

Abstract
Flavivirus NS5 protein encodes methyltransferase and RNA-dependent RNA polymerase (RdRp) activities. Structural analysis of flavivirus RdRp domains uncovered two conserved cavities (A and B). Both cavities are located in the thumb subdomains and represent potential targets for development of allosteric inhibitors. In this study, we used dengue virus as a model to analyze the function of the two RdRp cavities. Amino acids from both cavities were subjected to mutagenesis analysis in the context of genome-length RNA and recombinant NS5 protein; residues critical for viral replication were subjected to revertant analysis. For cavity A, we found that only one (Lys-756) of the seven selected amino acids is critical for viral replication. Alanine substitution of Lys-756 did not affect the RdRp activity, suggesting that this residue functions through a nonenzymatic mechanism. For cavity B, all four selected amino acids (Leu-328, Lys-330, Trp-859, and Ile-863) are critical for viral replication. Biochemical and revertant analyses showed that three of the four mutated residues (Leu-328, Trp-859, and Ile-863) function at the step of initiation of RNA synthesis, whereas the fourth residue (Lys-330) functions by interacting with the viral NS3 helicase domain. Collectively, our results have provided direct evidence for the hypothesis that cavity B, but not cavity A, from dengue virus NS5 polymerase could be a target for rational drug design.
Keywords: Drug Design, Enzyme Mechanisms, Flaviviruses, Protein-Protein Interactions, RNA Polymerase, Site-directed Mutagenesis, Viral Replication, Cavities for Allosteric Inhibitor, Dengue Virus, NS3/NS5 Interaction
Introduction
The genus Flavivirus from the family Flaviviridae contains more than 70 viruses, many of which are important human pathogens causing major public health threats worldwide. Dengue virus (DENV)2 is a mosquito-borne flavivirus responsible for 50–100 million human infections and ∼20,000 deaths each year (1). Besides DENV, West Nile virus (WNV), Japanese encephalitis virus, yellow fever virus, and tick-borne encephalitis virus also cause significant human diseases (1). No antiviral therapy is currently available for treatment of flavivirus infections. Therefore, development of antiviral therapy is urgently needed for flaviviruses.
The flavivirus genome is a single strand, plus-sense RNA of about 11 kb in length. The genomic RNA contains a 5′-untranslated region (UTR), a single open reading frame, and a 3′-UTR. The single open reading frame encodes a long polyprotein that is processed by viral and host proteases into 10 mature viral proteins (2). Three structural proteins (capsid, pre-membrane, and envelope) are primarily involved in virus particle formation, and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) are mainly responsible for viral replication (2). Among these, NS3 and NS5 possess enzymatic activities and have been targeted for antiviral development. NS3 functions as a protease (with NS2B as a cofactor), helicase, 5′-RNA triphosphatase, and nucleoside triphosphatase (3–5). NS5 is the largest and the most conserved viral protein. The N-terminal part of NS5 is a methyltransferase that methylates the N-7 and 2′-O positions of the viral RNA cap structure (6–9); the C-terminal part of NS5 has an RNA-dependent RNA polymerase (RdRp) activity (10, 11). Because the RdRp activity is essential for viral replication, and human host cells are devoid of RdRp, it is an attractive target for antiviral development.
Two approaches have been successfully used to develop RdRp inhibitors in clinical trials for hepatitis C virus (HCV) (12), a virus closely related to flavivirus. The first approach is based on a nucleoside analogue. Nucleoside analogues function as RNA chain terminators to block viral RNA synthesis. The second approach is based on an allosteric inhibitor. Allosteric inhibitors bind to pockets on polymerase to block conformational changes required for enzyme function. Both nucleoside analogues and allosteric inhibitors (targeting one of the four pockets in HCV RdRp) are currently in clinical trials (12). The success of HCV RdRp inhibitors suggests that the same approaches could be used to develop inhibitors of flavivirus RdRp.
Flavivirus RdRp initiates the RNA synthesis via a de novo mechanism, which differs from a primer-dependent mechanism used by other viruses, such as poliovirus and SARS-CoV (10, 13–15). Crystal structures of RdRp from DENV-3 and WNV have been reported (16, 17). Like all polymerases, the structure of flavivirus RdRps resembles a right hand with characteristic fingers, palm, and thumb subdomains. Malet et al. (18) recently proposed two cavities (named cavities A and B) that were conserved in the crystal structures of DENV and WNV RdRp. Both cavities are located in the thumb subdomain and could be potentially targeted for development of small molecule inhibitors. However, the biological relevance of the cavities has not been validated.
In this study, we performed a biochemical and genetic analysis of the two cavities conserved among flavivirus RdRp. Mutagenesis analysis was used to analyze the function of the cavities in viral replication. Residues critical for viral replication were subjected to revertant analysis. In addition, the critical residues were further examined for their roles in RdRp activity. Our results demonstrate that cavity B, but not cavity A, is essential for DENV replication. Mutations in cavity B could abrogate viral replication by decreasing the initiation of RdRp activity; alternatively, mutations in cavity B could suppress viral replication by interference with the interaction between the NS5 and NS3 helicase domains. The results have provided functional evidence that cavity B could be a rational target for drug discovery.
EXPERIMENTAL PROCEDURES
Cells and Viruses
Baby hamster kidney cells (BHK-21) and African green monkey kidney cells (Vero) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 at 37 °C. DENV-2 TSV01 strain (GenBankTM accession number AY037116) was first isolated from a patient in Australia in 1993 and then was passaged on C6/36 cells five times (19). The DENV-2 TSV01 from cell culture fluids was aliquoted and stored at −80 °C.
