

Abstract
The GES-2 β-lactamase is a class A carbapenemase, the emergence of which in clinically important bacterial pathogens is a disconcerting development as the enzyme confers resistance to carbapenem antibiotics. Tazobactam is a clinically used inhibitor of class A β-lactamases, which inhibits the GES-2 enzyme effectively, restoring susceptibility to β-lactam antibiotics. We have investigated the details of the mechanism of inhibition of the GES-2 enzyme by tazobactam. By the use of UV spectrometry, mass spectroscopy, and x-ray crystallography, we have documented and identified the involvement of a total of seven distinct GES-2·tazobactam complexes and one product of the hydrolysis of tazobactam that contribute to the inhibition profile. The x-ray structures for the GES-2 enzyme are for both the native (1.45 Å) and the inhibited complex with tazobactam (1.65 Å). This is the first such structure of a carbapenemase in complex with a clinically important β-lactam inhibitor, shedding light on the structural implications for the inhibition process.
Keywords: Antibiotics, Drug Resistance, Enzyme Inhibitors, Enzyme Kinetics, Enzyme Mechanisms, Mass Spectrometry (MS), UV Spectroscopy, X-ray Crystallography
Introduction
β-Lactam antibiotics represent more than 65% of the worlds antibiotic market and encompass a range of natural, semisynthetic, and synthetic compounds that include penicillins, cephalosporins, monobactams, carbapenems, and penems (1). They are extensively used in the treatment of a wide variety of infections caused by Gram-negative and Gram-positive bacteria (2). β-Lactams kill bacteria by selectively inhibiting enzymes involved in the synthesis of the microbial cell wall, specifically its peptidoglycan component, a uniquely bacterial structure (1). These enzymes, collectively known as penicillin-binding proteins, because of their ability to bind covalently to penicillins (3, 4), carry out cross-linking of the bacterial cell wall through polypeptide bridges via a transpeptidation reaction. Covalent and irreversible modification of penicillin-binding proteins, whose biosynthetic function is vitally important for bacteria, defines the mechanism of action of all β-lactam antibiotics.
The production of β-lactamases, enzymes capable of efficiently inactivating β-lactams, is the major mode of resistance to these antibiotics in Gram-negative pathogens (5). Four molecular classes of β-lactamases, A, B, C, and D, are recognized, based mainly on similarities in their amino acid sequences (5, 6). Class A, C, and D enzymes utilize an active-site serine residue to catalytically inactivate β-lactam drugs, whereas the class B enzymes include various zinc-dependent β-lactamases operating through a different mechanism. Within the β-lactamase family, class A enzymes are the most abundant. Initially recognized as relatively narrow spectrum enzymes capable of hydrolyzing penicillins and some early cephalosporins, class A β-lactamases are now represented by hundreds of members characterized by a wide β-lactam resistance spectrum (7, 8). To circumvent bacterial resistance produced by the broad spectrum class A β-lactamases, three β-lactam inhibitors, tazobactam, clavulanic acid, and sulbactam (Fig. 1) have been introduced into clinical use. These inhibitors are used in combination with β-lactam antibiotics to improve the efficacy of the drugs, and examples include Augmentin (GlaxoSmithKline), a combination of amoxicillin and clavulanic acid, and Zosyn (Wyeth), a combination of piperacillin and tazobactam. Extensive use of these inhibitors has resulted in selection of numerous inhibitor-resistant variants in the TEM, CTX, and SHV families of class A β-lactamases.
FIGURE 1.

Structures of clinically used β-lactamase inhibitors.
Carbapenems were subsequently introduced into the clinic as last resort antibiotics due to their high potency and exceptional broad spectrum of antimicrobial activity that includes both Gram-negative and Gram-positive aerobic and anaerobic bacteria (9–11). Another advantage of carbapenems over other β-lactams has been their low potential for selection of resistant mutants. Although extensive use of penicillins and cephalosporins has resulted in selection of numerous extended spectrum variants of the TEM-, CTX-, and SHV-type β-lactamases, no carbapenem-resistant mutants of these enzymes have been reported during more than two decades of clinical use. Instead, clinical use of carbapenems has resulted in the appearance, from yet unidentified sources, of new types of class A β-lactamases, the carbapenemases. These enzymes are capable of producing resistance to a wide variety of β-lactam antibiotics, including carbapenems, either by themselves and/or in combination with other resistance mechanisms, such as decreased permeability of the bacterial outer membrane or lowered affinity for penicillin-binding proteins (12). Phylogenetic analysis of class A β-lactamases indicates that all presently known class A carbapenemases can be classified into six branches, represented by GES-, KPC-, IMI/NMC-, SME-, BIC-, and SFC-type enzymes that share between 32 and 70% amino acid sequence identity (12, 13). Although IMI/NMC-, SFC-, BIC-, and SME-type carbapenemases are found only sporadically in a limited number of bacterial species, the KPC- and GES-type enzymes are widely distributed in clinical bacterial pathogens. Because of their ability to produce resistance to all β-lactam antibiotics, including carbapenems, these enzymes pose a very serious threat to our ability to treat life threatening infections.
The first GES-type β-lactamase, GES-1, was discovered in Klebsiella pneumoniae isolated in 1998 in France (14), which incidentally has no significant ability to hydrolyze carbapenem antibiotics. Twelve years hence, this growing family of β-lactamases now includes 15 members (GES-1 to GES-15), isolated from various Gram-negative bacteria of clinical origins in Europe, Asia, Africa, and the Americas. Although some GES β-lactamases produce antibiotic resistance profiles similar to those of classical extended spectrum enzymes (15–17), some GES variants (GES-2, -4, -5, and -6) also confer decreased susceptibility to imipenem, the first clinical carbapenem. The acquisition of carbapenemase activity by GES enzymes was attributed to a single amino acid substitution at position 170 (Ambler numbering is used) (18). The GES-4, -5, and -6 carbapenemases all have a G170S substitution, whereas GES-2 has a G170N substitution. Amino acid residue 170 is located within the active site and is a conserved asparagine in all class A β-lactamases, including the various carbapenemases.
In this report, we describe the kinetic and structural basis for inhibition of GES-2, the only GES-type carbapenemase that possesses the canonical asparagine at position 170, by tazobactam, a clinically used β-lactamase inhibitor. Using a combination of ultraviolet (UV) and mass spectrometries, we provide evidence for intermediates formed during inhibition of GES-2 by tazobactam. Our kinetic analyses also provide rate constants for the formation of some of these key intermediates. Furthermore, we have determined the structure of the GES-2 enzyme, in both the native form (at 1.45-Å resolution) and in complex with tazobactam (at 1.65-Å resolution). This structural information is the first for a class A carbapenemase in complex with a clinically used β-lactamase inhibitor. Elucidation of the structural and mechanistic details for interaction of the clinically used β-lactamase inhibitors with class A carbapenemases has important implications, as the ultimate emergence of resistance to inhibitors will dramatically narrow our already very limited therapeutic options.
EXPERIMENTAL PROCEDURES
Plasmids
A constitutive expression vector, pHF016, containing a synthetic gene for GES-2 (pHF:GES-2), cloned between the NdeI and HindIII sites, was used for minimal inhibitory concentration (MIC)3 determinations as previously described (19). For protein expression, pET24a(+) containing the synthetic gene for GES-2 (pET:GES-2), cloned between the unique NdeI and HindIII sites, was used as previously described (19).
MIC Determinations
The MICs of β-lactam antibiotics were determined by the broth microdilution method, as recommended by the Clinical and Laboratory Standards Institute (20). GES-2 was expressed in Escherichia coli JM83 using the plasmid pHF:GES-2. E. coli JM83 harboring pHF016 was used as a control. The MICs were determined in Mueller-Hinton II broth (Difco) using a bacterial inoculum of 5 × 105 colony forming units/ml. All plates were incubated at 37 °C for 16–20 h, before results were interpreted.
