

Abstract
SIRT6 is a member of the evolutionarily conserved sirtuin family of NAD+-dependent protein deacetylases and functions in genomic stability and transcriptional control of glucose metabolism. Early reports suggested that SIRT6 performs ADP-ribosylation, whereas more recent studies have suggested that SIRT6 functions mainly as a histone deacetylase. Thus, the molecular functions of SIRT6 remain uncertain. Here, we perform biochemical, kinetic, and structural studies to provide new mechanistic insight into the functions of SIRT6. Utilizing three different assays, we provide biochemical and kinetic evidence that SIRT6-dependent histone deacetylation produces O-acetyl-ADP-ribose but at a rate ∼1,000 times slower than other highly active sirtuins. To understand the molecular basis for such low deacetylase activity, we solved the first crystal structures of this class IV sirtuin in complex with ADP-ribose and the non-hydrolyzable analog of O-acetyl-ADP-ribose, 2′-N-acetyl-ADP-ribose. The structures revealed unique features of human SIRT6, including a splayed zinc-binding domain and the absence of a helix bundle that in other sirtuin structures connects the zinc-binding motif and Rossmann fold domain. SIRT6 also lacks the conserved, highly flexible, NAD+-binding loop and instead contains a stable single helix. These differences led us to hypothesize that SIRT6, unlike all other studied sirtuins, would be able to bind NAD+ in the absence of an acetylated substrate. Indeed, we found that SIRT6 binds NAD+ with relatively high affinity (Kd = 27 ± 1 μm) in the absence of an acetylated substrate. Isothermal titration calorimetry and tryptophan fluorescence binding assays suggested that ADP-ribose and NAD+ induce different structural perturbations and that NADH does not bind to SIRT6. Collectively, these new insights imply a unique activating mechanism and/or the possibility that SIRT6 could act as an NAD+ metabolite sensor.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Histone Deacetylase, Metabolic Regulation, NAD, Sirtuins, Transcription, Transcription Regulation, SIRT6
Introduction
Sirtuins comprise an ancient and diverse family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases that are evolutionarily conserved from bacteria to eukaryotes (1). Unlike other classes of histone deacetylases, which utilize an active site Zn2+ and involve direct attack of a water molecule on acetylated lysines, sirtuins transfer the acetyl group from the lysine side chain of a protein or peptide substrate to the co-factor NAD+, generating nicotinamide, 2′-O-acetyl-ADP-ribose (OAADPr)3 (2, 3), and a deacetylated substrate. This unique requirement of NAD+ suggests that sirtuins might act as sensors of the cellular metabolic state (4), relaying changes in cellular metabolism to reverse acetylation-mediated pathways, which include transcription, cell cycle progression, genome maintenance, apoptosis, and organism longevity. The founding member of the sirtuin family, yeast Sir2 (silent information regulator 2), has emerged as an important regulator in extending the life span of Saccharomyces cerevisiae (5). In other organisms, such as Caenorhabditis elegans and Drosophila melanogaster, sirtuin-activated pathways promote longevity (6, 7). It was recently demonstrated that mammalian SIRT3 deacetylates isocitrate dehydrogenase 2, reduces oxidative damage, and prevents age-related hearing loss in response to caloric restriction (8). This result provides a direct mechanistic link between mammalian sirtuins and aging.
There are seven members in the human sirtuin family, SIRT1 to -7; these exhibit diversity and complexity in their cellular localization patterns and targets (9). SIRT1, being the putative ortholog to yeast Sir2, has been the most studied. A recent study reported that SIRT6-deficient mice have a striking degenerative phenotype leading to shortened life span (10). At the cellular level, SIRT6 deficiency leads to marked metabolic and genomic instability caused by hypoglycemia and potential defects in base excision repair or DNA double strand break repair (10). However, the molecular mechanism of the protective role of SIRT6 in preventing these processes is not fully understood.
The early in vitro analysis suggested that SIRT6 was not an active deacetylase and instead could undergo intramolecular mono-ADP-ribosylation utilizing NAD+ as a substrate (11). More recently, however, it was reported that SIRT6 does harbor NAD+-dependent deacetylase activity toward the H3K9Ac (12) and H3K56Ac (13, 14) histone marks as well as toward the double strand break resection protein, CtIP (C-terminal binding protein (CtBP)-interacting protein) (15). Hundreds of genes are differentially expressed in SIRT6−/− mouse cells when compared with wild-type controls (16), and the modulation of NF-κB-dependent pathways (16) and HIF1α-dependent pathways (17) was reported to be achieved through H3K9Ac deacetylation by SIRT6.
Despite these recent advances in elucidating the biological functions of SIRT6, the lack of detailed biochemical and structural studies has hindered our mechanistic understanding of this class IV sirtuin. Three human sirtuins (SIRT2 (18), SIRT3 (19), and SIRT5 (20)) have crystal structures available. Along with structures from other sirtuin homologs (21–32), these results have provided important insights into the mechanism for activity, inhibition, and substrate specificity of individual sirtuins. Crystal structures of the class IV sirtuins, which include mammalian SIRT6 and SIRT7, have not been solved.
Here we explore the biochemical function and structural details of human SIRT6 protein. We provide the first quantitative assessment of SIRT6 deacetylation and give direct evidence for OAADPr as one of the end products of a very inefficient SIRT6-catalyzed reaction. We also report the first sets of crystal structures of human SIRT6: a complex structure of SIRT6 with ADP-ribose (ADPr) and another complex with a synthetic OAADPr analog, 2′-N-acetyl-ADP-ribose (NAADPr) (33). Comparison of SIRT6 with other sirtuin structures reveals several unique features that provide a rationale for the low deacetylase activity and the ability to bind NAD+ with high affinity in the absence of acetylated substrate. Furthermore, isothermal titration binding and tryptophan fluorescence assays suggest that NAD+ and ADPr may induce different structural changes upon binding. These novel features provide insight into the class IV sirtuins, which have poorly defined molecular functions.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
NAD+, NADH, ADP-ribose, dithiothreitol (DTT), TCEP, glutamate dehydrogenase from bovine liver, and α-ketoglutarate were purchased from Sigma. Tris-HCl, Tris base, sodium phosphate monobasic, sodium phosphate dibasic, and sodium chloride were purchased through Fisher. [3H]Acetic anhydride (50–100 mCi/mmol) was purchased from American Radiolabeled Chemicals, Inc. Site-directed mutagenesis kits from Stratagene were utilized to introduce a point mutation at His131 of SIRT6. 2′-NAADPr was synthesized as described previously (33).
Expression and Purification of Recombinant WT SIRT6, H131Y SIRT6, and Hst2 for Deacetylation Assays and Isothermal Titration Calorimetry (ITC)
pQE-80L (Qiagen, Valencia, CA) His-tagged WT or H131Y SIRT6 were transformed into the competent Escherichia coli strain, BL21 DE3. Overexpression was initiated by growing cells to an A600 of 0.6–0.8 at 37 °C. To induce expression, 0.5 mm isopropyl-1-thio-d-galactopyranoside was added, and cells were grown at room temperature for 18 h. Cells were harvested by centrifugation at 5,000 rpm for 10 min and stored at −20 °C. For purification, cells were resuspended in 50 mm sodium phosphate, pH 7.2, 250 mm NaCl, 5 mm imidazole, and 1 mm β-mercaptoethanol, lysed by sonication, and purified by nickel resin affinity chromatography. WT and H131Y SIRT6 were further purified via a HiTrap SP-Sepharose Fast Flow column (GE Healthcare) using a linear gradient from 50 to 750 mm NaCl in 50 mm sodium phosphate, pH 7.2, and 1 mm β-mercaptoethanol. Fractions containing purified WT or H131Y SIRT6 were pooled, concentrated, and dialyzed into 50 mm Tris, pH 8.0 (4 °C), 150 mm NaCl, 100 μm TCEP, and 5% (w/v) glycerol. Hst2 was expressed and purified as described previously (34). Protein concentrations were determined by the Bradford reagent assay.
Synthesis and Analysis of the H3K9 Peptide
A peptide corresponding to residues 5–13 of histone H3 (QTARKSTGG) was synthesized by the University of Wisconsin-Madison Biotechnology Center and purified over a preparative C18 HPLC column. The chromatographic purity of the peptide was determined to be ≥95%, and mass spectrometric analysis on a Bruker REFLEX II: MALDI-TOF instrument confirmed the identification of the peptide.