Plasmid Construction
To construct a full-length DENV-2 TSV01 cDNA clone, the virus stock was subjected to viral RNA extraction by RNeasy kit (Qiagen), and four cDNA fragments (A–D) covering the complete genome were amplified from viral RNA by RT-PCR using SuperScript III one-step RT-PCR kits (Invitrogen). The supplemental Fig. 1A depicts the cloning scheme to construct the cDNA of full-length genomic RNA of DENV-2. Fragment A contained an NheI restriction site, a T7 promoter sequence, and cDNA representing nucleotides 1–2,428 of the viral genome, followed by an extra XhoI restriction site. The RT-PCR product of fragment A was cloned into a predigested low copy number plasmid pACYC177 (New England Biolabs), resulting in pACYC TSV-A plasmid. Fragment B spanned from the EcoRI site (nucleoside position 2,340) to the XhoI site (nucleoside position 5,426). Fragment C spanned from the XhoI (nucleoside position 5,426) site to the AgeI site (nucleoside position 7,613). Fragment D spanned from the AgeI site (nucleoside position 7,613) to the 3′ end of the genome, followed by a hepatitis delta virus ribozyme sequence and an extra ClaI site. The RT-PCR products of fragments B–D were individually ligated into pCR2.1-TOPO with TOPO TA cloning kit according to the manufacturer's instructions (Invitrogen), resulting in constructs TA TSV-B, -C, and -D, respectively (supplemental Fig. 1A). Each subclone was validated by DNA sequencing before it was used for subsequent assembly. Next, fragment B was inserted into subclone pACYC TSV-A at the EcoRI and XhoI site, resulting in construct pACYC TSV-E. Fragment C was inserted into subclone TA TSV-D at the XhoI and AgeI sites, yielding plasmid TA TSV-F. Finally, the fragments C and D from plasmid TA TSV-F was inserted into construct pACYC TSV-E at the XhoI and ClaI sites, resulting in the full-length cDNA clone pACYC FLTSV. Escherichia coli HB101 (Promega) was used as the host for construction and propagation of cDNA clones. All restriction enzymes were purchased from New England Biolabs.
The genome-length cDNA clones with cavity mutations in NS5 RdRp were constructed by using the subclone TA TSV-F. The mutations were introduced into the plasmid TA TSV-F by the QuikChange II XL site-directed mutagenesis kit (Stratagene); the mutated DNA fragment was cut and inserted into the pACYC FLTSV at the NruI (located at nucleotide 7,737 of the viral genome) and ClaI sites. All constructs were verified by DNA sequencing.
In Vitro Transcription, RNA Transfection, Specific Infectivity Assay, and Immunofluorescence Assay (IFA)
The genome-length RNA of DENV-2 TSV01 was transcribed in vitro from the corresponding cDNA plasmids that were linearized with ClaI. A T7 mMESSAGE mMACHINE kit (Ambion) was used for RNA synthesis as described previously (6). The genome-length RNA was electroporated into BHK-21 cells as described previously (20). After transfection of the genome-length RNA, cells were cultured at 37 °C for the first 24 h and then transferred to 30 °C. We and others previously found that culturing the transfected cells at 30 °C increased the yield of DENV (21, 22). Supernatants were collected every 24 h until day 5 post-transfection. The culture medium containing viruses were aliquoted and stored at −80 °C. The specific infectivity assay was performed as described previously (6). The E protein expression in the transfected cells was monitored by IFA using mouse anti-E mAb 4G2 antibody as described previously (23).
Plaque Assay
Plaque assay was performed as described previously with minor modifications (24). Briefly, BHK-21 cells (ATCC) were seeded with a cell density of 1 × 105 per well in a 24-well plate (Nalge Nunc) 2 days in advance. A series of 1:10 dilutions were made by mixing 15 μl of virus sample with 135 μl of RPMI 1640 medium containing 2% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. One hundred microliters of undiluted and 10-fold dilutions of viral supernatant were seeded to individual wells of 24-well plates. The plates were incubated at 30 °C with 5% CO2 for 1 h with shaking every 15 min, and then the virus inocula were replaced with 0.6 ml of RPMI 1640 medium plus 0.8% methylcellulose (Aquacide II, Calbiochem) and 2% FBS. After 4 days of incubation at 37 °C with 5% CO2, the cells were fixed with 3.7% formalin and stained with 1% crystal violet. For some mutant viruses, the incubation period was more than 4 days to visualize the plaques.
Selection and Sequencing of Revertant Viruses
Five mutant viruses defective in viral replication were subjected to revertant analysis by continuously culturing on Vero cells for seven rounds. For each round of culturing, Vero cells (1 × 106 cells) in a T-25 flask were infected with 500 μl of culture supernatant derived from the previous passaging and cultured in DMEM with 2% FBS in 5% CO2 at 37 °C. The cytopathic effect was monitored under a microscope. The incubation period last 5 days unless an apparent cytopathic effect appeared earlier. Viral RNA extracted from culture supernatants of passages 2 and 7 was subjected to RT-PCR using SuperScript III one-step RT-PCR kits (Invitrogen). The entire NS5 region or the complete viral genome was sequenced.
RdRp Assay
Recombinant protein of full-length DENV-4 NS5 was prepared using an E. coli expression system and purified through affinity and gel filtration chromatography. Mutagenesis of the NS5 expression plasmid was performed using QuikChange II XL site-directed mutagenesis kit. The detailed protocols for mutagenesis, protein expression, and purification have been reported previously (25). Two types of RdRp assays were performed, de novo RdRp assay and elongation RdRp assays. The de novo RdRp reaction (25 μl) contained 50 mm HEPES, pH 8.0, 10 mm KCl, 5 mm MgCl2, 2 mm MnCl2, 10 mm DTT, 0.5 mm ATP, 0.5 mm UTP, 0.5 mm CTP, 2.5 μCi of [α-33P]GTP (10 μCi/μl, 3,000 Ci/mmol; PerkinElmer Life Science), 0.25 μg of NS5, 0.5 μg of RNA template. The RNA template was transcribed in vitro from a PCR product using a T7 MEGAscript kit (Applied Biosystems). The PCR template contained a T7 promoter followed by a cDNA fragment representing a DENV-2 subgenome with a deletion from nucleotide 169 to 10,263 (GenBankTM accession number AF038403.1). The de novo RdRp reactions were incubated at 23 °C for 30 min; the mixtures were passed through a MicroSpin G-25 column (GE Healthcare) to remove unincorporated NTPs; and the RNA elute was extracted with phenol/chloroform and precipitated with ethanol. The RNA pellet was dissolved in 10 μl of RNase-free water, mixed with an equal volume of denaturing Gel Loading Buffer II (Applied Biosystems), and loaded onto a 10% denaturing polyacrylamide gel with 7 m urea. A PhosphorImager was used to quantify the 33P-labeled RNA products. The elongation RdRp assay was performed using an RNA template and a 2′-[2-benzothiazoyl]-6′-hydroxybenzothiazole (BBT)-ATP as a substrate mimic (26). The BBT-ATP was chemically synthesized by attaching the BBT fluorophore group to the γ-phosphate of adenosine triphosphate. During the polymerization reaction, the adenosine monophosphate was incorporated into the RNA chain, releasing BBTppi as a by-product. BBTppi was then hydrolyzed by calf intestinal alkaline phosphatase, producing two inorganic phosphates (Pi) and BBT, which yields fluorescence. The details of assay conditions were recently reported (26).
