtubule assembly reactions and catastrophe frequency)) and (ii) several cases in which the two singly pseudophosphorylated Tau proteins exhibited quite different activity levels from one another and the doubly pseudophosphorylated protein closely resembled the activity level of one but not the other of the constituent singly pseudophosphorylated Tau proteins (such as T231D/S262D (shortening rate), S262D/S404D (shortening rate and shortening length), and S396D/S404D (shortening rate, shortening length, and growth rate)). Interestingly, among the doubly pseudophosphorylated Tau proteins in this latter class, position Ser404 appears to be dominant (i.e. the doubly pseudophosphorylated Tau protein exhibits the activity level of the singly pseudophosphorylated Ser404 Tau protein rather than that of the second pseudophosphorylated site). In contrast, pseudophosphorylation at position Ser262 or Ser396 does not exert such a “dominant” effect in any of the doubly pseudophosphorylated Tau proteins. Finally, there were several cases in which the two singly pseudophosphorylated Tau proteins exhibited similar levels of an activity and the doubly pseudophosphorylated Tau protein also exhibited a similar (but not additive) activity level.
Another unexpected and striking effect was the lack of widespread site-specific effects of single pseudophosphorylation events on the ability of Tau to regulate microtubule dynamics (i.e. the relatively high frequency with which different pseudophosphorylated Tau proteins exhibit very similar levels of an activity that differ from the activity level of control Tau). One might anticipate that different sites of pseudophosphorylation would exert very different effects on different parameters of microtubule dynamics. However, little site specificity was observed. Indeed, the only clear cases in which different singly pseudophosphorylated Tau proteins differentially affected microtubule dynamics markedly were in the regulation of shortening events. For example, pseudophosphorylation of Tau at positions 262 and 396 greatly compromised the ability of Tau to suppress the rate of microtubule-shortening events, whereas pseudophosphorylation of Tau at either 231 or 404 had little effect. Similarly, pseudophosphorylation of Tau at 231, 262, or 396 greatly compromised its ability to reduce the average length of shortening events, whereas pseudophosphorylation of Tau at 404 had little effect. However, by all other parameters of microtubule dynamics, the loss-of-function effects resulting from pseudophosphorylation were similar for the four different singly pseudophosphorylated Tau proteins despite having their pseudophosphates at different locations. The similarity of activity levels among Tau proteins harboring different sites of pseudophosphorylation was even more pronounced among the doubly pseudophosphorylated Tau proteins. For example, Tau pseudophosphorylated at Thr231/Ser262 showed very similar activity levels to Tau pseudophosphorylated at Ser262/Ser396 by all of our many assays. An even more remarkable case is the fact that Tau pseudophosphorylated at Thr231/Ser262 is very similar to Tau pseudophosphorylated at Ser396/Ser404 in numerous assays (shortening rate, growth length, rescue frequency, and catastrophe frequency), despite having no pseudophosphorylation sites in common. Many other examples of different doubly pseudophosphorylated Tau molecules acting similarly by 3–4 criteria can be identified (see Fig. 5–8 and Table 4).
Phosphorylation Regulates the Ability of Tau to Regulate Microtubule Assembly
Previous work, both in vitro and in cultured cells, has led to the conclusion that phosphorylation of Tau at a large number of individual sites reduces microtubule binding and assembly activities (74–81). Our analysis of Tau proteins harboring single pseudophosphorylation events at positions Thr231, Ser262, Ser396, and Ser404 are consistent with these conclusions (i.e. all exhibit reduced levels of microtubule assembly activity). It is therefore all the more remarkable to note that, at all Tau/tubulin ratios short of saturation, all doubly pseudophosphorylated Tau proteins promoted microtubule assembly more effectively than either singly pseudophosphorylated Tau or control, non-pseudophosphorylated Tau, despite their reduced Tau/tubulin packing ratios. This increased potency of the doubly pseudophosphorylated Tau is apparent from both the tubulin assembly data in Fig. 3A and the tubulin/Tau packing ratio data in Fig. 3B. These data clearly demonstrate the inadequacy of the simple additivity mechanism for combinatorial phosphorylation-mediated regulation of Tau action.