Generation of a 3H-Labeled H3K9Ac Peptide
To produce a 3H-labeled H3K9Ac peptide, 4 mm H3K9 peptide was reacted with 20 mm tritium-labeled acetic anhydride in 50 mm HEPES, pH 8.0, and incubated at 4 °C for 4 h. The reaction was quenched by the addition of 100 mm Tris-HCl, pH 7.4 (final concentration). During synthesis, the N terminus was left unblocked, and therefore acetic anhydride reacted with the ϵ-amine of the lysine residue as well as the N-terminal amine to produce a peptide that was acetylated at both the lysine residue and the N terminus. The presence of the doubly acetylated peptide was confirmed by mass spectrometric analysis on a Bruker REFLEX II: MALDI-TOF of a peptide acetylated with unlabeled acetic anhydride. The molar concentration of [3H]H3K9Ac (3.7 mm) was determined by the amount of tritium in 2–3 μl of the resuspended peptide and calculated as described previously (35).
Charcoal-binding Assay Determination of SIRT6 Activity
The charcoal-binding assay was performed as described previously (35). Reactions were performed in 400 μl containing 600 μm NAD+, 300 μm [3H]H3K9Ac, 1 mm DTT, and 4 μm WT SIRT6 or H131Y SIRT6 in 50 mm Tris, pH 7.5, at room temperature. Time points (0.5, 1, 1.5, 2, 2.5, and 3 h) were taken by adding 60 μl of the reaction mixture to 60 μl of activated charcoal, pH 9.5 (1:3 (w/v) of charcoal (Sigma, C3345) and 2:3 (v/v) of 2 m glycine, pH 9.5).
HPLC Assay to Monitor OAADPr Formation
Reactions were performed in 40 μl containing 2 mm NAD+, 300 μm [3H]H3K9Ac, 1 mm DTT, and 4 μm WT SIRT6, Hst2, or no enzyme in 50 mm Tris, pH 7.5, at room temperature. The reaction was quenched after 3 h by the addition of 1% (v/v) TFA to 20 μl of the reaction mixture. The reaction mixture was separated on an analytical C18 HPLC column by a gradient separation at 0.5 ml/min: 0–8% solvent B (0.02% TFA in acetonitrile) over 25 min and 8–100% solvent B over 3 min, followed by 100% solvent B for 10 min. Fractions (1 min) were collected, and radioactivity was quantified by scintillation counting 150 μl of each fraction and plotted versus the fractions. The peak corresponding to [3H]OAADPr was confirmed by alignment with purified OAADPr.
Sirtuin Enzyme-coupled Assay
SIRT6 activity was measured continuously using the method previously reported (36) with minor variations. The assay solution contained 2 μm maltose-binding protein fused-PncA (nicotinamidase), 0.2 mm NADH, 0.6 mm NAD+, 3.3 mm α-ketoglutarate, 1 mm DTT, 2 units of glutamate dehydrogenase from bovine liver, 300 μm H3K9Ac peptide, and 0 or 8 μm SIRT6 enzyme in 20 mm phosphate buffer (pH 7.5). Continuous readings were taken every 20 s for a total duration of 1 h and 30 min. The rate of nicotinamide formation was determined by the use of the coupled enzymes nicotinamidase and glutamate dehydrogenase. Nicotinamidase converts nicotinamide, formed by the cleavage of NAD+, into nicotinic acid and ammonia. The ammonia is then utilized by glutamate dehydrogenase to convert α-ketoglutarate to glutamate, consuming NADH in the process. The oxidation/consumption of NADH was monitored continuously at 340 nm, and subsequently the rate of nicotinamide formation could be determined.
Protein Cloning, Expression, and Purification for Crystallization
Human SIRT6(3–318) was amplified by PCR and subcloned into the pET28a-LIC vector downstream of the polyhistidine coding region. The recombinant protein was overexpressed in E. coli BL21(DE3)-R3-pRARE2 cells in Terrific Broth in the presence of 50 μg/ml kanamycin at 37 °C to an A600 of 0.8. Protein expression was induced by the addition of isopropyl-1-thio-d-galactopyranoside to a final concentration 0.5 mm, and the cells were then incubated overnight at 15 °C. Cells were harvested by centrifugation. The cell pellets were frozen in liquid nitrogen and stored at −80 °C. During purification, the cell paste was thawed and resuspended in lysis buffer (1× PBS, pH 7.5, 0.5 m NaCl, 5% glycerol) with protease inhibitor (0.1 mm PMSF). Cell lysis was accomplished by sonication (Virtis408912, Virsonic) on ice; the sonication protocol was a 10-s pulse at 80% maximal frequency (8.0) and a 10-s rest for a 5-min total sonication time. The cell lysate was clarified by centrifugation using a Beckman JLA-16.250 rotor at 15,500 rpm for 45 min at 4 °C. The clarified lysate was loaded onto a 5-ml HiTrap chelating column (Amersham Biosciences), charged with Ni2+. The column was washed with 10 column volumes (5) of 20 mm Hepes, pH 7.0, containing 250 mm NaCl and 5% glycerol, and the protein was eluted with elution buffer (20 mm Hepes buffer, pH 7.0, 250 mm NaCl, 5% glycerol, and 250 mm imidazole). The protein was loaded onto a Superdex200 column (2.6 × 60 cm) (Amersham Biosciences), equilibrated with 20 mm Pipes buffer, pH 6.5, and 150 mm NaCl, at a flow rate of 4 ml/min. Thrombin was added to combined fractions containing SIRT6 at 4 °C overnight to cleave off the N-terminal poly-His tag. The protein was further purified to apparent homogeneity by ion exchange chromatography on a Source 30S column (1.0 × 10 cm) (Amersham Biosciences), equilibrated with 20 mm Mes buffer, pH 6.5, and eluted with a linear gradient of NaCl up to 500 mm concentration (30 column volumes).
SIRT6 Protein Crystallization
The H3K9Ac peptide (TKQTARKAcSTGGKAPY; Tufts University Core Facility, Peptide Synthesis Service) was dissolved in a buffer containing 20 mm Mes, pH 6.5, and 150 mm NaCl and then adjusted to pH 7.0 with a final concentration of 100 mm. Subtilisin A (Sigma) was dissolved in 10 mm NaOAC and 5 mm Ca(OAC)2 to a final concentration of 1 mg/ml. The protein solution contained 10 mg/ml purified SIRT6 mixed with 10 mm NAD+/ADPr (Sigma)/NAADPr and 3 mm H3K9Ac and 0.01 mg/ml subtilisin A (37) in 20 mm Mes buffer, pH 6.5. SIRT6·ADPr and SIRT6·NAADPr crystals were obtained using hanging drop vapor diffusion at 20 °C by mixing 2 μl of the protein mix with 2 μl of the reservoir solution containing 1.8–1.9 m (NH4)2SO4, 2% PEG 400, BisTris, pH 5.6–6.2. All crystals were soaked in the corresponding mother liquor supplemented with 15% ethylene glycol as cryoprotectant before freezing in liquid nitrogen.
Data Collection and Structure Determination
X-ray diffraction data were collected at 100 K at beamline 19ID of the Advanced Photon Source at Argonne National Laboratory and a Rigaku FR-E home source. Data were processed using the HKL-2000 software suite (38). The SIRT6·ADP-ribose structure (PDB code 3K35) was solved by molecular replacement using the program Phaser (39). A mixed model of Sir2Tm (PDB code 1YC5) generated by SCWRL and Jackal modeling method on the FFAS03 search server (62) was used as template. Refinement was carried out with Refmac (40). SIRT6·ADPr (PDB code 3PKI) and SIRT6·NAADPr (PDB code 3PKJ) were solved by molecular replacement using the initial SIRT6 structure (PDB code 3K35; ADPr and all other small molecules were removed) as a search model. Subsequent refinement was performed using Refmac and Buster-TNT (41), respectively. The graphics program COOT (42) was used for model building and visualization. Data statistics are reported in Table 1.
TABLE 1.