Surface Plasmon Resonance (SPR) Assay
SPR measurements were performed on Biacore 3000 equipment (Biacore, GE Healthcare). All experiments were run with a constant flow rate of 30 μl/min HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3.4 mm EDTA, and 0.0005% P-20). Wild-type full-length DENV-4 NS5 and its mutants (single mutant, K330A and G840R; double mutant K330A/G840R) were amine-coupled onto the surface of a CM5 chip (Biacore, GE Healthcare). A 3-fold serially diluted DENV-4 full-length NS3 (0.1–24 μm) or its helicase domain (1–244 μm) was injected in duplicate across the chip for 1 min, and dissociation was monitored for 2.5 min. Recombinant full-length NS3 and its helicase domain proteins were prepared as reported previously (27). A short pulse (30 s) of 15 mm HCl was passed through the flow system to completely remove any residual bound protein to prepare for the next injection cycle. Raw sensorgrams obtained were aligned, solvent-corrected, and double-referenced using Scrubber 2 software (BioLogic Software, Campbell, Australia). Processed data were globally analyzed and fit to a simple 1:1 interaction model using numeric integration to yield the individual affinities.
RESULTS
Construction and Characterization of a Full-length cDNA Clone of DENV-2 Strain TSV01
The low copy number plasmid pACYC177 and E. coli HB101 were used to construct the cDNA clone of the DENV-2 TSV01 strain. The selection of pACYC177 as the cloning vector was based on our previous experience in WNV infectious clones (6). The supplemental Fig. 1A shows the overall scheme of the cloning strategy (see details under “Experimental Procedures”). The assembled full-length cDNA clone contained a T7 promoter at the 5′ end for in vitro transcription and a 67-bp hepatitis delta virus ribozyme sequence at the 3′ end for generating the authentic 3′ end viral sequence. The stability of the cDNA clone was tested by six rounds of plasmid extraction and re-transformation into E. coli HB101. DNA sequencing of the 6th round plasmid did not show any mutations throughout the cDNA insert (data not shown), demonstrating the stability of the clone.
BHK-21 cells transfected with the in vitro transcribed genome-length RNA exhibited an apparent cytopathic effect on day 5 post-transfection (p.t.). IFA of the transfected cells showed increasing numbers of cells expressing viral E protein from day 1 to 3; more than 80% of the cells were IFA-positive on day 3 p.t. (supplemental Fig. 1B). Plaque assay of the culture fluids showed an increase in viral titer from day 1 to 4, peaking at 1 × 106 to 1 × 107 pfu/ml on day 4 p.t. (supplemental Fig. 1C). Specific infectivity assay showed that transfection of BHK-21 cells with 1 μg of RNA generated about 1.2 × 105 pfu of virus. The results demonstrate that the RNA transcribed from the cDNA clone was highly infectious.
Structure and Sequence Analyses of Cavities A and B of DENV RdRp
Structure analysis uncovered two cavities conserved in DENV and WNV RdRps (18). The two cavities (A and B) are located on two opposite sides of the thumb subdomain of DENV and WNV RdRp. Fig. 1A shows the two cavities on the crystal structure of DENV-3 RdRp. On DENV-2 RdRp, cavity A is formed by amino acids Lys-756, Gln-760, Ser-763, Asn-777, Cys-780, Ser-785, Thr-806, Glu-807, Asp-808, Met-809, Leu-810, and Tyr-882, among which Lys-756, Thr-806, Glu-807, Asp-808, Met-809, and Leu-810 are completely conserved among various members of flaviviruses (Fig. 1B). Cavity B consists of amino acids Leu-327, Leu-328, Lys-330, Thr-858, Trp-859, Asn-862, Ile-863, and Ala-866, among which Leu-328, Trp-859, and Ile-863 are conserved among flaviviruses (Fig. 1B).
FIGURE 1.

Structure and sequence analyses of cavities A and B from DENV RdRp. A, cavities A and B in DENV-3 RdRp structure. The crystal structure of DENV-3 RdRp (Protein Data Bank entry 2J7U) was used to illustrate cavity A (left panel) and cavity B (right panel). Amino acids constituting the cavities are labeled in yellow or other colors. Residues selected for mutagenesis analysis are colored as follows: for cavity A, Lys-756 (red), Gln-760 (green), Cys-780 (blue), Thr-806 (magenta), Glu-807 (cyan), Asp-808 (orange), and Met-809 (tint); for cavity B, Leu-328 (red), Lys-330 (green), Trp-859 (blue), and Ile-863 (pink). The images of RdRp were produced using PyMOL. B, amino acid sequence alignment of partial RdRp region from the four serotypes of DENV and other flaviviruses. The RdRp amino acid sequences of DENV-1, DENV-2, DENV-3, DENV-4, WNV, yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV) are derived from GenBankTM accession numbers U88535, AY037116, M93130, AY947539, AF404756, X03700, and AF069066, respectively. Identical amino acids among all RdRps are shaded. The numbering of amino acid sequence is based on DENV-2. The residues involved in the formation of cavity A and B are indicated by ● and ▾ above the sequence, respectively. The mutated residues in this study are indicated by * below the sequence.
Role of Cavity A in Viral Replication
To determine the biological function of cavity A, we selected seven amino acids for mutagenesis analysis. As shown in Fig. 1A (left panel), Lys-756 (red), Gln-760 (green), Cys-780 (blue), and Met-809 (tint) are located deep inside the cavity. Thr-806 (magenta), Glu-807 (cyan), and Asp-808 (orange) form the left side wall of the cavity. Among the seven selected residues, five amino acids (Lys-756, Thr-806, Glu-807, Asp-808, and Met-809) are completely conserved among flaviviruses; two residues (Gln-760 and Cys-780) have variations in yellow fever virus and/or tick-borne encephalitis virus RdRp (Fig. 1B). It should be noted that residues Thr-806, Glu-807, Asp-808, and Met-809 also form the C-terminal part of the priming loop (16).