Phosphorylation Regulates the Ability of Tau to Regulate Microtubule Dynamic Instability
In contrast to the abundance of previous work on the effects of Tau phosphorylation upon its ability to bind to microtubules and to promote microtubule assembly, there has been much less work examining the effects of phosphorylation upon the ability of Tau to regulate microtubule dynamic instability. Drechsel et al. (15) examined the effects of MAP2 kinase on the ability of Tau to regulate microtubule dynamic instability in vitro under non-steady state conditions, observing an increased rate of microtubule growth, a decreased rate of shortening, and decreased catastrophe frequency. However, the extent of phosphorylation and site(s) of phosphorylation were not determined. Trinczek et al. (9) examined the effects of CDK5 and MARK kinase activities upon the ability of Tau to regulate microtubule dynamic instability in vitro (9), also under non-steady state conditions. Phosphorylation of Tau by CDK5, which phosphorylated Ser202, Ser235, and Ser404 to undetermined extents, increased the catastrophe frequency 4-fold compared with control, non-phosphorylated Tau. Phosphorylation of Tau by MARK (mainly at Ser262 but perhaps also Ser356) increased the catastrophe frequency quite significantly, thereby increasing the dynamicity about 40-fold compared with control Tau. It is important to note that both the Drechsel et al. (15) and Trinczek et al. (9) studies were performed at non-steady state conditions (which is probably a better approximation of conditions in a developing rather than a mature neuron) and at extraordinarily high Tau/tubulin ratios, in marked contrast to our work.
In the work presented here, we show that pseudophosphorylation of Tau at defined single sites (Thr231, Ser262, Ser396, or Ser404) can modulate its ability to regulate microtubule dynamic instability (Figs. 5–8 and Table 3). The most marked effects are on shortening events (shortening rate, average length of shortening events, and rescue frequency) as well as the percentage of time in different phases. Further, the data are consistent with the microtubule assembly data described above in that they do not support the simple additivity view of combinatorial Tau phosphorylation effects on Tau action.
A Novel View of Combinatorial Tau Phosphorylation
Given that there are ∼30 known phosphorylation sites on Tau (43, 82, 83), regulation of Tau activity via phosphorylation has often been compared with a rheostat as opposed to a series of toggle switches, suggesting that the many different phosphorylation sites each confer different structural and functional effects along a gradient of activity levels. This analogy would predict that there should be significant activity differences among the four different singly pseudophosphorylated constructs and the four different doubly pseudophosphorylated constructs. However, as noted above, this was not generally observed. Given these observations, perhaps both the rheostat and toggle switch analogies require revisiting. As an alternative, perhaps there are only a small number of different structure-function states of Tau, but these states need to be tightly regulated by a large number of different signaling pathways. In its simplest form, perhaps one phosphate located at any of a number of sites confers structure-function state “A”, whereas two phosphates located at any of a number of different possible locations on a Tau molecule confer structure-function state “B”. Thus, it could be that the primary mechanistic effects of phosphorylation are determined by the number of phosphates per molecule, with more subtle modulatory effects mediated in a site-specific manner. By this perspective, the large number of phosphorylatable sites in Tau could be a consequence of the many signaling pathways regulating Tau activity (and the sequence-specific substrate requirements of their many kinases) rather than a large number of structure-function states. This is certainly a testable hypothesis.
Statistical analyses also highlighted the fact that the activities of many doubly pseudophosphorylated Tau molecules closely resemble those of control Tau. Speculatively, this could provide a kind of molecular “history” or “memory” by which comparable levels of activity can be achieved in differently phosphorylated Tau molecules, which could be regulated differentially by subsequent effectors or circumstances.
Phosphorylation and Pathological Tau Action
A large body of biochemical and genetic data supports the conclusion that aberrant regulation of Tau phosphorylation can cause neurodegeneration (5, 6). Unfortunately, the underlying mechanism(s) remains elusive. One widely held model suggests that abnormal Tau phosphorylation increases the probability that Tau will oligomerize and then aggregate and that either one or both of these conditions is cytotoxic. Alternatively, integrating work in dividing cells with our recent in vitro (8, 21, 22, 30, 60) and cell culture work (10, 61), we and our collaborators have hypothesized that Tau-mediated neuronal cell death in the Tauopathies might be caused, at least in part, by aberrant Tau-mediated regulation of microtubule dynamic instability. In the original presentations of this hypothesis, the emphasis was upon errors in Tau action caused by mutations (61, 62). Here, with the confirmation of the fact that Tau phosphorylation can alter the regulation of microtubule dynamic instability, we can expand the earlier hypothesis to propose that errors in Tau action, caused by either mutations or aberrant phosphorylation, can induce neuronal cell death and dementia.