Data collection and refinement statistics
| Parameters | Values |
||
|---|---|---|---|
| PDB 3K35 | PDB 3PKI | PDB 3PKJ | |
| Data collection | |||
| Complex | SIRT6·ADP-ribose | SIRT6·ADP-ribose | SIRT6·N-acetyl-ADP-ribose |
| Beamline | RIGAKU FR-E | APS 19ID | RIGAKU FR-E |
| Wavelength (Å) | 1.54 | 0.97945 | 1.54 |
| Space group | P21 | P1 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 89.4, 136.3,89.3 | 77.4, 90.2, 90.8 | 89.0, 135.8, 89.1 |
| α, β, γ (degrees) | 90,0, 119.9, 90.0 | 118.1, 91.4, 115.8 | 90.0,120.0,90.0 |
| Resolution (Å)a | 20.0-2.0 (2.07-2.00) | 50.0-2.0 (2.07-2.03) | 44.5-2.1 (2.17-2.12) |
| Rsyma | 0.086 (0.702) | 0.120 (0.965) | 0.120 (0.930) |
| I/(σI)a | 18.96 (2.25) | 15.50 (1.64) | 15.90 (2.02) |
| Completeness (%)a | 97.1 (94.3) | 98.1 (97.0) | 96.2 (97.5) |
| Redundancya | 3.7 (3.6) | 4.5 (4.1) | 5.4 (5.4) |
| Refinement | |||
| Resolution (Å) | 19.95-2.00 | 29.9-2.04 | 20.5-2.12 |
| No. of reflections | 119,269 | 115,679 | 99,730 |
| Rworkb/Rfreec | 0.202/0.267 | 0.182/0.213 | 0.2366/0.2695 |
| Mean B-factors (Å2) | 30.84 | 33.39 | 37.82 |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.013 | 0.010 | 0.010 |
| Bond angles (degrees) | 1.506 | 0.94 | 0.940 |
| Ramachandran plot | |||
| Favored regions (%) | 99.0 | 99.03 | 99.09 |
| Additionally allowed regions (%) | 1.0 | 0.97 | 0.91 |
| Disallowed regions (%) | 0 | 0 | 0 |
a Values in parentheses are for the highest resolution shell.
b Rwork = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|, where Fobs and Fcalc are observed and calculated structure-factor amplitudes, respectively.
c Rfree is the R factor for the test set (1–10% of the data).
Isothermal Titration Calorimetry Studies of WT and H131Y SIRT6 with NAD+ Metabolites
ITC measurements were taken using a VP-ITC microcalorimeter from MicroCal, LLC (Northampton, MA). Binding experiments were all performed in 50 mm Tris, pH 7.5 (25 °C), 150 mm NaCl, 100 μm TCEP, and 5% (w/v) glycerol. Purified WT and H131Y SIRT6 were dialyzed against the previously described buffer, and ligands were suspended in the dialysis buffer. Experimental data were fitted to a one-site binding model using Origin scientific plotting software. Enthalpy (ΔH), binding constant (Ka), and number of binding sites (n) were all flexible parameters, whereas the free energy (ΔG) and entropy (ΔS) were calculated according to the following equation.
For each experiment, 37 automatic injections (1–8 μl) were titrated into the cell (initial cell volume 1.42 ml) while being stirred at 300 rpm. NAD+ (1.40 mm) was titrated into 27 μm WT SIRT6. NAD+ (1.63 mm) was titrated into 50 μm Hst2. NADH (450 μm) was titrated into 25 μm WT SIRT6. ADPr (300 μm) was titrated into 28 μm WT SIRT6. 2′-NAADPr (2.96 μm) was titrated into 38 μm WT SIRT6. NAD+ (767 μm) was titrated into 33 μm H131Y SIRT6, and 1.45 mm ADPr was titrated into 37 μm H131Y SIRT6. All titrations were also performed in the absence of enzyme to account for the heat caused by ligand dilution.
Intrinsic Tryptophan Fluorescence Emission Studies of WT and H131Y SIRT6 with NAD+ Metabolites
Fluorescence emission was monitored on a FluoroMax®-4 spectrofluorometer from HORIBA Jobin Yvon. Samples were excited at 295 nm (slit width 2 mm), and emission was monitored from 320 to 390 nm (slit width 3 mm). Experiments under native conditions were performed in 50 mm Tris, pH 7.5, at room temperature. Increasing concentrations of NAD+ from 0 to 600 μm or ADPr from 0 to 400 μm were added to 1 μm WT SIRT6, and the emission spectrum was monitored. Likewise, increasing concentrations of NAD+ (0–200 μm) or ADPr (0–1.2 mm) were added to 1 μm H131Y SIRT6. The peak maximum was determined for each concentration. To account for decreases in fluorescence from nonspecific quenching due to increasing concentration of ligand, the same experiments were carried out in 7 m urea. The peak maxima were obtained, and the correction factor was determined by (Fobs/F0) where Fobs is the maximum intensity at each concentration, and F0 is the maximum intensity at 0 μm ligand. The intensities from experiments under native conditions were divided by their correction factors, and the fraction of substrate bound (FSB) was determined by Equation 2,
 |
where Fs represents the maximum intensity under saturating ligand, and Fobs and F0 are the same as stated previously. The FSB values were plotted versus ligand concentration, and the data were fitted to a one-site binding equation to determine the Kd,
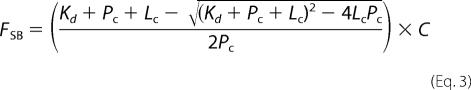 |
where Kd is the dissociation constant, Pc is the protein concentration (1 μm), Lc is the varying ligand concentration, and C is the y axis maximum.
RESULTS
SIRT6 Deacetylase Activity
It was initially reported that SIRT6 was not an active deacetylase but rather was capable of performing intramolecular auto-ADP-ribosylation using NAD+ as a co-substrate (11). More recently, Michishita et al. (12) used mass spectrometry and Western blot analysis to support SIRT6 as a histone H3K9 deacetylase. However, SIRT6 did not deacetylate H3K9 with high efficiency; noncatalytic amounts of SIRT6 (∼100 pmol) were needed to observe deacetylation of ∼65 pmol of acetylated histone by Western blot analysis, which detects loss of immunoreactivity from an anti-acetyl lysine antibody. In mass spectrometry assays, ∼100 pmol of SIRT6 was needed to convert ∼10% of 360 pmol of H3K9Ac peptide to the deacetylated form (12). It is still unknown whether SIRT6 can form 2′-O-acetyl-ADP-ribose as the product of the deacetylase reaction. Thus, the molecular function(s) of SIRT6 remains unclear. A detailed quantitative assessment of the SIRT6 reaction is essential toward our understanding of its cellular functions.
To monitor the deacetylase activity of purified recombinant SIRT6, we used an in vitro charcoal-binding assay (35) that measures acetate released from deacetylation of a tritium-labeled H3K9Ac peptide. [3H]Acetate, formed through the hydrolysis of [3H]OAADPr, was measured in the presence of 300 μm [3H]H3K9Ac, 600 μm NAD+ and 4 μm SIRT6. After subtracting the control reaction without SIRT6, reactions containing SIRT6 exhibited a linear increase in acetate formation over time (Fig. 1A). The rate of deacetylation was 3.2 ± 0.2 μm/h, with a specific activity of 0.80 ± 0.05 h−1 (0.00022 ± 0.00001 s−1). This low level of activity is in general agreement with the previous qualitative assays (12) and indicates that SIRT6 is ∼1000 times less active than the well characterized yeast Hst2 enzyme, which deacetylates the same H3K9Ac peptide with a kcat of 0.32 ± 0.08 s−1 (34).
FIGURE 1.

SIRT6 deacetylation assays. A, charcoal-binding assay measuring the production of OAADPr. All assays were carried out in the presence of 4 μm SIRT6 WT (●) or H131Y (■), 300 μm [3H]H3K9Ac peptide, and 600 μm NAD+. Shown is the average of three experiments for WT and H131Y. The rate of OAADPr production was 3.2 ± 0.2 μm/h based on linear regression analysis with an R2 of 0.98612. B, HPLC separation of 3H-labeled products monitoring deacetylation of a [3H]H3K9Ac peptide. Reactions were carried out in the presence of 300 μm [3H]H3K9Ac peptide, 2 mm NAD+, and either 4 μm SIRT6, 4 μm Hst2 or no enzyme. Shown are all counts for the Hst2 deacetylation reaction (green), SIRT6 (brown), and a no enzyme control (blue) as well as a zoomed in view displaying counts for [3H]acetate, [3H]OAADPr, and H3K9 ([3H]acetylated N terminus). C, continuous assay monitoring the release of nicotinamide over time. Shown is the average of three trials with assays carried out in the presence of 8 μm (gray) or 0 μm (white) SIRT6, 300 μm H3K9Ac, and 0.6 mm NAD+. The rate of nicotinamide release in the presence of SIRT6 was 10.5 ± 0.4 μm/h and the rate in its absence was 5.9 ± 0.8 μm/h. Error bars, S.D.
Such low activity raised the concern that the deacetylation activity could be due to a contaminating E. coli sirtuin co-purifying with SIRT6. To ensure that the activity measured was dependent on SIRT6, the invariant catalytic base histidine residue (3, 43) (His131) was mutated to a tyrosine (H131Y). If the low level of deacetylase activity emanated from SIRT6, the H131Y mutant purified in exactly the same manner (supplemental Fig. 1) should yield no [3H]H3K9 deacetylation. H131Y SIRT6 deacetylase activity was monitored by the charcoal-binding assay. No detectable activity was observed above a control reaction containing no SIRT6 (Fig. 1A), thereby providing further evidence that the low level of deacetylase activity observed in the WT reactions stemmed from catalytically active SIRT6.
