

Abstract
γ-Aminobutyric acid type A receptors (GABAARs) in the spinal cord are evolving as an important target for drug development against pain. Purinergic P2X2 receptors (P2X2Rs) are also expressed in spinal cord neurons and are known to cross-talk with GABAARs. Here, we investigated a possible “dynamic” interaction between GABAARs and P2X2Rs using co-immunoprecipitation and fluorescence resonance energy transfer (FRET) studies in human embryonic kidney (HEK) 293 cells along with co-localization and single particle tracking studies in spinal cord neurons. Our results suggest that a significant proportion of P2X2Rs forms a transient complex with GABAARs inside the cell, thus stabilizing these receptors and using them for co-trafficking to the cell surface, where P2X2Rs and GABAARs are primarily located extra-synaptically. Furthermore, agonist-induced activation of P2X2Rs results in a Ca2+-dependent as well as an apparently Ca2+-independent increase in the mobility and an enhanced degradation of GABAARs, whereas P2X2Rs are stabilized and form larger clusters. Antagonist-induced blocking of P2XRs results in co-stabilization of this receptor complex at the cell surface. These results suggest a novel mechanism where association of P2X2Rs and GABAARs could be used for specific targeting to neuronal membranes, thus providing an extrasynaptic receptor reserve that could regulate the excitability of neurons. We further conclude that blocking the excitatory activity of excessively released ATP under diseased state by P2XR antagonists could simultaneously enhance synaptic inhibition mediated by GABAARs.
Keywords: Fluorescence Resonance Energy Transfer (FRET), GABA Receptors, Purinergic Agonists, Purinergic Receptor, Receptor Endocytosis, Receptor Regulation, Trafficking, Pain, Single Particle Tracking, Spinal Cord
Introduction
GABAARs2 are the major inhibitory transmitter receptors in the central nervous system and the site of action of benzodiazepines, barbiturates, neuroactive steroids, anesthetics, and convulsants. They are ligand-gated chloride channels composed of five subunits that can belong to different subunit classes. The majority of these receptors are composed of one γ, two α, and two β subunits (1–3). GABAARs are widely distributed in the brain (4, 5) and spinal cord (6). Clusters of these receptors can be found at inhibitory synapses mediating phasic inhibition but also at extrasynaptic locations where they are mediating tonic inhibition (7). Using single particle tracking (SPT), it has been demonstrated that most neurotransmitter receptors, including GABAARs, are exchanged between synaptic and extrasynaptic domains by lateral diffusion (8–10). The lateral mobility of receptors can be modulated by interaction with scaffolding molecules such as gephyrin for GABAA (7, 11) and glycine receptors (12, 13). In addition, receptor insertion and removal are considered major determinants in the regulation of receptor number at the cell surface and the strength of GABAergic transmission (14, 15).
The P2X receptor superfamily includes seven different subunits (P2X1–P2X7) (16). P2X2 subunits mainly form homotrimeric receptors but also assemble with P2X3 subunits to form heterotrimeric P2X2/3 receptors (P2X2/3Rs) (17). In contrast to the anion conducting GABAARs, P2X2Rs are permeable to Ca2+, Na+, and K+. Among the various subtypes, P2X2R and P2X3R are enriched in spinal cord (18, 19), where they play a role in sensory transmission and modulation of synaptic function. GABAARs and P2X2Rs have been demonstrated to be co-localized and to functionally interact with each other in the spinal cord and dorsal root ganglion (20, 21). Simultaneous activation of GABAARs and P2X2Rs results in nonadditive currents or “cross-talk” of the receptors (22, 23). Similar cross-talk was also observed between P2X2Rs and other members of the cys-loop receptor family (24 and references therein).
As a starting point for clarifying the dynamics of interaction, here we investigated the interaction of GABAARs and P2X2Rs in more detail. We demonstrate that a small proportion of GABAARs and P2X2Rs already interact with each other in intracellular compartments and then presumably co-traffic to the cell membrane, where they are co-localized extrasynaptically. Activation of P2X2Rs by 2MeS-ATP results in the dissociation of these receptors. Whereas GABAARs are internalized and degraded, P2X2Rs are stabilized and form clusters. Overall, our studies identified a novel mechanism by which GABAAR distribution and dynamics can be modulated by P2X2Rs in the spinal cord.
EXPERIMENTAL PROCEDURES
Plasmids
Wild-type α1, β2, or γ2S subunits of GABAAR from rat brain were cloned into the mammalian expression vector pCI (Promega) as described previously (25) resulting in constructs α1-pCI, β2-pCI, and γ2-pCI. For generation of the constructs α1-ECFP or α1-EYFP, the fluorescence tags ECFP or EYFP were cloned between amino acids +343 and +344 (numbering according to the mature peptide) of the intracellular loop of the α1 subunit by PCR, respectively, using the “gene splicing by overlap extension” technique (26). Restriction sites NheI and AgeI were introduced in the N-terminal part (amino acids −27 to +343) of the α1 sequence, and XhoI and EcoRI were introduced in the C-terminal part (amino acids +344 to +428) of the α1 sequence for subcloning into the pECFP-C1 or EYFP-C1 vector (Clontech). In analogy, for β2-ECFP or β2-EYFP, fluorescence tags were cloned between amino acids +355 and +356 within the loop of the β2 subunit, and for γ2-ECFP or γ2-EYFP, fluorescence tags were cloned between amino acids +361 and +362 within the loop of the γ2 subunit. The fidelity of the final expression constructs was verified by DNA sequencing. Experiments were performed with each of these fluorescent constructs with comparable results. The mutated α1(A160C) construct was generated as described previously (27). The β3-GLV construct was cloned by the gene splicing by overlap extension technique (26) replacing the intracellular loop of the wild-type β3 subunit between amino acids 323 and 425 (of the mature protein) by the amino acid sequence (SQPARAAAIDRW) of the short intracellular loop from the Gloeobacter violaceus protein (GLIC) that is homologous to GABAA receptor subunits (28). P2X2-ECFP and P2X2-EYFP were kindly provided by F. Soto (Washington University, St. Louis). P2X2-FLAG-EGFP was a kind gift of R. D. Murrell-Lagnado (University of Cambridge, UK) (29).
Antibodies
The antibodies anti-α1(1–9), anti-β2/β3(1–13), and anti-γ2(1–33) were generated and affinity-purified as described previously (30–32). A similar approach was used for the generation of antibodies against EGFP protein. EGFP was cloned in pETBlue-2 vector (Novagen) followed by expression in Tuner (DE3) pLacI cells (Novagen). The animals were immunized with full-length EGFP protein, and antibodies were affinity-purified (33). Mouse monoclonal anti-β2/β3 subunit-specific and anti-GFP antibodies were purchased from Millipore and Roche Diagnostics, respectively. Rabbit anti-FLAG antibody was purchased from Sigma. For generation of P2X2R-specific antibodies, surface-exposed residues were selected based on a P2X2R homology model (kindly provided by T. Grutter, Université Louis Pasteur, Illkirch, France) (34). A peptide corresponding to amino acid sequence 205–213 (SQKSDYLKH) of the mature receptor subunit was selected and custom-synthesized (piCHEM, Graz, Austria) with an additional C-terminal cysteine and was coupled to keyhole limpet hemocyanin. Rabbits were immunized with this adduct, and antibodies were purified from the serum of the rabbits by affinity chromatography on a column consisting of the respective peptide coupled to thiopropyl-Sepharose (33).
Cell Culture and Transfection
HEK 293 cells (CRL 1573; American Type Culture Collection, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (BioWhittaker, Lonza), 2 mm glutamine, 50 μm β-mercaptoethanol, 100 units/ml penicillin G, and 100 μg/ml streptomycin in 75-cm2 culture dishes using standard cell culture techniques. HEK 293 cells (3 × 106) were transfected with a total amount of 20 μg of subunit cDNAs via the calcium phosphate precipitation method (35). For co-transfection with four different subunits, 5 μg of cDNA was used for each subunit. The expression of GABAA and P2X2-EYFP/EGFP receptors was kept constant by co-transfection with empty pEYFP-N1 vector or pCI vector. The cells were harvested 48 h after transfection. For live cell confocal imaging or FRET experiments, 150,000 cells were plated over 15- or 24-mm coverslips (Paul Marienfeld GmbH) pre-coated with poly-d-lysine (Sigma) in a 6-well culture dish, respectively. Cells were imaged 24 h after transfection.
Spinal cord neurons from homozygous mrfp-gephyrin knock-in mice were prepared at embryonic day 13 (E13) as described previously (36, 37). Briefly, cells were grown in neurobasal medium supplemented with B27, 2 mm glutamine, and antibiotics (Invitrogen) at 36 °C and 5% CO2. Neurons were transfected 8–9 days after plating using Lipofectamine 2000 (Invitrogen) with 0.5 μg of P2X2-FLAG-EGFP per coverslip. SPT experiment on transfected cells was performed 48 h after transfection.
Co-immunoprecipitation of Total Receptors and Cell Surface Receptors
The culture medium was removed from transfected HEK cells, and cells from four culture dishes were extracted with 1 ml of a C12E10 extraction buffer (1% polyoxyethylene 10 lauryl ether (Sigma), 0.18% phosphatidylcholine (Sigma), 150 mm NaCl, 5 mm EDTA, and 50 mm Tris-HCl, pH 7.4, containing one “Mini Complete protease inhibitor mixture” tablet (Roche Diagnostics)) per 10 ml of extraction buffer for 8–12 h at 4 °C. The extract was centrifuged for 20 min at 45,000 rpm at 4 °C. The protein concentration of the supernatant was determined (Pierce, BCA protein assay kit), and the supernatant was then incubated for 4 h under gentle shaking with 15 μg of α1 subunit-specific antibodies or 3 μg of mouse monoclonal GFP (Roche Diagnostics) antibodies.