Technical Issues; Choice of Sites to Investigate, Concerns with Taxol, and Pseudophosphorylation as a Strategy
Three technical issues regarding the relatively large literature regarding Tau action and phosphorylation are especially worthy of note. First, we have focused our efforts on four particularly important Tau phosphorylation sites (Fig. 1). Thr231 lies in the proline-rich regulatory region of Tau, and phosphorylation of this site is essential for prolyl isomerase Pin1 activity, which catalyzes the cis/trans isomerization of the Thr231-Pro232 bond and is critical for neuronal function (76, 84). Ser262 maps to the first imperfect repeat. The role of Ser262 phosphorylation in Tau action is controversial, with some investigators observing dramatic effects upon the microtubule binding activity of Tau, whereas others observe more muted effects (78, 79, 81). Both Ser396 and Ser404 map to the regulatory C-terminal tail. It has been suggested that the FTDP-17 R406W mutation might cause disease indirectly by altering the ability of nearby serines (including Ser396, Ser400, and Ser404) to act as kinase substrates (61, 85). Further, Thr231, Ser262, Ser396, and Ser404 are all consistently hyperphosphorylated in neurofibrillary tangles and widely believed to be important players in pathological Tau action (42, 43). Finally, it is important to note that essentially nothing is known about the combinatorial pattern of Tau phosphorylation under either normal or pathological conditions. The combinations used here may or may not exist in vivo; they have been selected for analysis based primarily upon the importance of the individual sites and placed in the same protein to assess their combinatorial effects.
Second, many previous studies have assessed the effects of site-specific phosphorylation upon the ability of Tau to bind to taxol-stabilized microtubules in vitro. However, taxol binds very close to one of two Tau binding sites on microtubules (86), raising the possibility that the presence of taxol might affect Tau binding to microtubules. Indeed, studies from others and from our laboratory show that taxol reduces Tau binding to microtubules (86–88). To avoid this complication, the work in this paper has assayed Tau activities in the absence of taxol.
Finally, a brief comment is warranted regarding the means of generating phosphorylated Tau proteins for analysis. In most earlier studies, this has been achieved with purified kinases. However, generating Tau proteins with site-specific phosphorylation using kinases in vitro can be problematic because (i) kinases generally phosphorylate Tau at multiple sites, and (ii) the efficiency of phosphorylation reactions at any given site can be low and difficult to control. These issues can greatly complicate data analysis and interpretation. To avoid these problems, we have employed “pseudophosphorylation” (i.e. the substitution of aspartic or glutamic acid residues to mimic phosphorylated amino acids). The major advantages of this widely used strategy over enzymatically phosphorylating a protein with a particular kinase are (i) “phosphorylation” at the site of interest is 100% efficient, and (ii) there is no phosphorylation at unintended sites to complicate data interpretation (51, 89–92). The main caveat of pseudophosphorylation is that it does not precisely mimic the chemistry of a phosphate group. Nonetheless, pseudophosphorylation has been widely and successfully used in vitro and in cultured cells to assess various aspects of phosphorylation-mediated events, including the regulation of normal and pathological Tau action (51, 92–96). Thus, although some differences may be found between the mechanistic effects of phosphorylated Tau versus that of pseudophosphorylated Tau, precedent suggests that pseudophosphorylation provides a reasonable prediction of how site-specific phosphorylation will affect Tau function.
Acknowledgments
We are very grateful to Herb Miller for generously providing superb tubulin and axonemes. We are also grateful to Jack Reifert for generating the T231D construct and proteins, to Doug Thrower for assistance with the dynamics protocols, to Brian Matsumoto for help with microscopy, and to Bob Jacobs for sharing the ultracentrifuge.
This work was supported, in whole or in part, by National Institutes of Health Grants NS-35010 (to S. C. F.), NS-13560 (to L. W.), and CA-57291 (to M. A. J.). This work was also supported by National Science Foundation Grant ITR-0331697 (to B. S. M., K. R., S. C. F., and L. W.).
