

Abstract
Background
Regulator of G protein signaling 4 (RGS4) is one of the smaller members of the RGS family of proteins, which are known to control signaling amplitude and duration via interactions with G protein α subunits or other signaling molecules. Earlier evidence suggests dynamic regulation of RGS4 levels in neuronal networks mediating actions of opiates and other drugs of abuse, but the consequences of RGS4 actions in vivo are largely unknown.
Methods
In this study, we use constitutive and nucleus accumbens-inducible RGS4 knockout mice, as well as mice overexpressing RGS4 in the nucleus accumbens via viral mediated gene transfer, to examine the influence of RGS4 on behavioral responses to opiates. We also use electrophysiology and immunoprecipitation assays to further understand the mechanisms underlying the tissue-specific actions of RGS4.
Results
Inducible knockout or selective overexpression of RGS4 in the nucleus accumbens reveals that, in this brain region, RGS4 acts as a negative regulator of morphine reward, while in the locus coeruleus RGS4 opposes morphine physical dependence. In contrast, we show that RGS4 does not affect morphine analgesia or tolerance, but is a positive modulator of certain opiate analgesics, such as methadone and fentanyl.
Conclusions
These findings provide fundamentally novel information concerning the role of RGS4 in the cellular mechanisms underlying the diverse actions of opiate drugs in the nervous system.
Keywords: Nucleus accumbens, Locus coeruleus, Place Preference, Fentanyl, Morphine, Tolerance, Withdrawal, AAV-Cre, HSV-RGS4
INTRODUCTION
Regulators of G protein signaling (RGS) are critical modulators of G protein-coupled receptor-mediated signal transduction via multiple interactions with G protein α subunits, scaffolds, and effector molecules (1–4). At least ten of the 25 mammalian RGS proteins are expressed in the CNS (5) and modulate essential physiological functions such as vision (6,7), locomotion (8) and working memory (9). In addition, several neuropsychiatric disorders including Parkinson’s disease (10) addiction (11,12), and schizophrenia (13) are linked to dysfunctions of particular RGS proteins.
RGS4, is a 28 kDa member of the R2 subfamily of RGS proteins, which is expressed widely in brain, including prefrontal cortex, striatum, locus coeruleus (LC), and hippocampus (5). RGS4 consists of a 120 aa domain responsible for the GTPase-activating protein (GAP) activity that regulates G protein function and defines the RGS superfamily, and an N terminal element containing a cystein rich domain (N-end rule) which triggers arginylation and promotes ubiquitination and proteasomal degradation (14–16). A gene array analysis linked decreased levels of RGS4 in prefrontal cortex with schizophrenia and triggered a large number of clinical and preclinical studies on this subject (17). Most of these studies point to RGS4 as a vulnerability factor for schizophrenia (4,13,17–21), whereas evidence also supports a role of RGS4 in antipsychotic drug action (18,21,22). In striatum, RGS4 has a wide range of modulatory actions on muscarinic M2 autoreceptors (23) as well as on dopamine D1 and D2 receptors (4,24,25). Stress, corticosteroids and drugs of abuse modulate RGS4 levels in several brain sites (11,26–28).
Previous work established the striatal enriched RGS9-2 as a key regulator of opioidergic and dopaminergic responses (8,12,29–32). The presence of RGS4 in striatal and areas mediating opiate actions, and the evidence for the involvement of this RGS member in opiate physical dependence (4,11), led us to hypothesize that, in addition to RGS9-2, RGS4 may also be involved in opiate addiction. Opiates produce reward, physical dependence, and analgesia via activation of the G-protein coupled μ opioid receptor (MOR) (33,34). Although it is generally accepted that opiate addiction is associated with adaptations in MOR signal transduction, the cell specific events remain incompletely understood (35–37). Here, we use constitutive and inducible knockout mouse models to examine the role of RGS4 in acute and chronic opiate actions. Our behavioral, electrophysiological, and biochemical findings establish that RGS4 exerts differential effects on distinct actions of opiates in the nervous system.
METHODS
Animals
Constitutive and inducible RGS4 mutant mice were generated as described in Supplemental Methods (See Suppl. Fig. 1). All constitutive mutant mice used in this study were generated from breedings of heterozygous RGS4 mice. For all behavioral assays, we used 2–3 month old male knockout mice and their wildtype littermates. For electrophysiological assays, we used 3–4 week old male mice. For overexpression studies, 2–3 month old C57/Bl6 mice were infected with Herpes simplex virus (HSV)-LacZ or HSV-RGS4 as described (12). For local knockout of RGS4, 2–4 month old floxed RGS4 male mice were bilaterally infected with adeno-associated virus (AAV)-Cre or AAV-GFP. Animals were housed in a 12 hr dark/light cycle room according to the animal care and use committees of UT Southwestern Medical Center and the University of Crete. For place preference, analgesia, and opiate withdrawal paradigms, we used two way ANOVAs and Bonferroni post hoc tests for within group comparisons whenever analysis revealed a genotype affect. For co-IP assays, we used one way analysis of variance and Dunnett’s post hoc test. For western blotting and cAMP inhibition assays, we used t-tests.
Behavioral Tests
A published unbiased conditioned place preference (CPP) procedure was used (8). For morphine locomotor activity assays, mice were placed in chambers as described (8) and ambulatory activity was monitored for 30 min after s.c. saline (days 1–3) or morphine (days 4–9) injections. Analgesia was measured using a 52° C hot plate apparatus (IITC Life Sciences, CA), as described (12). For opiate withdrawal, mice were implanted with 25 mg morphine pellets, withdrawal was precipitated 3 days later with naloxone (1 mg/kg, s.c., Sigma, MO) and withdrawal signs (jumps, wet dog shakes, tremor, ptosis, diarrhea, weight loss) were monitored for 25 min. Fear conditioning was carried out according to published procedures (see Supplemental Methods).