When GABAA receptors were precipitated from spinal cord tissue, spinal cords from three animals were pooled, homogenized by an Ultra-Turrax® in artificial cerebrospinal fluid (118 mm NaCl, 3 mm KCl, 1 mm MgCl2, 1.5 mm CaCl2, 1 mm NaH2PO4, 25 mm NaHCO3, and 30 mm glucose) containing 1 tablet of “complete protease inhibitor mixture” (Roche Diagnostics) and 1 phosphatase inhibitor mixture tablet (Roche Diagnostics) per 50 ml of artificial cerebrospinal fluid, saturated with carbogen (95% oxygen, 5% carbon dioxide), and centrifuged for 40 min at 50,000 rpm at 4 °C. Membranes were washed twice by suspension and recentrifugation and extracted for 12 h at 4 °C using a C12E10 extraction buffer containing 1 tablet of complete protease inhibitor mixture (Roche Diagnostics) per 50 ml of extraction buffer. The extract was centrifuged for 40 min at 50,000 rpm at 4 °C. The supernatant was then incubated for 4 h on a roller shaker with a mixture of α1, β2, and γ2 subunit-specific antibodies (15 μg of each).
Proteins bound to antibodies were then precipitated by addition of Pansorbin (formalin-fixed Staphylococcus aureus cells, purchased from Calbiochem) and 0.5% nonfat dry milk powder and shaking for an additional 2 h at 4 °C. The precipitate was washed three times with a low salt buffer for immunoprecipitation (IP low buffer) (50 mm Tris-HCl, 0.5% Triton X-100, 150 mm NaCl, and 1 mm EDTA, pH 8.0). The precipitated proteins were dissolved in sample buffer (NuPAGE LDS sample buffer, Invitrogen). To determine receptors present in the extracts by subsequent Western blots, all proteins present in extracts from spinal cord were precipitated using the chloroform/methanol procedure (38).
Co-immunoprecipitation of receptors expressed at the cell surface was performed according to a protocol described previously (25, 31). For experiments with drug exposure, cells were preincubated with 2MeS-ATP (30 μm, Sigma) or TNP-ATP (10 μm, Sigma) for 60 min. The culture medium was removed from transfected HEK cells, and the cells were washed once with 1× PBS (2.7 mm KCl, 1.5 mm KH2PO4, 140 mm NaCl, and 4.3 mm Na2HPO4, pH 7.3). Cells were then detached from the culture dishes by incubating with 2.5 ml of 5 mm EDTA in PBS for 5 min at room temperature. The resulting cell suspension was diluted in 6 ml of cold Dulbecco's modified Eagle's medium and centrifuged for 5 min at 1500 rpm. The cell pellet from four dishes was incubated with α1(1–9) (35 μg) or FLAG (18 μg, rabbit polyclonal, Sigma) antibodies in 3 ml of the same medium for 45 min at 37 °C. Cells were again pelleted, and free antibodies were removed by washing twice with 6 ml of 1× PBS buffer. The receptors were extracted with IP low buffer containing 1% Triton X-100 for 1 h under gentle shaking conditions that did not lead to a significant dissociation of antibodies from the receptors (25, 31). Cell debris was removed by centrifugation (45,000 rpm for 20 min at 4 °C). Following protein concentration determination, Pansorbin and 0.5% nonfat dry milk powder was added, and after shaking for 2 h at 4 °C, the precipitate was washed three times and dissolved in sample buffer (NuPAGE LDS sample buffer, Invitrogen).
To investigate a possible redistribution of the antibodies during the extraction procedure, in other experiments HEK cells were transfected with wild-type α1, β3, and γ2 subunits as well as a truncated form of γ2 subunits. After cell surface labeling by α1(1–9) antibodies, the extracts containing the cell surface-labeled receptors was divided in two fractions. One fraction was kept at 4 °C for 2 h, and the other fraction was incubated with additional α1(1–9) antibodies at 4 °C for the same time period. Pansorbin was added to both fractions, and the resulting precipitates were centrifuged, washed, dissolved in sample buffer, and subjected to SDS-PAGE and Western blot analysis. In both precipitates, full-length subunits forming complete receptors could be detected, whereas truncated subunits could only be detected in the fraction where additional α1(1–9) antibodies had been added after cell lysis (31).
Western blot analysis was performed using the NuPAGE electrophoresis system (Invitrogen), and precipitated proteins were detected using digoxigenin-labeled antibodies (Roche Diagnostics, DIG protein labeling kit) and sheep anti-digoxigenin antibodies conjugated with alkaline phosphatase (Roche Diagnostics). Secondary antibodies were visualized by the reaction of alkaline phosphatase with CDP Star (Applied Biosystems, Bedford, MA) as described previously (25). To compensate for a possible heterogeneity of expression in different dishes, HEK cells from four culture dishes were pooled for each sample. In addition, four independent experiments were performed.
The chemiluminescent signal of the protein bands on blots of the same gel and exposure time were quantified by densitometry using the Fluor-S MultiImager (Bio-Rad) and evaluated using Quantity One® quantitation software (Bio-Rad). The linear range of the detection system was established by determining the antibody response to a range of antigen concentrations following immunoblotting. The experimental conditions were designed such that immunoreactivities obtained in the assay were within this linear range, thus permitting a direct comparison of the amount of antigen applied per gel lane between the samples. Different exposures of the same membrane were used to ensure that the measured signal was in the linear range.
Live Cell Confocal Imaging and FRET Imaging
The expression of ECFP- and EYFP-tagged receptors in HEK cells were visualized by confocal microscopy using a Zeiss Axiovert 200-LSM 510 confocal microscope (argon laser, 30 milliwatts; helium/neon laser, 1 milliwatt) equipped with an oil immersion objective (Zeiss Plan-NeoFluar ×63/1.3) as described previously (39). Fluorescent protein-tagged constructs were detected with a band pass filter (475–525 nm) using the 458-nm (CFP) or 488-nm (YFP) laser lines. Images were captured sequentially, and overlay images were produced with Zeiss imaging software. Fluorescence resonance energy transfer was measured as described previously (39–41). Briefly, FRET was performed using an epifluorescence microscope (Carl Zeiss Axiovert 200) using the “three-filter method” (39). The images were taken using a ×63 oil immersion objective and Ludl filter wheels to allow for rapid switching between the fluorescence excitation and emission filters for CFP (ICFP, excitation 436 nm and emission 480 nm and dichroic mirror 455 nm), YFP (IYFP, excitation 500 nm and emission 535 nm and dichroic mirror 515 nm), and FRET (IFRET, excitation 436 nm and emission 535 nm and dichroic mirror 455 nm). The images were captured by a CCD camera and analyzed using PixFRET plugin of ImageJ (rsbweb.nih.gov) (42, 43). This program allows the determination of the spectral bleed through of the images generated using ECFP and EYFP filters. A threshold value of 2 was selected, and the FRET images were generated using the following formula: NFRET = (IFRET − BTCFP × ICFP − BTYFP × IYFP)/(ICFP × IYFP)1/2, where BT indicates bleed through and I indicates intensity (44). The computed FRET images are visualized on 256 bit color level; the minimum value displayed is in black and maximum value is in white.
Immunocytochemistry, Image Acquisition, and Analysis
The 11–12 DIV neurons were fixed for 15 min in 4% (w/v) paraformaldehyde in PBS. Cells were then incubated for 30 min in 5% (w/v) bovine serum albumin (BSA, Sigma) to block nonspecific staining and then incubated for 2 h with primary antibodies in 5% BSA. After washing, cells were incubated for 45 min with secondary antibodies conjugated to appropriate fluorophores. Following washes, the coverslips were mounted on slides with Vectashield (Vector Laboratories). For experiments involving drug treatment, cells were incubated with 2MeS-ATP (100 μm) or TNP-ATP (100 μm) for 2 h at 37 °C before fixation. The primary antibodies used were mouse monoclonal anti-β2/β3 subunit-specific (extracellular, clone bd17, 1:100, Millipore) and rabbit anti-P2X2 receptor-specific (extracellular, 5 μg/ml). Inhibitory synapses were labeled by mrfp-gephyrin clusters. Secondary antibodies were Cy5-conjugated goat anti-mouse or FITC-conjugated donkey anti-rabbit (1:400, Jackson ImmunoResearch). Fluorescent images were acquired under identical conditions using Leica DM5000B spinning disk microscope using 491-nm laser (Cobolt CalypsoTM, 50 milliwatts), 561-nm laser (Cobolt JiveTM, 50 milliwatts), and 633-nm laser (Coherent Cube, 25 milliwatts).
Images were processed using Metamorph software (Meta Imaging, Downington, PA). For quantitative analysis, GABAAR, P2X2R, and gephyrin images were processed with multidimensional image analysis interface that employs two-dimensional object segmentation by wavelet transformation (11). Fluorescence intensity was normalized by determining the pixel with highest intensity under control conditions. All other pixels either under control conditions or after drug treatment were divided by this value. Objects composed of ≥3 pixels were defined as clusters. GABAAR clusters were considered synaptic when at least 1 pixel overlaps with mrfp-gephyrin clusters.
Live Cell Staining and Quantum Dot Imaging
Labeling of receptors for SPT of GABAARs was performed as described previously (11). Neurons were incubated with anti-GABAAR γ2 subunit-specific antibodies (1:100; Alomone Labs) for 5 min and then washed and incubated for 5 min in biotinylated anti-rabbit Fab antibody (1:200; Jackson ImmunoResearch). Following washes, coverslips were then incubated for 1 min with 1 nm streptavidin-coated QDs emitting at 655 nm (Invitrogen) in borate buffer (45). Incubation with antibodies and washes was performed at 37 °C in the imaging medium. Cells were washed and imaged in the presence of appropriate drugs. For SPT analysis of P2X2Rs, P2X2-FLAG-EGFP-transfected neurons were incubated with low concentrations of anti-FLAG antibody (1:1500, rabbit polyclonal, Sigma) for 5 min followed by secondary and QD labeling similar to GABAAR labeling.