Each of the selected residues was mutated to Ala in the infectious clone of DENV-2 TSV01. Equal amounts of wild-type (WT) and mutant genome-length RNAs were electroporated into BHK-21 cells. Viral protein synthesis, plaque morphology, and virus production were compared. For viral protein synthesis, IFAs (detecting viral E protein) showed that similar percentages of IFA-positive cells were detected for the WT and mutants T806A, E807A, and D808A RNAs at 72 h p.t.; mutants Q760A, C780A, and M809A showed a slight decrease in the percentage of IFA-positive cells, and mutant K756A remained less than 1% IFA-positive cells at 72 h p.t. (Fig. 2A). For different RNA mutants, the relative IFA-positive cells were similar when the transfected cells were analyzed at 24 and 48 h p.t. (data not shown). No infectious virus was recovered from the K756A RNA-transfected cells, whereas other mutant RNAs yielded similar amounts of infectious viruses with no significant difference in plaque morphology (Fig. 2, A and B). These results suggest that only K756A among the selected mutations in cavity A is essential for viral replication.
FIGURE 2.

Mutagenesis of cavities A and B using an infectious cDNA clone of DENV-2. A and C, analysis of viral E protein synthesis and plaque morphology for cavity A and B mutant viruses, respectively. BHK-21 cells were transfected with WT and mutant genome-length RNAs (10 μg), and analyzed for viral E protein expression by IFA at 72 h post-transfection (upper panel). Plaque morphologies of WT and mutant viruses were shown (lower panel). N.D., not detectable. B and D, production of cavity A and B mutant viruses after transfection, respectively. Viruses from culture supernatants were collected every 24 h. Viral titers were determined by plaque assay on BHK-21 cells. Error bars indicate the standard deviations from two independent experiments; dashed line, limit of sensitivity of the plaque assay.
Role of Cavity B in Viral Replication
Four residues were selected for mutagenesis analysis of cavity B. As depicted in Fig. 1A (right panel), Leu-328 (red) and Ile-863 (pink) are located at the bottom of the cavity; Trp-859 (blue) is at the side of cavity wall; and Lys-330 (green) is on the surface of the cavity. The four residues were individually mutated to Ala in the genome-length RNA of DENV-2 TSV01. Unlike cavity A mutants, genome-length RNAs containing the mutations in cavity B showed a significant decrease in viral protein synthesis in the transfected BHK-21 cells. At 72 h p.t., ∼5% of the cells transfected with I863A RNA expressed viral protein; less than 1% of IFA-positive cells were detected for L328A and K330A; no IFA-positive cells was detected in cells transfected with W859A RNA (Fig. 2C). In agreement with the IFA results, only the I863A RNA-transfected cells yielded viruses (Fig. 2D) with tiny plaques (Fig. 2C); no plaque was detected from the L328A, K330A, and W859A RNA-transfected cells, up to 120 h p.t. (Fig. 2, C and D). Overall, the results indicate that cavity B plays a critical role in viral replication.
Effects of Mutations on RdRp Activity
To determine whether the cavities play a direct role in RdRp activity, we prepared a panel of full-length DENV-4 NS5 proteins and analyzed the mutational effects on polymerase activity. The reason to choose DENV-4 NS5 was that this protein was more stable than DENV-2 NS5 (data not shown). All five replication-defective NS5 mutations (L328A, K330A, K756A, W859A, and I863A) were engineered into recombinant full-length NS5 proteins. The proteins were analyzed in two types of RdRp assays. First, a de novo RdRp assay was performed using a viral subgenomic RNA as template. As shown in Fig. 3A, mutants L328A, W859A, and I863A exhibited only 15, 10, and 7% of the WT activity, respectively, whereas mutants K330A and K756A retained 80 and 82% of the WT activity, respectively.
FIGURE 3.
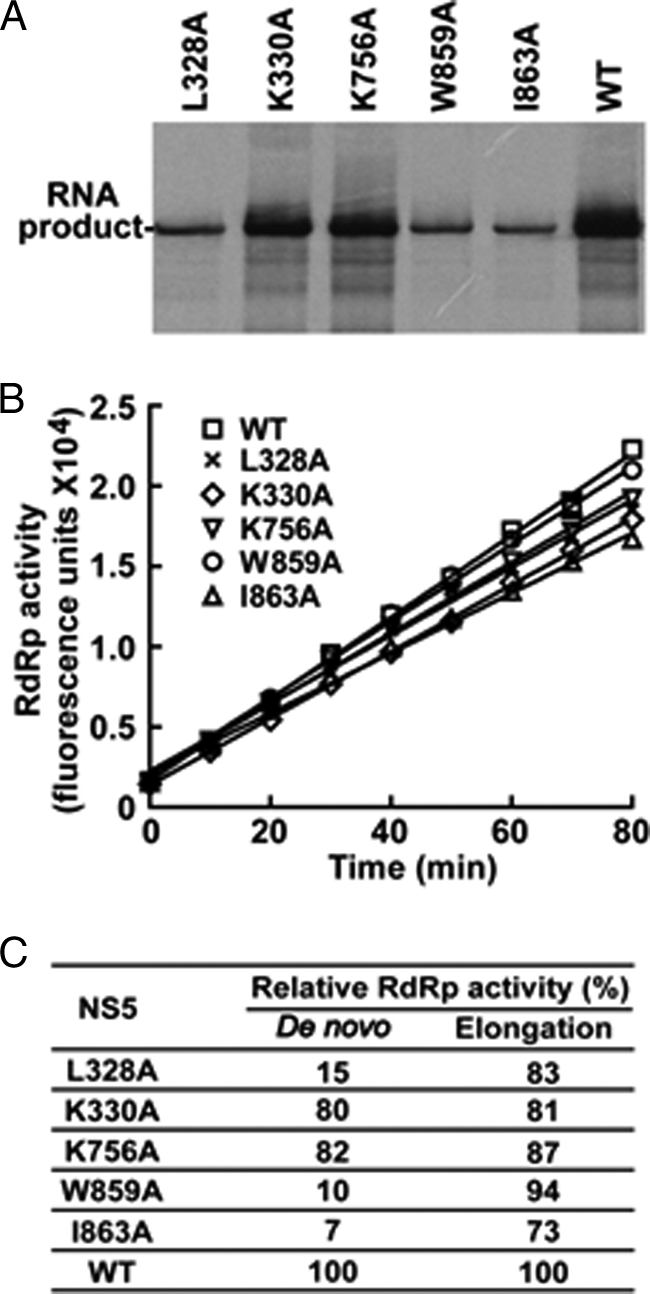
Effects of cavity mutations on de novo and elongation RdRp activities. A, de novo RdRp activities of mutant NS5 proteins. B, elongation RdRp activities. The elongation assay measured the fluorescence generated by using a substrate mimic BBT-ATP and RNA template, as reported previously (26). Results are the average of two independent experiments performed in duplicate. C, summary of de novo and elongation RdRp activities. The relative de novo RdRp activities were calculated by comparing the products generated using the mutant NS5 proteins with that produced using the WT protein (set as 100%). The relative RNA elongation activities were calculated by comparing the slope from the mutant NS5 proteins with that of the WT protein (set as 100%).