Laser Capturing and PCR
Laser capture was performed as described (38). Floxed RGS4 mice were injected with either AAV-GFP or AAV-CreGFP into the NAc (41,42). Several weeks later, brains were coronally cryosectioned at 8 μm and mounted onto membrane slides (Leica). Infected regions were laser-dissected and processed with PicoPure RNA extraction kit (Arcturus). RNA was amplified with the RiboAmp kit (Arcturus) and reverse transcribed using superscript III (Invitrogen). Quantitative PCR (qPCR) was performed as described previously (39) using SYBR Green (Applied Biosystems) and primers for RGS4 (Fwd: GGCTGAATCGTTGGAAAACCT, Rvs: TGTTGCTTGCACTGAGATGAA) and glyceraldehyde-6-phosphate dehydrogenase (GAPDH) as a control.
Co-Immunoprecipitation and Western Blotting
Striatum from control or treated mice were rapidly dissected (40). The following antibodies were used for immunoprecipitation (IP) and western blotting: rabbit anti-MOR (Immunostar, CA.), rabbit anti-Gαq (P. Sternweiss, UT Southwestern), a rabbit anti-RGS2 (provided by D. Siderovski, UNC, Chapel Hill) and rabbit anti-RGS4 (S. Mumby, UT Southwestern). For RGS4, in addition to a protease inhibitor cocktail (Sigma, MO), samples contained a proteasome inhibitor (MG132, Sigma, MO), to prevent degradation of the protein.
Electrophysiology
Recordings were obtained from LC neurons in brain slices obtained from drug-naïve and morphine-dependent mice (treated as stated earlier). Please see Supplemental Methods for a description of the protocols used, all of which are published (43). Recordings were made 2 hrs after maintaining slices in a recording chamber to allow morphine to wash out fully from the slices (43).
RESULTS
RGS4 and Morphine Reward: Actions in the Nucleus Accumbens
To assess the role of RGS4 in morphine reward, we induced a local knockout of RGS4 in the nucleus accumbens (NAc, part of the ventral striatum), a key brain reward region, of adult animals. This inducible, localized deletion of RGS4 was achieved by stereotaxic injection of an AAV vector expressing GFP-tagged Cre recombinase into the NAc of mice homozygous for a floxed RGS4 gene. Control animals were injected with AAV-GFP. Fig. 1a,b shows low and high power magnifications of AAV-CreGFP infected areas. To confirm recombination, we isolated the GFP labeled region of the NAc by laser capture microdissection and measured RGS4 mRNA levels using qPCR. RGS4 expression is reduced by >90% in the NAc of mice injected with AAV-CreGFP as compared to mice injected with AAV-GFP (Fig. 1c).
Figure 1. Selective deletion of RGS4 in the NAc increases sensitivity to morphine reward.

Low (a, Zeiss 10x) and high (b, Leica confocal 40x) power magnifications of immunofluorescence for GFP in the NAc in AAV-CreGFP-injected floxed RGS4 mice. To verify recombination, the infected area was isolated using laser capture microdissection, and the extracted RNA was analyzed by qPCR to measure RGS4 and GFP expression. (c) shows RGS4 levels (normalized to GAPDH) in AAV-CreGFP and AAV-GFP infected brains (p<0.01, t-test). Mice lacking RGS4 in the NAc CPP at 3 mg/kg morphine, whereas their wildtype controls CPP at 5 mg/kg (d, n=5–6 per group). Conversely, overexpression of RGS4 in the NAc of C57Bl/6 mice, via infection with an HSV-RGS4 vector, prevents CPP to morphine (5 mg/kg s.c.) compared to control animals which were injected with an HSV-LacZ vector (e, n=7–8 per group). Selective deletion of RGS4 from the NAc increases sensitivity to the locomotor activating effects of repeated morphine exposure (10 mg/kg) (f, n=6 per group). For all behavioral studies, data are expressed as means ± S.E.M., p<0,01, two way ANOVA followed by Bonferroni test.
We next used CPP to determine how loss of RGS4 in the NAc affects morphine reward. As shown in Fig. 1d, the selective knockout of RGS4 from this brain region increases sensitivity to the rewarding effects of morphine, as the AAV-CreGFP injected mice exhibit a CPP at a 3 mg/kg dose of morphine, whereas AAV-GFP-injected controls require a higher morphine dose (5 mg/kg) to show a significant preference. A lower (1 mg/kg) morphine dose fails to establish a CPP in both groups. Sensitivity to morphine reward was also assessed using an overexpression model, where we injected an HSV vector expressing RGS4 (or LacZ as a control) into the NAc of wildtype C57Bl/6 mice. In contrast to the RGS4 local knockouts, mice overexpressing RGS4 selectively in this brain region are significantly less sensitive to morphine (5 mg/kg s.c.) reward compared to LacZ expressing controls (Fig. 1e). At higher morphine doses, both genotypes show the same CPP score (not shown). Together, these data support a role of RGS4 in the NAc in morphine reward.
In striking contrast to these local manipulations of RGS4 levels within the NAc, mice with constitutive and ubiquitous knockout of RGS4 do not exhibit morphine CPP even at high drug doses (CPP score in sec for wildtype animals: 1 mg/kg morphine = 29 ± 45, 5 mg/kg morphine = 143 ± 32; for RGS4 knockout mice: 1 mg/kg morphine = 46 ± 105, 5 mg/kg morphine = 4.0 ± 52). This finding likely reflects loss of RGS4 from other brain regions where it is highly expressed or at early times during development, and emphasizes the importance of using inducible and brain region-specific knockout strategies.