To provide direct evidence that the acetate formed through the charcoal-binding assay was due to deacetylation by SIRT6 and subsequent hydrolysis of OAADPr, an HPLC assay was utilized to monitor the formation of [3H]OAADPr. Reactions containing 300 μm [3H]H3K9Ac, 2 mm NAD+, and either 4 μm SIRT6, 4 μm Hst2 or no enzyme were incubated for 3 h and quenched by the addition of 1% (v/v) TFA. As a positive control for OAADPr formation, deacetylation of the H3K9 peptide by Hst2 (34) was monitored. Hst2 deacetylated the H3K9Ac peptide after 3 h, as demonstrated by the loss of a peak corresponding to [3H]H3K9Ac peptide and generation of peaks corresponding to [3H]OAADPr and [3H]acetate, which was formed through the hydrolysis of [3H]OAADPr (Fig. 1B).
Compared with Hst2, SIRT6 (4 μm) was able to weakly deacetylate the [3H]H3K9Ac peptide (Fig. 1B), producing 13 μm OAADPr during the 3-h incubation. Thus, SIRT6 deacetylated H3K9Ac at a rate of 4.3 μm/h and had a specific activity of 1.1 h−1 (0.0003 s−1), which is consistent with the results from the charcoal-binding assay (Fig. 1A). Along with the charcoal-binding assay, the results provide the first evidence that SIRT6 can perform multiple turnovers and can generate OAADPr through NAD+-dependent protein deacetylation.
To provide an independent method for determining SIRT6 activity toward acetylated peptides, a continuous assay was utilized (36). The continuous assay measures nicotinamide formation, which is the first product released from either the NAD+-dependent deacetylation or an ADP-ribosylation reaction. Therefore, the assay provides a good indication of the coupling between nicotinamide cleavage and OAADPr formation. H3K9Ac peptide (300 μm) was incubated with 0.6 mm NAD+ and either 0 or 8 μm SIRT6, and a linear increase of nicotinamide was observed over 1.5 h. The rate of nicotinamide formation was greater in the presence of SIRT6 compared with spontaneous NAD+ hydrolysis (10.5 ± 0.4 μm/h versus 5.9 ± 0.8 μm/h), indicating that the SIRT6-dependent rate of NAD+ cleavage is 4.6 ± 0.9 μm/h with a specific activity of 0.6 ± 0.1 h−1 (Fig. 1C). The three different assays (Fig. 1, A–C) are in excellent agreement and confirm the extremely low level of deacetylase activity displayed by SIRT6. Importantly, the fact that the rate of nicotinamide cleavage is similar to that for OAADPr formation indicates that NAD+ cleavage is tightly coupled to deacetylation and argues against a significant contribution from protein ADP-ribosylation that does not require deacetylation.
SIRT6 Protein Construct for Crystallization
It is unclear why SIRT6 displays such low deacetylase activity (∼1000-fold lower) compared with other sirtuins. To investigate the molecular basis for this low activity, the crystal structures of human SIRT6 bound to ADPr and 2′-NAADPr were solved. Full-length human SIRT6 has 355 amino acids and consists of a putative catalytic sirtuin core with N- and C-terminal flanking extensions. In our attempts to obtain SIRT6 protein crystals, we generated several minimal truncations of SIRT6 as well as full-length protein. Among the various constructs examined, the SIRT6(3–318) construct was expressed and purified from E. coli in the 10–20 milligram per liter range, compared with lower yields obtained with the full-length construct. Therefore, SIRT6(3–318) was selected for crystallization trials. This construct has both the N-terminal extension and the core catalytic domain, but lacks the C-terminal extension. It was previously reported that a SIRT6 construct lacking the C-terminal extension is enzymatically active but is impaired in its ability to associate with chromatin (44).
In Situ Proteolysis and the Two ADPr-bound SIRT6 Structures
We initially attempted to co-crystallize SIRT6 with H3K9Ac and the cofactor NAD+, but no crystals were observed. We then attempted to promote crystallization using in situ proteolysis (37). After adding trace amounts of the protease, subtilisin A, into the protein mix, protein crystals appeared within 24 h after setting up the crystallization experiment. The crystals were refined to achieve x-ray diffraction quality. In the solved structure (PDB code 3K35), we did not observe the nicotinamide moiety of NAD+. The nicotinamide ribose ring adopted a C3′-endo conformation and remained in the β-form. We repeated the crystallization experiment, substituting ADPr for NAD+ in the protein mix, and obtained crystals within 24 h. Although the space group changed from P21 to P1, the number of molecules in the asymmetric unit remained the same, and monomer structural features were nearly identical. The only difference was that the ADPr now assumed an α-form. Because both the α and β anomers of the ribose ring interconvert in aqueous solution, we do not believe that there is any significant biological differences between the two SIRT6·ADPr structures. However, at similar resolution (∼2 Å), the model (PDB code 3PKI) was built on a more complete data set with higher redundancy and had lower Rwork and Rfree values (Table 1). In describing the structure of SIRT6·ADPr, we will be referring to the model/data set in which ADPr was used for co-crystallization.
Overall Structure of SIRT6 in Complex with ADP-ribose
To gain insight into the structural basis for SIRT6 enzymatic function, we initially solved a 2.0 Å crystal structure of the human SIRT6 protein in complex with ADPr. The asymmetric unit consists of six molecules, and each monomer contains one zinc atom, one molecule of ADPr, and several sulfate molecules, which were present in the crystallization buffer. Residues 11–295 of SIRT6(3–318) were visible in the crystal structure. It is possible that the N-terminal (positions 3–10) and C-terminal (positions 295–318) residues were cleaved by trace amounts of the subtilisin that was added to promote crystallization (37), or alternatively these regions are disordered. Both the N- and C-terminal residues visible in the crystal structure form long unstructured coils, providing support that the unseen residues of the SIRT6 constructs are highly flexible and disordered.
Consistent with other solved sirtuin structures, SIRT6 contains two globular domains composed of eight α-helices and nine β-strands: a large Rossmann fold for NAD+ binding (residues 25–128 and 191–266) and a smaller domain, which contains a zinc-binding motif (residues 129–190) (Fig. 2A). The large Rossmann fold domain is formed by a six-stranded (β1, β2, β3, β7, β8, and β9) parallel β-sheet sandwiched between two helices (α6 and α7) on one side and four helices (α1, α4, α5, and α8) on the other side. The small domain is formed by two extending loops (connecting β3 and α6) from the large domain and consists of a three-stranded antiparallel β-sheet (β4, β5, and β6). Interestingly, although most sirtuins contain a Cys-X-X-Cys-X15–20-Cys-X-X-Cys sequence motif for Zn2+-binding, SIRT6 contains a 10-residue insertion between the second set of cysteines, resulting in an extended long loop (supplemental Fig. 2). The six molecules in the asymmetric unit appear to be almost identical (Fig. 2B), with variations in the extended loop of the zinc-binding motif as well as in the N and C termini. The extended loop is highly flexible, and only one of the six molecules in the asymmetric unit has an ordered extended loop.
FIGURE 2.

Structure of human SIRT6 in complex with ADP-ribose. A, overall structural features of SIRT6 monomer. B, superimposition of the six molecules in the asymmetric unit. Red, chain A; green, chain B; dark blue, chain C; orange, chain D; cyan, chain E; yellow, chain F. C, schematic illustration of the hydrogen bonding network surrounding ADPr; hydrogen bonds are indicated as dashed lines, and water molecules are shown as spheres. D, left, SIRT6·ADPr 2Fo − Fc electron density map (blue mesh, 1.5σ) of the residues within 4 Å of ADPr. Right, Fo − Fc omit electron density map (green mesh, 2σ) of the ADPr molecule; the putative peptide binding site contains an unidentified electron density.
The NAD+ binding pocket of sirtuins has been divided into three regions: sites A, B, and C (24). Similar to all other solved sirtuin structures, the adenosine moiety of ADPr is bound in the A site of SIRT6, and the nicotinamide ribose moiety is bound in the B site. Many of the residues involved in ADPr binding are conserved between SIRT6 and the other sirtuins (supplemental Fig. 2). The ADPr interactions in the SIRT6 active site are shown in Fig. 2C. For all six molecules in the asymmetric unit, the ribose moiety of ADPr assumes the C2′ exo conformation in its α-form (Fig. 2D).
Although acetylated H3K9Ac peptide was included during co-crystallization, we did not observe the peptide in the solved SIRT6 structure. However, almost perpendicular to the ADPr electron density, we observed elongated density (Fig. 2D). Given that this location is where peptide binding is predicted (19), we attempted to model a lysine side chain or a PEG molecule into this density. However, we were unable to unambiguously fit the density, and therefore we left these atoms unassigned.