E. Kiris, D. Ventimiglia, and S. C. Feinstein, unpublished data.
- 3R
- three-repeat Tau
- 4R
- four-repeat Tau
- FTDP-17
- frontotemporal dementia with parkinsonism linked to chromosome 17.
REFERENCES
- 1. Esmaeli-Azad B., McCarty J. H., Feinstein S. C. (1994) J. Cell. Sci. 107, 869–879 [DOI] [PubMed] [Google Scholar]
- 2. Caceres A., Potrebic S., Kosik K. S. (1991) J. Neurosci. 11, 1515–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knops J., Kosik K. S., Lee G., Pardee J. D., Cohen-Gould L., McConlogue L. (1991) J. Cell. Biol. 114, 725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977) J. Mol. Biol. 116, 207–225 [DOI] [PubMed] [Google Scholar]
- 5. Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 6. Goedert M., Spillantini M. G. (2006) Science 314, 777–781 [DOI] [PubMed] [Google Scholar]
- 7. Goode B. L., Chau M., Denis P. E., Feinstein S. C. (2000) J. Biol. Chem. 275, 38182–38189 [DOI] [PubMed] [Google Scholar]
- 8. Panda D., Samuel J. C., Massie M., Feinstein S. C., Wilson L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9548–9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trinczek B., Biernat J., Baumann K., Mandelkow E. M., Mandelkow E. (1995) Mol. Biol. Cell. 6, 1887–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bunker J. M., Wilson L., Jordan M. A., Feinstein S. C. (2004) Mol. Biol. Cell. 15, 2720–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feinstein S. C., Wilson L. (2005) Biochim. Biophys. Acta. 1739, 268–279 [DOI] [PubMed] [Google Scholar]
- 12. Butner K. A., Kirschner M. W. (1991) J. Cell. Biol. 115, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee G., Neve R. L., Kosik K. S. (1989) Neuron 2, 1615–1624 [DOI] [PubMed] [Google Scholar]
- 14. Goode B. L., Feinstein S. C. (1994) J. Cell. Biol. 124, 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drechsel D. N., Hyman A. A., Cobb M. H., Kirschner M. W. (1992) Mol. Biol. Cell 3, 1141–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jordan M. A., Wilson L. (2004) Nat. Rev. Cancer. 4, 253–265 [DOI] [PubMed] [Google Scholar]
- 17. Desai A., Mitchison T. J. (1997) Annu. Rev. Cell Dev. Biol. 13, 83–117 [DOI] [PubMed] [Google Scholar]
- 18. Roy S., Zhang B., Lee V. M., Trojanowski J. Q. (2005) Acta Neuropathol. 109, 5–13 [DOI] [PubMed] [Google Scholar]
- 19. Duncan J. E., Goldstein L. S. (2006) PLoS Genet. 2, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morfini G. A., Burns M., Binder L. I., Kanaan N. M., LaPointe N., Bosco D. A., Brown R. H., Jr., Brown H., Tiwari A., Hayward L., Edgar J., Nave K. A., Garberrn J., Atagi Y., Song Y., Pigino G., Brady S. T. (2009) J. Neurosci. 29, 12776–12786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonçalves A., Braguer D., Kamath K., Martello L., Briand C., Horwitz S., Wilson L., Jordan M. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 11737–11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yvon A. M., Wadsworth P., Jordan M. A. (1999) Mol. Biol. Cell. 10, 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Himmler A., Drechsel D., Kirschner M. W., Martin D. W., Jr. (1989) Mol. Cell. Biol. 9, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goedert M., Spillantini M. G., Potier M. C., Ulrich J., Crowther R. A. (1989) EMBO J. 8, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson G. V. (2006) J. Alzheimers Dis. 9, Suppl. 3, 243–250 [DOI] [PubMed] [Google Scholar]
- 26. Ennulat D. J., Liem R. K., Hashim G. A., Shelanski M. L. (1989) J. Biol. Chem. 264, 5327–5330 [PubMed] [Google Scholar]