To further assess the role of RGS4 in the NAc in regulating behavioral responses to morphine, we tested mice with local RGS4 knockouts from this brain region, and AAV-GFP-injected control mice, in a locomotor sensitization paradigm. Consistent with the CPP data, deletion of RGS4 from NAc accelerates the development of locomotor sensitization to repeated morphine exposure (Fig. 1f).
RGS4 and Morphine Dependence: Actions in the Locus Coeruleus
We next examined the role of RGS4 in morphine physical dependence. Constitutive RGS4 knockout mice were implanted with morphine pellets and, three days later, an acute withdrawal syndrome was induced by injection of the MOR antagonist naloxone (1 mg/kg s.c.). As shown in Fig. 2a, RGS4 knockout mice undergo a much more severe withdrawal syndrome, as several signs (jumping, tremor, diarrhea, ptosis, weight loss) are significantly increased compared to their wildtype littermate controls. This phenotype is not related to the function of RGS4 in NAc, because local deletion of the RGS4 gene from this brain region has no overall effect on withdrawal behavior, apart from a decrease in ptosis and a trend for an increase in jumps (Fig. 2b).
Figure 2. RGS4 knockout mice exhibit more severe opiate withdrawal.

(a) RGS4 knockout (KO) mice show a greater degree of morphine physical dependence as compared to wildtype (WT) littermates; several signs of naloxone precipitated opiate withdrawal are more intense in mutant mice compared to WT littermates. Data are expressed as mean ± S.E.M. (n=8 per group). (b) In contrast, selective deletion of the RGS4 gene in the NAc (as described in Fig. 1) has little effect on opiate withdrawal; it leads to a decrease in ptosis only and a trend for increase in jumping behaviour which was not statistically significant. Data are expressed as mean ± S.E.M. (n=8 per group) *p<0.01 for genotype versus treatment, two way ANOVA followed by Bonferroni post hoc test.
RGS4 appears to be the most abundant RGS protein expressed in noradrenergic neurons of the LC, and its levels are dynamically regulated during chronic opiate administration and withdrawal (11). Since morphine dependence and withdrawal are associated with changes in LC firing activity (46–48), we hypothesized that RGS4 modulates opiate withdrawal in part via actions on the LC. To examine this hypothesis, we obtained whole cell recordings from LC neurons in brain slices from RGS4 knockout mice and their wildtype littermates. Acute administration of the MOR agonist DAMGO had similar effects on inhibition of LC firing in slices from both genotypes (Fig. 3a). Forskolin, an activator of adenylyl cyclase, is known to increase LC firing in slices from naïve animals, and to further induce neuronal firing in slices from morphine dependent animals (47,48). This increased activity of the cAMP pathway has been implicated in withdrawal activation of LC neurons and in morphine withdrawal behaviors (49,50,51). Since RGS4 knockout mice undergo a more severe withdrawal syndrome than their controls, we hypothesized that their LC neurons may show greater than normal excitation by forskolin. Indeed, while forskolin produces the same effect in morphine naïve animals (Fig. 3b), it increases LC firing to a greater extent in neurons from RGS4 knockout mice than in wildtype controls over a wide dose range of the drug in morphine dependent animals (Fig. 3c). Note that recordings were obtained after morphine washes out of the slices, see Methods). Together, these data suggest that RGS4 is part of a mechanism that opposes the excitability of LC neurons during morphine dependence, such that loss of RGS4 leads to exaggerated dependence and withdrawal.
Figure 3. RGS4 modulates LC firing via a cAMP-dependent mechanism.
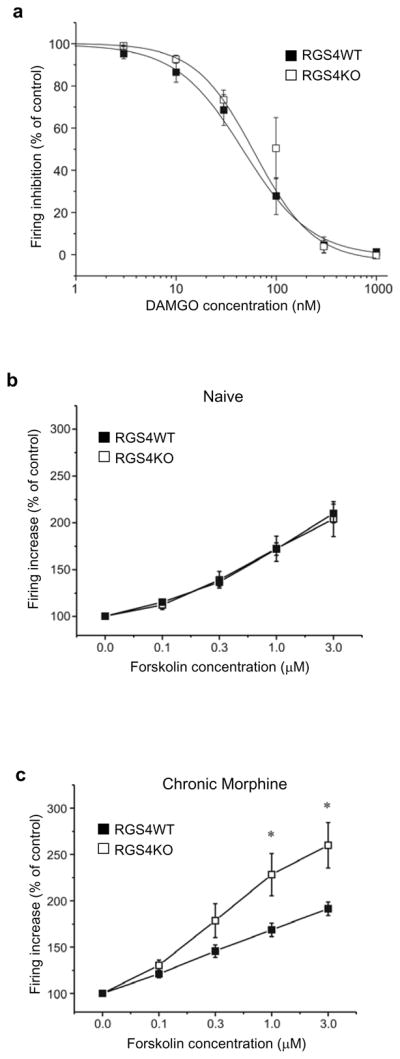
(a) No significant difference was observed in the sensitivity of DAMGO between LC neurons obtained from RGS4 wildtype (WT) and knockout (KO) mice (n=11–12 per data point; 4–5 mice each group). The sensitivity of LC neurons to forskolin (an activator of adenylyl cyclase) was determined in RGS4 KO mice and their wildtype littermates. (b) Effect of forskolin on LC firing in brain slices from drug-naïve RGS4 KO and WT mice. There is no difference in the effect of forskolin on LC firing rate between genotypes (n=13–16 per data point; 4–5 mice each group). (c) In contrast, after chronic morphine treatment, there was a significant difference in the sensitivity of LC neurons to forskolin: LC neurons became significantly more sensitive to forskolin in the KO group as compared to WT mice (n=12–13 per data point, 4–5 mice each group * p<0.05, two way ANOVA).