Expression and Purification of the GES-2 Enzyme
To express GES-2, E. coli BL21(DE3) was transformed with pET:GES-2 and cells containing the construct were selected on LB agar supplemented with 60 μg/ml of kanamycin. Selected cells were grown in LB medium supplemented with 60 μg/ml of kanamycin at 37 °C, with 220 rpm agitation, until the A600 reached 0.4. Isopropyl β-d-thiogalactoside was added to a final concentration of 1 mm and the cells were further grown at 25 °C, with 220 rpm agitation, for 24 h. The cells were pelleted by centrifugation at 20,000 × g at 4 °C and the media were concentrated by centrifugal filtration at 3,000 × g at 4 °C using a Centricon Plus 70 (Millipore) concentrator with a 10-kDa molecular mass cut-off filter. The concentrated medium was then dialyzed against 20 mm Tris (pH 7.5) and fractionated on a DEAE (Bio-Rad) column (2.5 × 22 cm) using a linear gradient of NaCl (0–0.3 m) in 20 mm Tris (pH 7.5). The fractions containing GES-2 were pooled and stored at 4 °C. The enzyme concentration was determined spectrophotometrically using the bicinchoninic acid protein assay kit (Pierce), using bovine serum albumin as a standard.
Data Collection and Analysis
All spectrophotometric data were collected on a Cary 50 spectrophotometer (Varian) at 22 °C. Analyses were performed using the nonlinear regression program Prism 5 (GraphPad Software, Inc.) employing data obtained from experiments performed at least in triplicate.
Determination of the Dissociation Constant for Tazobactam and the Rate Constant for Inactivation by Tazobactam
The inhibitor dissociation constant (Ki) for tazobactam and rate constant describing inactivation by tazobactam (ktazo) with GES-2 was determined using nitrocefin (Δϵ500 = +15,900 cm−1 m−1) as a reporter substrate. The rate constant ktazo describes formation of both transient and irreversibly inactivated species. Reactions containing 50 mm NaPi (pH 7.0), 100 mm NaCl, 10 or 20 μm nitrocefin, and varying concentrations of tazobactam were initiated by the addition of the enzyme (20 nm) using a SFA-20 stopped-flow apparatus (Hi-Tech Scientific, Salisbury, UK). The absorbance was monitored at 500 nm and the time courses were fit with Equation 1.
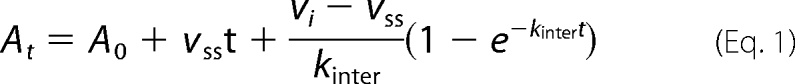 |
where At is the absorbance at time t, A0 is the initial absorbance, vss is the steady-state velocity, vi is the initial velocity, and kinter is the rate constant for the interconversion between vi and vss. The initial steady-state velocity data were plotted as a function of tazobactam concentration and the dissociation constant was determined using the method of Dixon (21). The values for kinter were plotted as a function of tazobactam concentration and were fit with Equation 2,
 |
where kinter and ktazo are as defined above, I is the concentration of tazobactam, and KI is the apparent concentration of tazobactam required to reach kinter = ktazo/2.
Determination of the Partition Ratio
The partition ratio was determined using the titration method (22). Varying molar ratios of tazobactam and GES-2 (up to 20,000:1) were incubated overnight at 4 °C in 50 mm NaPi (pH 7.0), 100 mm NaCl. The reactions were then dialyzed against 50 mm NaPi (pH 7.0), 100 mm NaCl using Slide-A-Lyzer MINI Dialysis units (Pierce). The remaining activity was measured after a 1:50-fold dilution of the enzyme (4–10 nm final) into 50 mm NaPi (pH 7.0), 100 mm NaCl containing excess nitrocefin (100–200 μm final). The absorbance was monitored at 500 nm and the steady-state velocities were determined from the linear phase of the reaction time courses. The ratio of tazobactam:enzyme resulting in ≥90% inactivation was designated as the partition ratio, as previously defined (23, 24).
Detection and Kinetic Characterization of Enamine Species by UV Spectroscopy
The cis- and trans-enamine reaction intermediates, formed during inhibition by β-lactamase inhibitors, are characterized by chromophores absorbing at wavelengths greater than 250 nm (25–28). Reactions containing 50 mm NaPi (pH 7), 100 mm NaCl, and 500 μm tazobactam were initiated by the addition of 5 μm GES-2. The absorbance from 200 to 350 nm was monitored every 0.3 min over 30 min. Subtracting the spectrum of GES-2 from the GES-2/tazobactam spectra generated the desired difference spectra.
The first-order rate constants describing formation and/or disappearance of the cis- and trans-enamine were determined using a SFA-20 stopped-flow apparatus (Hi-Tech Scientific, Salisbury, UK). Reaction mixtures contained 50 mm NaPi (pH 7.0), 100 mm NaCl, 500 μm tazobactam, and 5 μm GES-2. The cis-enamine species was detected at 297 nm and the time course was fit with Equation 3. The trans-enamine species was detected at 277 nm and the time course was fit with Equation 4.
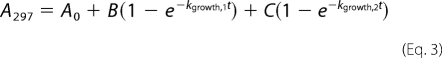 |
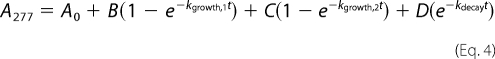 |
Where A297 and A277 are the absorbance at 297 and 277 nm, respectively, at time t, A0 is the initial absorbance, B and C are the amplitudes of the exponential growth phases, kgrowth,1 and kgrowth,2 are the rate constants of the exponential growth phases, D is the amplitude for the exponential decay phase, and kdecay is the rate constant of the exponential decay phase.
Detection of GES-2/Tazobactam Reaction Intermediates by Electrospray Ionization Mass Spectrometry
Reactions containing 50 mm NaPi (pH 7.0), 100 mm NaCl, and 70 μm GES-2 were preincubated in the presence and absence of 1 mm tazobactam. A 15-μl aliquot of each reaction was diluted 1:4 into 0.1% formic acid, 2% acetonitrile (Fisher) immediately prior to LC/MS analysis. A 25-μl aliquot of the quenched reaction, containing 12.5 μg of GES-2, was injected onto a 2.1 mm × 75-mm Poroshell C3 column (Agilent) at 350 μl/min using a Dionex RSLC HPLC system (A = H2O with 0.1% formic acid, B = acetonitrile with 0.1% formic acid). The gradient was held at 15% B for 3.1 min, followed by a linear gradient from 15 to 90% B over 5.4 min. The gradient was held constant at 90% B for 3 min and then re-equilibrated in 15% B for 6.5 min. The sample was diverted from the MS source during the first 3-min post-injection, during which time a calibration solution was passed into the instrument.
Mass spectrometry was performed on a Bruker MicroQTOF Q-TOF instrument. Single MS spectra from 300 to 3000 m/z were acquired at a spectral sum rate of 1 Hz. Two spectra were averaged together per displayed/saved spectrum. Calibration was provided by a six-point parameterized fit (MicroTOFControl) using a Agilent electrospray ionization spectroscopy tune mixture from m/z 322.0481 to 2721.8948. Spectra were acquired in positive mode with a bias of 4500 V on the capillary, a −500 V End Plate offset, 3.5 Bar of nebulizer gas, dry gas at 8 liter/min, and dry gas temperature of 180 °C.
The charge/mass deconvolution was performed using Bayesian Reconstruction (ABSciex) from exported ASCII data (FlexAnalysis). The following mass reconstruction parameters were used: mass considered was 20,000 to 35,000 Da, S/N 5:1, step size = 0.25 Da, iterations = 20. To confirm the findings, the data were also charge-deconvolved within Flex Analysis. Mass agreement with charge deconvolution was identical (data not shown). The average mass error for the GES-2 measurements was 0.006% (predicted mass average is 29,271.1 Da for the GES-2 polypeptide).
