

Abstract
Vertebrate skeletogenesis consists in elaborating an edifice of more than 200 pieces of bone and cartilage. Each skeletal piece is crafted at a distinct location in the body, is articulated with others, and reaches a specific size, shape, and tissue composition according to both species instructions and individual determinants. This complex, customized body frame fulfills multiple essential tasks. It confers morphological features, allows controlled postures and movements, protects vital organs, houses hematopoiesis, stores minerals, and adsorbs toxins. This review provides an overview of the multiple facets of this ingenious process for experts as well as non-experts of skeletogenesis. We explain how the developing vertebrate uses both specific and ubiquitously expressed genes to generate multipotent mesenchymal cells, specify them to a skeletogenic fate, control their survival and proliferation, and direct their differentiation into cartilage, bone and joint cells. We review milestone discoveries made towards uncovering the intricate networks of regulatory factors that are involved in these processes, with an emphasis on signaling pathways and transcription factors. We describe numerous skeletal malformation and degeneration diseases that occur in humans as a result of mutations in regulatory genes, and explain how these diseases both help and motivate us to further decipher skeletogenic processes. Upon discussing current knowledge and gaps in knowledge in the control of skeletogenesis, we highlight ultimate research goals, and propose research priorities and approaches for future endeavors.
INTRODUCTION
The development of a skeleton made of cartilage, bone, and joints is a novel process that has critically contributed to the emergence of vertebrates (Ota and Kuratani, 2009). Its importance is reflected in the word “vertebrate”, which means “having a vertebral column” or “having joints”. Both meanings are justified. The vertebral column is indeed the skeletal feature shared by all vertebrates, as other skeletal elements were acquired later, were never acquired, or were lost in some vertebrate species. Joints are also characteristic features of the vertebrate skeleton, and the vertebral column uses the notochord, a primitive skeletal structure in vertebrate ancestors and embryos, to form the core part (nucleus pulposus) of its joints (intervertebral discs). The rigid, articulated elements of the vertebrate skeleton permit vertebrates deliberate postures and movements. The subsequent acquisition of a skull, jaw, and appendicular skeleton allowed vertebrates to develop a defined brain, face and limbs. The thoracic cage and marrow space evolved to protect the brain, hematopoietic tissue and other organs. Furthermore, bones became mineral reserves and toxin clearance centers. Interestingly, aware that the skeleton has key physical roles, ancient cultures even endowed it with spiritual meaning and thought that it housed the soul. Skeletogenesis is thus an essential process in the development of vertebrates.
Skeletogenesis starts in the vertebrate embryo once multipotent mesenchymal cells arise from ectoderm and mesoderm, migrate to specific locations in the body, and commit to a skeletal fate. Most skeletogenic cells later develop into cartilage cells (chondrocytes), bone cells (osteoblasts), or joint cells (mainly articular chondrocytes and synovial cells), while some may persist as mesenchymal stem cells throughout life. The primary skeleton is entirely cartilaginous. It grows quickly and most of it is progressively replaced by bone throughout fetal and postnatal growth. The process is called endochondral ossification. Concomitantly, joints and additional bones form. The latter develop upon a mesenchymal template, without cartilage intermediate, through a process called intramembranous ossification. Bone, cartilage, and joints differ in composition and regulation, but their associated developments are tightly coordinated. Our knowledge of the cellular and molecular events that govern skeletogenesis has greatly increased over the last two decades thanks to the identification of disease-causing mutations, gene manipulations in animals, and novel molecular and cellular approaches. It is now clear that an amazingly large number of factors are involved in skeletogenesis. In fact, no other process, except perhaps brain development, may recruit as many factors. These factors are hormones, growth factors, receptors, signaling mediators, transcription factors, extracellular matrix components, and enzymes. Factors determining the identity of skeletal cells are called differentiation factors, and factors specifying the number, size and shape of skeletal elements are called patterning factors. The latter greatly outnumber the former, and contribute to the incredible skeletal variations that exist between individuals both within and between species.