RGS4 and Opiate Analgesia
We next examined the role of RGS4 in morphine analgesia and tolerance. RGS4 knockout mice show normal analgesic responses to morphine in the 52°C hot plate test, and they develop tolerance at the same rate as their wildtype littermate controls (Fig. 4a, b). Moreover, the onset and duration of morphine analgesia was not different between wildtype and mutant mice. RGS4 knockout mice and their wildtype controls were also tested in the fear conditioning paradigm to evaluate possible learning differences between genotypes, as well as differential sensitivity to foot shocks. RGS4 mutants show normal performance in both cue and contextual fear conditioning tests (Suppl. Fig. 2).
Figure 4. Agonist selective regulation of hot plate analgesia by RGS4.

RGS4 knockout (KO) mice show normal responses to morphine in the 52°C hot plate assay (a). In addition, there is no genotype effect on the rate of analgesic tolerance to morphine (b, n=9–10 per group). Data are expressed as % of maximal possible effect (MPE=[Latency-baseline]/[cutoff-latency]). (c) In contrast to morphine, RGS4 KO mice are less sensitive to the analgesic actions of fentanyl and methadone (n=8–10 per group). (d) shows analgesic responses to fentanyl at different time points within 1.5 hr after morphine administration (n=7 per group) and (e) shows a dose response to fentanyl in the hot plate assay for RGS4 KO and wildtype (WT) mice (n=5–8 per group). Deletion of RGS4 from the NAc leads to a rightward shift in fentanyl dose response in the hot plate assay (f, 9–10 per group) but does not affect responses to morphine in this test (g, n=9–10 per group). For all behavioral experiments, data are expressed as mean ± S.E.M. *p<0.01 for genotype versus treatment, two way ANOVA followed by Bonferroni post hoc test.
Since no analgesia phenotype was revealed in the hot plate assay after morphine administration, we examined hot plate responses to two other MOR agonists, fentanyl and methadone. As shown in Fig. 4c, RGS4 knockout mice show dramatically reduced responses to fentanyl and methadone. This effect is not related to a change in the onset of analgesic response, since no analgesia to a high fentanyl dose (0,125mg/kg) is observed for 1.5 hrs post drug administration (Fig. 4d), and is observed over a wide dose range of fentanyl (Fig. 4e). Although the NAc is best studied for its role in morphine reward, we have shown that manipulation of several signaling molecules in NAc influences opiate analgesia and tolerance (13,42). We therefore examined if loss of RGS4 in the NAc affects opiate actions in the 52°C hot plate assay. Consistent with the result from the constitutive RGS4 knockout line, inducible deletion of RGS4 in the NAc reduces sensitivity to the analgesic actions of fentanyl, but does not affect responses to morphine in this assay (Fig. 4f,g). This phenotype is milder compared to constitutive knockouts (Fig 4e), suggesting the involvement of additional brain regions in RGS4-mediated control of fentanyl’s effects in the hot plate test.
Regulation of RGS4 by MOR agonists
Given the influence of RGS4 on opiate actions, and the differential effects of RGS4 knockout on morphine versus fentanyl analgesia, we used western blot analysis to examine the pattern of RGS4 regulation in the NAc by MOR agonists. As shown in Fig. 5a, RGS4 protein levels are decreased 2 hrs after morphine or fentanyl administration. A well established difference between morphine and other MOR agonists is the relative delay in MOR internalization following activation of the receptor by morphine (33). This delay in MOR endocytosis following morphine application is thought to contribute to the more rapid development of tolerance to morphine compared to other opiate analgesics. We recently showed that RGS9-2 is a negative modulator of MOR endocytosis (31), which raised the question of whether RGS4 also regulates this process. We used mouse embryonic fibroblasts from RGS4 wildtype and knockout mice and examined MOR cellular localization under basal conditions and following morphine exposure. Our data indicate that RGS4 does not affect the rate of MOR endocytosis in MEF cells (See Suppl. Figure 3). Similar negative data were obtained with transfected HEK cells (data not shown).
Figure 5. RGS4 and MOR signal transduction in striatum.

Quantitation of RGS4 and RGS2 levels in NAc 2 hrs after acute morphine or fentanyl administration by western blotting. Data are expressed as means ± S.E.M, n=3–4 per group, *, p<0.05 t-test.
In an effort to better understand the agonist-selective analgesia phenotype of RGS4 knockout mice, we used co-IP assays to examine the composition of RGS4 signal transduction complexes formed in striatum 30 min after morphine or fentanyl administration. First, we examined the complexes formed between MOR and Gα subunits. Although both morphine and fentanyl are reported to promote the association of MOR with several Gαi subunits in various tissues, we found that only fentanyl promotes the formation of complexes between MOR and Gαq in striatum (Fig 6a). We also found no interactions between RGS4 and Gαi1, Gαi2, or Gαi3 subunits in striatum, while RGS9-2 interacts with all three proteins under basal conditions and following MOR activation (V. Zachariou, unpublished observations). Since RGS4 is known to interact with Gαq subunits in other systems, we sought to determine how activation of MOR affects associations between RGS4 and Gαq. Interestingly, both fentanyl and morphine promote the formation of RGS4/Gαq complexes in striatum (Fig 6b). The next set of IPs investigated the effect of morphine or fentanyl on MOR-RGS4 complexes. We found that, although RGS4 is part of MOR containing complexes under basal conditions, this association is decreased following fentanyl, but not following morphine, administration. Together, these findings suggest that RGS4 and Gαq may compete for association with MOR in the presence of fentanyl, an effect not observed with morphine.