Crystallization and Data Collection
Initial crystallization trials with the native GES-2 enzyme (6.1 mg/ml) were set up using PEG/Ion screens 1 and 2 (Hampton Research). Small crystals were observed in several conditions, with the best crystals obtained from unbuffered 0.2 m sodium iodide, 20% (w/v) PEG 3350 after incubation at 15 °C. Several of these crystals were flash-cooled in a cryoprotectant comprising the well solution and 28% ethylene glycol. The crystals, belonging to the monoclinic space group P21 with cell dimensions a = 42.94 Å, b = 81.47 Å, c = 71.99 Å, β = 101.9°, diffracted to ∼1.45-Å resolution. The Matthews coefficient (29), assuming 2 molecules in the asymmetric unit, each with molecular mass of 29,500 Da, is 2.09 Å3/Da (41% solvent content). A complete data set, comprising 1000 images with an oscillation angle of 0.1°, was collected from this crystal on beam line BL12-2 at the Stanford Synchrotron Radiation Laboratory using x-rays at 12,658 eV (0.9785 Å). The images were processed with XDS and XSCALE (30). The final data set, containing 82,915 reflections, had an Rmerge of 0.104 to 1.45-Å resolution. Additional data collection statistics are given in Table 1.
TABLE 1.
Data collection and refinement statistics for the native GES-2 enzyme and the GES-2·tazobactam complex structures
| Native GES-2 | GES-2·tazobactam complex | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P43212 |
| Unit cell dimensions, a, b, c (Å) | 42.94, 81.47, 71.99, β = 101.9° | 83.44, 83.44, 70.97 |
| Wavelength (Å) | 0.97945 | 0.97945 |
| Resolution range (Å) | 70.5–1.45 | 27.2-1.65 |
| Observed reflections | 307,344 | 281,188 |
| Unique reflections | 82,915 | 30,744 |
| Rmergea (%) | 10.4 (49.0)b | 9.5 (52.2)b |
| I/sI | 9.78 (2.97) | 19.93 (3.55) |
| Completeness (%) | 97.3 (99.1) | 99.9 (99.2) |
| Refinement | ||
| R-factor/Rfree (%)c | 16.5/19.9 | 15.5/19.3 |
| Total atoms: protein / solvent / ligands | 4168/464d/30e | 2104/350/28 |
| ADF (Å2) | ||
| Protein/solvent/ligands | 13.9, 13.2f/26.8d/25.4e | 11.94/25.31/14.81 |
| R.m.s. deviation from ideality | ||
| Bonds (Å) | 0.006 | 0.006 |
| Angles (°) | 1.073 | 1.135 |
a Rmerge = S|I − 〈I〉|/SI × 100, where I = the observed intensity and 〈I〉 is the mean intensity.
b Numbers in parentheses relate to the highest resolution shell; for the native GES2 enzyme data, 1.50-1.45 Å, for the GES-2·tazobactam complex data, 1.70-1.65 Å.
c r = S‖Fo | − k|Fc ‖/S|Fo | × 100. Rfree was calculated with 5% of the reflections.
d Solvent comprises 449 water molecules and 15 iodide ions. The average ADFs for the water and iodide were 26.7 Å2 and 28.6 Å2, respectively.
e Two HEPES molecules.
f The first two values are for the A and B protein chains, respectively.
The GES-2 enzyme (1.4 mg/ml) was incubated overnight in the presence of 9.6 mm tazobactam at 4 °C. The GES-2·tazobactam complex was concentrated to 8.7 mg/ml and initial crystallization trials were set up using PEG/Ion screens 1 and 2 (Hampton Research). Crystals were observed in 0.2 m sodium thiocyanate with 20% (w/v) PEG 3350 and 0.2 m potassium thiocyanate with 20% (w/v) PEG 3350 after incubation at 4 °C. The crystals appeared suitable for diffraction experiments and several were flash-cooled in a cryoprotectant comprising the well solution and 25% ethylene glycol. The crystals belonged to space group P43212 with cell dimensions a = b = 83.44 Å, c = 70.97 Å, and diffracted to 1.65-Å resolution. The Matthews coefficient (29), assuming 1 molecule in the asymmetric unit, is 2.1 Å3/Da (41% solvent content). A complete dataset, comprising 240 images with an oscillation angle of 0.5°, was collected from one of these crystals on beam line BL7-1 at the Stanford Synchrotron Radiation Laboratory. The images were processed with XDS and XSCALE (30). The final data set contained 30,744 reflections with an Rmerge of 0.095 to 1.65-Å resolution. Additional data collection statistics are given in Table 1.
Structure Solution and Refinement
The refined GES-1 structure (Protein Data Bank code 2qpn) was used as the search model in the determination of the native GES-2 enzyme structure by molecular replacement. Initially, all four primitive orthorhombic space groups were tested, and two independent molecules were located with the program MOLREP, from the CCP4 suite (31), in space group P21, with an R-factor of 0.35 and a score of 0.66. The native GES-2 structure was initially refined using REFMAC (32), where the initial R-factor and Rfree were 0.380 and 0.382 for data between 70.5- and 1.45-Å resolution. Two rounds of manual model building with COOT (33), where the Asn170 side chain was added along with 190 water molecules, followed by restrained positional and isotropic temperature factor refinement, lowered the R-factor and Rfree to 0.232 and 0.263 for all data up to 1.45-Å resolution. Refinement was then transferred to PHENIX (34) and simulated annealing was used to remove any potential model bias. Subsequent cycles of positional and individual atomic displacement parameter refinement gave a final model with R-factor and Rfree of 0.165 and 0.199, respectively. Additional refinement statistics are given in Table 1.
The refined GES-1 structure was also used as the starting model for the structure determination of the GES-2·tazobactam complex. The resulting model from molecular replacement (R-factor = 0.38 and score = 0.66) was initially refined with REFMAC (32) using all data between 27.2 and 1.65 Å. Inspection of the electron density in the active site after 10 cycles of refinement (R-factor = 0.243, Rfree = 0.280) showed very strong electron density connected to the Ser70 side chain and extending through the active site to the surface of the molecule. A linear tazobactam ligand was built into the density and refined. Refinement was transferred to PHENIX (34) and additional cycles of refinement and manual model building, where water molecules were added to the structure, were carried out. In the latter stages of the refinement, a large piece of residual density, near the Ser130 side chain, was observed and modeled as species 6. The final R-factor and Rfree were 0.155 and 0.193, respectively. Additional refinement statistics are given in Table 1.
The atomic coordinates and the structure factors for the native GES-2 enzyme and the GES-2·tazobactam complex were deposited in the Protein Data Bank (35) with PDB codes 3NI9 and 3NIA, respectively. Superimpositions were performed using the SSM procedure (36) as implemented in COOT (33), and the program LSQKAB in the CCP4 suite (31). Figures were generated using PyMOL (37).
RESULTS
Susceptibility of GES-2 to Tazobactam
We had previously shown that the GES-2 enzyme confers clinical resistance against piperacillin according to the Clinical and Laboratory Standards Institute breakpoints (≥128 μg/ml is considered resistant) (19). In addition, it has been reported that GES-2 has a high affinity for the β-lactam inhibitor tazobactam (IC50 = 0.5 μm) (16). Using our constitutive expression vector, we evaluated the effect of tazobactam on the MIC for GES-2 against piperacillin. The presence of 4 μg/ml of tazobactam decreases the MIC for piperacillin from 128 to 1 μg/ml, thus rendering the organism clinically susceptible.
Kinetic Parameters Describing Inhibition of GES-2 by Tazobactam
The GES-2 enzyme was purified to apparent homogeneity to evaluate the kinetics of inhibition by tazobactam. Inhibition of class A β-lactamases by tazobactam can proceed through a series of species, both transient and irreversible (Scheme 1). Because of the importance of tazobactam and clavulanic acid as clinically used inhibitors, their mechanisms of action have been scrutinized (38). The inhibition mechanism is stepwise and, as indicated above, involves a number of species, many of which have been presumed to exist, but not unequivocally documented. The rate at which the GES-2 inhibition occurs is rapid, with complete inhibition within 5 min. A stopped-flow apparatus was used to determine the initial velocity of the reaction. The dissociation constant for the noncovalent complex between tazobactam and GES-2 (the preacylation complex) was in the nanomolar range (Ki = 700 ± 50 nm), indicative of high affinity of the enzyme for the inhibitor and consistent with the previously reported IC50 value (500 nm) (16). The rate constant describing inhibition of GES-2 by tazobactam (ktazo), which includes contributions from both the transient and irreversible species, was determined directly using a continuous assay. This parameter is often reported as kinact in the literature, however, this parameter should only represent formation of the irreversible species, and cannot be evaluated by the continuous method. The value for ktazo of 0.035 ± 0.002 s−1 would impart a half-life for inhibition of only 20 s, which is obviously rapid.