Neurons were imaged at 37 °C using an inverted microscope (IX71, Olympus) equipped with an oil immersion objective (Olympus, 60×, NA 1.45), a xenon lamp, and cooled CCD camera Cascade+128 (Roper Scientific). Fluorescent signals were detected using appropriate filter sets (QD: D455/70x and HQ655/20; GFP, HQ500/20 and HQ535/30; mrfp, D535/50 and E590lpv2). The movement of QDs on the dendrites was recorded with an integration time of 75 ms with 500 consecutive frames (37.5 s). The recording was done maximum up to 20 min after drug addition.
Single Particle Tracking and Analysis
Tracking and analysis of QDs has been well described recently (11, 46). Briefly, QDs were detected by cross-correlating the image with a Gaussian model of the point spread function, and the diffusion parameters were calculated using custom software (13, 47, 48) using Matlab (The Mathworks Inc., Natick, MA). Single QDs were identified by intermittent fluorescence (i.e. blinking). The spots in a given frame were connected with the maximum likely trajectories estimated on previous frames of the image sequence. Only trajectories with at least 15 consecutive frames were used for further analysis. Synaptic area was defined by processing mrfp-gephyrin images with the multidimensional image analysis interface. GABAAR QDs were classified as “synaptic” when the trajectories overlapped with synaptic area. The trajectories were considered “extrasynaptic” when they are ≥2 pixels away from the synapse. P2X2R QDs were rarely observed at/near mrfp-gephyrin clusters, so the analysis was performed independent of inhibitory synapse localization. The mean square displacement (MSD) was calculated using Equation 1,
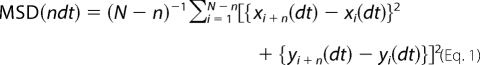 |
where xi and yi are the coordinates of an object on frame i; N is the total number of steps in the trajectory; dt is the time interval between two successive frames; and ndt is the time interval over which displacement is averaged. The diffusion coefficient D was calculated by fitting the first two to five points of the MSD plot versus time with Equation 2,
where σx is the spot localization accuracy in one direction (13). Given the resolution, trajectories with D <104 μm2/s for QDs were classified as immobile. The size of the average confinement area was calculated fitting the average MSD plot with the equation proposed in Ref. 46. Dwell time was calculated as described previously (48, 49).
Statistics and Image Preparation
Statistical analysis was performed using GraphPad Prism 4 (GraphPad Software Inc.) and Microsoft Excel (Microsoft Corp.). Images were prepared using Microsoft PowerPoint 2007 (Microsoft Corp.), Adobe Photoshop CS2 (Adobe Systems),and CorelDraw X3 (Corel Corp.). The supplemental movies were prepared using After Effects CS5 (Adobe System).
RESULTS
Intracellular Oligomerization and Co-trafficking of GABAARs and P2X2Rs
We first investigated whether GABAARs and P2X2Rs are able to interact directly. For that, receptors were extracted from HEK cells transfected as indicated in Fig. 1A and were subjected to immunoprecipitation using antibodies against the α1 subunit of GABAARs. Precipitated receptors were subjected to SDS-PAGE and Western blot analysis. Because the α1 antibodies were not able to directly precipitate β2 or γ2 subunits (experiments not shown), co-precipitation of β2 and γ2 subunits indicated their assembly with α1 subunits (Fig. 1A, 1st lane). The α1 antibodies did not directly precipitate EYFP-labeled P2X2Rs (Fig. 1A, 2nd lane). Their precipitation from cells co-transfected with GABAARs and EYFP-tagged P2X2Rs (Fig. 1A, 3rd lane) thus indicates an association of these receptors with GABAARs. Interestingly, however, the total amount of GABAARs precipitated by α1 antibodies was increased on co-expression with P2X2Rs (Fig. 1A, 1st and 3rd lanes).
FIGURE 1.

Co-immunoprecipitation and co-trafficking of GABAARs and P2X2Rs. HEK cells were co-transfected with GABAAR α1, β2, and γ2 subunits and/or EYFP-tagged P2X2R subunits (P2X2-EYFP) or a P2X2-FLAG-EGFP receptors having the FLAG tag in the extracellular region and EGFP at the C terminus, as indicated. GABAARs or P2X2Rs were immunoprecipitated with rabbit anti-α1 subunit-specific antibodies or mouse anti-GFP antibodies, respectively. This was followed by SDS-PAGE and Western blot analysis using digoxigenin-labeled α1-Dig, β2-Dig, γ2-Dig, and EGFP-Dig antibodies. A, co-immunoprecipitation of GABAARs and P2X2Rs from total cell extracts. Results are from a typical experiment analyzed on a single SDS gel and performed four times with comparable results. Co-expression of GABAARs and P2X2Rs caused an increase in total GABAARs but not in P2X2Rs as determined from the same gel and blots using the same exposure time. Because of the different precipitation and detection efficiencies of the antibodies used, however, staining intensity cannot be used for estimating the extent of co-association of receptors. Because of the low extent of co-localization of the receptors, Western blots sometimes also had to be exposed for different time periods to allow visualization of weakly stained co-precipitated bands. Different exposures were then cut and recombined to generate the figure shown. B, co-immunoprecipitation of cell-surface GABAARs and P2X2Rs. The quantification of surface receptors was performed in blots from the same gel and exposure time. The surface expression of GABAARs (left upper three lanes, left bar graphs) but not that of P2X2Rs (right lower three lanes, right bar graphs) decreased on co-expression of GABAARs with P2X2Rs. Co-transfection with the trafficking-deficient α1(A160C) GABAAR subunit caused a 91% reduction of GABAA and a 21% reduction of P2X2Rs at the cell surface (***, p < 0.001, t test, n = 4 independent experiments). ns, not significant.
In parallel experiments, the extracted receptors were immunoprecipitated with mouse monoclonal anti-GFP antibodies. These antibodies did not directly precipitate GABAAR subunits (Fig. 1A, 4th lane). Co-precipitation of α1, β2, and γ2 subunits (Fig. 1A, 6th lane) thus again indicates association of GABAARs with EYFP-tagged P2X2Rs. Interestingly, the amount of P2X2Rs precipitated was comparable in the absence or presence of GABAARs (Fig. 1A, 5th and 6th lanes). It is also important to note that only a very small fraction of GABAARs was associated with P2X2Rs and vice versa (Fig. 1A, 3rd and 6th lanes).
To investigate whether the two receptors also interact at the cell surface, receptors were first labeled with antibodies directed against the extracellular N terminus of the GABAAR α1 subunit or against the FLAG tag in the extracellular loop of P2X2Rs (P2X2-FLAG-EGFP) followed by protein extraction and precipitation of the antibody-labeled receptors by Pansorbin (see under “Experimental Procedures”). Results from a typical experiment are shown in Fig. 1B. Western blotting indicated that expression of GABAARs was reduced at the cell surface by 25.6 ± 0.7% (mean ± S.E., p < 0.0001, n = 4 independent experiments; Fig. 1B, 1st and 2nd lanes) when P2X2Rs were co-expressed, even though we observed an increase in total GABAARs under these conditions (Fig. 1A).
P2X2Rs and GABAARs could also be co-precipitated at the cell surface when antibodies against the extracellular FLAG tag were used (Fig. 1B, 5th lane). In contrast to GABAARs, we observed no significant change in the surface expression of P2X2Rs on co-expression of GABAARs (surface level reduced by 5.3 ± 3.3%, mean ± S.E., p = 0.14, n = 4 independent experiments; Fig. 1B, 4th and 5th lanes). As observed for total receptors, only a small fraction of GABAARs and P2X2Rs associate with each other.
The observed down-regulation of surface GABAARs in the presence of P2X2Rs (Fig. 1B) might have been caused by an overexpression-induced altered maturation of GABAAR in the endoplasmic reticulum. To investigate this possibility, we aimed to co-express P2X2Rs with another membrane protein that does not interact with this receptor. For that, we generated a mutated β3 subunit (β3-GLV) in which the large intracellular loop between transmembrane domains 3 and 4 was replaced by the loop of the related G. violaceus subunit sequence (28). Because the intracellular loop between the third and fourth transmembrane domain of β-subunits has been reported to prevent cross-talk between the two receptors (22), it might represent the site of interaction of GABAAR with P2X2R. We thus argued that its absence might prevent the interaction of these receptors. As β3 wild-type subunits (β3-WT) form homopentameric receptors, we compared the change in surface expression of β3-WT and β3-GLV GABAARs in the presence of P2X2Rs (supplemental Fig. 1). Co-expression of β3-WT GABAARs with P2X2Rs resulted in a reduced surface expression of these GABAARs as observed for co-expression with α1β2γ2 receptors, whereas this effect was not observed with β3-GLV GABAARs. This indicates that the reduction of GABAARs at the cell surface was not caused by an overexpression-induced slow maturation of proteins in the endoplasmic reticulum but by a direct interaction between the two receptors.
To investigate whether the two receptors co-traffic to the cell surface, we generated a trafficking-deficient GABAAR where the α1 subunit had an alanine to cysteine mutation (α1-A160C). This mutant assembles with other subunits of GABAARs (supplemental Fig. 2) but does not reach the cell surface (surface level reduced by 91.4 ± 0.4%, p < 0.0001, n = 4 independent experiments; Fig. 1B, 1st and 3rd lanes). We hypothesized that if the two receptors are co-trafficking, the intracellular retention of GABAARs should also retain the associated P2X2Rs. In fact, trafficking-deficient GABAARs reduced the cell surface expression of P2X2Rs by 21 ± 2.7% (p < 0.0001, n = 4 independent experiments; Fig. 1B, 4th and 6th lanes). Under these conditions, no associated GABAARs and P2X2Rs were detectable at the cell surface (Fig. 1B, 3rd and 6th lanes). Together, we conclude that GABAARs and P2X2Rs associate with each other in intracellular compartments and co-traffic to the cell surface.