Figure 6. Morphine and fentanyl promote the formation of distinct RGS4 complexes in striatum.

(a) Mice received s.c. injections of saline, fentanyl (0.125 mg/kg), or morphine (15 mg/kg) and striata were extracted 30 min later. Striatal extracts were IP’d with an anti-MOR antibody and the immunoprecipitate was analyzed by western blot (WB) for Gαq. Fentanyl promotes the formation of complexes between MOR and Gαq. p<0.01 for fentanyl versus saline and morphine, one way ANOVA followed by Dunnett’s post hoc test. (b) Mice received acute injections of saline, fentanyl, or morphine as in (a) and striata were dissected 30 min later. Striatal extracts were IP’d with an anti-Gαq antibody and the immunoprecipitate was immunoblotted for RGS4. Fentanyl and morphine promote the formation of complexes between Gαq and RGS4. p<0.01 between treatment, one way ANOVA followed by Dunnett post hoc test. (c) Mice were treated as in (a) and striatal extracts were immunoprecipitated with an anti-MOR antibody and the immunoprecipitate was immunoblotted for RGS4. Fentanyl treatment decreases MOR-RGS4 complex levels in striatum. p<0.01 for fentanyl versus saline and morphine, one way ANOVA followed by Dunnett’s post hoc test.
DISCUSSION
Our studies reveal an important brain region-specific role for RGS4 in opiate responses. Since several RGS proteins are expressed in neurons that influence drug reward, dependence, and analgesia, it is important to understand the function of each of these proteins in particular networks and cell types. A large body of clinical and preclinical work links RGS4 to neuropsychiatric disorders such as schizophrenia, a syndrome linked by some to striatal dysfunction. Although RGS4 is moderately expressed in striatum, compared to other regions such as prefrontal cortex, it appears to exert important modulatory effects in this brain region on several GPCRs including muscarinic M2 autoreceptors and D1 and D2 dopamine receptors (4,23,24,25). While evidence from in vitro studies suggests that RGS4 may associate with MOR (52), and electrophysiological studies implicate RGS4 in opiate actions in LC slices (11), there is to date no information about the way RGS4 modulates opiate actions in vivo. In the present study, we use inducible and constitutive gene knockout models to elucidate the role of RGS4 in opiate actions. Given the lack of pharmacological tools for the study of RGS4 in vivo, and the wide distribution of this protein in the CNS, inducible knockout models permit local alterations in RGS activity in particular brain regions of the adult mouse. Indeed, we show that AAV-CreGFP injection into the NAc of floxed RGS4 mice mediates a near complete, but selective, loss of RGS4 from this brain region. Conversely, we selectively overexpressed RGS4 in NAc by use of viral vectors. These data are further supported by biochemical assays in striatal tissue, showing that RGS4 participates in MOR-dependent signal transduction complexes.
Our findings demonstrate that RGS4 action in the NAc controls behavioral and biochemical responses to morphine. In particular, we show that local knockout of RGS4 from the NAc of adult mice increases sensitivity to the rewarding and locomotor activating effects of morphine. Therefore, like RGS9-2 (12), RGS4 is a negative regulator of morphine action in the NAc. However, RGS9-2 has a more potent effect on reward sensitivity, as deletion of the RGS9 gene results in a tenfold increase in sensitivity to morphine in the CPP paradigm, whereas knockout of RGS4 causes a less dramatic shift. Interestingly, RGS4 overexpression in the NAc of RGS9 knockout mice cannot compensate for the loss of RGS9, which supports the involvement of independent pathways (12). It remains to be elucidated whether RGS9-2 and RGS4 act in the same cell types, and if they participate in the same biochemical signaling complexes. The use of constitutive RGS4 knockout mice in the CPP test revealed the opposite phenotype. The decreased sensitivity to morphine CPP in RGS4 knockout mice presumably reflects RGS4 actions in brain regions outside the NAc (e.g., prefrontal cortex, amygdala, hippocampus) that also regulate morphine reward or influence associations with cues required for CPP. Although the global loss of RGS4 had no effect on fear conditioning, different aspects of cognitive function could be affected. On the other hand, both constitutive and NAc-specific RGS4 knockout mice are less sensitive to the analgesic actions of fentanyl in the hot plate test, but the global knockout shows a more dramatic impairment likely due to RGS4 actions in other CNS regions.
Another important finding from our study concerns the actions of RGS4 in the LC. A large literature implicates changes in LC firing activity in contributing to aspects of opiate physical dependence and withdrawal (46,47,48). We have shown that alterations in gene expression in LC following chronic morphine are distinct from those observed in reward related networks like the NAc (53,54). Morphine-induced changes in LC firing activity occur in part from adaptive responses in signal transduction pathways downstream of the MOR. One of the most robust adaptations is upregulation of adenylyl cyclase (AC) activity, particularly the AC1 and AC8 isoforms (55,56), molecules highly regulated by G protein βγ subunits (57). It is, therefore, expected that proteins like RGS4, which regulate α and βγ subunit availability to effectors, play a prominent role in modulation of LC activity. Here, we report that, although inhibition of LC firing upon acute exposure to MOR agonists is unaffected by the loss of RGS4, firing provoked by activation of the cAMP pathway is greatly enhanced by the absence of this protein. These findings support earlier studies on the role of RGS4 in the cellular adaptations of LC neurons to chronic morphine (11), and provide a better understanding of the neuron-specific signaling events that contribute to opiate dependence. In accord with the electrophysiology findings, RGS4 knockout mice exhibit more severe withdrawal compared to their wildtype littermates. These experiments were only performed using constitutive RGS4 knockouts, as it was not feasible to reliably target the LC with our AAV vectors. Analysis of NAc-specific RGS4 knockout mice suggested that the morphine withdrawal phenotype is not related to RGS4 actions in the NAc.