SCHEME 1.

Proposed mechanism for the inhibition of GES-2 by tazobactam.
The partition ratio (kcat/kinact) provides a measure of the number of molecules of inhibitor that experience turnover prior to the enzyme being irreversibly inactivated. The lower the value, the more effective the enzyme inhibition process. As determined in this study, this value is 7500 for GES-2.
Detection and Kinetic Characterization of Enamine Intermediates by UV Difference Spectroscopy
The aforementioned preacylation complex leads to acyl-enzyme species 2 (Scheme 1), as is typical of any β-lactam substrate. This species forms on a millisecond time scale, however, we were unable to directly detect its formation due to the low extinction coefficient associated with opening of the β-lactam ring (23). The smaller chromophore was masked by the trailing end of the much larger one of the α,β-unsaturated systems of species 4 and 5, which form on the same time scale. The intermediate 2 then undergoes further processing to the imine 3, which tautomerizes to both the cis- (4) and trans-enamine (5). We used UV spectroscopy to detect these species as it has previously been shown that cis- and trans-enamine intermediates can be detected at wavelengths greater than 250 nm due to the presence of the β-amino-α,β-unsaturated carbonyl chromophore (25, 26, 39). The UV difference spectra of GES-2 in the presence of tazobactam revealed two species whose concentrations changed with time (Fig. 2A). The lower wavelength peak, with a λmax at 277 nm, is consistent with a trans-enamine intermediate and the higher wavelength peak, with a λmax at 297 nm, is consistent with a cis-enamine intermediate (25, 26). Using a stopped-flow apparatus, we were able to observe the simultaneous formation of both isomers upon mixing of the enzyme and inhibitor (Fig. 2, B and C). The chromophore corresponding to the trans-enamine diminishes with time, whereas the signal representing the cis-enamine increases. After 5 min, there is no further change in the UV difference spectra and it remains unchanged (data not shown). Hence, on initial mixing of tazobactam and GES-2, both 4 and 5 are formed, but the trans-enamine (5) isomerizes to the cis-enamine (4) rapidly, a process that would involve the transient reformation of the imine (3). The initial formation of both enamine species is likely related to the dynamic nature of the imine, which would predispose the system to the formation of one or the other enamine. But given time, it would appear that the active site is better able to stabilize the cis-enamine (4).
FIGURE 2.

UV detection of enamine intermediates during inhibition of GES-2 by tazobactam. A, the UV difference spectra of GES-2 (5 μm) inhibition by tazobactam (500 μm) over 10 min. The arrows indicate the disappearance of the trans-enamine species (λmax = 277 nm) and the appearance of the cis-enamine species (λmax = 297 nm). B, the time course for formation of the cis-enamine species monitored at 297 nm. The line represents the best fit for Equation 3, which describes two exponential processes. The inset shows the initial exponential growth phase. C, the time course for formation of the trans-enamine species monitored at 277 nm. The line represents the best fit for Equation 4, which describes three exponential processes. The inset shows the initial exponential growth phase. The plot of residuals for the nonlinear fit of data for the indicated equation can be found in supplemental Fig. S1.
From the time courses obtained using the stopped-flow apparatus, we determined the rate constants describing the formation and/or disappearance of the enamine intermediates (Table 2). The time course representing the cis-enamine intermediate shows two exponential growth phases (Fig. 2B). There is an initial rapid phase (kgrowth,1 = 1.8 s−1), which is followed by a slower phase (kgrowth,2 = 0.023 s−1). The time course for the trans-enamine intermediate consists of three phases (Fig. 2C). There are two exponential growth phases (kgrowth,1 = 3.4 s−1 and kgrowth,2 = 0.17 s−1), followed by a slow exponential decay (kdecay = 0.021 s−1). The rate constant for the decay of the trans-enamine is the same as that for the second growth phase of the cis-enamine, within the limits of error for such measurements, indicative that this is likely the cis-trans isomerization of the two intermediates via the imine species (3).
TABLE 2.
Kinetic parameters for the formation of enamine intermediates during inhibition of GES-2 by tazobactam
| Parameter | cis-Enamine | trans-Enamine |
|---|---|---|
| kgrowth,1 (s−1) | 1.8 ± 0.3 | 3.4 ± 0.7 |
| kgrowth,2 (s−1) | 0.023 ± 0.001 | 0.17 ± 0.02 |
| kdecay (s−1) | 0.021 ± 0.001 |
Detection of Reaction Intermediates by Electrospray Ionization Mass Spectrometry
Species 8, 10, and 11 contain a UV chromophore that would be predicted to absorb near 225 nm, however, this overlaps the range for the peptide bonds from the enzyme. To identify these GES-2/tazobactam intermediates, as well as species 9, we employed LC/MS with electrospray ionization. The deconvoluted mass spectra for GES-2 and GES-2 inhibited by tazobactam are shown in Fig. 3. Immediately after mixing GES-2 and tazobactam (Fig. 3B), three major species are formed: 29,323.4 Da (+Δ51.2 ± 1.8), 29,340.7 Da (+Δ68.5 ± 1.8), and 29,359.4 Da (+Δ87.2 ± 1.8). In addition, three minor species are detected: 29,272.2 ± 1.8 Da (native GES-2), 29,375.6 Da (+Δ103.4 ± 1.8), and 29,394.0 Da (+Δ121.8 ± 1.8). After 20 min of incubation, the deconvoluted mass spectrum did not differ from the spectrum obtained immediately after mixing, indicating that these species are stable over this time frame (Fig. 3C). Some of the mass differences detected during inhibition of GES-2 by tazobactam are similar to those previously detected with the PC1 and TEM-1 β-lactamases (40) (+Δ52, +Δ70, and +Δ80) and the SHV-1 β-lactamase (+Δ88) (41) upon inhibition with tazobactam. As these results are consistent with the previous mass spectral studies, we propose that the +Δ52 intermediate is a cross-linked species between Ser70 and Ser130 (10), the +Δ70 is either an aldehyde intermediate (8) or an irreversibly inactivated GES-2 complex (11), and the +Δ88 is the hydrated aldehyde intermediate (9), as shown in Scheme 1. The minor species, corresponding to mass differences of +Δ103.4 and +Δ121.8, have not previously been detected, however, due to the spacing of approximately +Δ18, these are likely hydration products of the other species, as was seen with the dominant peak of the native GES-2 enzyme (Fig. 3A).
FIGURE 3.

Mass spectra of GES-2 inactivated by tazobactam. The mass spectra of the deconvoluted LC/MS after electrospray ionization of native GES-2 (A) and the GES-2/tazobactam intermediates formed immediately after the initiation of inactivation (B) and after 20 min of inactivation (c) are shown. The arrow indicates the position of the native GES-2 enzyme and all mass changes are relative to this peak.