Intracellular and Surface Co-localization of GABAARs and P2X2Rs, Indicated by Confocal Microscopy and FRET
To further characterize this interaction, we generated fluorescent constructs of GABAARs having ECFP or EYFP tags in the large intracellular loop of subunits (supplemental Fig. 3). P2X2Rs having ECFP or EYFP tags in the intracellular C-terminal domain have been described previously (50). HEK cells were then co-transfected with P2X2-ECFP and P2X2-EYFP subunits or with GABAAR α1-ECFP, β2 and γ2 subunits, and P2X2-EYFP subunits. 24 h after transfection, receptor expression and distribution in living cells were imaged using a confocal microscope (supplemental Fig. 4). As expected, P2X2-ECFP and P2X2-EYFP subunits were strongly co-localized at the cell membrane, as well as in intracellular compartments. For cells expressing GABAA-ECFP and P2X2-EYFP receptors, we also observed co-localization in the intracellular compartment as well as at the cell surface. Apparently, co-transfection of single subunits of GABAARs (α1-ECFP, β2-ECFP, or γ2-ECFP) with P2X2-EYFP subunits resulted in a differential localization of the two fluorophores. Whereas, P2X2-EYFP receptors were mainly localized at the cell surface, single subunits were confined to the endoplasmic reticulum (supplemental Fig. 4). In addition, cell surface precipitation experiments indicated that single GABAAR subunits do not traffic to the cell surface in the absence or presence of P2X2R (experiments not shown). This indicates that only fully assembled GABAARs seem to associate with P2X2Rs and are co-transported to the cell surface.
To investigate a possible direct interaction of GABAARs and P2X2Rs, we performed FRET experiments on appropriately transfected HEK cells. FRET images obtained were processed using pixFRET plugin of ImageJ to visualize the FRET signal in pseudo-color (Fig. 2A) (42, 43). Co-transfection of P2X2-ECFP and P2X2-EYFP subunits generated homotrimeric P2X2Rs where the donor (ECFP) and the acceptor (EYFP) are sufficiently close to resulting in an intense FRET signal (50). Similarly, an intense FRET signal was observed for the GABAA-ECFP/P2X2-EYFP pair, whereas a negligible signal was observed when ECFP and EYFP were co-transfected without being bound to receptor subunits (Fig. 2A). Similar to previous observations from confocal imaging, FRET between P2X2-ECFP/P2X2-EYFP pair was not only observed at cell membranes (identified by their intense signal at the border of the cells) but also in intracellular regions (identified by a diffuse signal distributed within the cell). The average FRET intensity (±S.E.) measured for P2X2-ECFP/P2X2-EYFP pair in the cytosol (130.9 ± 5.1 arbitrary units, n = 48 cells) and at cell membranes (131.6 ± 5.4 arbitrary units, n = 48 cells) was similar (p = 0.9, t test). Similarly, FRET between donor protein, GABAA-ECFP, and acceptor P2X2-EYFP was observed at cell membranes as well as in intracellular compartments. The FRET intensities for GABAA-ECFP/P2X2-EYFP receptors in the cytosol (104.4 ± 5.3 arbitrary units, n = 46 cells) and at the cell surface (104.9 ± 4.2 arbitrary units, n = 46 cells) were comparable (p = 0.9, t test) (Fig. 2B). The FRET intensity measured for cells expressing ECFP/EYFP was negligible (data not shown, Fig. 2A).
FIGURE 2.

Intracellular and surface FRET between GABAARs and P2X2Rs. HEK cells were co-transfected with P2X2-ECFP and P2X2-EYFP, pECFP and pEYFP, or α1-ECFP, β2, γ2, and P2X2-EYFP subunits. A, FRET images obtained are depicted in pseudo-color code. Results indicate a clear FRET signal between P2X2-ECFP and P2X2-EYFP or GABAA-ECFP and P2X2-EYFP subunits. Examples of region of interest (white) on cell surface to compute FRET are shown. ***, p < 0.05. B, average FRET intensity (±S.E.) for P2X2-ECFP/P2X2-EYFP receptors is not significantly different at the cell membrane and in the intracellular compartment (ns, p = 0.922, n = 48 cells, t test). Similarly, a strong FRET signal was measured for GABAA-ECFP/P2X2-EYFP receptors at the cell membrane and in the intracellular compartment (ns, p = 0.992, n = 48 cells, t test). C and D, cell-by-cell analysis indicated that the FRET intensity at the membrane of individual cells and in their intracellular compartment did not vary significantly for cells transfected with P2X2-ECFP/P2X2-EYFP receptors (ns, p = 0.429, n = 38 cells, paired t test) and GABAA-ECFP/P2X2-EYFP receptors (ns, p = 0.197, n = 38 cells, paired t test).
To rule out that the similar FRET intensity values resulted from averaging data from different cells, we performed cell-by-cell FRET intensity analysis. This allowed us to calculate possible changes in FRET intensity between cytosol and membrane receptors due to a change in distance between fluorophores during co-trafficking. However, cell-by-cell intensity analysis revealed no significant difference between intracellular and membrane FRET for both P2X2-ECFP/P2X2-EYFP pair (p = 0.429, n = 38, paired t test) and GABAA-ECFP/P2X2-EYFP pair (p = 0.197, n = 38, paired t test) (Fig. 2, C and D). Altogether, these results suggest that GABAARs associate with P2X2Rs before reaching the cell surface, possibly in the endoplasmic reticulum. Moreover, the comparable FRET intensity in the cytosol and at the cell membrane suggests that there was no significant change in the distance between the donor and acceptor during trafficking from the cytosol to the cell membrane.
Extrasynaptic Co-localization and Co-immunoprecipitation of GABAARs and P2X2Rs in Spinal Cord Neurons
P2X2Rs are highly expressed in spinal cord either as homotrimeric P2X2Rs or as heterotrimeric P2X2/3Rs. To study whether these receptors interact with endogenous GABAARs, we performed immunolabeling of receptors at the surface of spinal cord neurons. Such neurons were cultured from mrfp-gephyrin knock-in mice (36, 37) where the inhibitory synapses can be identified by visualizing mrfp-gephyrin clusters. 10–11 DIV neurons were stained using rabbit antibodies against the extracellular region of P2X2Rs (supplemental Fig. 5) and mouse monoclonal anti-β2/β3 subunit-specific antibodies to label the extracellular domain of β2/β3 subunits of GABAARs. Immunostaining was performed in the absence of detergent to label only surface receptors. The images were acquired by a spinning disk confocal microscope using a ×63 magnification objective. Cells showing good fluorescence signal for both P2X2Rs and GABAARs were imaged, and acquisition conditions were kept constant during the experiment. P2X2Rs show a clear labeling over the surface of the cell body as well as over dendrites. In contrast, GABAARs are highly enriched in dendrites and much less over the cell body. Merged images demonstrate that indeed the two receptors co-localize with each other as indicated by the tightly associated red and green dots shown in Fig. 3A, top panel. Quantitative analysis (±S.E.) shows that 7.1 ± 0.5% of β2/β3 subunit containing GABAAR clusters co-localize with P2X2R clusters, whereas 20.6 ± 0.9% of P2X2R clusters overlap with GABAAR clusters (n = 55 cells, four independent experiments from four different cultures). Multidimensional image analysis images (see under “Experimental Procedures”) for a section of dendrite is shown for P2X2Rs, GABAARs, and overlaid channels (Fig. 3A, bottom panel). The mrfp-gephyrin (shown in Fig. 3, B–D, blue) images were also acquired simultaneously along with the two receptors. Quantitative analysis of the merged images for gephyrin/P2X2 (Fig. 3B), gephyrin/GABAA (Fig. 3C), and gephyrin/P2X2/GABAA (Fig. 3D) demonstrate that only 3.4 ± 0.3% (±S.E.) of P2X2Rs exist at inhibitory synapses (n = 40 cells, four independent experiments from four different cultures). Altogether, we demonstrate co-existence of GABAAR/P2X2R clusters in cultured spinal cord neurons. Furthermore, as P2X2Rs are very rare at inhibitory synapses, we conclude that GABAARs and P2X2Rs co-localize mainly at extrasynaptic localizations.
FIGURE 3.

Extrasynaptic co-localization of P2X2Rs and GABAARs in spinal cord neurons. Cultured spinal cord neurons (10–11 DIV) from mrfp-gephyrin knock-in mice were stained for P2X2Rs and GABAARs (β2/β3-subunit) without permeabilization. P2X2Rs (green, FITC), GABAARs (red, cy5), and gephyrin (blue, mrfp) were visualized using a spinning disk confocal microscope. A, P2X2Rs are expressed both over the surface of the cell body as well as over the dendrites, whereas GABAARs are mainly enriched over dendrites. Lower panel represents a section of a dendrite (boxed region) after multidimensional image analysis (MIA) (see “Experimental Procedures”). Two representative co-localized clusters depict GABAARs and P2X2Rs that co-localize at synaptic (line arrow) and extrasynaptic (block arrow) locations. B–D, overlaid images for P2X2Rs and gephyrin, GABAARs and gephyrin, and all three channels. Line arrow represents all three proteins co-localized, and other arrows depict co-localization of only two proteins. Scale bar, 10 μm. E, co-immunoprecipitation (IP) of P2X2Rs along with GABAARs from spinal cord protein extract. Spinal cord extract was subjected to precipitation using a mixture of rabbit anti-α1, anti-β2, and anti-γ2 antibodies followed by precipitation of the antibody-bound receptors using Pansorbin cells (4th lane). For negative control, only Pansorbin cells but no primary antibodies were used (3rd lane). P2X2Rs were detected using digoxygenized rabbit anti-P2X2R (P2X2-Dig) antibodies. Dioxygenation drastically reduced the affinity of the rabbit P2X2R-antibodies, explaining the weak signal of these receptors on co-precipitation with GABAA receptors (4th lane) as well as in total extract (2nd lane).
In the experiments of Fig. 3, GABAA receptors were not extensively co-localized with gephyrin. To further investigate this low co-localization, we performed double labeling of spinal cord neurons from mrfp-gephyrin knock-in mice with GABAARs and GlyRs (supplemental Fig. 6). In agreement with previous reports (7, 51), results indicate that the majority of inhibitory post-synaptic gephyrin clusters in spinal cord neurons are associated with GlyRs and not with GABAAR. A large fraction of GABAARs was localized extrasynaptically, where they are partially co-localized with P2X2Rs.