Although RGS4 negatively modulates morphine reward and physical dependence, it does not affect morphine analgesia or the development of morphine analgesic tolerance. These findings are in agreement with earlier studies of a different line of RGS4 mutant mice (58). However, the study by Grillet and colleagues found no effect of RGS4 deletion on morphine withdrawal. This discrepancy might be a result of genetic background or related to the use of a morphine treatment protocol in the earlier study that leads to maximal withdrawal intensity in wildtype animals. In striking contrast to RGS4, RGS9-2 negatively modulates morphine analgesia and analgesic tolerance. This difference between RGS9-2 and RGS4 could be explained by the distinct localization of these proteins in the CNS, but may also reflect their distinct functions. Specifically, the difference between RGS9-2 and RGS4 with respect to morphine analgesia may lie in distinct selectivity for Gα subunits (59,60,61). In vitro data indicate that RGS4 functions as part of a G protein receptor kinase 2 (GRK2)/Gαq complex without preventing GRK2 action (61). Our data indicate that RGS4 is a necessary component of signaling complexes mediating the analgesic actions of those opiates that, in addition to activating Gαi subunits, also activate Gαq subunits (62, 63), since RGS4 knockout mice show decreased sensitivity to the analgesic actions of fentanyl and methadone in the hot plate assay. Earlier in vitro studies have indicated an interaction between RGS4 and opioid receptors (52) but there is no information about RGS4 containing complexes in the brain, as this type of assay is not easy to perform due to the modest density of MOR and RGS4 in striatum. Here, we used IP’s to better understand the mechanisms underlying the agonist selective interactions between MOR and RGS4 in striatum. These interactions appear to have a great impact on hot plate analgesia, as behavioral responses to fentanyl are diminished in RGS4 knockout mice. We hypothesize that loss of RGS4 permits greater association between MOR and Gαq. These data further support the notion that although many opiate analgesics activate MOR, and then recruit several Gα subunits including Gαi and Gαq, morphine does not recruit Gαq and, therefore, its actions are not affected by the absence of RGS4. Future studies are needed, however, to conclusively determine the role of Gαq signaling in morphine analgesia. It should also be mentioned that as morphine acts at both MOR and DOR, while fentanyl is MOR selective, and RGS4 does not affect DOR actions (64), the lack of a morphine analgesia phenotype may be related to the fact that DOR responses were unaltered in RGS4 knockout mice.
This study thereby provides a better understanding of the signal transduction mechanisms underlying the different actions of opiate agonists. Earlier studies revealed that deletion of the β-arrestin-2 gene affects opiate analgesia and tolerance (65) but not physical dependence, while deletion of the spinophilin gene increases reward sensitivity (40), promotes the development of morphine tolerance, and decreases responsiveness to all MOR agonists in the hot plate assay. On the other hand, RGS9-2 has a very potent role in morphine reward and dependence, and may also delay the development of tolerance in the hot plate assay (12). All of these findings point to a very precise role of signal transduction molecules downstream of MOR in different opiate actions. The fact that RGS9-2 is a negative modulator of morphine’s rewarding and analgesic actions makes it a difficult pharmacological target, as improving analgesia by decreasing RGS9-2 activity might also increase abuse potential. In contrast, RGS4 represents a better target for analgesia, since increasing RGS4 function would promote opiate analgesic responses while reducing reward and dependence liability.
In conclusion, RGS4 is a negative regulator of opiate reward and physical dependence via actions in the NAc and LC, respectively, and likely other brain regions as well. Moreover, as a Gαq-associated protein, RGS4 promotes responses to opiate analgesics such as fentanyl and methadone that recruit Gαq. These findings provide a better understanding of the molecular mechanisms of the diverse actions of opiates on the nervous system and reveal specific actions of RGS4 in opiate reward, dependence, and analgesia.
Supplementary Material
Acknowledgments
Supported by NIDA grants DA008227 (E.J.N.) and DA008863 (L.A.D.) and by the Greek Secretariat for Research and Technology-PENED03 (V.Z.)
Footnotes
The authors reported no biomedical financial interests or potential conflicts of interest.