Crystallographic Studies of GES-2 with Tazobactam
The crystal structures of the native GES-2 and the acyl-enzyme intermediate complex with tazobactam were determined to 1.45-Å and 1.65-Å resolution, respectively. The native GES-2 structure has two independent molecules in the asymmetric unit (residues 24–293 in both molecules) arranged in a loose dimer (∼900 Å2 of surface buried per monomer), and are related by a non-crystallographic 2-fold rotation axis. Superimposition of the two molecules shows that they are essentially identical in structure, with r.m.s. deviations in atomic positions of 0.27 Å for 267 matching Cα atoms. Inspection of the dimer interface in the native GES-2 enzyme shows that helix H3 from one molecule fits into a slight depression formed by the loop connecting helices H9 and H10 in the adjacent molecule, which creates an open cavity at the center of the interface. The two molecules are subsequently held together by a series of ionic and hydrogen bonding interactions at both points of contact of the H3 helices with the H9–H10 loop, and also across the center cavity (Fig. 4). The dimer observed for the native GES-2 enzyme is essentially identical to the non-crystallographic dimer in the P21 asymmetric unit of the GES-1 structure (42). Superimposition of the GES-2 dimer onto the GES-1 dimer gives a r.m.s. deviation of 0.51 Å (for 531 matching Cα positions), only a little greater than the r.m.s. deviations following superimposition of the individual chains (0.35–0.36 Å). The GES-2/tazobactam structure has one molecule in the asymmetric unit, but inspection of the P43212 symmetry-related molecules shows the presence of the same dimeric interaction, this time as a crystallographic dimer with the 2-fold rotation axis parallel to the ab diagonal. Superimposition of the native GES-2 dimer onto this crystallographic P43212 dimer gives a r.m.s. deviation of 0.45 Å for 534 matching Cα atoms. The observation of this dimer in two unrelated crystal forms implies an inherent propensity for GES-2 (and presumably GES-1) to dimerize, although there is currently no evidence that such a dimer is the active catalytic species in solution.
FIGURE 4.

The three-dimensional structure of GES-2. Stereoview of the GES-2 monomer bound to tazobactam. The covalently bound tazobactam molecule is shown as a yellow ball-and-stick representation and the thin white sticks represent the hydrolysis fragment 6. The structural elements in blue and gray are the main points of contact in the dimer and the Ω-loop, which forms one wall of the active site, is shown as a magenta loop. A number of secondary structure elements discussed in the text are also labeled.
The GES-2 monomer is similar in structure to other class A β-lactamases. Briefly, the enzyme is composed of two structural domains, the first (residues 26–62 and 218–293) consisting of a five-stranded antiparallel β-sheet, with two helices (H1 and H11) packed against the outer face of the sheet (Fig. 4). The second domain (residues 63–217) is an all α-helical domain consisting of seven α-helices. Superimposition of GES-2 onto some representative penicillinases and carbapenemases (Table 3) gives overall r.m.s. deviations between 1.1 and 1.4 Å. Omitting external loops from the superimposition to give a “core” region composed of the five β-strands in the first domain and the α-helices in the second domain give r.m.s. deviations between 0.7 and 1.0 Å. The active site is located at the interface of the two domains, formed primarily by β-strand B3 (residues 230–238) and a highly conserved structural motif known as the Ω-loop (residues 160–180). The catalytic serine residue (Ser70) is located on a short 310 helix at the N terminus of helix H2. Lys73, a catalytic residue in class A enzymes, is one turn further on in helix H2, and is hydrogen-bonded through the Nζ atom to the Oγ atom of Ser70. This lysine promotes serine for acylation by the substrate (43). On one side of Ser70 sits the invariant Glu166, a residue that activates a water molecule in the deacylation half-reaction. The hydrolytic water molecule is located between Ser70 and Glu166, within hydrogen bonding distance of both, and is also hydrogen bonded to the side chain of Asn170 (Fig. 5A). This residue is the site of the amino acid substitution where GES-2 differs from GES-1 (which has a glycine in this position). In the initial 2Fo − Fc and Fo − Fc electron density maps following molecular replacement, the presence of the asparagine side chain was clearly evident (Fig. 5A). On the opposite side of Ser70, a large piece of residual Fo − Fc density was observed and subsequently modeled as a HEPES buffer molecule in both of the native GES-2 enzyme monomers. Adjacent to the active site of GES-2 is a disulfide bond between Cys69 and Cys238. This disulfide bond is only known to be present in class A carbapenemases (SME-1, NMC-A, GES-1, and KPC-2), but lacking in all other class A β-lactamases.
TABLE 3.
Comparison of GES-2 with other representative class A β-lactamases
The carbapenemases NMC-A was from Enterobacter cloacae (58), SME-1 from Serratia marcescens (59), and KPC-2 from E. cloacae (60), and the penicillinases TEM-1 from E. coli (54) and SHV-1 from K. pneumoniae (61).
| Enzyme | PDB | Resolution | Rfree | Sequence identitya | R.m.s. deviation values (Å) |
|
|---|---|---|---|---|---|---|
| All Cαb | Core Cα | |||||
| Å | ||||||
| GES-2c | This work | 1.65 | 0.193 | |||
| GES-1 | 2qpn | 1.10 | 0.182 | –d | 0.36 (266) | 0.28 |
| NMC-A | 1bue | 1.64 | 0.214 | 34% | 1.3 (246) | 0.81 |
| SME-1 | 1dy6 | 2.13 | 0.244 | 36% | 1.3 (251) | 0.81 |
| KPC-2 | 3dw0 | 1.60 | 0.211 | 36% | 1.2 (240) | 1.02 |
| TEM-1 | 1zg4 | 1.55 | 0.240 | 34% | 1.1 (238) | 0.71 |
| SHV-1 | 1vm1 | 2.02 | 0.257 | 32% | 1.4 (250) | 0.87 |
a Not including the leader sequence.
b Values in parentheses give the total number of Cα atoms matched. The core (155 Cα atoms) comprises residues 42–48, 61–90, 119–193, 199–217, 230–238, 243–250, and 259–265.
c The coordinates used are for the GES-2·tazobactam complex.
d The difference between GES-1 and GES-2 is the amino acid residue at position 170 (glycine in GES-1 and asparagine in GES-2).
FIGURE 5.

The active site of the GES-2·tazobactam complex. A, stereoview of the electron density in the active site of the GES-2·tazobactam complex, showing the final 2Fo − Fc electron density for the protein (blue, contoured at 1.5 σ) and the initial Fo − Fc electron density prior to refinement (magenta, contoured at 3.5 σ) modeled as either the trans-enamine (5) or imine (3) species (yellow ball-and-stick) covalently attached to Ser70. The Fo − Fc electron density for Asn170 (brown) is also shown. Water molecules are represented as red spheres, black dashed lines indicate hydrogen bonds, and hydrolysis fragment 6 is shown in white. B, stereoview of the residual Fo − Fc electron density (magenta) near Ser130 and the partially occupied hydrolysis fragment 6 (white sticks). Hydrogen bonding interactions with this fragment are indicated.
In the GES-2·tazobactam complex, a linear serine-linked ligand was clearly visible in the initial 2Fo − Fc and Fo − Fc electron density maps following the first cycles of REFMAC refinement (Fig. 5A). The electron density is connected to the Ser70 side chain density and extends out of the active site cleft into the solvent. Even in the early stages of the refinement the electron density was very well defined and lobes for the carboxylate, sulfinate, methyl, and triazole moieties were easily assigned. Once the model based on the linear tazobactam-derived species was built into the density and refined, residual Fo − Fc density was observed for the sulfinate group, and this was modeled in two configurations with a SOF ratio of 0.8:0.2. To test the occupancy of the covalently bound ligand, the SOFs of the atoms (except for the sulfinate group) were set to 1.0 and the ADPs, or B-factors, were refined. The refined ADPs ranged from 6.9 to 29.0 Å2 (average 14.2 Å2) and were found to be similar to the B-factors of the surrounding protein atoms (average 10.2 Å2). The triazole group, which makes no interaction with the protein and is predominantly solvent exposed, had the highest ADPs.
At 1.65-Å resolution, it is not possible to definitively state which tautomeric form, the imine (3) or trans-enamine (5), was adopted. Apart from the covalent linkage between C7 and the Oγ atom of Ser70, there are very few direct contacts between the inhibitor species and GES-2. The O8 atom of the inhibitor accepts two hydrogen bonds from the main chain amide nitrogen atoms of Ser70 and Thr237. The Nδ2 atom of Asn170 donates hydrogen bonds to the O12 and O13 atoms of the sulfinate group. All other contacts involve water-mediated hydrogen bonds with the carboxylate and sulfinate. The triazole moiety makes no interactions with either the protein or water molecules, and consequently has, on average, ADPs almost twice as large (23.0 Å2) relative to the rest of the tazobactam molecule (12.0 Å2). In addition, there is a potential intermolecular hydrogen bonding interaction between the N4 atom and one of the oxygen atoms of the sulfinate group.