A corollary of the importance and complexity of the vertebrate skeleton is a high frequency and diversity of severe skeleton malformation diseases in humans (Spranger, 2006). The incidence of these diseases is estimated at 1/4,000 births, with half of them being early lethal. Their true incidence, however, may be twice as high because many develop only years after birth. Osteochondrodysplasias are generalized skeleton malformation diseases, whereas dysostoses affect only a specific subset of skeletal elements. As genes controlling skeletogenesis are often involved in several processes, their alterations can cause complex syndromes, of which skeletal disease is only one component. In addition, it must be noted that skeletal variations at the edge of normalcy are often inconsequential at a young age, but constitute high-risk factors for skeleton degeneration diseases later in life, such as osteoarthritis (articular cartilage degeneration) and osteoporosis (bone loss disease).
Vertebrate skeletogenesis is thus a fascinating process to study for developmental and evolutionary biologists who want to understand how the skeleton and its variations are generated in developing vertebrates. Furthermore, it is also a mandatory process to study for geneticists and clinician scientists who want to decipher the molecular basis of skeletal diseases in humans and develop greatly needed therapies for these diseases. We review here key aspects of the vertebrate skeleton composition and development. We analyze milestone discoveries made towards understanding mechanisms underlying skeletogenic cell fate determination, chondrogenesis, osteogenesis, joint formation, and individual skeletal variations. We discuss current knowledge and gaps in knowledge, and end with suggestions for important goals and approaches for future research.
STRUCTURAL ORGANIZATION AND ADVANTAGES OF THE VERTEBRATE SKELETON
Invertebrates have no skeleton or a skeleton made exclusively of minerals (e.g., calcium carbonate or silica) or carbohydrates (e.g., chitin). The vertebrate skeleton, in contrast, is made of specialized connective tissues, i.e., structures composed of cells embedded in an abundant extracellular matrix. The invertebrate and vertebrate skeletons are thus very different from each other and this can be explained by independent evolutions (Ota and Kuratani, 2009).
Cartilage and bone are built and populated by chondrocytes and osteoblasts, respectively. Cartilage is capable of growing rapidly and is therefore profuse in embryos and youngsters. It is also highly resilient and thus maintained in adult skeletal sites subject to load, such as airways and synovial joints. Bone is very strong mechanically and thus preponderant in the adult skeleton. Both tissues are constructed upon a collagenous fibrillar network that confers structure and tensile strength. Collagen-2 is a very abundant and specific protein in cartilage, and collagen-1 is the major protein in bone. The cartilage collagen network entraps a highly charged gel of aggrecan and other proteoglycans, which confer tissue resiliency and control the diffusion of growth factors. Bone has a low content of proteoglycan, but contains specific proteins, such as osteocalcin and bone sialoprotein. These proteins control the deposition of hydroxyapatite, the main calcium phosphate mineral of the bone matrix. Its high collagen and mineral content confer on bone its unique mechanical and mineral homeostatic functions.
As they invented bone and cartilage elements, vertebrates also devised joint structures to interconnect these elements. They created synovial joints between limb elements to be able to display great ranges of motions; intervertebral discs to confer pliability to the vertebral column; and fibrous joints to minimize movements between skull bones. These different types of joints are made of distinct tissues. Synovial joints, for instance, consist of articular cartilage coating bone surfaces, a fibrous capsule surrounding the joint and sealing its cavity, a synovial membrane lining the capsule internally and producing a lubricating fluid, tendons transmitting muscle force to bones, and ligaments stabilizing the joints by connecting bones. The vertebrate skeleton is thus a complex and highly advantageous edifice.