References
- 1.Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272:3871–3874. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 2.Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gates. J Bio Chem. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- 3.Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 4.Terzi D, Stergiou E, King SL, Zachariou V. Regulators of G protein signaling and neuropsychiatric disorders. Progress in Molecular Biology and Translational Science. 2009;86 doi: 10.1016/S1877-1173(09)86010-9. in press. [DOI] [PubMed] [Google Scholar]
- 5.Gold SJ, Ni YG, Dohlman HG, Nestler EJ. Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat Brain. J Neurosci. 1997;17:8024–8037. doi: 10.1523/JNEUROSCI.17-20-08024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen CK, Burns ME, He W, Wensel T, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- 7.Nishiguchi KM. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 2004;427:75–78. doi: 10.1038/nature02170. [DOI] [PubMed] [Google Scholar]
- 8.Rahman Z, et al. RGS9 modulates dopamine signaling in the basal ganglia. Neuron. 2003;38:941–952. doi: 10.1016/s0896-6273(03)00321-0. [DOI] [PubMed] [Google Scholar]
- 9.Buckholtz JW, et al. Allelic variation in RGS4 impacts functional and structural connectivity in the human brain. J Neurosci. 2007;27:1584–1593. doi: 10.1523/JNEUROSCI.5112-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tekumalla PK, et al. Elevated levels of ΔFosB and RGS9 in striatum in Parkinson’s disease. Biol Psych. 2001;50:813–816. doi: 10.1016/s0006-3223(01)01234-3. [DOI] [PubMed] [Google Scholar]
- 11.Gold SJ, et al. Regulation of RGS proteins by chronic morphine in rat locus coeruleus. Eur J Neurosci. 2003;17:971–980. doi: 10.1046/j.1460-9568.2003.02529.x. [DOI] [PubMed] [Google Scholar]
- 12.Zachariou V, et al. Essential role for RGS9 in opiate action. Proc Natl Acad Sci USA. 2003;100:13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitt P, Ebert P, Mirnics K, Nimgaonkar VL, Lewis DA. Making the case for a candidate vulnerability gene in schizophrenia: convergent evidence for regulator of G-protein signaling 4 (RGS4) Biol Psych. 2006;60:534–537. doi: 10.1016/j.biopsych.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 14.Zeng W, et al. The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J Biol Chem. 1998;273:34687–34690. doi: 10.1074/jbc.273.52.34687. [DOI] [PubMed] [Google Scholar]
- 15.Davydov IV, Varshavsky A. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J Biol Chem. 2000;275:22931–22941. doi: 10.1074/jbc.M001605200. [DOI] [PubMed] [Google Scholar]
- 16.Lee MJ, et al. RGS4 and RGS5 proteins are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci USA. 2005;102:15030–15035. doi: 10.1073/pnas.0507533102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- 18.Erdely HA, Tamminga CA, Roberts RC, Vogel MW. Regional alterations in RGS4 protein in schizophrenia. Synapse. 2006;59:472–479. doi: 10.1002/syn.20265. [DOI] [PubMed] [Google Scholar]
- 19.Bowden NA, Scott RJ, Tooney PA. Altered expression of regulator of G-protein signalling 4 (RGS4) mRNA in the superior temporal gyrus in schizophrenia. Schizophr Res. 2007;89:165–8. doi: 10.1016/j.schres.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Ding L, Hegde AN. Expression of RGS4 Splice Variants in Dorsolateral Prefrontal Cortex of Schizophrenic and Bipolar Disorder Patients. Biol Psychiatry. 2008;65:541–545. doi: 10.1016/j.biopsych.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Gu Z, Jiang Q, Yan Z. RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Mol Pharmacol. 2007;71:1030–1039. doi: 10.1124/mol.106.032490. [DOI] [PubMed] [Google Scholar]
- 22.Campbell DB, Ebert PJ, Skelly T, Stroup TS, Lieberman J, Levitt P, Sulivan PF. Ethnic stratification of the association of RGS4 variants with antipsychotic treatment response in schizophrenia. Biol Psychiatry. 2008;63:32–41. doi: 10.1016/j.biopsych.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding J, et al. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- 24.Taymans JM, Leysen JE, Langlois X. Striatal gene expression of RGS2 and RGS4 is specifically mediated by dopamine D1 and D2 receptors: clues for RGS2 and RGS4 functions. J Neurochem. 2003;84:1118–1127. doi: 10.1046/j.1471-4159.2003.01610.x. [DOI] [PubMed] [Google Scholar]
- 25.Taymans JM, Kia HK, Claes R, Cruz K, Leysen H, Langlois X. Dopamine Receptor mediated regulation of RGS2 and RGS4 mRNA differentially depends on ascending dopamine projections and time. Eur J Neurosci. 2004;19:2219–60. doi: 10.1111/j.0953-816X.2004.03336.x. [DOI] [PubMed] [Google Scholar]
- 26.Ni YG, et al. Region-specific regulation of RGS4 (regulator of G protein signalling type 4) in brain by stress and glucocorticoids: in vivo and in vitro studies. J Neurosci. 1999;19:3674–3680. doi: 10.1523/JNEUROSCI.19-10-03674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bishop GB, Cullinan WE, Curran E, Gutstein HB. Abused drugs modulate RGS4 mRNA levels in rat brain: comparison between acute drug treatment and a drug challenge after chronic treatment. Neurobiol Disease. 2002;10:334–343. doi: 10.1006/nbdi.2002.0518. [DOI] [PubMed] [Google Scholar]
- 28.Schwendt M, Gold SJ, McGinty JF. Acute amphetamine down-regulates RGS4 mRNA and protein expression in rat forebrain: distinct roles of D1 and D2 dopamine receptors. J Neurochem. 2006;96:1606–1615. doi: 10.1111/j.1471-4159.2006.03669.x. [DOI] [PubMed] [Google Scholar]
- 29.Kovoor A, et al. D2 dopamine receptors colocalize regulator of G protein signalling 9-2 via the RGS9 DEP domain, and RGS9 knockout mice develop dyskinesias associated with dopamine pathways. J Neurosci. 2005;280:5133–5136. doi: 10.1523/JNEUROSCI.2840-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traynor JR, Neubig RR. Regulators of G protein signaling and drugs of abuse. Mol Interv. 2005;5:30–41. doi: 10.1124/mi.5.1.7. [DOI] [PubMed] [Google Scholar]