Throughout the course of the refinement two large pieces of residual Fo − Fc density were seen adjacent to the side chain of Ser130. The density closest to the serine had a distinct Y-shape (Fig. 5B) with a second fragment of roughly spherical density ∼2.5 Å from the center of the Y-shaped density. Sulfate anions and other small molecules, including the sulfonate buffers HEPES and MES, have been seen in this location in numerous β-lactamase structures but in this case none of these molecules could account for the observed density. Furthermore, neither the crystallization components (thiocyanate and PEG), the cryoprotectant (ethylene glycol), nor the small molecule break down products of 8 and 9 from the proposed mechanism of enzyme-catalyzed tazobactam hydrolysis (40, 44, 45) fit the available density nor could they account for the adjacent spherical peak. It was initially thought that the Y-shaped density may represent a vinyl carboxylate moiety, similar to that seen in the SHV-1·tazobactam complex (PDB code 1vm1), where the vinyl carboxylate is covalently attached to the Oγ atom of Ser130 (11). Although a vinyl carboxylate could be modeled into the available density with the carboxylate group forming a number of strong hydrogen bonds with the side chains of Lys234, Thr235, Thr237, and Arg244, the terminal carbon of the vinyl group was not covalently bound to Ser130 and instead made a close contact with a very well ordered water molecule at a distance of ∼2.6 Å, implying a hydrogen bond donor or acceptor at this location. Given the necessity for hydrogen bond donors and acceptors at both ends of the molecule, the density and the surrounding interactions seemed to fit a glycinyl moiety derived from the displaced fragment containing the unusual amino acid 6, which would arise from hydrolysis of the imine bound species 3 or the turnover product 7 (Scheme 1). When 6 was modeled into the density, simple rotation of the N4-C3 torsion angle placed the sulfinate group into the second fragment of unaccounted density with the triazole group extending into the solvent. The atoms beyond C2 were assigned an SOF of 0.5 and the fragment refined. The resultant temperature factors were double that of the surrounding protein, indicating a high degree of disorder in the triazole moiety. The atoms refined with a SOF of 1.0 had ADPs consistent with the residues to which this end of the molecule was hydrogen bonded.
Superimposition of the native GES-2 enzyme onto the tazobactam complex gives a r.m.s. deviation of 0.32 Å for 267 matching Cα positions. Although there are no significant changes in the overall structure of the enzyme, tazobactam binding does give rise to some differences in the vicinity of the active site, including a change in the rotamer conformation of Glu104, which has to swing away to accommodate the triazole moiety, and a major restructuring of the water molecules. The hydrolytic water molecule, which is promoted in the deacylation step by Glu166, has moved ∼1.2 Å away from the Ser70 side chain yet remains in hydrogen bonding contact with Ser70, Glu166, and Asn170, although the side chain of the latter has rotated slightly to maintain contact. Three water molecules in the native GES-2 active site have been displaced, including one previously occupying the oxyanion hole, between the main chain amide nitrogen atoms of Ser70 and Thr237, and the positions occupied by the sulfinate group and the methyl carbon (C20) of the tazobactam. Six new water molecules are associated with the inhibitor, three with the sulfinate group and three with the carboxylate, and one of the latter also forms a hydrogen bond with the N4 atom of the tazobactam fragment (6) as noted above.
DISCUSSION
The catalytic mechanism of class A β-lactamases involves acylation of Ser70 by the β-lactam ring of the antibiotic substrate to produce a transient acyl-enzyme intermediate. This intermediate is subsequently hydrolyzed to complete the catalytic cycle. The interaction of tazobactam with the GES-2 enzyme leads to enzyme acylation, similar to that of β-lactam antibiotics. This acyl-enzyme species (2) experiences deacylation, just as would the acyl-enzyme species from a typical substrate. Indeed, as reported here, there are ∼7500 turnover events prior to the onset of complete enzyme inhibition by tazobactam. This turnover number is much higher than that observed with other class A carbapenemases (185 for NMC-A and 500 for KPC-2) showing that tazobactam is not only a better substrate for GES-2, but that GES-2 is less susceptible to irreversible inhibition by tazobactam (46, 47). However, acyl-enzyme species 2 enjoys sufficient longevity and has the opportunity to undergo a set of reactions (Scheme 1) that initiate the process of enzyme inhibition/inactivation. A multitude of the processes that can and do take place with comparable rates, hence they are competing with one another, has been reported (38). Our analyses of these events for inhibition of the GES-2 enzyme by tazobactam have identified eight species that contribute to inhibition (Scheme 1).
The sulfone group in species 2 is predisposed for elimination to give the linear acyl-enzyme species 3. This elimination reaction has been known to occur with all clinically used inhibitors for β-lactamases, including tazobactam. That it takes place was documented in our study by determination of the x-ray structure for species 3 or 5. Furthermore, we showed by stopped-flow UV spectroscopy that species 5, the so-called trans-enamine, interconverts with 4, the cis-enamine. This tautomerization step was also seen in the class A enzyme PC1 when inhibited by clavulanic acid with a rate constant of 0.006 s−1, which is 3-fold slower than what we observed with GES-2 (25). Intermediates 4 and 5 are referred to as “transiently inhibited” species in the early literature on β-lactamase inhibition in recognition of the fact that they convert to other irreversibly inhibited species, as documented in the TEM-type enzymes. This transient inhibition is due to the fact that species 4 and 5 are α,β-unsaturated esters that ultimately should experience hydrolytic deacylation from the enzyme (hence, “transient” inhibition). Regardless, these esters would appear to enjoy a sufficient longevity in the GES-2 enzymes that we could entrap species 3 or 5 by x-ray study. An interesting, and unexpected, finding was that we also observed compound 6 within the active site. Hence, whereas its formation as a matter of hydrolysis of the imine in compound 7 or species 3 is understood, that it could remain bound to the enzyme is not precedented in the literature. Once compound 6 is released from 3, we detect four additional species (8-11) by mass spectrometry, each of which contributes to inhibition of the enzyme. Whereas 8 and 9 could be construed also as transiently inhibited species, species 10 and 11 are decidedly contributors to irreversible inhibition.
GES-2 is inhibited by tazobactam in living bacteria, as the presence of tazobactam reduces the MIC of piperacillin from 128 to 1 μg/ml, and renders the strain clinically susceptible to piperacillin (20). This is important for treatment of bacterial infections caused by strains harboring GES-2, as it provides a potential treatment option to overcome resistance. The susceptibility of GES-2 to tazobactam can also be seen in the kinetic parameters associated with inhibition. GES-2 has a high affinity for tazobactam (Ki = 700 nm) and the on-set of inhibition is rapid (ktazo = 0.035 s−1). The mass spectral data also support the rapid inhibition of GES-2 by tazobactam, as little free enzyme and three major intermediates (+Δ52, +Δ70, and +Δ80) are detected immediately upon mixing of the enzyme with inhibitor. These species also appear to enjoy longevity as the relative abundance of 4 and 5, by UV spectroscopy, and 8-11, by mass spectroscopy (Fig. 3), do not appear to change over a 20-min time frame. This time frame also corresponds to the relative doubling time of E. coli.
To our knowledge, the kinetic parameters associated with formation of the GES-2 complex with the cis- and trans-enamine species of tazobactam have not previously been determined. Prior studies with SHV-1 were unable to distinguish the two isomers, thus they were only able to monitor formation of the total enamine species (23, 48). The two isomers in complex with GES-2 show a difference in λmax of 20 nm, allowing us to monitor each species separately (Fig. 2A). Stopped-flow analysis highlights the rapid formation of both isomers with first-order rate constant of 2–3 s−1 (Table 2). This is followed by a slower phase of trans-enamine formation (k = 0.17 s−1). After the rapid phase of formation of both isomers, there is a slow phase in which the trans-enamine disappears, whereas the cis-enamine increases in abundance. The first-order rate constants describing both of these phases are comparable (k = 0.02 s−1, Table 2), which we propose indicates the isomerization of the two species via the imine (3). It is generally assumed that the species responsible for tazobactam inhibition of β-lactamases is the trans-enamine (48–50). These studies are all based on x-ray crystallography and Raman spectroscopy using single crystals. Likewise, the GES-2·tazobactam complex from our study shows tazobactam bound as either the trans-enamine or imine (Fig. 5A). It is thus surprising that in solution, the trans-enamine appears to isomerize to the cis-enamine, and it is this isomer that is stable over time (Fig. 2B).