DEVELOPMENT OF SKELETOGENIC CELLS
The first step in skeletogenesis consists in generating skeletogenic cells. The origin of these cells can be tracked back to the onset of organogenesis, when the vertebrate embryo is comprised of three germ layers: ectoderm, mesoderm, and endoderm (Fig. 1A). These layers transform themselves into multiple early derivatives. These include the ectoderm-derived neural tube, mesoderm-derived notochord, paraxial mesoderm, and lateral plate mesoderm, which give rise to the skeleton as well as other organs. The neural crest is a population of cells that delaminates from the neural tube, undergoes epithelial-to-mesenchymal transformation, and migrates to numerous locations in the embryo (Fig. 1B). Once there, the cells develop into various types, such as neuronal cells, melanocytes, and skeletogenic cells. The latter give rise to several throat and craniofacial skeletal elements (Fig. 1c). The lateral plate mesoderm gives rise to the other craniofacial skeletal structures, the limb skeletal elements (appendicular skeleton), the sternum (part of the axial skeleton), and non-skeletal structures. The paraxial mesoderm gives rise to somites, which develop into dermomyotomes and sclerotomes. The latter form most of the axial skeleton, i.e., the ribs and vertebrae. As they develop around the notochord, the vertebrae force the notochord cells to change phenotype, migrate to the intervertebral spaces and develop the nuclei pulposi of intervertebral discs (Fig. 1D). Many factors control neural crest and mesoderm cells before they reach skeletal sites. We will not review them, but suggest excellent recent reviews on this topic (Sauka-Spengler et al., 2008; Limura et al., 2009).
Figure 1. Origin of skeletal cells in the vertebrate embryo.

A. Schematic of a cross section through a mouse embryo soon after gastrulation at day 8 of development (equivalent to gestation day 17 in humans). The three germ layers are seen: ectoderm, endoderm, and mesoderm. Ectoderm-derived neural folds are rising. The mesoderm has formed the notochord and is starting to form lateral plate and paraxial derivatives on either sides of the midline.
B. Schematic showing the delamination of neural crest cells from the neural folds at the time of neural tube closure. These cells are starting to migrate inside the embryo (blue arrows), where they will participate in the formation of various structures.
C. Schematic showing the contribution of the neural crest, lateral plate mesoderm, paraxial mesoderm, and notochord to the three major parts of the skeleton.
D. Mid-sagittal sections through the notochord of mouse embryos at the gestation days 12.5 (E12.5, top) and E15.5 (bottom). The E12.5 notochord is a rod-like structure that becomes surrounded by the mesenchymal cell condensations of the prospective vertebral bodies (VB) and intervertebral discs (IVD). E15.5 vertebral bodies are cartilaginous and notochord cells have migrated into the intervertebral disc spaces, where they have formed nuclei pulposi (NP). Sections are stained with nuclear fast red and with Alcian blue, which is specific of the notochord and cartilage extracellular matrix.
Upon settling in skeletal sites, neural crest- and mesoderm-derived cells produce a matrix rich in collagen-1, fibronectin, and hyaluronan, and they proliferate or die in a tightly controlled spatial and temporal manner (Li et al., 2007; reviewed by Shum et al., 2003). They thereby establish mesenchymal structures that prefigure the future skeletal elements. They are often called osteochondroprogenitors because most of them give rise to osteoblasts and chondrocytes, and are able to switch fate under specific conditions (Fig. 2). Some, however, give rise to synovial cells, tenocytes, bone marrow stromal cells, endothelial cells, and presumably mesenchymal stem cells. We therefore prefer to call them skeletogenic cells.
Figure 2.
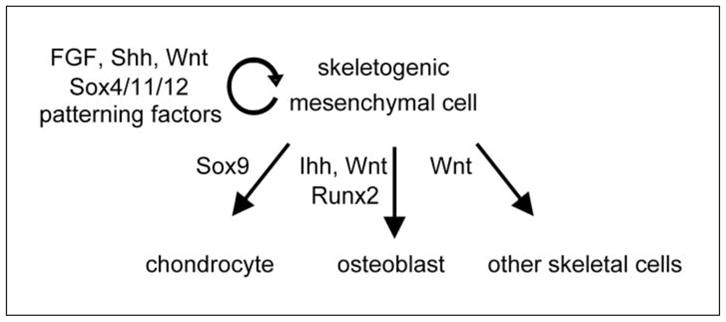
Fate and molecular control of skeletogenic mesenchymal cells.