To additionally confirm that GABAARs and P2X2Rs interact in vivo, we performed co-immunoprecipitation experiments of the two receptors from spinal cord tissue (Fig. 3E). For that, we immunoprecipitated spinal cord extracts with a combination of rabbit antibodies directed against the α1, β2, and γ2 subunits to pull down the majority of GABAARs containing these subunits. The antibodies used had been demonstrated previously to not directly precipitate P2X2Rs (see also Fig. 1). Co-precipitation of P2X2Rs was then demonstrated in Western blots using digoxigenin-labeled P2X2R antibodies (Fig. 3E, 4th lane). The protein band labeled was identical in molecular mass to that of P2X2R identified by these antibodies in brain extracts (Fig. 3E, 2nd lane). The results of the 4th lane in Fig. 3E were not due to unspecific adsorption to the Pansorbin used in these immunoprecipitation experiments, because no protein band was detected in the absence of the GABAAR subunit antibodies during immunoprecipitation experiments (3rd lane). Because of the poor precipitation capability of our P2X2Rs antibodies, we could not perform the reverse co-precipitation.
Modulation of GABAAR and P2X2R Distribution by Purinergic Drugs in Spinal Cord Neurons
We were interested to see if activation or deactivation of P2X2Rs has any effect on the strength of this association. 10–11 DIV spinal cord neurons were first incubated with the P2XR agonist 2MeS-ATP, or the antagonist TNP-ATP, for 2 h followed by immunostaining. A section of dendrites stained for GABAARs β2/β3 subunit (red) and P2X2Rs (green) is shown for all conditions (Fig. 4A). 2MeS-ATP treatment had no visible effect on GABAAR fluorescence intensity, but at the same time P2X2R clusters showed increased fluorescence intensity (Fig. 4A, 2nd row). Quantitative analysis indicated no significant change in the intensity of both synaptic and extrasynaptic GABAARs but a significant up-regulation of the fluorescence intensity of total P2X2Rs (Fig. 4B).
FIGURE 4.

Regulation of GABAA and P2X2Rs immunoreactivity by purinergic drugs. Spinal cord neurons (10–11 DIV) were treated for 2 h with 2MeS-ATP (100 μm) or TNP-ATP (100 μm) and stained for GABAARs (β2/β3 subunit, red, cy5) and P2X2Rs (green, FITC). Inhibitory synapses were identified based on gephyrin clusters. A, representative dendrite is shown for different conditions. B, normalized fluorescence intensity value (±S.E.) for total GABAARs: control, 0.81 ± 0.02, n = 43; 2MeS-ATP, 0.85 ± 0.02, n = 44 (ns, p = 0.205); TNP-ATP, 0.90 ± 0.02, n = 44 (**, p < 0.001); for synaptic GABAARs: control, 0.85 ± 0.02, n = 15; 2MeS-ATP, 0.85 ± 0.03 (n = 15; ns, p = 0.851); TNP-ATP, 0.93 ± 0.03, n = 15 (*, p < 0.05); for extrasynaptic GABAARs: control, 0.90 ± 0.02, n = 15; 2MeS-ATP, 0.91 ± 0.03, n = 15 (ns, p = 0.948), TNP-ATP, 1.04 ± 0.02, n = 15 (***, p < 0.0001); for total P2X2Rs: control, 0.76 ± 0.05, n = 24, 2MeS-ATP, 1.25 ± 0.06, n = 30 (***, p < 0.0001); TNP-ATP, 1.24 ± 0.05, n = 31 (***, p < 0.0001). Scale bar, 10 μm.
However, the competitive P2XR antagonist TNP-ATP strongly elevated the fluorescence signal for both GABAARs and P2X2Rs (Fig. 4A, 3rd row). Quantitative analysis of fluorescence intensity of GABAAR and P2X2R clusters shows that TNP-ATP treatment significantly enhanced the fluorescence intensity of both receptors (Fig. 4B). Even though pharmacological modulation resulted in re-distribution of GABAARs and P2X2Rs, we observed no significant change in the percentage of co-localized receptor clusters (data not shown). Together, these results suggest that purinergic receptors can directly modulate the distribution of not only P2X2Rs but also of GABAARs.
Regulation of GABAAR Diffusion Dynamics by Drugs Acting on Purinergic Receptors
We performed single particle tracking using the quantum dot technique to evaluate the effects of P2XR agonists or antagonists on the lateral diffusion of GABAARs. The experiments were performed on spinal cord neurons from mrfp-gephyrin knock-in mice allowing the direct visualization of inhibitory synapses. The drugs were added to the imaging medium after labeling the receptors with quantum dots (GABAAR-QD). The effect of drugs on receptor diffusion could be evaluated from the area explored by GABAAR-QD trajectories. The activation of P2XRs by 2MeS-ATP (100 μm) increased, whereas the antagonist TNP-ATP (100 μm) decreased the surface explored by GABAARs (Fig. 5A and supplemental movie 1). The cumulative frequency distribution of diffusion coefficient (D) for extrasynaptic (p < 0.05) and synaptic (not significant) receptors (Fig. 5B) indicated an overall but weak increase in the diffusion rate in the presence of the P2XR agonist 2MeS-ATP. In contrast, the antagonist strongly reduced the diffusion of both synaptic (***, p < 0.001) and extrasynaptic (***, p < 0.001) GABAAR-QD (Fig. 5B, green).
FIGURE 5.

Modulation of GABAAR membrane dynamics by purinergic drugs. The γ2 subunit of GABAARs was labeled with QD in spinal cord neurons (DIV 11–12) from mrfp-gephyrin mice. A, examples of surface exploration by GABAAR-QDs (green) for 38.4 s in the presence of 2MeS-ATP (100 μm) and TNP-ATP (100 μm). mrfp-gephyrin (red clusters) represents inhibitory synapses. Notably, GABAAR-QDs explored a larger surface area in the presence of agonist, although their surface exploration is greatly diminished in the presence of antagonist (supplemental movie 1). B, cumulative frequency distribution of diffusion coefficient D for GABAAR-QD trajectories in the synapse (control, n = 293; 2MeS-ATP, n = 279; TNP-ATP, n = 150) and outside the synapse (control, n = 858; 2MeS-ATP, n = 1016; TNP-ATP, n = 1206). Agonist accelerated extrasynaptic receptors, although antagonist slowed down both synaptic and extrasynaptic receptors. Values are from four independent experiments from four different cultures (*, p < 0.05; ***, p < 0.005; Kolmogorov-Smirnov test). C, average (±S.E.) MSD over time for GABAAR-QDs at synaptic and extrasynaptic sites. D, average confinement size (μm) of synaptic and extrasynaptic GABAAR-QDs indicates that TNP-ATP-treated extrasynaptic receptors were more confined in a microdomain. E, percentage of immobile (D <10−4 μm2/s) synaptic and extrasynaptic receptors. The number of immobile (±S.E.) synaptic receptors was 8.3 ± 3.4% for control (n = 134), 9.4 ± 3.8% for 2MeS-ATP (n = 139), and 5.2 ± 2.7% for TNP-ATP (n = 97)-treated cells, and extrasynaptic receptors was 11.8 ± 0.9% for control (n = 858), 11.7 ± 1.9% for 2MeS-ATP (n = 1016), and 18.1 ± 1.9% for TNP-ATP (n = 1377)-treated cells. Note that the agonist had no distinct effect on the proportion of immobile GABAAR-QDs, whereas the antagonist significantly increased the number of immobile extrasynaptic GABAAR-QDs. (***, p < 0.001, t test.) Scale bar, 5 μm.
Synaptic and extrasynaptic trajectories of GABAARs were further analyzed using the MSD plotted as a function of time (Fig. 5C). The negative bent of the MSD curve indicates the level of confinement of receptors in a given subdomain (11, 52). 2MeS-ATP treatment slightly increased the slope of the average MSD for both synaptic and extrasynaptic receptors (Fig. 5C, red). On the other hand, TNP-ATP decreased the average slope of the MSD for synaptic receptors, and this effect was even more dramatic for extrasynaptic GABAARs (Fig. 5C, green). These changes in the GABAAR QDs MSD curves are consistent with the modulations observed for the diffusion coefficients (Fig. 5B). The size of the microdomains in which receptors are confined can be calculated from the MSD curves (see under “Experimental Procedures”) (11, 49). At synapses, the size of the GABAAR confinement domain was not modified by drug treatment (control: 0.18 ± 0.02 μm, n = 93; 2MeS-ATP: 0.19 ± 0.02 μm, n = 111; TNP-ATP: 0.16 ± 0.02 μm, n = 69) (Fig. 5D, black). At extrasynaptic locations, agonist treatment had no effect on the confinement domain, but it is significantly reduced by the antagonist indicating that the receptors were more confined (control: 0.26 ± 0.01 μm, n = 545; 2MeS-ATP: 0.29 ± 0.01 μm, n = 615; TNP-ATP: 0.12 ± 0.01 μm, n = 718) (Fig. 5D, gray). These observations are in line with the modification of the shape of the MSD plots (Fig. 5C). The reduction in the size of the confinement domain suggested that some extrasynaptic receptors may be stabilized in the presence of TNP-ATP. This can also be estimated by the proportion of immobile (D <10−4 μm2/s) receptors. As expected, only TNP-ATP treatment increased the number of immobile extrasynaptic receptors, which almost doubled (Fig. 5E, gray).