To date, the crystal structures of tazobactam bound to a β-lactamase have been limited to the SHV-1 β-lactamase, where the inhibitor has been observed in multiple configurations, including an unbound form with an intact β-lactam ring (1) (44), the stable trans-enamine (5) (50, 51) and cis-enamine (4) (45) isomers, and the less-stable imine form (3), along with the inactivation product (11) (44). The current structural analysis of GES-2 represents the first structure of a carbapenemase enzyme with a clinically used β-lactamase inhibitor bound in the active site, in this case as either the trans-enamine or imine isomer. Furthermore, this is the first time that hydrolysis product 6 has been observed bound to the active site.
The conformation of the intermediate bound to GES-2 differs somewhat from the conformations seen in the tazobactam complexes of SHV-1 (PDB codes 1rcj, 2h10, 1tdg, and 1vm1) (44, 45, 50, 51). The five structures were superimposed based upon the short 310 helix preceding the active site serine, and the first two turns of helix H2. The superimposition between GES-2 and the two trans-enamine complexes (PDB 1rcj and 2h10) is shown in Fig. 6A and GES-2 with the cis-enamine (PDB 1tdg) and imine forms (PDB 1vm1) in Fig. 6B. In all complexes, with the exception of the cis-enamine, the carbonyl oxygen of the β-lactam group (O8) is located within the oxyanion hole formed by the main chain amide nitrogen atoms of Ser70 and Thr237. Comparison of the three trans-isomers shows that although the O8 atoms are in roughly the same location (the average distance between them is ∼0.35 Å), the enamine moieties begins to diverge after the N4 atoms. At this point a rotation of ∼180° around the C5–N4 bond puts the remainder of the inhibitor in a completely different orientation in the two enzymes. In SHV-1, the carboxylate group projects in the same direction as the sulfinate in GES-2, although, due to the C5–N4 rotation, the C9 atom is ∼2.4 Å from the S1 position. The carboxylate group accepts a hydrogen bond from the side chain of Asn132 and two water molecules, and makes a long interaction with the side chain of Asn170. The triazole group in the SHV-1 complex extends across the binding site and sits in approximately the same location as the sulfonate group of the HEPES molecule in the native GES-2 enzyme, yet makes no interactions with the protein. This pocket, formed by additional invariant residues including Ser130, Lys234, and Thr235, and the partially conserved Thr237, is frequently occupied in the class A β-lactamases by small anionic molecules including sulfate (42, 52–54) and sulfonate-containing buffer molecules, such as HEPES (55–57).
FIGURE 6.

Comparison of the tazobactam species in GES-2 and SHV-1. A, the trans-enamine or imine intermediate in GES-2 (yellow sticks), PDB 1rcj (cyan sticks), and PDB 2h10 (pink sticks). B, the trans-enamine or imine intermediate in GES-2 (yellow sticks), the cis-enamine intermediate in PDB 1tdg (cyan sticks), and imine intermediate from PDB 1vm1 (pink sticks). The position of the covalently bound vinyl carboxylate (VC) moiety is also shown in pink from the PDB 1vm1 structure. The location of hydrolysis fragment 6 observed in GES-2 is indicated (gray sticks). Part of the GES-2 protein structure is shown in green in both panels.
In the tazobactam complex of GES-2, this pocket is occupied by hydrolysis fragment 6 and in the imine complex of SHV-1 (PDB code 1vm1) by a vinyl carboxylate (11) covalently attached to Ser130 (Fig. 6B). The vinyl carboxylate molecule is anchored to the Ser130 side chain, and the carboxylate group forms two hydrogen bonds to the side chains of Thr235 and Arg244, which firmly anchor the molecule. Thus, the vinyl carboxylate would most likely have significantly less freedom to move compared with the GES-2 fragment, which is bound via hydrogen bonds to the glycinyl moiety, with the remainder of the molecule able to adopt multiple conformations. It was argued in the SHV-1 structure that the presence of the inactivation complex caused significant steric strain in the binding pocket that forced the Ser70-bound tazobactam to adopt the less favored imine isomer (44, 50). This does not appear to be the case in GES-2. Hydrolysis fragment 6 is significantly larger than the vinyl carboxylate, yet in the GES-2 enzyme, the tazobactam molecule bound at Ser70 is readily able to adopt either the trans-enamine or imine configuration similar to the sterically unhindered SHV-1 complexes (PDB codes 1rcj and 2h10). Just why this should be is difficult to ascertain. One possibility is that the size of the binding pocket in the two enzymes might be different, but based upon the superimposition of the two complexes (Fig. 6B) there does not appear to be any significant difference in size. As a rough approximation of the width of the pocket, the distance between two sets of residues on both sides of the cleft was measured (Ser130 to Asn170, and Glu166 to Thr237) and was found to be almost identical in GES-2 and SHV-1. A more likely possibility is the conformational flexibility of the larger fragment 6 in GES-2, relative to the tightly bound vinyl carboxylate group in SHV-1, allows the fragment to move away from the incoming tazobactam and creates enough space to allow the stable complex to form.
CONCLUSION
Resistance of clinical isolates to β-lactam antibiotics is often overcome by co-administration with β-lactamase inhibitors. The GES-2 β-lactamase is a carbapenemase, but is also susceptible to tazobactam. Our studies show that inhibition by tazobactam is not manifested through irreversible inactivation, but via the accumulation of multiple stable intermediates. Enamine species were detected using UV spectroscopy, whereas x-ray crystallography analysis is consistent with detection of either the trans-enamine or imine intermediates. The hydrated aldehyde, aldehyde, and cross-linked species were detected by mass spectrometry. By combining multiple techniques we were able to both detect multiple intermediates along the reaction pathway, but also characterize the rate of their formation to understand the physiologically relevant inhibition species in vivo.
Acknowledgments
The Stanford Synchrotron Radiation Laboratory is a national user facility operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Science. The Stanford Synchrotron Radiation Laboratory Structural Molecular Biology Program is supported by the Department of Energy (Basic Energy Sciences, Biological and Environmental Research) and National Institutes of Health (National Center for Research Resources, Biotechnology Training Program, National Institute of General Medical Sciences).
This work was supported by National Science Foundation Grant CHE-0741793.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 3NI9 and 3NIA) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- MIC
- minimal inhibitory concentration
- ADP
- atomic displacement factor
- SOF
- site occupancy factor
- r.m.s. deviations
- root mean square deviation
- PDB
- Protein Data Bank.