Because the de novo RdRp assay includes steps of initiation and elongation, we next analyzed the mutant NS5 in an elongation RdRp assay (26). In contrast to the de novo synthesis results, all mutants retained greater than 70% of the WT RNA elongation activity (Fig. 3B). Fig. 3C summarizes the effects of mutations on de novo and elongation RdRp activities. The comparable elongation activities between the WT and mutant NS5 suggest the following: (i) the engineered mutations do not change the global conformation of the NS5 protein; and (ii) the observed defect in de novo RNA synthesis for mutants L328A, W859A, and I863A is not due to variations in protein preparations. These results allowed us to conclude that L328A, W859A, and I863A reduce viral replication on or before de novo initiation, whereas K330A and K756A decrease RNA replication through a nonenzymatic mechanism.
Revertant Analysis of Cavity A
Among the mutated amino acids from cavities A and B, five of them resulted in replication-defective viruses (L328A, K330A, K756A, W859A, and I863A; Fig. 2). Only K756A belongs to cavity A; L328A, K330A, W859A, and I863A belong to cavity B (Fig. 1A). We performed revertant analysis by continuous passaging of culture supernatants on Vero cells for seven rounds (P1 to P7). The culture fluids from passages 2 (P2) and 7 (P7) were subjected to plaque assay and sequencing analysis. For cavity A, both the P2 and P7 supernatants of the K756A mutant produced plaque morphology similar to the WT; sequencing of the viruses revealed a true reversion to the WT sequence (data not shown). Together with the result that the K756A RNA-transfected cells (P0) did not yield any infectious virus (Fig. 2, A and B), we conclude that residue Lys-756 in cavity A is essential for viral replication.
Revertant Analysis of Cavity B
For cavity B, both P2 and P7 culture fluids derived from the L328A, K330A, and I863A RNAs produced plaques. Sequencing of the recovered viruses showed various reversions. For L328A mutant, the P2 virus had a mixed population of Val (GTA) and Leu (CTA), whereas the P7 virus became a single population of Leu (CTA) (Fig. 4A). These results suggest that the L328A mutant underwent a stepwise reversion of Ala (GCA) → Val (GTA) → Leu (CTA) (mutant nucleotides are underlined), each step with a single nucleotide change.
FIGURE 4.

Revertant analysis of cavity B mutant viruses, L328A and K330A. The mutant viruses were continuously cultured on Vero cells for seven rounds (P1 to P7). The plaque morphology and sequencing chromatogram for P2 and P7 are presented for the L328A (A) and K330A (B) viruses. The engineered mutant nucleotides are underlined. C, validation of the K330A adaptive mutation. Revertant analysis of the P2 K330A virus revealed a second site mutation G840R in the RdRp domain (data not shown). For validation of G840R in restoring the replication of K330A mutation, two mutant genome-length RNAs (G840R and K330A/G840R) were transfected into BHK-21 cells. The transfected cells were monitored for viral E protein expression at 24, 48, and 72 h post-transfection. Plaque morphologies were shown for recovered viruses.
For the K330A mutant, transfection of this mutant RNA into BHK-21 cells did not yield any infectious virus (Fig. 2, C and D). However, the culture fluids from P2 and P7 yielded infectious viruses with plaques smaller than the WT virus (Fig. 4B). The sequencing result showed that both the P2 and P7 viruses retained the engineered mutation (Fig. 4B); complete genome-length sequencing of the P2 virus revealed a second site nucleotide mutation G→A at position 10,087, which resulted in a Gly → Arg amino acid change at position 840 of NS5. To demonstrate whether the G840R substitution rescued the replication defect of K330A RNA, we prepared two mutants genome-length RNAs containing the G840R alone or the K330A/G840R double mutations. Transfection of BHK-21 cells with equal amounts of RNA showed that the G840R RNA generated fewer IFA-positive cells than the WT RNA did, but the double mutant K330A/G840R RNA produced more IFA-positive cells than the G840R RNA did (Fig. 4C). At 72 h p.t., equivalent amounts of IFA-positive cells were observed between the WT and the K330A/G840R double mutant RNA-transfected cells. The plaque size of G840R virus was smaller than K330A/G840R viruses, and the plaques of both viruses are fainter and slightly smaller than that of the WT virus (Fig. 4C). These results demonstrate that the G840R adaptation is responsible for the restoration of the replication of mutant K330A (see further mechanistic analysis below).
For the I863A mutant, the P0 culture fluids showed tiny plaques (Fig. 2C). The P2 virus showed plaques of different sizes, and the P7 virus showed only large plaques (Fig. 5A). In agreement with the plaque morphology, sequencing analysis showed that the P2 virus had a mixed population of Val (GTC) and Ala (GCC), whereas the P7 virus became a single population of Val (GTC) (Fig. 5A). These results suggest that the I863A mutant underwent a reversion of Ala (GCC) → Val (GTC) through a single nucleotide change (mutant nucleotides are underlined). Engineering the I863V substitution into genome-length RNA restored viral replication. After transfection into BHK-21 cells, the I863V RNA and WT RNA generated equivalent amounts of IFA-positive cells from 24 to 72 h p.t. (compare Figs. 5B with 4C). The I863V RNA generated an infectious virus with plaque morphology similar to that of the WT virus (Fig. 5B). In addition, comparable viral titers were detected for WT and I863V mutant virus at various time points post-transfection; sequencing of the recovered virus showed that the engineered I863V mutation was retained without any other mutations (data not shown). Next, we compared the RdRp activity of recombinant NS5 containing the I863A or I863V mutation. In the de novo synthesis assay, the I863V and I863A NS5 exhibited 100 and 5% of the WT RdRp activity, respectively (Fig. 5C). In the elongation assay, the I863V and I863A NS5 exhibited 101 and 73% of the WT RdRp activity, respectively (Fig. 5D). These results strongly indicate that the reversion of I863A → I863V improves viral replication through enhancement of RdRp activity, especially at the step of initiation of RNA synthesis.