Structural Comparison of SIRT6·ADP-ribose with SIRT6·2′-N-Acetyl-ADP-ribose
As we have demonstrated in Fig. 1, SIRT6 forms 2′-OAADPr as one product of the reaction, and therefore to gain insight into the interactions between 2′-OAADPr and SIRT6, we co-crystallized SIRT6 with 2′-NAADPr (Fig. 3A), a non-hydrolyzable analog of OAADPr in which the O-acetyl moiety is substituted with an N-acetyl group (33). The overall structural features are identical between the SIRT6·ADPr and SIRT6·NAADPr crystal structures (Fig. 3B). This is not surprising because NAADPr only contains an extra N-acetyl group compared with ADPr. There are no significant changes in the orientation of residues surrounding the NAD+ binding site (Fig. 3C). It is interesting to note that the unknown density observed in the SIRT6·ADPr structure was not present in the SIRT6·NAADPr structure (Fig. 3D). The N-acetyl group protrudes into the space that is otherwise occupied by the unassigned chemical entity in the SIRT6·ADPr structure.
FIGURE 3.

Comparison of SIRT6·ADP-ribose with SIRT6·2′-N-acetyl-ADP-ribose structure. A, left, SIRT6·NAADPr 2Fo − Fc electron density map (purple mesh, 1.5σ) of the residues within 4 Å of NAADPr. Right, Fo − Fc omit electron density map (green mesh, 1σ) of the NAADPr molecule. B, superimposition of SIRT6·ADPr (blue) onto SIRT6·NAADPr (purple); the unknown density (Fo − Fc omit electron density map contoured at 2σ, green mesh) observed in the SIRT6·ADPr structure is not present in the SIRT6·NAADPr structure, and the N-acetyl group occupies part of the unidentified density space. C, residues immediately surrounding the N-acetyl group of NAADPr (purple) and their corresponding residues (blue) in the SIRT6·ADPr structure.
Structural Comparison with Other Solved Human Sirtuins (SIRT2, SIRT3, and SIRT5)
Sirtuins have been phylogenetically divided into five subclasses: I–IV and U (45–47). Among the seven human sirtuins, SIRT1 to -3 belong to class I; SIRT4 and -5 belong to class II and III, respectively; and SIRT6 and -7 are both class IV sirtuins. The three previously characterized human sirtuins provide structural insight into classes I and III. The SIRT6 structure constitutes the first solved sirtuin structure in class IV. Structural features revealed by SIRT6 provide additional information on how the different evolutionary lines developed in the sirtuin family.
Although SIRT6 shares the overall domain architecture with SIRT2 (18), SIRT3 (19), and SIRT5 (20), there are several differences on the surface of the protein. In all previously solved sirtuin structures, a conserved “cofactor binding loop” (supplemental Fig. 2) (48) is involved in NAD+ binding; this loop adopts several different conformations depending on the ligand(s) bound in the active site. A small region of the cofactor-binding loop is disordered in the ADPr-bound SIRT5 structure, but in the suramin·SIRT5 complex structure, this loop becomes ordered and even forms β strands and a small helix, which directly interact with suramin, an anti-parasitic drug that also binds sirtuins with micromolar affinity (20). The changes in the cofactor-binding loop are also observed in the set of SIRT3 structures (19). SIRT6 lacks the cofactor-binding loop, which is replaced by a single helix (α3) containing several NAD+ binding residues (Fig. 4A). Unlike the cofactor-binding loop in the ADPr-bound SIRT5 structure and the SIRT3 apo structure, helix α3 appears to be ordered in the SIRT6·ADPr and SIRT6·NAADPr structures. This is an indication that the SIRT6 substrate-binding pocket may be less flexible and does not significantly vary its conformation when different ligands are bound. In addition, helix α1 in SIRT6 is much longer, and the N-terminal unstructured coil folds back toward and may structurally stabilize the NAD+ binding site (Fig. 4B), although there is no direct interaction between the random coil and ADPr.
FIGURE 4.

SIRT6·ADPr structure compared with other solved human SIRTs. A, SIRT6 structure (PDB code 3PKI, orange) superimposed onto SIRT3 (PDB code 3GLR, green). The left panel highlights differences in the zinc-binding domain, and the right panel shows a missing helix bundle. B, comparison of the N-terminal loop of SIRT6 (3PKI, orange), SIRT2 (1J8F, pink), SIRT3 (3GLR, green), and SIRT5 (2B4Y, blue); SIRT6 contains a long loop covering the NAD+ binding site. C, SIRT6 lacks the conserved salt bridge. Left, SIRT3 (3GLR) structure with conventional salt bridge in the sirtuin family. Right, the green pair (Arg180 and Asp188) is where the bridge would be predicted to form on the SIRT6 structure according to other sirtuin crystal structures. Pink pair (Arg124 and Gln145), the actual hydrogen bonding pair found in SIRT6 structure. D, stereo diagram of flipped FGEXL (WEDsL) loop of SIRT6 (PDB code 3PKI, orange) complexed with ADPr (orange) compared with SIRT3 (PDB code 3GLT, green) with an ADPr·thioAceCS2 (green) complex.
In contrast to class I, II, and III human sirtuins, class IV sirtuins contain a deletion in the sequence immediately following the α3 helix (supplemental Fig. 2). The result is that SIRT6 does not have a helix bundle in its small domain (Fig. 4A). The helix bundle is replaced by a short loop, which interacts with the loop between α2 and α3 and contacts a small region on the zinc-binding module. The lack of a helix bundle to form extensive interactions with the β-sheets in the zinc-binding module provides one explanation for why SIRT6 has a splayed small domain. This unique structural feature is likely to be adopted by SIRT7, another class IV sirtuin that also contains a deletion in this region (supplemental Fig. 2) but that has not yet been structurally and functionally characterized. This unique feature may be responsible for the observed lower activity (1000 times) of SIRT6. To date, there are no reports of detectable deacetylase activity for SIRT7.
In addition to the missing helical bundle, another reason for the splayed small domain in SIRT6 is the loss of a conserved salt bridge, which has been reported to contribute to the positioning of the zinc-binding motif with respect to the Rossmann fold domain and substrate-binding site (20, 24). Based on precedent from other sirtuins, a salt bridge should be formed between Arg180 and Asp188 (Fig. 4C) in SIRT6. However, this interaction is not observed. The side chain of Arg180 is directed toward the surface of the protein rather than toward the Asp188 residue. Instead, Arg124 on the loop connecting α5 and β3 and Gln145 on β5 form a hydrogen bond to tether the zinc-binding motif and the large domain together (Fig. 4C). The loss of the previously described salt bridge removes the constraint on the two loops, which would otherwise result in a more closed conformation. The new Arg124–Gln145 hydrogen bond in SIRT6 pulls the two domains together on the opposite side, and therefore, the small domain is not directly above the Rossmann fold but rather in an open conformation tilted in a 45° angle.
It is generally observed that peptide substrates interact with sirtuins via nonspecific backbone interactions (19). There is a conserved FGEXL loop that forms hydrogen bonds with the substrate peptide (27). However, in the case of SIRT6, the sequence is WEDSL (supplemental Fig. 2). The amide nitrogen of Trp186 appears to be flipped, whereas its analog in other sirtuins is involved in a hydrogen bond with a carbonyl group from the peptide substrate backbone. Upon superimposition of the SIRT6 structure with the SIRT3-acetylated peptide structure (PDB code 3GLT) (19), we observed that the backbone amide between the tryptophan and glutamate side chains is further away and cannot form a hydrogen bond with the carbonyl in the amide backbone of the lysine (Fig. 4D). This poses an issue for the peptide-protein anti-parallel β-sheet formation. It is possible that a β-sheet configuration might still form, but a kink near the active site would adversely affect the interactions between the peptide substrate and the protein. Therefore, a less stable substrate-protein interaction might be the cause of the poor deacetylase activity for SIRT6 and the reason why the H3K9Ac peptide does not exhibit binding saturation by ITC (supplemental Fig. 3).
Sirtuins catalyze deacetylation through a sequential mechanism in which the substrate peptide binds first to form an ordered NAD+-binding pocket for the subsequent co-substrate binding. SIRT1, SIRT2, and SIRT3 were all found to require a substrate/ligand binding prior to NAD+ binding (19, 34, 49). The unique SIRT6 structural features suggest that SIRT6 utilizes a different binding mechanism, which does not require peptide substrate binding prior to NAD+ binding. These features include an ordered helix regardless of substrate binding, a large open channel in the substrate and NAD+ binding site due to the missing helix bundle, and a NAD+ site potentially reinforced by the N-terminal loop. To test this hypothesis, we performed a series of equilibrium binding experiments with SIRT6.