REFERENCES
- 1. Testero S. A., Fisher J. F., Mobashery S. (2010) in Burger's Medicinal Chemistry, Drug Discovery and Development (Abraham D. J., Rotella D. P. eds) pp. 259–404, Wiley-Blackwell, Oxford [Google Scholar]
- 2. Poole K. (2004) Cell. Mol. Life Sci. 61, 2200–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fisher J. F., Mobashery S. (2010) J. Med. Chem. 53, 4813–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goffin C., Ghuysen J. M. (2002) Microbiol. Mol. Biol. Rev. 66, 702–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher J. F., Meroueh S. O., Mobashery S. (2005) Chem. Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 6. Bush K., Jacoby G. A. (2010) Antimicrob. Agents Chemother. 54, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradford P. A. (2001) Clin. Microbiol. Rev. 14, 933–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perez F., Endimiani A., Hujer K. M., Bonomo R. A. (2007) Curr. Opin. Pharmacol. 7, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baughman R. P. (2009) J. Intensive Care Med. 24, 230–241 [DOI] [PubMed] [Google Scholar]
- 10. Shahid M., Sobia F., Singh A., Malik A., Khan H. M., Jonas D., Hawkey P. M. (2009) Crit. Rev. Microbiol. 35, 81–108 [DOI] [PubMed] [Google Scholar]
- 11. Kattan J. N., Villegas M. V., Quinn J. P. (2008) Clin. Microbiol. Infect. 14, 1102–1111 [DOI] [PubMed] [Google Scholar]
- 12. Walther-Rasmussen J., Høiby N. (2007) J. Antimicrob. Chemother. 60, 470–482 [DOI] [PubMed] [Google Scholar]
- 13. Girlich D., Poirel L., Nordmann P. (2010) Antimicrob. Agents Chemother. 54, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poirel L., Le Thomas I., Naas T., Karim A., Nordmann P. (2000) Antimicrob. Agents Chemother. 44, 622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moubareck C., Brémont S., Conroy M. C., Courvalin P., Lambert T. (2009) Antimicrob. Agents Chemother. 53, 3579–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poirel L., Weldhagen G. F., Naas T., De Champs C., Dove M. G., Nordmann P. (2001) Antimicrob. Agents Chemother. 45, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vourli S., Giakkoupi P., Miriagou V., Tzelepi E., Vatopoulos A. C., Tzouvelekis L. S. (2004) FEMS Microbiol. Lett. 234, 209–213 [DOI] [PubMed] [Google Scholar]
- 18. Ambler R. P., Coulson A. F., Frère J. M., Ghuysen J. M., Joris B., Forsman M., Levesque R. C., Tiraby G., Waley S. G. (1991) Biochem. J. 276, 269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frase H., Shi Q., Testero S. A., Mobashery S., Vakulenko S. B. (2009) J. Biol. Chem. 284, 29509–29513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clinical and Laboratory Standards Institute (2006) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 7th Ed., Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 21. Dixon M. (1953) Biochem. J. 55, 170–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silverman R. B. (1988) Mechanism-based Enzyme Inactivation: Chemistry and Enzymology, CRC Press, Boca Raton, FL [Google Scholar]
- 23. Bush K., Macalintal C., Rasmussen B. A., Lee V. J., Yang Y. (1993) Antimicrob. Agents Chemother. 37, 851–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Padayatti P. S., Sheri A., Totir M. A., Helfand M. S., Carey M. P., Anderson V. E., Carey P. R., Bethel C. R., Bonomo R. A., Buynak J. D., van den Akker F. (2006) J. Am. Chem. Soc. 128, 13235–13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rizwi I., Tan A. K., Fink A. L., Virden R. (1989) Biochem. J. 258, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cartwright S. J., Coulson A. F. (1979) Nature 278, 360–361 [DOI] [PubMed] [Google Scholar]
- 27. Sulton D., Pagan-Rodriguez D., Zhou X., Liu Y., Hujer A. M., Bethel C. R., Helfand M. S., Thomson J. M., Anderson V. E., Buynak J. D., Ng L. M., Bonomo R. A. (2005) J. Biol. Chem. 280, 35528–35536 [DOI] [PubMed] [Google Scholar]
- 28. Bonomo R. A., Liu J., Chen Y., Ng L., Hujer A. M., Anderson V. E. (2001) Biochim. Biophys. Acta 1547, 196–205 [DOI] [PubMed] [Google Scholar]
- 29. Matthews B. W. (1968) J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 30. Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 31. Collaborative Computational Project, N. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krissinel E., Henrick K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 37. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 38. Drawz S. M., Bonomo R. A. (2010) Clin. Microbiol. Rev. 23, 160–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ostercamp D. L. (1970) J. Org. Chem. 35, 1632–1641 [Google Scholar]
- 40. Yang Y., Janota K., Tabei K., Huang N., Siegel M. M., Lin Y. I., Rasmussen B. A., Shlaes D. M. (2000) J. Biol. Chem. 275, 26674–26682 [DOI] [PubMed] [Google Scholar]
- 41. Pagan-Rodriguez D., Zhou X., Simmons R., Bethel C. R., Hujer A. M., Helfand M. S., Jin Z., Guo B., Anderson V. E., Ng L. M., Bonomo R. A. (2004) J. Biol. Chem. 279, 19494–19501 [DOI] [PubMed] [Google Scholar]
- 42. Smith C. A., Caccamo M., Kantardjieff K. A., Vakulenko S. (2007) Acta Crystallogr. D Biol. Crystallogr. 63, 982–992 [DOI] [PubMed] [Google Scholar]
- 43. Meroueh S. O., Fisher J. F., Schlegel H. B., Mobashery S. (2005) J. Am. Chem. Soc. 127, 15397–15407 [DOI] [PubMed] [Google Scholar]
- 44. Kuzin A. P., Nukaga M., Nukaga Y., Hujer A., Bonomo R. A., Knox J. R. (2001) Biochemistry 40, 1861–1866 [DOI] [PubMed] [Google Scholar]
- 45. Sun T., Bethel C. R., Bonomo R. A., Knox J. R. (2004) Biochemistry 43, 14111–14117 [DOI] [PubMed] [Google Scholar]
- 46. Papp-Wallace K. M., Bethel C. R., Distler A. M., Kasuboski C., Taracila M., Bonomo R. A. (2010) Antimicrob. Agents Chemother. 54, 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mariotte-Boyer S., Nicolas-Chanoine M. H., Labia R. (1996) FEMS Microbiol. Lett. 143, 29–33 [DOI] [PubMed] [Google Scholar]
- 48. Kalp M., Bethel C. R., Bonomo R. A., Carey P. R. (2009) Biochemistry 48, 9912–9920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kalp M., Totir M. A., Buynak J. D., Carey P. R. (2009) J. Am. Chem. Soc. 131, 2338–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Padayatti P. S., Helfand M. S., Totir M. A., Carey M. P., Hujer A. M., Carey P. R., Bonomo R. A., van den Akker F. (2004) Biochemistry 43, 843–848 [DOI] [PubMed] [Google Scholar]
- 51. Totir M. A., Padayatti P. S., Helfand M. S., Carey M. P., Bonomo R. A., Carey P. R., van den Akker F. (2006) Biochemistry 45, 11895–11904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ibuka A., Taguchi A., Ishiguro M., Fushinobu S., Ishii Y., Kamitori S., Okuyama K., Yamaguchi K., Konno M., Matsuzawa H. (1999) J. Mol. Biol. 285, 2079–2087 [DOI] [PubMed] [Google Scholar]
- 53. Tranier S., Bouthors A. T., Maveyraud L., Guillet V., Sougakoff W., Samama J. P. (2000) J. Biol. Chem. 275, 28075–28082 [DOI] [PubMed] [Google Scholar]
- 54. Jelsch C., Mourey L., Masson J. M., Samama J. P. (1993) Proteins 16, 364–383 [DOI] [PubMed] [Google Scholar]
- 55. Brown N. G., Shanker S., Prasad B. V., Palzkill T. (2009) J. Biol. Chem. 284, 33703–33712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nukaga M., Mayama K., Hujer A. M., Bonomo R. A., Knox J. R. (2003) J. Mol. Biol. 328, 289–301 [DOI] [PubMed] [Google Scholar]
- 57. Toth M., Smith C., Frase H., Mobashery S., Vakulenko S. (2010) J. Am. Chem. Soc. 132, 816–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swarén P., Maveyraud L., Raquet X., Cabantous S., Duez C., Pédelacq J. D., Mariotte-Boyer S., Mourey L., Labia R., Nicolas-Chanoine M. H., Nordmann P., Frère J. M., Samama J. P. (1998) J. Biol. Chem. 273, 26714–26721 [DOI] [PubMed] [Google Scholar]
- 59. Sougakoff W., L'Hermite G., Pernot L., Naas T., Guillet V., Nordmann P., Jarlier V., Delettré J. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 267–274 [DOI] [PubMed] [Google Scholar]
- 60. Ke W., Bethel C. R., Thomson J. M., Bonomo R. A., van den Akker F. (2007) Biochemistry 46, 5732–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Knox J. R., Moews P. C. (1991) J. Mol. Biol. 220, 435–455 [DOI] [PubMed] [Google Scholar]