FIGURE 5.

Revertant analysis of cavity B mutant virus, I863A. A, plaque morphology and sequencing chromatogram from the I863A P2 and P7 viruses. The engineered mutant nucleotides are underlined. B, validation of the I863V revertant mutation. The I863V genome-length RNA was electroporated into BHK-21 cells. The transfected cells were monitored for E protein expression at 24, 48, and 72 h post-transfection. Plaque morphology of the I863V virus (P0) is shown. C, comparison of de novo RdRp activities among WT, I863V, and I863A NS5 proteins. The relative de novo RdRp activities (WT set as 100%) are indicated below the denaturing gel. D, comparison of elongation RdRp activities among WT, I863V, and I863A NS5 proteins. The relative elongation activities were shown in parentheses (WT set as 100%). Results are the average of two independent experiments performed in duplicate.
For the W859A mutant, no infectious virus was recovered (Fig. 2, C and D), even after seven rounds of passaging (data not shown), indicating that Trp-859 is essential for viral replication. To examine whether amino acids other than Trp at position 859 could support viral replication, we substituted Trp-859 with Phe, Lys, Asp, His, or Leu in the context of genome-length RNA. Equal amounts of WT and mutant RNAs were transfected into BHK-21 cells. Only the cells transfected with W859F RNA showed a few number of IFA-positive cells (less than 1%) at 72 h p.t. (Fig. 6). None of the Trp-859 variant RNAs produced any infectious viruses, as judged by plaque assays (data not shown). These results demonstrate that Trp-859 in cavity B is essential for viral replication, even an aromatic amino acid substitution (W859F) could not sufficiently support viral replication.
FIGURE 6.

Site-directed mutagenesis of Trp-859 in cavity B. BHK-21 cells were transfected with WT and mutant genome-length RNAs (10 μg) and analyzed for viral E protein synthesis by IFA at 72 h post-transfection. The engineered mutant nucleotides are underlined.
K330A Mutation Affects Viral Replication by Decreasing NS3/NS5 Interaction
K330A mutation abolished viral replication (Fig. 2, C and D) but did not affect RdRp activity (Fig. 3C), suggesting that the mutation affects viral replication through a nonenzymatic mechanism. Revertant analysis showed that a second site mutation (G840R) in the RdRp domain could rescue the replication defect of the K330A mutation (Fig. 4C). These results prompted us to test the hypothesis that K330A mutation affects replication through decreasing the interaction NS5 with other replication component(s). Because the NS3/NS5 interaction has been reported for DENV and other flaviviruses (28, 29), we measured the binding affinity between the recombinant full-length NS3 and NS5 proteins using an SPR assay. As shown in Fig. 7C, the K330A mutation increased the KD value of the NS3/NS5 interaction by 12.4-fold; the compensatory mutation G840R alone also increased the KD by 8.1-fold; remarkably, the double mutant K330A/G840R restored the KD value to 1.8-fold of the WT KD. The experimental curve of SPR fits closely to a 1:1 binding model of NS3 and NS5. The slight discrepancy between the experimental and theoretical curves (particularly in the dissociation phase) is most likely due to NS3 aggregation at high concentrations. Such aggregation was observed when a high concentration of NS3 was fractionated through size-exclusion chromatography (data not shown).
FIGURE 7.

Compensatory mutation G840R rescued the replication defect of K330A through restoration of the NS3/NS5 interaction. A and B, SPR measurements of the interaction between the wild-type and mutants of NS5 and the full-length NS3 and its helicase domain, respectively. 3-Fold serially diluted DENV-4 full-length NS3 (0.1–24 μm) or its helicase domain (1–244 μm) were injected across immobilized DENV-4 NS5 (and its mutants) in duplicate (only one of the duplicates is shown in figure for clarity) for 1 min and allowed to dissociate in running buffer for another 2.5 min. The aligned and double-referenced sensorgrams were globally fit to a simple 1:1 interaction model (thin red lines). To verify the accuracy of our rate parameters, we also fit the same data to a bivalent model (BIAEvaluation version 4.1, Biacore, GE Healthcare) and the results tallied with the 1:1 model. Hence, we chose to present the 1:1 model fit to avoid complication and to provide a better comparison between the full-length NS3 data and the NS3 helicase domain data. C, summary of the resultant affinities for interaction between full-length DENV-4 NS3 and its helicase domain with wild-type NS5, its single mutant (K330A and G840R), and its double mutant (K330A/G840R). The resultant affinity for interaction is reflected by KD (equilibrium dissociation constants), which was calculated using the ratio of kd/ka from the SPR assay. RU, response units.
Because the NS3/NS5 interaction was previously mapped to the helicase domain of NS3 (30, 31), we performed SPR assays using recombinant helicase domain (without protease domain) and various NS5 proteins. The results showed that the KD value of helicase/NS5 was 4.4-fold higher than that of the full-length NS3/NS5 (Fig. 7C), indicating that the protease domain contributes to the NS3/NS5 interaction. Similar to the full-length NS3 results, mutation K330A increased the KD value of helicase/NS5 interaction by 10.2-fold, and the double mutation K330A/G840R restored the KD value to 1.4-fold of the WT KD. However, the model fitting for the helicase domain is not as good as the full-length NS3 (Fig. 7, A and B) due to the fact that the helicase domain is more prone to aggregation than the full-length NS3 at a high concentration (data not shown). Nevertheless, these results indicate that the compensatory mutation G840R rescued the replication defect of K330A by restoration of the NS3/NS5 interaction.