NAD+ Metabolite Binding
Isothermal titration calorimetry was used to monitor the binding of NAD+ in the absence of an acetyl lysine substrate. Consistent with previous kinetic results, Hst2, SIRT1 to -3, and SIRT5 did not bind NAD+ in the absence of the acetyl lysine substrate (Fig. 5A and supplemental Fig. 4). Strikingly, NAD+ bound to SIRT6 with a Kd of 27 ± 1 μm (Fig. 5B), providing the first physical evidence that SIRT6, unlike other characterized sirtuins, can bind NAD+ efficiently in the absence of an acetylated substrate. We detected no binding when NADH was titrated into SIRT6 (supplemental Fig. 5A), suggesting that SIRT6 cannot accommodate the puckered conformation of the nicotinamide ring of NADH. Interestingly, ADPr bound to SIRT6 with a Kd of 4.7 ± 0.5 μm (supplemental Fig. 5B and Fig. 5B), suggesting that the binding pocket of SIRT6 distinguishes structural differences between NAD+ and ADPr. The construct (residues 3–318) used for SIRT6 crystallization was able to bind NAD+ and ADPr with similar affinities compared with the full-length SIRT6 construct (supplemental Fig. 6), suggesting that there is negligible variability between the full-length construct used for kinetic assays and the crystallized constructs.
FIGURE 5.

Isothermal titration calorimetry studies. A, representative ITC trace. The top graph shows data obtained for 37 automatic injections (1–8 μl) of 1.4 mm NAD+ titrated into 27 μm SIRT6. The bottom graph represents integrated curves of the experimentally generated heats. The data were fitted to a one-site binding curve (solid line). NAD+ (1.40 or 1.63 mm) was titrated into 27 μm WT SIRT6 (■) or 50 μm Hst2 (●), respectively. B, dissociation constants and column graph displaying thermodynamic parameters for WT and H131Y SIRT6 binding to NAD+, ADPr, and 2′-NAADPr. NAD+ (1.40 mm or 767 μm) was titrated into 27 μm or 33 μm WT or H131Y SIRT6, respectively. ADPr (390 μm or 1.45 mm) was titrated into 28 or 37 μm WT or H131Y SIRT6, respectively. 2′-NAADPr (2.96 mm) was titrated into 38 μm WT SIRT6. The change in Gibbs free energy (ΔG) is shown in black. Change in enthalpy (ΔH) is shown is gray, and −TΔS is displayed in light gray. Error bars, S.D.
Consistent with the crystal structure, the non-hydrolyzable product analog 2′-NAADPr bound to SIRT6 with a Kd of 22 ± 2 μm (supplemental Fig. 5C and Fig. 5B). Interestingly, unlike NAD+ and ADPr, 2′-NAADPr bound with a negative −TΔS (Fig. 5, A and B), suggesting that there is more disorder when 2′-NAADPr binds. This result is consistent with the crystal structure, which showed a high degree of variability and poor electron density for the 2′-N-acetyl-ribose moiety of 2′-NAADPr.
The invariant catalytic base histidine residue of Hst2 is important for both NAD+ binding and activation of the 2′-hydroxyl of the α-1′-O-alkylamidate intermediate during the deacetylation reaction (43). To investigate the role of SIRT6 His131 in NAD+ and ADPr binding, ITC was used to monitor the effects of the H131Y substitution on the ability of SIRT6 to bind NAD+ metabolites. The substitution to tyrosine led to a dramatic effect in the ability of SIRT6 to bind ADPr. The Kd of 69 ± 4 μm was ∼15 times higher than the Kd for WT SIRT6 (supplemental Fig. 5D and Fig. 5B). The enthalpy increased 6 ± 1 kcal/mol, consistent with the loss of hydrogen bonds to the 2′- and 3′-hydroxyl groups of the ribose, observed in the crystal structure of SIRT6 bound to ADPr (Fig. 2C). Interestingly, the H131Y mutant bound NAD+ with a Kd of 8.4 ± 0.5 μm, or ∼3 times lower than the Kd for WT SIRT6 (supplemental Fig. 5E and Fig. 5B). This is a 47-fold change in specificity for NAD+ over ADPr from WT SIRT6 to H131Y SIRT6. The enthalpy of binding NAD+ decreased 15 ± 1 kcal/mol, whereas −TΔS became less favorable as it increased 14 ± 1 kcal/mol from WT to H131Y (Fig. 5B). These changes probably reflect increased hydrogen bonding and van der Waal's contacts between the nicotinamide moiety of NAD+ and H131Y of SIRT6, causing the ΔH to become more negative, and subsequent ordering of both NAD+ and SIRT6, leading in turn to the increase in the entropy noted in the −TΔS value.
As an independent method for analyzing NAD+ metabolite binding, we monitored changes in tryptophan fluorescence as a function of ligand binding. SIRT6 contains two tryptophan residues near the active site (Trp69 and Trp186). We observed a decrease in the tryptophan fluorescence signal of SIRT6 with increasing concentrations of NAD+ (supplemental Fig. 7A), suggesting a change in the local environment surrounding the tryptophan residues upon NAD+ binding. The fluorescence intensities were adjusted for nonspecific quenching of tryptophan residues located outside of the active site and the data were fitted to a one-site binding curve (Equation 3). The Kd of 24 ± 7 μm is in excellent agreement with the Kd determined by ITC (Kd of 27 μm). However, at increasing concentrations of ADPr, no change in fluorescence was observed (supplemental Fig. 7B), despite the fact that by ITC, ADPr binds to SIRT6 with a Kd of 4.7 μm. The results suggest that there are different structural rearrangements in the active site, depending on the NAD+ metabolite.
In agreement with the ITC results, a decrease in the fluorescence of H131Y SIRT6 was observed at increasing concentrations of NAD+, yielding a Kd of 3.5 ± 0.6 μm for NAD+ (supplemental Fig. 7C). Interestingly, an increase in fluorescence was observed with H131Y SIRT6 upon binding ADPr (supplemental Fig. 7D), suggesting that the environment surrounding the tryptophan residues is more non-polar when ADPr binds. The Kd of 51 ± 8 μm is in agreement with the previous ITC results (Kd of 69 μm; Fig. 5B). These results further highlight the conclusion that NAD+ and ADPr induce different structural perturbations on SIRT6 upon binding.
DISCUSSION
Several physiological and cellular investigations have suggested that SIRT6 plays a role in genome maintenance and metabolic regulation (10, 12, 15–17, 50–53). Although these studies have provided insight into the biological role of SIRT6, the molecular functions of SIRT6 remain unclear. Here we have reported the first quantitative assessment of SIRT6 activity, providing direct evidence that OAADPr is formed as a product of an extremely inefficient deacetylase reaction. Unlike other sirtuins, SIRT6 displays tight binding of NAD+ in the absence of an acetylated substrate. Furthermore, tryptophan fluorescence results suggest that NAD+ and ADPr induce different structural arrangements upon binding. We have also solved the first crystal structures of SIRT6, a member of the class IV sirtuins. The crystal structures provide insight into the biochemical functions presented in this study and reveal several significant differences between SIRT6 and the other solved sirtuin structures.
SIRT6 was reported to act as an H3K9 deacetylase (12). Here, we utilized three quantitative assays to establish that SIRT6 forms OAADPr, but at a specific activity of ∼0.0002 s−1 toward H3K9Ac, which is ∼1,000 times slower than other highly active sirtuins (34). Furthermore, the rate of nicotinamide cleavage is similar to the rate of OAADPr formation, suggesting that SIRT6 functions as a NAD+-dependent deacetylase rather than as an ADP-ribosyltransferase. It is unlikely that the low deacetylase activity is caused by inhibition of the undefined molecule observed in the acetyl lysine-binding site of the SIRT6-ADPr structure. The SIRT6·NAADPr structure shows no evidence of this molecule, suggesting that it was either readily displaced by NAADPr or was not present during crystallization. It is important to note that the acetyl group of NAADPr occupied the same space as that of the undefined molecule in the SIRT6·ADPr structure. If this molecule were responsible for the low activity of SIRT6, we would expect to observe major differences in binding affinity among NAADPr, NAD+, and ADPr. The fact that NAADPr and NAD+ displayed nearly identical binding affinity suggests that this molecule, if present, did not impede NAADPr binding and therefore would not significantly contribute to inhibiting SIRT6 activity. In addition, the results are consistent with the previous analyses, which demonstrated that SIRT6 is not an efficient H3K9 deacetylase (12). The fact that SIRT6 has been implicated in important processes, including several metabolic pathways (17, 50, 51, 53), double strand break repair (15, 52), base excision repair (10), and telomere maintenance (12, 13), raises the question of why a seemingly important sirtuin would display such low deacetylase activity.