Modulation of P2X2R Diffusion Properties by Purinergic Drugs
We then explored how the membrane dynamics of P2X2Rs itself could be modulated by purinergic drugs. SPT experiments were performed using P2X2-FLAG-EGFP receptor transfected in spinal cord neurons (see under “Experimental Procedures”). P2X2R-QDs were only rarely observed at gephyrin-positive synapses (data not shown); this is why we have analyzed the global pool of P2X2Rs on the neuronal membrane independent of inhibitory synaptic localization. The surface explored by QDs over the acquisition period emphasizes the effects of the drug on lateral diffusion (Fig. 6A and supplemental movie 2) and showed that the overall explored surface area was reduced with both the agonist and the antagonist (Fig. 6A). The distribution of diffusion coefficient, D, was not significantly different between the 2MeS-ATP and the control experiments (not significant, p = 0.226, n = 495 for control and n = 481 for 2MeS-ATP, Kolmogorov-Smirnov test) (Fig. 6B, blue and red). However, as indicated by the MSD plot, the confinement of P2X2R-QDs increased in the presence of 2MeS-ATP compared with the control (Fig. 6C, blue and red). The antagonist treatment lowered the diffusion of P2X2R-QDs (***, p < 0.005, n = 495 for control and n = 475 for TNP-ATP) (Fig. 6B, blue and green) and also increased the confinement (Fig. 6C, blue and green). Thus, the mechanisms leading to reduced diffusion of P2X2Rs in the presence of 2MeS-ATP or TNP-ATP are different (53). In both cases there is an increase in confinement, but the decrease in diffusion coefficient was observed only for the latter. In the case of 2MeS-ATP, this indicates that the diffusion rate was not affected by the binding of agonist but that the surface area in which diffusion take place was reduced. This is in favor of multiple binding events of short dwell time with unbiased diffusion between them. In the case of TNP-ATP, the antagonist rather led to long binding events that reduced the overall lateral diffusion and increased the confinement.
FIGURE 6.

Regulation of P2X2R diffusion dynamics by purinergic drugs. Spinal cord neurons (DIV 9) were transfected with P2X2-FLAG-EGFP receptors having the FLAG tag in the extracellular region and EGFP at the C terminus. EGFP was used to identify transfected cells, and single particle QD tracking of P2X2Rs was performed by labeling the FLAG tag of the receptor. A, examples of surface explored by P2X2R-QDs (red) over transfected cell (green) for different conditions (supplemental movie 2). B, cumulative frequency distribution of diffusion for P2X2R-QDs trajectories shows no significant change in receptor diffusion on agonist treatment, whereas antagonist treatment significantly slowed down the receptors (***, p < 0.001, Kolmogorov-Smirnov test). C, average MSD plot for trajectories of P2X2R-QDs shows that 2MeS-ATP as well TNP-ATP increased the confinement of the P2X2Rs. These are typical results from three independent experiments.
Calcium-dependent and an Apparently Calcium-independent Regulation of GABAAR Dynamics by P2X2Rs
In the hippocampus, GABAAR diffusion dynamics is known to be regulated by Ca2+ influx through N-methyl-d-aspartic acid receptors (11). To investigate whether in spinal cord neurons P2XR-mediated Ca2+ influx can regulate GABAAR diffusion dynamics, we performed SPT experiments in the absence or presence of the Ca2+ chelator EGTA (0.5 mm). The increased mobility of GABAAR induced by 2MeS-ATP (Fig. 5) could be reduced in the presence of EGTA (Fig. 7), indicating that this effect is Ca2+-dependent. In the absence of 2MeS-ATP, EGTA even further reduced the mobility of GABAARs, possibly demonstrating an additional Ca2+-independent effect of this compound on GABAARs. We cannot exclude, however, that this additional Ca2+-independent effect was also caused by a P2X2R-mediated influx of Ca2+ that was not chelated by EGTA.
FIGURE 7.

Role of Ca2+ in regulation of GABAAR dynamics. The γ2 subunit of GABAARs was labeled with QD in spinal cord neurons (DIV 11–12) from mrfp-gephyrin mice. Cumulative frequency distribution of diffusion coefficient D for GABAAR-QD trajectories are shown independent of localization (2MeS-ATP, n = 126; EGTA, n = 119; 2MeS-ATP + EGTA, n = 123). ***, p < 0.005; Kolmogorov-Smirnov test.
Modulation of GABAAR and P2X2R Interaction by Purinergic Drugs
SPT experiments suggested that an agonist of P2X2Rs increased but an antagonist decreased the mobility of extrasynaptic GABAARs at the cell surface. To further study this effect, we investigated whether the pharmacological regulation of P2X2Rs by purinergic drugs alters the amount of associated GABAARs at the surface of HEK cells. Cell surface co-immunoprecipitation was performed as described in Fig. 1. Cells expressing GABAAR α1, β2, and γ2 subunits as well as P2X2-EYFP subunits were incubated with either 2MeS-ATP (30 μm) or TNP-ATP (10 μm) for 1 h. Surface GABAARs were immunolabeled by α1 subunit-specific antibodies followed by extraction and precipitation of antibody-labeled receptors by adding Pansorbin cells. The remaining intracellular GABAARs were subsequently precipitated by incubating with α1 subunit-specific antibodies. Changes in surface expression were then measured by Western blotting. 2MeS-ATP treatment reduced the level of surface GABAARs by 17% (control: 100 ± 0.3; 2MeS-ATP: 83.0 ± 14.6; mean ± S.E., not significant, Student's t test, n = 3), but due to experimental variability, presumably caused by various amounts of endogenous ATP present in the culture, these changes were not significant. At the same time, we observed increased degradation of intracellular receptors (Fig. 8, 1st and 2nd lanes). The co-associated P2X2Rs were detected using digoxigenin-labeled anti-EGFP antibodies. Similar to previous observation (Fig. 1B, 2nd lane), we observed a very weak association of P2X2Rs with GABAARs for control and 2MeS-ATP-treated cells. (Fig. 8, 1st and 2nd lanes). TNP-ATP treatment had no effect on the surface level of GABAARs (control: 100 ± 0.3; TNP-ATP: 110.9 ± 23.6; not significant, t test, n = 3) but up-regulated the associated P2X2Rs by 100% (Fig. 8, 1st and 3rd lanes (control: 100 ± 0.3; TNP-ATP: 199.6 ± 36.7, p < 0.05, t test, n = 3). These data seem to indicate that agonist binding on P2X2Rs shows a tendency to reduce the surface expression of GABAARs and targets it for degradation, whereas antagonist binding highly stabilizes the interaction, probably by preventing the action of ATP present in the culture medium.
FIGURE 8.

Modulation of direct association of GABAARs and P2X2Rs by purinergic drugs. HEK cells were co-transfected with GABAAR α1, β2, and γ2 subunits and EYFP-tagged P2X2R subunits. 48 h after transfection, cells were treated with either 2MeS-ATP (30 μm) or TNP-ATP (10 μm) for 1 h followed by surface precipitation of GABAAR using α1 subunit-specific antibodies. Precipitated GABAARs were detected using digoxigenin (Dig)-labeled α1 antibodies, and co-precipitated P2X2Rs were detected using digoxigenin-labeled EGFP antibodies. 2MeS-ATP, but not TNP-ATP treatment, showed a tendency to decrease the surface level of GABAARs. Although we detected very weak association of P2X2Rs with GABAARs for control and agonist-treated cells, we observed a very strong association when the cells were treated with TNP-ATP.
DISCUSSION
Intracellular Association and Co-trafficking of GABAARs and P2X2Rs Ensures Specific Targeting of P2X2Rs
Several lines of evidence indicate that GABAARs and P2X2Rs directly associate with each other intracellularly. First, both receptors could be co-immunoprecipitated from the cell surface or from a total extract of appropriately transfected HEK cells, using antibodies directed against either one of these receptors (Fig. 1). Second, both receptors are co-localized in membranes and the cytoplasm of HEK cells co-transfected with ECFP-tagged GABAARs and EYFP-tagged P2X2Rs as demonstrated by confocal microscopy (supplemental Fig. 4). Third, FRET experiments performed in these cells resulted in a similar FRET signal in the cytosol and at the membrane, suggesting that the same type of association of the GABAA-P2X2R complex is observed in these compartments (Fig. 2). Fourth, transfecting HEK cells with P2X2Rs and a trafficking-deficient GABAAR mutant resulted in 91% reduction of GABAARs and a 21% reduction of P2X2Rs at the cell surface, providing direct evidence for co-trafficking of these receptors (Fig. 1B).
Interestingly, co-expression of P2X2Rs resulted in an up-regulation in the expression of GABAARs in HEK cells (Fig. 1A), whereas the number of P2X2Rs expressed was not significantly influenced by the co-expression of GABAARs. This seems to indicate that P2X2Rs might have a stabilizing or chaperone function on a GABAAR subpopulation, preventing them from degradation. Such an intracellular function of P2X2Rs is supported by the fact that these receptors are highly enriched in the cytoplasm of the cell body of spinal cord neurons (data not shown). Once associated, the complex is then co-trafficked to the cell surface where it is predominantly co-localized extrasynaptically (Fig. 3). Previously, it was demonstrated that co-expression of P2X2Rs resulted in distal targeting of GABAC and GABAARs (22, 54). Together with our results, this indicates that GABAARs stabilized by P2X2 receptors help to traffic these receptors to specific localizations at the cell surface.