- 31.Psifogeorgou K, et al. RGS9-2 is a negative modulator of mu opioid receptor function. J Neurochem. 2007;103:617–625. doi: 10.1111/j.1471-4159.2007.04812.x. [DOI] [PubMed] [Google Scholar]
- 32.Gold SJ, et al. RGS9-2 negatively modulates L-3,4-dihydroxyphenylalanine-induced dyskinesia in experimental Parkinson’s disease. J Neurosci. 2007;52:14338–14348. doi: 10.1523/JNEUROSCI.4223-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans CJ. Secrets of the opium poppy revealed. Neuropharm. 2004;47(Suppl 1):293–299. doi: 10.1016/j.neuropharm.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 36.Kreek MJ. Drug addictions. Molecular and cellular endpoints. Ann NY Acad Sci. 2001;937:27–49. [PubMed] [Google Scholar]
- 37.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 38.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 39.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 40.Charlton JJ, et al. Multiple actions of spinophilin modulate mu opioid receptor function. Neuron. 2008;58:238–247. doi: 10.1016/j.neuron.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med. 2003;12:1539–44. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- 42.Zachariou V, et al. An Essential Role for ΔFosB in the Nucleus Accumbens in Morphine Action. Nat Neurosci. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]
- 43.Han MH, et al. Role of cAMP response element-binding protein in the rat locus coeruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosc. 2006;26:4624–4629. doi: 10.1523/JNEUROSCI.4701-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kogan JH, Aghajanian GK. Long-term glutamate desensitization in locus coeruleus neurons and its role in opiate withdrawal. Brain Res. 1995;14:111–121. doi: 10.1016/0006-8993(95)00545-2. [DOI] [PubMed] [Google Scholar]
- 45.Pineda J, Aghajanian GK. Carbon dioxide regulates the tonic activity of locus coeruleus neurons by modulating a proton-and polyamine-sensitive inward rectifier potassium current. Neuroscience. 1997;77:723–743. doi: 10.1016/s0306-4522(96)00485-x. [DOI] [PubMed] [Google Scholar]
- 46.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 47.Aston-Jones G, Hirata A, Akaoka H. Local opiate withdrawal in locus coeruleus in vivo. Brain Res. 1997;765:331–336. doi: 10.1016/s0006-8993(97)00682-3. [DOI] [PubMed] [Google Scholar]
- 48.Nestler EJ, Aghajanian GK. Molecular and Cellular basis of Addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 49.Taylor JR, et al. Clonidine infusions into the locus coeruleus attenuate behavioral and neurochemical changes associated with naloxone-precipitated withdrawal. Psychopharm. 1998;96:121–134. doi: 10.1007/BF02431544. [DOI] [PubMed] [Google Scholar]
- 50.Kogan JH, Nestler EJ, Aghajanian GK. Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 51.Maldonado R, Koob GF. Destruction of the locus coeruleus decreases physical signs of opiate withdrawal. Brain Res. 1993;605:128–138. doi: 10.1016/0006-8993(93)91364-x. [DOI] [PubMed] [Google Scholar]
- 52.Georgoussi Z, et al. Selective interactions between G protein subunits and RGS4 with the C-terminal domains of the mu- and delta-opioid receptors regulate opioid receptor signaling. Cell Signal. 2006;18:771–782. doi: 10.1016/j.cellsig.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 53.McClung CA, Nestler EJ, Zachariou V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus coeruleus and ventral tegmental area. J Neurosci. 2005;25:6005–6015. doi: 10.1523/JNEUROSCI.0062-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zachariou V, et al. Distinct roles of adenylyl cyclases 1 and 8 in opiate dependence: behavioral, electrophysiological, and molecular studies. Biol Psychiatry. 2008;63:1013–1021. doi: 10.1016/j.biopsych.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–9. [PubMed] [Google Scholar]
- 56.Lane-Ladd SB, et al. CREB in the locus coeruleus: Biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang J, Gilman AG. Type-specific regulation of adenylyl cyclase by G protein beta gamma subunits. Science. 1991;254:1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- 58.Grillet N, et al. Generation and characterization of RGS4 mutant mice. Mol Cell Biol. 2005;25:4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu X, et al. RGS proteins determine signaling specificity of Gq-coupled receptors. J Biol Chem. 1999;274:3549–3556. doi: 10.1074/jbc.274.6.3549. [DOI] [PubMed] [Google Scholar]
- 60.Huang C, Hepler JR, Gilman AG, Mumby SM. Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc Natl Acad Sci USA. 1997;94:6159–6163. doi: 10.1073/pnas.94.12.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJ. Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science. 2005;310:1686–1690. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 62.Lee JW, Joshi S, Chan JS, Wong YH. Differential coupling of mu, delta and kappa opioid receptors to Ga16 mediated coupling of phospholipase C. J Neurochem. 1998;70:2203–2211. [PubMed] [Google Scholar]
- 63.Rubovitch T, Gafni M, Sarne Y. The mu opioid agonist DAMGO, stimulates cAMP production in SK-N-SH cells through a PLC-PKC-Ca++ Pathway. Mol Brain Res. 2003;110:261–266. doi: 10.1016/s0169-328x(02)00656-3. [DOI] [PubMed] [Google Scholar]
- 64.Xie Z, Li Z, Guo L, Ye C, Li J, Yu X, Yang H, Wang Y, Chen C, Zhang D, Liu-Chen LY. Regulator of G protein signaling proteins differentially modulate signaling of mu and delta opioid receptors. Eur J Pharmacol. 2007;565:45–53. doi: 10.1016/j.ejphar.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raehal KM, Bohn LM. Mu opioid receptor regulation and opiate responsiveness. AAPS J. 2005:E587–91. doi: 10.1208/aapsj070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.