As of today, it is unknown whether master transcription factors control the identity and maintenance of these cells in all sites, but Sox4, Sox11, and Sox12, which form the group C of Sry-related HMG box transcription factors, are strong candidates, as they are expressed in all embryonic mesenchymal cells and are required for the survival of these cells (Dy et al., 2008; Bhattaram et al., 2010). Numerous factors are known, in contrast, that participate in the formation of skeletogenic templates in a site- and time-specific manner. These patterning regulators include transcription factors from many families and most were identified through dysostosis-causing mutations (reviewed by Hermanns and Lee, 2001). Our knowledge of their exact roles and targets remains incomplete, as studies are complicated by a high degree of redundancy existing between co-expressed factors. Patterning factors are too numerous to be reviewed in detail here. Instead, we will focus on the Hox proteins, which were among the first ones to be discovered and which spectacularly illustrate the importance of patterning factors (reviewed by Wellik, 2009).
Hox proteins are encoded by a family of 39 genes in humans and most other vertebrates, and these genes are distributed in 4 paralogous clusters. The position of each gene in a cluster correlates with its expression pattern along the anteroposterior axis of the trunk, limb, and head. Hox proteins feature a DNA-binding domain called the homeodomain, in reference to the fact that Hox mutations often cause homeotic transformations. For instance, inactivation of the mouse Hox9 genes results in transformations of the posterior thoracic vertebrae such that up to 14 pairs of full-length ribs form and attach to the sternum instead of 7 (McIntyre et al., 2007). The fact that all four Hox9 genes (a, b, c, and d) need to be inactivated to generate this phenotype demonstrates the high degree of redundancy existing between Hox paralogues. Despite redundancy, single HOX gene mutations can cause severe skeletal malformations in humans. For instance, amplification of an alanine stretch in HOXD13 causes syndactyly (fusion of digits), polydactyly (extranumerary digits), brachydactyly (short digits), and transformation of metacarpals and metatarsals to carpals and tarsals, respectively (reviewed by Zhao et al., 2007). A similar phenotype was observed in mice with an equivalent mutation and was proposed to result from a dominant-negative action of the mutant protein. These examples thus highlight how locally but yet powerfully patterning transcription factors participate in the design of skeletal structures.
Complex networks of morphogens control the expression and activity of patterning transcription factors. These morphogens include but are not limited to Sonic Hedgehog (Shh), fibroblast growth factors (FGFs), Wnt ligands, bone morphogenetic proteins (BMPs), and retinoic acid. Their roles and interactions are reviewed in an accompanying paper with a special emphasis on the developing limb (reviewed by Butterfield et al., 2010).
The lineage potential of skeletogenic cells is established early, as revealed by expression of master transcription factors, such as the chondrogenic factor Sox9 and the osteogenic Runt-domain transcription factor Runx2 (Ducy et al., 1997; Ng et al., 1997; Eames et al., 2004). By definition, however, skeletogenic cells are undifferentiated. Mechanisms must therefore be in place to keep them as such. Several have been identified. For instance, Wnt/beta-catenin signaling is able to block the activity of Sox9 (Akiyama et al., 2004; Day et al., 2005; Hill et al., 2005), and both Sox9 and the Twist1/2 homeodomain transcription factor can physically interact with Runx2 to block its activity (Bialek et al., 2004; Zhou et al., 2006). Moreover, and as further described later, lack of expression of Sox5 and Sox6, two potent partners of Sox9 in chondrogenesis, helps keep chondrogenic cells undifferentiated, while lack of expression of Osx, a zinc-finger transcription factor required for osteoblast differentiation, keeps osteogenic cells undifferentiated. The generation of skeletogenic cells and the proper control of their fate are thus critical early steps in the formation of the vertebrate skeleton.