Purinergic Transmission Decreases GABAergic Inhibition in Spinal Cord Neurons
It is possible that associated GABAARs and P2X2Rs become enriched at inhibitory synapses. Actually, however, we observed only a very small fraction of co-localized receptors at inhibitory synapses (Fig. 3). This is consistent with previous studies indicating that P2X2Rs are mainly enriched at glutamatergic synapses in the brain (55, 56). These observations indicate that the two associated receptors either function at extrasynaptic regions or they dissociate and subsequently exhibit functions independent from each other. In fact, several lines of evidence indicate that upon activation of P2X2Rs by an agonist the co-associated GABAARs dissociate and are internalized and degraded, whereas P2X2Rs are stabilized. First, application of the P2X2R agonist 2MeS-ATP increased the mobility of extrasynaptically located GABAARs (Fig. 5) but did not change the mobility of P2X2Rs and instead increased their confinement (Fig. 6) and cluster size (Fig. 4). This is consistent with a previous study where it was reported that ATP treatment resulted in hot spots of P2X2-GFP receptors (57). Second, immunoprecipitation of receptors at the surface and intracellular compartments of HEK cells co-transfected with GABAA and P2X2Rs indicated that incubation with 2MeS-ATP reduced the number of GABAARs at the cell surface and increased the formation of GABAAR degradation products in the cytosol (Fig. 8). The amount of P2X2Rs was not changed under these conditions (Fig. 8), indicating that the increased clustering of these receptors (Fig. 4) on 2MeS-ATP treatment was not caused by newly incorporated receptors but by an increased confinement (Fig. 6) of freely diffusing P2X2Rs at pre-existing P2X2R clusters. Third, co-transfection of HEK cells with GABAARs and P2X2Rs increased the expression of GABAARs in intracellular compartments but caused a reduction of GABAARs at the cell surface (Fig. 1). Because there was no change in the number of P2X2Rs in intracellular compartments and at the cell surface under these conditions, the increased amount of GABAARs in the intracellular compartments did not result in an increase of P2X2R incorporation into the cell membrane. The reduced number of GABAARs at the cell membrane then probably was caused by a dissociation of GABAARs from the associated P2X2Rs mediated by endogenous ATP present in the cell culture medium, followed by their internalization and degradation. In a similar line, TNP-ATP-induced increase in clustering of GABAA receptors can be explained by a blockade of the actions of ATP endogenously present in the cultures and by trapping of nonclustered and freely diffusing receptor pairs by the available clusters.
Dissociation of GABAARs from associated P2X2Rs can either be elicited by an ATP-induced conformational change in P2X2Rs or by the subsequent P2X2R-mediated Ca2+ influx into the cell. SPT experiments indicated that the effects of 2MeS-ATP on GABAAR mobility were drastically reduced in presence of the Ca2+-chelator EGTA, suggesting a Ca2+-dependent regulation of GABAAR dynamics (Fig. 7). In addition, GABAAR mobility was even further reduced by EGTA when no 2MeS-ATP was present, possibly indicating an additional Ca2+-independent regulation of GABAAR dynamics. We cannot exclude, however, that this effect was at least partially due to P2XR-mediated influx of Ca2+ that was not chelated by EGTA. Interestingly, an N-methyl-d-aspartic acid receptor-induced Ca2+ influx was not sufficient to increase GABAAR dynamics in spinal cord neurons (7). This discrepancy might be explained by the apparently Ca2+-independent effect of 2MeS-ATP on GABAAR dynamics and/or by the tight association of GABAAR and P2X2Rs. Agonist-induced Ca2+ influx might drastically enhance the local Ca2+ concentration in the immediate surroundings of the intracellular domains of the associated receptors, thus allowing their dissociation that would not be possible when Ca2+ influx occurs via opening of more distant channels.
Rapid dissociation and internalization of GABAARs of course could be one of the mechanisms playing a role in the cross-talk of GABAA and P2X2Rs. But this for certain is not the only mechanism involved, and it has to be emphasized that the present results describe the effects of 2MeS-ATP on GABAA and P2X2R distribution in neurons or HEK cells after 2 h of treatment with high drug concentrations. In addition, the single particle tracking experiments were also performed over a time period of 5–20 min during drug application. These data therefore describe long term effects of drugs and cannot necessarily provide explanations for a possible mechanism of the cross-talk between GABAA and P2X2Rs as determined by electrophysiological measurements.
As members from the P2XR family have been demonstrated to show cross-talk with not only GABAARs (20, 22, 23, 58) but also with several other members of Cys loop receptors, which includes 5-hydroxytryptamine/serotonin type 3 receptors (59, 60), GABAC receptors (54), and nicotinic acetylcholine receptors (61, 62), association of P2X2Rs with other receptors and co-trafficking to specific locations at the cell surface in fact may be a general phenomenon. Indeed, such Cys loop receptor-mediated recruitment to the cell surface is possibly not restricted to P2X2Rs as several other members of P2XR family are now known to interact with GABAARs (P2X1, P2X4, and P2X5, experiments not shown; P2X3 (20, 58)) and nicotinic receptors (P2X2, P2X3, and P2X4 (61)). Such a receptor-mediated targeting of P2XRs might be important due to the lack of specifically defined synapses for these receptors. In any case, in the absence of P2X2R-selective drugs, we cannot exclude that part of the agonist- or antagonist-induced changes observed were elicited via other P2XR subtypes present in the spinal cord.
Blocking P2XRs as a Dual Therapeutic Strategy for Spinal Pain Processing
Inflammatory diseases and neuropathic injury are frequently accompanied by severe and debilitating pain. Loss of synaptic inhibition by GABAergic and glycinergic spinal dorsal horn neurons has been proposed as one of the key pathways in propagation of nociceptive and neuropathic information (63–65). A key molecule involved in spinal pain processing is ATP. It was established previously that exogenously applied ATP can induce pain sensation (66, 67) by activating P2XRs in lamina II of spinal cord (68–70). Our study demonstrates that the two pathways mediated by P2X and GABAARs overlap, and a fine balance between the two systems is essential to maintain homeostasis. Excess ATP released in diseased states will not only activate P2XR but also result in a dissociation, internalization, and degradation of P2XR-associated surface GABAARs, thus further strengthening P2XR activity by spinal dis-inhibition. Future experiments with spinal cord slices will have to strengthen this hypothesis.
The present finding that treatment with the competitive P2XR antagonist, TNP-ATP (71), resulted in increased clustering and slowing down of both GABAARs and P2X2Rs (Figs. 4–6) suggests a dual mechanism of action as follows: blockade of the excitatory actions of P2XRs and strengthening of GABAergic inhibition by preventing degradation (Fig. 8). In fact, TNP-ATP has been shown to suppress the ATP-induced effect in acute inflammatory or visceral pain following injury (72, 73). The short half-life of TNP-ATP, however, does not make it suitable for therapeutic studies (74). P2X2R antagonists exhibiting a longer half-life, however, might be useful candidates for preventing spinal excitation as well as GABAergic disinhibition.
Acknowledgments
We thank Florentina Soto for P2X2-ECFP, P2X2-EYFP, and P2X4-EGFP plasmids; Ruth Murrell-Lagnado for P2X2-FLAG-EGFP plasmids; Wolfgang Junger for P2X1-EGFP and P2X5-EGFP constructs; Thomas Grutter for P2X2 receptor homology model, and Stefan Böhm and Michael Freissmuth for access to their FRET and confocal setup and additional valuable suggestions during the study. We also acknowledge the technical suggestions from Margot Ernst, Karoline Fuchs, Marianne Renner, and Géraldine Gouzer.
This work was supported by the International Ph.D. Program “Cell Communication in Health and Disease” of the Medical University of Vienna and the Austrian Science Fund, INSERM, Ecolé Normale Supérieure, and a grant from the Pierre-Gilles de Gennes Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–6 and Movies 1 and 2.
- GABAAR
- GABAA receptor
- P2X2R
- P2X2 receptor
- SPT
- single particle tracking
- EGFP
- enhanced GFP
- TNP-ATP
- 2′,3′-O-(2,4,6-trinitrophenyl)adenosine 5′-triphosphate
- DIV
- days in vitro
- QD
- quantum dot
- MSD
- mean square displacement
- ns
- not significant
- ECFP
- enhanced cyan fluorescent protein
- EYFP
- enhanced YFP
- 2MeS-ATP
- 2-methylthioadenosine-5′-O-triphosphate.