The newly solved crystal structures of SIRT6 in complex with ADPr and NAADPr provide some insight. SIRT6 shares an overall similar architecture with the previously solved sirtuin structures and contains the conserved catalytic residues. However, the SIRT6 structure revealed many unique features. SIRT6 exists in a more open conformation, containing a zinc-binding motif in the small domain, which is splayed from the larger Rossmann fold domain. Several structural features contribute to the displacement of the small domain relative to its position on other sirtuins, including the lack of a conserved salt bridge between the zinc-binding motif and the substrate-binding loop. This conserved interaction was previously demonstrated to be important for maintaining the proper orientation of the small domain with respect to the large domain and is important for forming the acetylated substrate-binding pocket (20, 24). In SIRT6, however, a new hydrogen bond connects the zinc-binding motif to the lower portion of the Rossmann fold, stabilizing SIRT6 in an open conformation. Furthermore, the class IV sirtuins, which include SIRT6, lack a conserved helix bundle region that forms contacts between the small domain and the Rossmann fold domain. The consequence of this feature contributes to the open conformation observed in the SIRT6 crystal structure. The open conformation might disrupt the structural integrity of the substrate-binding pocket, providing one explanation for why H3K9Ac does not exhibit binding site saturation, which in turn leads to poor deacetylase efficiency.
Instead of containing a highly conserved cofactor binding loop (48) that aids in NAD+ binding, SIRT6 contains a single helix that forms interactions with ADPr and NAADPr. In several sirtuin structures, the orientation of the loop and the ordering of the residues depend on the substrate(s) bound in the active site (26, 29). The single helix in SIRT6, however, appears to be ordered in both structures, indicating that the substrate-binding pocket is less flexible and does not widely vary its conformation. This led us to hypothesize that, unlike all other studied sirtuins, SIRT6 would be able to bind NAD+ in the absence of an acetylated substrate.
We found that SIRT6 can bind NAD+ with a relatively high affinity (Kd = 27 μm) in the absence of an acetylated substrate. Furthermore, we determined that SIRT6 can bind both ADPr (4.7 μm) and NAADPr (22 μm) with relatively similar affinities compared with NAD+. However, the fact that a change in tryptophan fluorescence was observed when the concentration of NAD+ was increased and not when ADPr was increased suggests that there may be differing structural changes near the tryptophan residues when the ligands bind; this provides evidence that SIRT6 is able to distinguish between the ligands that are bound in the active site. To investigate NAD+ binding more closely, we mutated the catalytic histidine residue (His131), which was previously reported to be important not only for catalysis but also for NAD+ binding (43). We found that H131Y SIRT6 can still bind NAD+ (8.4 μm) but has a decreased ability to bind ADPr (69 μm). In addition, although a decrease in the tryptophan fluorescence signal was seen with increasing NAD+, an increase in fluorescence was seen with increasing ADPr. Together, these results further support the conclusion drawn for WT SIRT6, that there is an inherent structural difference near the tryptophan residues in SIRT6 between NAD+ and ADPr binding. Therefore, this structural difference may play an important role in controlling SIRT6 activity.
The fact the NAD+ binds to SIRT6 in the absence of an acetyl lysine substrate is a unique feature of this sirtuin. Also, the low basal activity and the unique structural features suggest that regulatory mechanisms may be needed to activate SIRT6 in cells. The open structure of SIRT6 might provide a surface for regulatory protein binding and stimulation of activity. Similar mechanisms have been demonstrated for other histone-modifying proteins, including the yeast acetyltransferase RTT109, which requires the histone chaperones Asf1 (54) and Vps75 (55) for efficient activity. SIRT6 interacts with several transcription factors, including NF-κB (16) and Hif1α (17); these interactions might stabilize an active conformation of SIRT6 and subsequently stimulate histone deacetylation at the site of action. It is also possible that SIRT6 is indeed a robust enzyme, but we and others have not supplied the appropriate protein substrate. Alternatively, the deacetylase activity of SIRT6 might not be regulated by activation, but instead this intrinsically low deacetylase activity might suggest that SIRT6 has a different molecular function.
Interestingly, SIRT6 has been implicated in many of the same pathways as SIRT1, including the targeting of H3K9 for deacetylation (56). SIRT1 is a highly active deacetylase, begging the question of why purified SIRT6 would also deacetylate H3K9 but very inefficiently. The ability of SIRT6 to bind NAD+ in the absence of an acetylated substrate is unique, and the possibility that SIRT6 may adopt different structural arrangements depending on which ligand is bound suggests that an alternative hypothesis for SIRT6 molecular function is plausible: that SIRT6 has evolved away from a functioning NAD+-dependent deacetylase and toward a function as a NAD+ metabolite sensor. There are many examples of enzymes that have evolved away from their catalytic role and toward a regulatory function that takes advantage of the ability to bind ligand. These include signaling proteins that belong to the large family of protein kinases (57) and phosphatases (58).
SIRT6 controls flux into glycolysis by inhibiting Hif1α-dependent transcription of several glycolytic genes to promote mitochondrial respiration (17). In addition, SIRT6 has also been reported to negatively regulate PPARγ-mediated transcription to prevent triglyceride formation (59). These are two pathways controlled by cellular NAD+ levels and furthermore, they are also pathways in which SIRT1 has been reported to play an important role (60, 61). SIRT6 is found at the promoters of several metabolic genes involved in these pathways, and its interaction with transcription factors suggests that NAD+ levels might control SIRT6 function. Presumably, under high levels of NAD+, SIRT6 could bind to transcription factors at promoters of metabolic genes preventing triglyceride formation and flux into glycolysis. Alternatively, below Kd levels of NAD+, the interaction would be destabilized, allowing transcription factors to access the targeted genes. Therefore, SIRT6 might ensure metabolic homeostasis and promote more efficient energy usage through its ability to sense the levels of NAD+ (and related metabolites) and regulate protein-protein interactions with transcription factors. Future in vivo studies will be needed to explore these possibilities.
Acknowledgments
We thank members of the Denu laboratory, especially Kristin Dittenhafer-Reed and Brittany Albaugh, for valuable discussions and insight.
This work was supported, in whole or in part, by National Institutes of Health Grant GM065386 (to J. M. D.). This research was also supported by the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–7.
The atomic coordinates and structure factors (codes 3K35, 3PKI, and 3PKJ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- OAADPr
- O-acetyl-ADP-ribose
- NAADPr
- N-acetyl-ADP-ribose
- ADPr
- ADP-ribose
- ITC
- isothermal titration calorimetry
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank.