DEVELOPMENT OF CARTILAGE ANLAGEN
Chondrogenesis, the fate of most skeletogenic cells, results in the construction of a multitude of cartilage anlagen, which altogether constitute the primary skeleton of the vertebrate embryo (Fig. 3A). This process occurs in two steps: precartilaginous condensation and chondrocyte early differentiation (Fig. 3B).
Figure 3. Chondrocyte early differentiation and development of cartilage primordia.

A. Alcian blue staining of a mouse embryo at E14.5 demonstrates that chondrocyte differentiation of skeletogenic cells leads to the formation of a primary skeleton that is entirely cartilaginous.
B. Sections through the developing paws of mouse embryos illustrate the major steps of early chondrogenesis. At E10.5, the limb bud is filled with skeletogenic cells. By E12.5, some of these cells have formed precartilaginous condensations that prefigure the future digits. By E14.5, condensed prechondrocytes have undergone chondrocyte early differentiation. The sections are stained with Alcian blue and nuclear fast red.
Precartilaginous condensation requires that skeletogenic cells stop proliferating and expressing collagen-1 and hyaluronan and that they start expressing N-cadherin, tenascin-C, and other adhesion proteins that allow them to tightly aggregate (reviewed by Shum et al., 2003; Hall and Miyake, 2000). In vitro studies have suggested that transforming growth factor-beta (Tgf-beta) and Wnt/beta-catenin signaling govern this process, but in vivo validation is still lacking (Tuli et al., 2003). In vivo data have indicated that Sox9 is somehow required for precartilaginous cell condensation and survival, but exact roles in this step, probably beyond TGF-beta and Wnt/beta-catenin signaling, remain unknown (Bi et al., 1999; Akiyama et al., 2002). Our knowledge of the regulation of precartilaginous condensation is thus still scarse.
Condensed cells undergoing chondrocyte early differentiation stop expressing adhesion molecules, resume proliferation, and start producing profuse amounts of cartilage extracellular matrix. This event first kicks off in the center of precartilaginous condensations, but quickly spreads towards the periphery. A few layers of peripheral cells, however, remain skeletogenic and form a so-called perichondrium. They express patterning factors and concomitantly give rise to new chondrocytes for appositional growth of cartilage primordia. Chondrocyte early differentiation is driven at the transcriptional level by Sox5, Sox6, and Sox9 (reviewed by Lefebvre and Smits, 2005; and Akiyama, 2008). The Sox5 and Sox6 proteins are very similar to each other and act in redundancy, but they differ from Sox9 in DNA-binding specificity and lack of intrinsic transactivation function. The importance of Sox9 in chondrogenesis was revealed when heterozygous mutations within and around SOX9 were found to cause Campomelic Dysplasia, a severe form of human chondrodysplasia (Foster et al., 1994; Wagner et al., 1994). Sox9 is expressed in skeletogenic cells prior to Sox5 and Sox6, and the three genes are co-expressed in all precartilaginous condensations and cartilage elements. Homozygous inactivation of mouse Sox9 in early chondrocytes blocks cell differentiation (Bi et al., 1999; Akiyama et al., 2002), whereas inactivation of Sox5 and Sox6 severely impairs, but does not block chondrocyte early differentiation (Smits et al., 2001). Gain-of-function experiments in vivo and in vitro have revealed that the three proteins are capable together of inducing chondrocyte differentiation of mesenchymal cells (Ikeda et al., 2004). Molecular studies have demonstrated that they cooperatively bind and activate the genes for many cartilage-specific extracellular matrix components (Lefebvre et al., 1997 and 1998; Han and Lefebvre, 2008). Sox5/Sox6 and Sox9 thus constitute a trio of transcription factors that is both needed and sufficient for chondrocyte early differentiation.