REFERENCES
- 1. Sieghart W. (1995) Pharmacol. Rev. 47, 181–234 [PubMed] [Google Scholar]
- 2. Olsen R. W., Sieghart W. (2009) Neuropharmacology 56, 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olsen R. W., Sieghart W. (2008) Pharmacol. Rev. 60, 243–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sperk G., Schwarzer C., Tsunashima K., Fuchs K., Sieghart W. (1997) Neuroscience 80, 987–1000 [DOI] [PubMed] [Google Scholar]
- 5. Pirker S., Schwarzer C., Wieselthaler A., Sieghart W., Sperk G. (2000) Neuroscience 101, 815–850 [DOI] [PubMed] [Google Scholar]
- 6. Todd A. J., Watt C., Spike R. C., Sieghart W. (1996) J. Neurosci. 16, 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lévi S., Schweizer C., Bannai H., Pascual O., Charrier C., Triller A. (2008) Neuron 59, 261–273 [DOI] [PubMed] [Google Scholar]
- 8. Choquet D., Triller A. (2003) Nat. Rev. Neurosci. 4, 251–265 [DOI] [PubMed] [Google Scholar]
- 9. Triller A., Choquet D. (2005) Trends Neurosci. 28, 133–139 [DOI] [PubMed] [Google Scholar]
- 10. Triller A., Choquet D. (2008) Neuron 59, 359–374 [DOI] [PubMed] [Google Scholar]
- 11. Bannai H., Lévi S., Schweizer C., Inoue T., Launey T., Racine V., Sibarita J. B., Mikoshiba K., Triller A. (2009) Neuron 62, 670–682 [DOI] [PubMed] [Google Scholar]
- 12. Meier J., Vannier C., Sergé A., Triller A., Choquet D. (2001) Nat. Neurosci. 4, 253–260 [DOI] [PubMed] [Google Scholar]
- 13. Ehrensperger M. V., Hanus C., Vannier C., Triller A., Dahan M. (2007) Biophys. J. 92, 3706–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moss S. J., Smart T. G. (2001) Nat. Rev. Neurosci. 2, 240–250 [DOI] [PubMed] [Google Scholar]
- 15. Bogdanov Y., Michels G., Armstrong-Gold C., Haydon P. G., Lindstrom J., Pangalos M., Moss S. J. (2006) EMBO J. 25, 4381–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burnstock G., Knight G. E. (2004) Int. Rev. Cytol. 240, 31–304 [DOI] [PubMed] [Google Scholar]
- 17. Gever J. R., Cockayne D. A., Dillon M. P., Burnstock G., Ford A. P. (2006) Pflügers Arch. 452, 513–537 [DOI] [PubMed] [Google Scholar]
- 18. Collo G., North R. A., Kawashima E., Merlo-Pich E., Neidhart S., Surprenant A., Buell G. (1996) J. Neurosci. 16, 2495–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wirkner K., Sperlagh B., Illes P. (2007) Mol. Neurobiol. 36, 165–183 [DOI] [PubMed] [Google Scholar]
- 20. Sokolova E., Nistri A., Giniatullin R. (2001) J. Neurosci. 21, 4958–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labrakakis C., Tong C. K., Weissman T., Torsney C., MacDermott A. B. (2003) J. Physiol. 549, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boué-Grabot E., Toulmé E., Emerit M. B., Garret M. (2004) J. Biol. Chem. 279, 52517–52525 [DOI] [PubMed] [Google Scholar]
- 23. Karanjia R., García-Hernández L. M., Miranda-Morales M., Somani N., Espinosa-Luna R., Montaño L. M., Barajas-López C. (2006) Eur. J. Neurosci. 23, 3259–3268 [DOI] [PubMed] [Google Scholar]
- 24. Köles L., Gerevich Z., Oliveira J. F., Zadori Z. S., Wirkner K., Illes P. (2008) Naunyn Schmiedebergs Arch. Pharmacol. 377, 1–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarto-Jackson I., Furtmueller R., Ernst M., Huck S., Sieghart W. (2007) J. Biol. Chem. 282, 4354–4363 [DOI] [PubMed] [Google Scholar]
- 26. Horton R. M., Ho S. N., Pullen J. K., Hunt H. D., Cai Z., Pease L. R. (1993) Methods Enzymol. 217, 270–279 [DOI] [PubMed] [Google Scholar]
- 27. Sarto-Jackson I., Ramerstorfer J., Ernst M., Sieghart W. (2006) J. Neurochem. 96, 983–995 [DOI] [PubMed] [Google Scholar]
- 28. Hilf R. J., Dutzler R. (2009) Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 29. Bobanovic L. K., Royle S. J., Murrell-Lagnado R. D. (2002) J. Neurosci. 22, 4814–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tretter V., Ehya N., Fuchs K., Sieghart W. (1997) J. Neurosci. 17, 2728–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klausberger T., Fuchs K., Mayer B., Ehya N., Sieghart W. (2000) J. Biol. Chem. 275, 8921–8928 [DOI] [PubMed] [Google Scholar]
- 32. Sarto I., Klausberger T., Ehya N., Mayer B., Fuchs K., Sieghart W. (2002) J. Biol. Chem. 277, 30656–30664 [DOI] [PubMed] [Google Scholar]
- 33. Zezula J., Sieghart W. (1991) FEBS Lett. 284, 15–18 [DOI] [PubMed] [Google Scholar]
- 34. Guerlet G., Taly A., Prado de Carvalho L., Martz A., Jiang R., Specht A., Le Novère N., Grutter T. (2008) Biochem. Biophys. Res. Commun. 375, 405–409 [DOI] [PubMed] [Google Scholar]
- 35. Chen C. A., Okayama H. (1988) BioTechniques 6, 632–638 [PubMed] [Google Scholar]
- 36. Machado P., Rostaing P., Guigonis J. M., Renner M., Dumoulin A., Samson M., Vannier C., Triller A. (2011) J. Neurosci. 31, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Charrier C., Machado P., Tweedie-Cullen R. Y., Rutishauser D., Mansuy I. M., Triller A. (2010) Nat. Neurosci. 13, 1388–1395 [DOI] [PubMed] [Google Scholar]
- 38. Wessel D., Flügge U. I. (1984) Anal. Biochem. 138, 141–143 [DOI] [PubMed] [Google Scholar]
- 39. Bartholomäus I., Milan-Lobo L., Nicke A., Dutertre S., Hastrup H., Jha A., Gether U., Sitte H. H., Betz H., Eulenburg V. (2008) J. Biol. Chem. 283, 10978–10991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schicker K., Hussl S., Chandaka G. K., Kosenburger K., Yang J. W., Waldhoer M., Sitte H. H., Boehm S. (2009) Biochim. Biophys. Acta 1793, 325–334 [DOI] [PubMed] [Google Scholar]
- 41. Milan-Lobo L., Gsandtner I., Gaubitzer E., Rünzler D., Buchmayer F., Köhler G., Bonci A., Freissmuth M., Sitte H. H. (2009) Mol. Pharmacol. 76, 1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feige J. N., Gelman L., Tudor C., Engelborghs Y., Wahli W., Desvergne B. (2005) J. Biol. Chem. 280, 17880–17890 [DOI] [PubMed] [Google Scholar]
- 43. Feige J. N., Sage D., Wahli W., Desvergne B., Gelman L. (2005) Microsc. Res. Tech. 68, 51–58 [DOI] [PubMed] [Google Scholar]
- 44. Xia Z., Liu Y. (2001) Biophys. J. 81, 2395–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bannai H., Lévi S., Schweizer C., Dahan M., Triller A. (2006) Nat. Protoc. 1, 2628–2634 [DOI] [PubMed] [Google Scholar]
- 46. Renner M., Choquet D., Triller A. (2009) J. Neurosci. 29, 2926–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bonneau S., Dahan M., Cohen L. D. (2005) IEEE Trans. Image Process 14, 1384–1395 [DOI] [PubMed] [Google Scholar]
- 48. Charrier C., Ehrensperger M. V., Dahan M., Lévi S., Triller A. (2006) J. Neurosci. 26, 8502–8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kusumi A., Sako Y., Yamamoto M. (1993) Biophys. J. 65, 2021–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khakh B. S., Fisher J. A., Nashmi R., Bowser D. N., Lester H. A. (2005) J. Neurosci. 25, 6911–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harvey R. J., Depner U. B., Wässle H., Ahmadi S., Heindl C., Reinold H., Smart T. G., Harvey K., Schütz B., Abo-Salem O. M., Zimmer A., Poisbeau P., Welzl H., Wolfer D. P., Betz H., Zeilhofer H. U., Müller U. (2004) Science 304, 884–887 [DOI] [PubMed] [Google Scholar]
- 52. Alcor D., Gouzer G., Triller A. (2009) Eur. J. Neurosci. 30, 987–997 [DOI] [PubMed] [Google Scholar]
- 53. Holcman D., Triller A. (2006) Biophys. J. 91, 2405–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boué-Grabot E., Emerit M. B., Toulmé E., Séguéla P., Garret M. (2004) J. Biol. Chem. 279, 6967–6975 [DOI] [PubMed] [Google Scholar]
- 55. Rubio M. E., Soto F. (2001) J. Neurosci. 21, 641–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Masin M., Kerschensteiner D., Dümke K., Rubio M. E., Soto F. (2006) J. Biol. Chem. 281, 4100–4108 [DOI] [PubMed] [Google Scholar]
- 57. Khakh B. S., Smith W. B., Chiu C. S., Ju D., Davidson N., Lester H. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5288–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Toulmé E., Blais D., Léger C., Landry M., Garret M., Séguéla P., Boué-Grabot E. (2007) J. Neurochem. 102, 1357–1368 [DOI] [PubMed] [Google Scholar]
- 59. Zhou X., Galligan J. J. (1998) J. Physiol. 513, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boué-Grabot E., Barajas-López C., Chakfe Y., Blais D., Bélanger D., Emerit M. B., Séguéla P. (2003) J. Neurosci. 23, 1246–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Decker D. A., Galligan J. J. (2009) Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1267–G1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khakh B. S., Zhou X., Sydes J., Galligan J. J., Lester H. A. (2000) Nature 406, 405–410 [DOI] [PubMed] [Google Scholar]
- 63. Knabl J., Witschi R., Hösl K., Reinold H., Zeilhofer U. B., Ahmadi S., Brockhaus J., Sergejeva M., Hess A., Brune K., Fritschy J. M., Rudolph U., Möhler H., Zeilhofer H. U. (2008) Nature 451, 330–334 [DOI] [PubMed] [Google Scholar]
- 64. Zeilhofer H. U., Zeilhofer U. B. (2008) Neurosci. Lett. 437, 170–174 [DOI] [PubMed] [Google Scholar]
- 65. Zeilhofer H. U., Möhler H., Di Lio A. (2009) Trends Pharmacol. Sci. 30, 397–402 [DOI] [PubMed] [Google Scholar]
- 66. Bleehen T., Keele C. A. (1977) Pain 3, 367–377 [DOI] [PubMed] [Google Scholar]
- 67. Nakagawa T., Wakamatsu K., Zhang N., Maeda S., Minami M., Satoh M., Kaneko S. (2007) Neuroscience 147, 445–455 [DOI] [PubMed] [Google Scholar]
- 68. Bardoni R., Goldstein P. A., Lee C. J., Gu J. G., MacDermott A. B. (1997) J. Neurosci. 17, 5297–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jo Y. H., Schlichter R. (1999) Nat. Neurosci. 2, 241–245 [DOI] [PubMed] [Google Scholar]
- 70. Souslova V., Cesare P., Ding Y., Akopian A. N., Stanfa L., Suzuki R., Carpenter K., Dickenson A., Boyce S., Hill R., Nebenuis-Oosthuizen D., Smith A. J., Kidd E. J., Wood J. N. (2000) Nature 407, 1015–1017 [DOI] [PubMed] [Google Scholar]
- 71. Trujillo C. A., Nery A. A., Martins A. H., Majumder P., Gonzalez F. A., Ulrich H. (2006) Biochemistry 45, 224–233 [DOI] [PubMed] [Google Scholar]
- 72. Jarvis M. F., Wismer C. T., Schweitzer E., Yu H., van Biesen T., Lynch K. J., Burgard E. C., Kowaluk E. A. (2001) Br. J. Pharmacol. 132, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Honore P., Mikusa J., Bianchi B., McDonald H., Cartmell J., Faltynek C., Jarvis M. F. (2002) Pain 96, 99–105 [DOI] [PubMed] [Google Scholar]
- 74. Ding Y., Cesare P., Drew L., Nikitaki D., Wood J. N. (2000) J. Auton. Nerv. Syst. 81, 289–294 [DOI] [PubMed] [Google Scholar]