- 27. Gustke N., Trinczek B., Biernat J., Mandelkow E. M., Mandelkow E. (1994) Biochemistry 33, 9511–9522 [DOI] [PubMed] [Google Scholar]
- 28. Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989) Neuron 3, 519–526 [DOI] [PubMed] [Google Scholar]
- 29. Goode B. L., Denis P. E., Panda D., Radeke M. J., Miller H. P., Wilson L., Feinstein S. C. (1997) Mol. Biol. Cell. 8, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Panda D., Goode B. L., Feinstein S. C., Wilson L. (1995) Biochemistry 34, 11117–11127 [DOI] [PubMed] [Google Scholar]
- 31. Kosik K. S., Orecchio L. D., Bakalis S., Neve R. L. (1989) Neuron 2, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 32. Ksiezak-Reding H., Liu W. K., Yen S. H. (1992) Brain. Res. 597, 209–219 [DOI] [PubMed] [Google Scholar]
- 33. Köpke E., Tung Y. C., Shaikh S., Alonso A. C., Iqbal K., Grundke-Iqbal I. (1993) J. Biol. Chem. 268, 24374–24384 [PubMed] [Google Scholar]
- 34. Kenessey A., Yen S. H. (1993) Brain. Res. 629, 40–46 [DOI] [PubMed] [Google Scholar]
- 35. Hanger D. P., Betts J. C., Loviny T. L., Blackstock W. P., Anderton B. H. (1998) J. Neurochem. 71, 2465–2476 [DOI] [PubMed] [Google Scholar]
- 36. Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Titani K., Ihara Y. (1995) J. Biol. Chem. 270, 823–829 [DOI] [PubMed] [Google Scholar]
- 37. Gong C. X., Liu F., Grundke-Iqbal I., Iqbal K. (2006) J. Biomed. Biotechnol. 2006, 31825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanger D. P., Byers H. L., Wray S., Leung K. Y., Saxton M. J., Seereeram A., Reynolds C. H., Ward M. A., Anderton B. H. (2007) J. Biol. Chem. 282, 23645–23654 [DOI] [PubMed] [Google Scholar]
- 39. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. (1992) Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 40. Goedert M. (2004) Semin. Cell. Dev. Biol. 15, 45–49 [DOI] [PubMed] [Google Scholar]
- 41. Geschwind D. H. (2003) Neuron 40, 457–460 [DOI] [PubMed] [Google Scholar]
- 42. Wang J. Z., Grundke-Iqbal I., Iqbal K. (2007) Eur. J. Neurosci. 25, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J. Z., Liu F. (2008) Prog. Neurobiol. 85, 148–175 [DOI] [PubMed] [Google Scholar]
- 44. Kelleher I., Garwood C., Hanger D. P., Anderton B. H., Noble W. (2007) J. Neurochem. 103, 2256–2267 [DOI] [PubMed] [Google Scholar]
- 45. Drewes G. (2004) Trends Biochem. Sci. 29, 548–555 [DOI] [PubMed] [Google Scholar]
- 46. Flaherty D. B., Soria J. P., Tomasiewicz H. G., Wood J. G. (2000) J. Neurosci. Res. 62, 463–472 [DOI] [PubMed] [Google Scholar]
- 47. Hooper C., Killick R., Lovestone S. (2008) J. Neurochem. 104, 1433–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., Feany M. B. (2001) Science 293, 711–714 [DOI] [PubMed] [Google Scholar]
- 49. Hoshi M., Sato M., Matsumoto S., Noguchi A., Yasutake K., Yoshida N., Sato K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6370–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Steinhilb M. L., Dias-Santagata D., Fulga T. A., Felch D. L., Feany M. B. (2007) Mol. Biol. Cell. 18, 5060–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fath T., Eidenmüller J., Brandt R. (2002) J. Neurosci. 