DISCUSSION
Flavivirus RdRp is an attractive target for antiviral development. Both nucleoside analogues and non-nucleoside inhibitors of DENV RdRp have recently been reported (25, 32–34). The crystal structures of WNV and DENV RdRps revealed two conserved cavities that could be used for development of allosteric inhibitors (18). Using both genetic and biochemical approaches, we probed the biological relevance of the two cavities. Fig. 8A summarizes the results about all critical residues identified in this study. For cavity A, we found that among the seven mutated amino acids, only K756A mutation was lethal for DENV replication; the other six mutated residues did not significantly affect viral replication (Fig. 2, A and B). It is worth noting that four of the mutated residues (Thr-806, Glu-807, Asp-808, and Met-809) reside in the C-terminal region of the priming loop (colored in yellow, Fig. 8B). For the K756A mutation, only true reversion could restore viral replication. In terms of mode-of-action, we found that the K756A mutation affect neither de novo RNA synthesis nor RNA elongation activity (Fig. 3). The latter result indicates that the K756A mutation reduces viral replication through a nonenzymatic mechanism and possibly by affecting interaction with other replicase component(s). However, structure analysis showed that residue Lys-756 is located deep inside cavity A; the residue is behind the C terminus of the priming loop (Fig. 8B). It is conceivable that during RNA synthesis, a local conformational change exposes residue Lys-756 to the protein surface, allowing Lys-756 to interact with other replicase component(s). Nevertheless, our results argue that cavity A is not an ideal target for designing small molecular inhibitors.
FIGURE 8.

A, summary of mutagenesis analysis of cavities A and B. Based on the IFA results, the replication levels of WT and mutants RNAs are categorized into strong (++++), medium (++), weak (+/−), and nonreplicative (−). B, crystal structure of DENV RdRp showing residues Thr-806, Glu-807, Asp-808, Met-809 (all in yellow), and Lys-756 (in red) from cavity A. The priming loop is shown in yellow. The GDD active site of RdRp is shown in pink. C, crystal structure of DENV RdRp showing residues Leu-328, Lys-330, Trp-859, and Ile-863 (all in red) from cavity B. Amino acid Gly-840, a compensatory mutation for K330A, is labeled in blue. The illustrations for B and C were prepared using PyMOL.
In contrast to cavity A, all four mutated residues in cavity B significantly reduced viral replication (Fig. 2, C and D). None of the mutations decreased RNA elongation activity by >27%; however, three mutations (L328A, W859A, and I863A) reduced de novo RNA synthesis by ≥85% (Fig. 3). Correlated with the de novo RNA synthesis results, revertant analysis showed the following: (i) only true reversion could restore the replication of L328A RNA (Fig. 4A); (ii) no revertant virus could be selected for W859A RNA; (iii) only amino acid Val, similar to the wild-type Ile-863, was selected to restore the replication of I863A RNA (Fig. 5). These results indicate that L328A, W859A, and I863A mutations reduced viral replication by decreasing the initiation of RNA synthesis. Although these residues are away from the active site and RNA tunnel (Fig. 8C), they may participate in the formation of the RNA template-RdRp-NTP complex during the initiation of RNA synthesis. In support of this notion, the Δ1 loop on the surface of thumb subdomain of HCV RdRp was previously shown to regulate the conformations needed for de novo initiation and for elongation of RNA synthesis; mutations that affect the Δ1 loop conformation selectively reduced de novo initiation (35). In this study, the three mutations that selectively decreased RNA synthesis initiation (L328A, W859A, and I863A) are also located at the same interface between the Δ1 loop and the rest of thumb subdomain of DENV RdRp.
Unlike the three residues in cavity B discussed above, mutation K330A abolished viral replication but retained 80 and 81% of the wild-type activities of de novo and elongation RNA synthesis, respectively (Fig. 8A). These results indicate that the K330A mutation abrogated RNA replication through a nonenzymatic mechanism. In support of this hypothesis, our SPR results showed that the K330A mutation reduced the NS3/NS5 interaction. Furthermore, revertant analysis showed that a second site mutation G840R in the RdRp domain could restore the replication of the K330A mutant. In agreement with the genetic data, our SPR results showed that, although the compensatory mutation G840R alone is also defective in NS3/NS5 interaction, the double mutant K330A/G840R restored the NS3/NS5 interaction. On the crystal structure, Gly-840 (blue in Fig. 8C) is 24.1 Å away from Lys-330 but is located on the surface of the thumb subdomain. The switch from the wild-type Lys-330 + Gly-840 to the revertant K330A/G840R maintained one positively charged residue, indicating that the positive charge on the surface of NS5 is important for the NS3/NS5 interaction. In support of our results, Johansson et al. (30) previously suggested that a region between residues 320 and 368 of DENV NS5 interacts with NS3 helicase domain.
In summary, our results have demonstrated that cavity B is essential for viral replication and could be targeted for development of the allosteric inhibitor of flavivirus NS5 polymerase. Pharmacological blockage of cavity B could potentially lead to suppression of initiation of viral RNA synthesis and/or inhibition of NS3/NS5 interaction. The SPR assay described in this study is amendable for high throughput screening to identify inhibitors of NS3/NS5 interaction.
Acknowledgments
We thank Ying Tan (Duke-National University of Singapore) and Sui Sum Yeong (Nanyang Technological University) for technical support during the course of the study. We also thank Shamala Devi for providing DENV-4 virus that was used for cloning the full-length ns5 gene.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- DENV
- dengue virus
- RdRp
- RNA-dependent RNA polymerase
- WNV
- West Nile virus
- HCV
- hepatitis C virus
- IFA
- immunofluorescence assay
- SPR
- surface plasmon resonance
- p.t.
- post-transfection
- BBT
- 2′-[2-benzothiazoyl]-6′-hydroxybenzothiazole.