REFERENCES
- 1. Michan S., Sinclair D. (2007) Biochem. J. 404, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackson M. D., Denu J. M. (2002) J. Biol. Chem. 277, 18535–18544 [DOI] [PubMed] [Google Scholar]
- 3. Sauve A. A., Celic I., Avalos J., Deng H., Boeke J. D., Schramm V. L. (2001) Biochemistry 40, 15456–15463 [DOI] [PubMed] [Google Scholar]
- 4. Haigis M. C., Guarente L. P. (2006) Genes Dev. 20, 2913–2921 [DOI] [PubMed] [Google Scholar]
- 5. Kaeberlein M., McVey M., Guarente L. (1999) Genes Dev. 13, 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tissenbaum H. A., Guarente L. (2001) Nature 410, 227–230 [DOI] [PubMed] [Google Scholar]
- 7. Rogina B., Helfand S. L. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15998–16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., Prolla T. A. (2010) Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michishita E., Park J. Y., Burneskis J. M., Barrett J. C., Horikawa I. (2005) Mol. Biol. Cell 16, 4623–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M. M., Mills K. D., Patel P., Hsu J. T., Hong A. L., Ford E., Cheng H. L., Kennedy C., Nunez N., Bronson R., Frendewey D., Auerbach W., Valenzuela D., Karow M., Hottiger M. O., Hursting S., Barrett J. C., Guarente L., Mulligan R., Demple B., Yancopoulos G. D., Alt F. W. (2006) Cell 124, 315–329 [DOI] [PubMed] [Google Scholar]
- 11. Liszt G., Ford E., Kurtev M., Guarente L. (2005) J. Biol. Chem. 280, 21313–21320 [DOI] [PubMed] [Google Scholar]
- 12. Michishita E., McCord R. A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T. L., Barrett J. C., Chang H. Y., Bohr V. A., Ried T., Gozani O., Chua K. F. (2008) Nature 452, 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michishita E., McCord R. A., Boxer L. D., Barber M. F., Hong T., Gozani O., Chua K. F. (2009) Cell Cycle 8, 2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang B., Zwaans B. M., Eckersdorff M., Lombard D. B. (2009) Cell Cycle 8, 2662–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaidi A., Weinert B. T., Choudhary C., Jackson S. P. (2010) Science 329, 1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Kawahara T. L., Michishita E., Adler A. S., Damian M., Berber E., Lin M., McCord R. A., Ongaigui K. C., Boxer L. D., Chang H. Y., Chua K. F. (2009) Cell 136, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhong L., D'Urso A., Toiber D., Sebastian C., Henry R. E., Vadysirisack D. D., Guimaraes A., Marinelli B., Wikstrom J. D., Nir T., Clish C. B., Vaitheesvaran B., Iliopoulos O., Kurland I., Dor Y., Weissleder R., Shirihai O. S., Ellisen L. W., Espinosa J. M., Mostoslavsky R. (2010) Cell 140, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finnin M. S., Donigian J. R., Pavletich N. P. (2001) Nat. Struct. Biol. 8, 621–625 [DOI] [PubMed] [Google Scholar]
- 19. Jin L., Wei W., Jiang Y., Peng H., Cai J., Mao C., Dai H., Choy W., Bemis J. E., Jirousek M. R., Milne J. C., Westphal C. H., Perni R. B. (2009) J. Biol. Chem. 284, 24394–24405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schuetz A., Min J., Antoshenko T., Wang C. L., Allali-Hassani A., Dong A., Loppnau P., Vedadi M., Bochkarev A., Sternglanz R., Plotnikov A. N. (2007) Structure 15, 377–389 [DOI] [PubMed] [Google Scholar]
- 21. Avalos J. L., Bever K. M., Wolberger C. (2005) Mol. Cell 17, 855–868 [DOI] [PubMed] [Google Scholar]
- 22. Cosgrove M. S., Bever K., Avalos J. L., Muhammad S., Zhang X., Wolberger C. (2006) Biochemistry 45, 7511–7521 [DOI] [PubMed] [Google Scholar]
- 23. Hawse W. F., Hoff K. G., Fatkins D. G., Daines A., Zubkova O. V., Schramm V. L., Zheng W., Wolberger C. (2008) Structure 16, 1368–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Min J., Landry J., Sternglanz R., Xu R. M. (2001) Cell 105, 269–279 [DOI] [PubMed] [Google Scholar]
- 25. Chang J. H., Kim H. C., Hwang K. Y., Lee J. W., Jackson S. P., Bell S. D., Cho Y. (2002) J. Biol. Chem. 277, 34489–34498 [DOI] [PubMed] [Google Scholar]
- 26. Avalos J. L., Boeke J. D., Wolberger C. (2004) Mol. Cell 13, 639–648 [DOI] [PubMed] [Google Scholar]
- 27. Avalos J. L., Celic I., Muhammad S., Cosgrove M. S., Boeke J. D., Wolberger C. (2002) Mol. Cell 10, 523–535 [DOI] [PubMed] [Google Scholar]
- 28. Zhao K., Chai X., Marmorstein R. (2004) J. Mol. Biol. 337, 731–741 [DOI] [PubMed] [Google Scholar]
- 29. Zhao K., Harshaw R., Chai X., Marmorstein R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8563–8568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanders B. D., Zhao K., Slama J. T., Marmorstein R. (2007) Mol. Cell 25, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao K., Chai X., Clements A., Marmorstein R. (2003) Nat. Struct. Biol. 10, 864–871 [DOI] [PubMed] [Google Scholar]
- 32. Zhao K., Chai X., Marmorstein R. (2003) Structure 11, 1403–1411 [DOI] [PubMed] [Google Scholar]
- 33. Comstock L. R., Denu J. M. (2007) Org. Biomol. Chem. 5, 3087–3091 [DOI] [PubMed] [Google Scholar]
- 34. Borra M. T., Langer M. R., Slama J. T., Denu J. M. (2004) Biochemistry 43, 9877–9887 [DOI] [PubMed] [Google Scholar]
- 35. Borra M. T., Denu J. M. (2004) Methods Enzymol. 376, 171–187 [DOI] [PubMed] [Google Scholar]
- 36. Smith B. C., Hallows W. C., Denu J. M. (2009) Anal. Biochem. 394, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong A., Xu X., Edwards A. M., Chang C., Chruszcz M., Cuff M., Cymborowski M., Di Leo R., Egorova O., Evdokimova E., Filippova E., Gu J., Guthrie J., Ignatchenko A., Joachimiak A., Klostermann N., Kim Y., Korniyenko Y., Minor W., Que Q., Savchenko A., Skarina T., Tan K., Yakunin A., Yee A., Yim V., Zhang R., Zheng H., Akutsu M., Arrowsmith C., Avvakumov G. V., Bochkarev A., Dahlgren L. G., Dhe-Paganon S., Dimov S., Dombrovski L., Finerty P., Jr., Flodin S., Flores A., Gräslund S., Hammerström M., Herman M. D., Hong B. S., Hui R., Johansson I., Liu Y., Nilsson M., Nedyalkova L., Nordlund P., Nyman T., Min J., Ouyang H., Park H. W., Qi C., Rabeh W., Shen L., Shen Y., Sukumard D., Tempel W., Tong Y., Tresagues L., Vedadi M., Walker J. R., Weigelt J., Welin M., Wu H., Xiao T., Zeng H., Zhu H. (2007) Nat. Methods 4, 1019–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 40. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 41. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 42. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 43. Smith B. C., Denu J. M. (2006) Biochemistry 45, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tennen R. I., Berber E., Chua K. F. (2010) Mech. Ageing Dev. 131, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frye R. A. (1999) Biochem. Biophys. Res. Commun. 260, 273–279 [DOI] [PubMed] [Google Scholar]
- 46. Tanner K. G., Landry J., Sternglanz R., Denu J. M. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 14178–14182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frye R. A. (2000) Biochem. Biophys. Res. Commun. 273, 793–798 [DOI] [PubMed] [Google Scholar]
- 48. Sanders B. D., Jackson B., Marmorstein R. (2010) Biochim. Biophys. Acta 1804, 1604–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milne J. C., Lambert P. D., Schenk S., Carney D. P., Smith J. J., Gagne D. J., Jin L., Boss O., Perni R. B., Vu C. B., Bemis J. E., Xie R., Disch J. S., Ng P. Y., Nunes J. J., Lynch A. V., Yang H., Galonek H., Israelian K., Choy W., Iffland A., Lavu S., Medvedik O., Sinclair D. A., Olefsky J. M., Jirousek M. R., Elliott P. J., Westphal C. H. (2007) Nature 450, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiao C., Kim H. S., Lahusen T., Wang R. H., Xu X., Gavrilova O., Jou W., Gius D., Deng C. X. (2010) J. Biol. Chem. 285, 36776–36784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim H. S., Xiao C., Wang R. H., Lahusen T., Xu X., Vassilopoulos A., Vazquez-Ortiz G., Jeong W. I., Park O., Ki S. H., Gao B., Deng C. X. (2010) Cell Metab. 12, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCord R. A., Michishita E., Hong T., Berber E., Boxer L. D., Kusumoto R., Guan S., Shi X., Gozani O., Burlingame A. L., Bohr V. A., Chua K. F. (2009) Aging 1, 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanfi Y., Shalman R., Peshti V., Pilosof S. N., Gozlan Y. M., Pearson K. J., Lerrer B., Moazed D., Marine J. C., de Cabo R., Cohen H. Y. (2008) FEBS Lett. 582, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsubota T., Berndsen C. E., Erkmann J. A., Smith C. L., Yang L., Freitas M. A., Denu J. M., Kaufman P. D. (2007) Mol. Cell 25, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berndsen C. E., Tsubota T., Lindner S. E., Lee S., Holton J. M., Kaufman P. D., Keck J. L., Denu J. M. (2008) Nat. Struct. Mol. Biol. 15, 948–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vaquero A., Scher M., Lee D., Erdjument-Bromage H., Tempst P., Reinberg D. (2004) Mol. Cell 16, 93–105 [DOI] [PubMed] [Google Scholar]
- 57. Anamika K., Abhinandan K. R., Deshmukh K., Srinivasan N. (2009) Comp. Funct. Genomics 2009, Article ID 365637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pils B., Schultz J. (2004) Mol. Biol. Evol. 21, 625–631 [DOI] [PubMed] [Google Scholar]
- 59. Kanfi Y., Peshti V., Gil R., Naiman S., Nahum L., Levin E., Kronfeld-Schor N., Cohen H. Y. (2010) Aging Cell 9, 162–173 [DOI] [PubMed] [Google Scholar]
- 60. Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005) Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 61. Picard F., Kurtev M., Chung N., Topark-Ngarm A., Senawong T., Machado De Oliveira R., Leid M., McBurney M. W., Guarente L. (2004) Nature 429, 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jaroszewski L., Rychlewski L., Li Z., Li W., Godzik A. (2005) Nucleic Acids Res. 33, W284–W288 [DOI] [PMC free article] [PubMed] [Google Scholar]