22, 9733–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clark L. N., Poorkaj P., Wszolek Z., Geschwind D. H., Nasreddine Z. S., Miller B., Li D., Payami H., Awert F., Markopoulou K., Andreadis A., D'Souza I., Lee V. M., Reed L., Trojanowski J. Q., Zhukareva V., Bird T., Schellenberg G., Wilhelmsen K. C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13103–13107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J. M., Nowotny P., Che L. K., Norton J., Morris J. C., Reed L. A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P. R., Hayward N., Kwok J. B., Schofield P. R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B. A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 54. Spillantini M. G., Murrell J. R., Goedert M., Farlow M. R., Klug A., Ghetti B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 7737–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hong M., Zhukareva V., Vogelsberg-Ragaglia V., Wszolek Z., Reed L., Miller B. I., Geschwind D. H., Bird T. D., McKeel D., Goate A., Morris J. C., Wilhelmsen K. C., Schellenberg G. D., Trojanowski J. Q., Lee V. M. (1998) Science 282, 1914–1917 [DOI] [PubMed] [Google Scholar]
- 56. Kraemer B. C., Zhang B., Leverenz J. B., Thomas J. H., Trojanowski J. Q., Schellenberg G. D. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9980–9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spittaels K., Van den Haute C., Van Dorpe J., Bruynseels K., Vandezande K., Laenen I., Geerts H., Mercken M., Sciot R., Van Lommel A., Loos R., Van Leuven F. (1999) Am. J. Pathol. 155, 2153–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Probst A., Götz J., Wiederhold K. H., Tolnay M., Mistl C., Jaton A. L., Hong M., Ishihara T., Lee V. M., Trojanowski J. Q., Jakes R., Crowther R. A., Spillantini M. G., Bürki K., Goedert M. (2000) Acta. Neuropathol. 99, 469–481 [DOI] [PubMed] [Google Scholar]
- 59. Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T. L., Lewis J., Hutton M., Janus C. (2007) J. Neurosci. 27, 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Levy S. F., Leboeuf A. C., Massie M. R., Jordan M. A., Wilson L., Feinstein S. C. (2005) J. Biol. Chem. 280, 13520–13528 [DOI] [PubMed] [Google Scholar]
- 61. Bunker J. M., Kamath K., Wilson L., Jordan M. A., Feinstein S. C. (2006) J. Biol. Chem. 281, 11856–11863 [DOI] [PubMed] [Google Scholar]
- 62. LeBoeuf A. C., Levy S. F., Gaylord M., Bhattacharya A., Singh A. K., Jordan M. A., Wilson L., Feinstein S. C. (2008) J. Biol. Chem. 283, 36406–36415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vulliet R., Halloran S. M., Braun R. K., Smith A. J., Lee G. (1992) J. Biol. Chem. 267, 22570–22574 [PubMed] [Google Scholar]
- 64. Toso R. J., Jordan M. A., Farrell K. W., Matsumoto B., Wilson L. (1993) Biochemistry 32, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 65. Miller H. P., Wilson L. (2010) Methods Cell Biol. 95, 3–15 [DOI] [PubMed] [Google Scholar]
- 66. Walker R. A., O'Brien E. T., Pryer N. K., Soboeiro M. F., Voter W. A., Erickson H. P., Salmon E. D. (1988) J. Cell. Biol. 107, 1437–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Panda D., Jordan M. A., Chu K. C., Wilson L. (1996) J. Biol. Chem. 271, 29807–29812 [DOI] [PubMed] [Google Scholar]
- 68. Hiller G., Weber K. (1978) Cell 14, 795–804 [DOI] [PubMed] [Google Scholar]
- 69. Zhai Y., Borisy G. G. (1994) J. Cell. Sci. 107, 881–890 [DOI] [PubMed] [Google Scholar]
- 70. Drubin D. G., Feinstein S. C., Shooter E. M., Kirschner M. W. (1985) J. Cell. Biol. 101, 1799–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gamblin T. C., Berry R. W., Binder L. I. (2003) Biochemistry 42, 15009–15017 [DOI] [PubMed] [Google Scholar]
- 72. Kuret J., Chirita C. N., Congdon E. E., Kannanayakal T., Li G., Necula M., Yin H., Zhong Q. (2005) Biochim. Biophys. Acta. 1739, 167–178 [DOI] [PubMed] [Google Scholar]