REFERENCES
- 1. Gubler D., Kuno G., Markoff L. (2007) in Fields Virology (Knipe D. M., Howley P. M. eds) 5th Ed. pp. 1153–1253, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 2. Lindenbach B. D., Thiel H. J., Rice C. M. (2007)) in Fields Virology (Knipe D. M., Howley P. M. eds) 5th Ed., pp. 1101–1151, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 3. Falgout B., Miller R. H., Lai C. J. (1993) J. Virol. 67, 2034–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wengler G., Wengler G. (1991) Virology 184, 707–715 [DOI] [PubMed] [Google Scholar]
- 5. Wengler G., Wengler G. (1993) Virology 197, 265–273 [DOI] [PubMed] [Google Scholar]
- 6. Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K. A., Shi P. Y., Li H. (2007) J. Virol. 81, 3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ray D., Shah A., Tilgner M., Guo Y., Zhao Y., Dong H., Deas T. S., Zhou Y., Li H., Shi P. Y. (2006) J. Virol. 80, 8362–8370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dong H., Ren S., Zhang B., Zhou Y., Puig-Basagoiti F., Li H., Shi P. Y. (2008) J. Virol. 82, 4295–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Egloff M. P., Benarroch D., Selisko B., Romette J. L., Canard B. (2002) EMBO J. 21, 2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ackermann M., Padmanabhan R. (2001) J. Biol. Chem. 276, 39926–39937 [DOI] [PubMed] [Google Scholar]
- 11. Tan B. H., Fu J., Sugrue R. J., Yap E. H., Chan Y. C., Tan Y. H. (1996) Virology 216, 317–325 [DOI] [PubMed] [Google Scholar]
- 12. Deore R. R., Chern J. W. (2010) Curr. Med. Chem. 17, 3806–3826 [DOI] [PubMed] [Google Scholar]
- 13. Paul A. V., van Boom J. H., Filippov D., Wimmer E. (1998) Nature 393, 280–284 [DOI] [PubMed] [Google Scholar]
- 14. te Velthuis A. J., Arnold J. J., Cameron C. E., van den Worm S. H., Snijder E. J. (2010) Nucleic Acids Res. 38, 203–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lescar J., Canard B. (2009) Curr. Opin. Struct. Biol. 19, 759–767 [DOI] [PubMed] [Google Scholar]
- 16. Yap T. L., Xu T., Chen Y. L., Malet H., Egloff M. P., Canard B., Vasudevan S. G., Lescar J. (2007) J. Virol. 81, 4753–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malet H., Egloff M. P., Selisko B., Butcher R. E., Wright P. J., Roberts M., Gruez A., Sulzenbacher G., Vonrhein C., Bricogne G., Mackenzie J. M., Khromykh A. A., Davidson A. D., Canard B. (2007) J. Biol. Chem. 282, 10678–10689 [DOI] [PubMed] [Google Scholar]
- 18. Malet H., Massé N., Selisko B., Romette J. L., Alvarez K., Guillemot J. C., Tolou H., Yap T. L., Vasudevan S., Lescar J., Canard B. (2008) Antiviral Res. 80, 23–35 [DOI] [PubMed] [Google Scholar]
- 19. Schul W., Liu W., Xu H. Y., Flamand M., Vasudevan S. G. (2007) J. Infect. Dis. 195, 665–674 [DOI] [PubMed] [Google Scholar]
- 20. Shi P. Y., Tilgner M., Lo M. K., Kent K. A., Bernard K. A. (2002) J. Virol. 76, 5847–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qing M., Liu W., Yuan Z., Gu F., Shi P. Y. (2010) Antiviral Res. 86, 163–171 [DOI] [PubMed] [Google Scholar]
- 22. Ansarah-Sobrinho C., Nelson S., Jost C. A., Whitehead S. S., Pierson T. C. (2008) Virology 381, 67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsieh S. C., Zou G., Tsai W. Y., Qing M., Chang G. J., Shi P. Y., Wang W. K. (2011) Virology 410, 170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poh M. K., Yip A., Zhang S., Priestle J. P., Ma N. L., Smit J. M., Wilschut J., Shi P. Y., Wenk M. R., Schul W. (2009) Antiviral Res. 84, 260–266 [DOI] [PubMed] [Google Scholar]
- 25. Niyomrattanakit P., Chen Y. L., Dong H., Yin Z., Qing M., Glickman J. F., Lin K., Mueller D., Voshol H., Lim J. Y., Nilar S., Keller T. H., Shi P. Y. (2010) J. Virol. 84, 5678–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niyomrattanakit P., Abas S. N., Lim C. C., Beer D., Shi P. Y., Chen Y. L. (2011) J. Biomol. Screen. 16, 201–210 [DOI] [PubMed] [Google Scholar]
- 27. Luo D., Xu T., Hunke C., Grüber G., Vasudevan S. G., Lescar J. (2008) J. Virol. 82, 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kapoor M., Zhang L., Ramachandra M., Kusukawa J., Ebner K. E., Padmanabhan R. (1995) J. Biol. Chem. 270, 19100–19106 [DOI] [PubMed] [Google Scholar]
- 29. Chen C. J., Kuo M. D., Chien L. J., Hsu S. L., Wang Y. M., Lin J. H. (1997) J. Virol. 71, 3466–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johansson M., Brooks A. J., Jans D. A., Vasudevan S. G. (2001) J. Gen. Virol. 82, 735–745 [DOI] [PubMed] [Google Scholar]
- 31. Wu J., Bera A. K., Kuhn R. J., Smith J. L. (2005) J. Virol. 79, 10268–10277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y. L., Yin Z., Duraiswamy J., Schul W., Lim C. C., Liu B., Xu H. Y., Qing M., Yip A., Wang G., Chan W. L., Tan H. P., Lo M., Liung S., Kondreddi R. R., Rao R., Gu H., He H., Keller T. H., Shi P. Y. (2010) Antimicrob. Agents Chemother. 54, 2932–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y. L., Yin Z., Lakshminarayana S. B., Qing M., Schul W., Duraiswamy J., Kondreddi R. R., Goh A., Xu H. Y., Yip A., Liu B., Weaver M., Dartois V., Keller T. H., Shi P. Y. (2010) Antimicrob. Agents Chemother. 54, 3255–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yin Z., Chen Y. L., Schul W., Wang Q. Y., Gu F., Duraiswamy J., Kondreddi R. R., Niyomrattanakit P., Lakshminarayana S. B., Goh A., Xu H. Y., Liu W., Liu B., Lim J. Y., Ng C. Y., Qing M., Lim C. C., Yip A., Wang G., Chan W. L., Tan H. P., Lin K., Zhang B., Zou G., Bernard K. A., Garrett C., Beltz K., Dong M., Weaver M., He H., Pichota A., Dartois V., Keller T. H., Shi P. Y. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20435–20439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chinnaswamy S., Yarbrough I., Palaninathan S., Kumar C. T., Vijayaraghavan V., Demeler B., Lemon S. M., Sacchettini J. C., Kao C. C. (2008) J. Biol. Chem. 283, 20535–20546 [DOI] [PMC free article] [PubMed] [Google Scholar]