- 73. Thomas S. M., Brugge J. S. (1997) Annu. Rev. Cell Dev. Biol. 13, 513–609 [DOI] [PubMed] [Google Scholar]
- 74. Cho J. H., Johnson G. V. (2004) J. Neurochem. 88, 349–358 [DOI] [PubMed] [Google Scholar]
- 75. Lu P. J., Wulf G., Zhou X. Z., Davies P., Lu K. P. (1999) Nature 399, 784–788 [DOI] [PubMed] [Google Scholar]
- 76. Zhou X. Z., Kops O., Werner A., Lu P. J., Shen M., Stoller G., Küllertz G., Stark M., Fischer G., Lu K. P. (2000) Mol. Cell. 6, 873–883 [DOI] [PubMed] [Google Scholar]
- 77. Bramblett G. T., Goedert M., Jakes R., Merrick S. E., Trojanowski J. Q., Lee V. M. (1993) Neuron 10, 1089–1099 [DOI] [PubMed] [Google Scholar]
- 78. Biernat J., Gustke N., Drewes G., Mandelkow E. M., Mandelkow E. (1993) Neuron 11, 153–163 [DOI] [PubMed] [Google Scholar]
- 79. Seubert P., Mawal-Dewan M., Barbour R., Jakes R., Goedert M., Johnson G. V., Litersky J. M., Schenk D., Lieberburg I., Trojanowski J. Q. (1995) J. Biol. Chem. 270, 18917–18922 [DOI] [PubMed] [Google Scholar]
- 80. Singh T. J., Wang J. Z., Novak M., Kontzekova E., Grundke-Iqbal I., Iqbal K. (1996) FEBS Lett. 387, 145–148 [DOI] [PubMed] [Google Scholar]
- 81. Devred F., Douillard S., Briand C., Peyrot V. (2002) FEBS Lett. 523, 247–251 [DOI] [PubMed] [Google Scholar]
- 82. Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Watanabe A., Titani K., Ihara Y. (1995) Neurobiol. Aging 16, 365–371 [DOI] [PubMed] [Google Scholar]
- 83. Watanabe A., Hasegawa M., Suzuki M., Takio K., Morishima-Kawashima M., Titani K., Arai T., Kosik K. S., Ihara Y. (1993) J. Biol. Chem. 268, 25712–25717 [PubMed] [Google Scholar]
- 84. Lim J., Lu K. P. (2005) Biochim. Biophys. Acta 1739, 311–322 [DOI] [PubMed] [Google Scholar]
- 85. Delobel P., Flament S., Hamdane M., Jakes R., Rousseau A., Delacourte A., Vilain J. P., Goedert M., Buée L. (2002) J. Biol. Chem. 277, 9199–9205 [DOI] [PubMed] [Google Scholar]
- 86. Kar S., Fan J., Smith M. J., Goedert M., Amos L. A. (2003) EMBO J. 22, 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Choi M. C., Raviv U., Miller H. P., Gaylord M. R., Kiris E., Ventimiglia D., Needleman D. J., Kim M. W., Wilson L., Feinstein S. C., Safinya C. R. (2009) Biophys. J. 97, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Peck A., Sargin M. E., LaPointe N. E., Rose K., Manjunath B. S., Feinstein S. C., Wilson L. (2011) Cytoskeleton 68, 44–55 [DOI] [PubMed] [Google Scholar]
- 89. Rankin C. A., Sun Q., Gamblin T. C. (2005) Brain. Res. Mol. Brain Res. 138, 84–93 [DOI] [PubMed] [Google Scholar]
- 90. Seifert F., Ciszak E., Korotchkina L., Golbik R., Spinka M., Dominiak P., Sidhu S., Brauer J., Patel M. S., Tittmann K. (2007) Biochemistry 46, 6277–6287 [DOI] [PubMed] [Google Scholar]
- 91. Liang J., Shao S. H., Xu Z. X., Hennessy B., Ding Z., Larrea M., Kondo S., Dumont D. J., Gutterman J. U., Walker C. L., Slingerland J. M., Mills G. B. (2007) Nat. Cell. Biol. 9, 218–224 [DOI] [PubMed] [Google Scholar]
- 92. Haase C., Stieler J. T., Arendt T., Holzer M. (2004) J. Neurochem. 88, 1509–1520 [DOI] [PubMed] [Google Scholar]
- 93. Eidenmüller J., Fath T., Hellwig A., Reed J., Sontag E., Brandt R. (2000) Biochemistry 39, 13166–13175 [DOI] [PubMed] [Google Scholar]
- 94. Eidenmüller J., Fath T., Maas T., Pool M., Sontag E., Brandt R. (2001) Biochem. J. 357, 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Necula M., Kuret J. (2004) J. Biol. Chem. 279, 49694–49703 [DOI] [PubMed] [Google Scholar]
- 96. Yi J. J., Barnes A. P., Hand R., Polleux F., Ehlers M. D. (2010) Cell 142, 144–157 [DOI] [PMC free article] [PubMed] [Google Scholar]