Since this trio controls cell differentiation in other lineages besides chondrocytes, it is likely that specific factors determine its chondrogenic activity. Such factors, however, have not been identified yet. The control of the expression of the Sox trio in chondrocytes remains scantily known. Sox9 is expressed upstream of Sox5 and Sox6 (Smits et al., 2001; Akiyama et al., 2002), and the Sox9 protein was proposed in vitro to be able to control its own gene expression via a positive feedback loop (Kumar et al., 2009). BMP signaling is required for expression of the three genes in skeletal sites in vivo (Yoon et al., 2005), and Shh signaling was shown in vitro to confer competence in somitic tissue for subsequent BMP signals to induce overt chondrogenesis (Zeng et al., 2002). FGF signaling increases Sox9 expression in cultured mesenchymal cells (Murakami et al., 2000). Shh, BMP, and FGF signaling are active in most skeletogenic sites and may thus cooperatively activate Sox9 expression. In contrast, Wnt/beta-catenin signaling blocks the chondrogenic activity of Sox9 and its gene expression in vivo and may thus help restrict Sox9 expression to early chondrocytes (Hartmann et al., 2001; Akiyama et al., 2004; Hill et al., 2005). Major progress has thus been made in recent years by identifying the Sox5/6/9 trio, but major progress remains to be accomplished to identify the factors that control precartilaginous condensation and to better understand the mechanisms underlying the expression and specific action of the chondrogenic trio.
DEVELOPMENT OF CARTILAGE GROWTH PLATES
Soon after they form, most cartilage primordia embark into rapid growth and shaping into a long shaft (diaphysis) flanked by globular ends (epiphyses). The growth of the shafts is achieved by so-called growth plates, i.e., layers of chondrocytes proceeding in a staggered manner through a series of maturation steps (Fig. 4A). Proliferating chondrocytes precede prehypertrophic, hypertrophic, and matrix-mineralizing terminal chondrocytes. Chondrocyte proliferation and hypertrophy are the two drivers of tissue elongation. Chondrocytes express an early phenotype through prehypertrophy, but also express stage-specific markers, such as matrilin-1 (Matn1) and the FGF receptor 3 (Fgfr3) at the proliferative stage, and the receptor for the parathyroid hormone and parathyroid hormone-related peptide (Ppr), Indian hedgehog (Ihh), and collagen-10 (Col10a1) at the prehypertrophic stage. Col10a1 is the only one of these markers that is still expressed at the hypertrophic stage. Terminal chondrocytes no longer express Col10a1, but express osteoblast markers, including the gene for the matrix metalloproteinase 13 (Mmp13). Like osteoblasts, they also mineralize their surrounding extracellular matrix and many are found positive for cell death by the TUNEL assay. It is thus believed that all terminal chondrocytes die. To our knowledge, however, conclusive lineage tracing experiments have not been published yet to definitively prove that no cells survive and directly participate as osteoblasts to endochondral ossification. We also like to stress that many studies on growth plate chondrocytes do not distinguish prehypertrophic from hypertrophic chondrocytes. In reviewing these studies, we paid attention to the actual phenotype of maturing chondrocytes and therefore sometimes reached a conclusion different from that of the authors.
Figure 4. Chondrocyte maturation and development of cartilage growth plates.

A. Sections through a mouse embryo tibia illustrate the development of growth plates and endochondral bone. At E13.5, early chondrocytes in the center of cartilage primordia undergo prehypertrophic and hypertrophic maturation. They reach terminal maturation and are replaced by endochondral bone by E15.5. Later on, growth plates maintain themselves and elongate developing bones. Chondrocytes keep proliferating and give rise, layer by layer, to maturing chondrocytes. These cells which eventually die and are replaced by bone. The sections are stained with Alcian blue and nuclear fast red.
B. Schematic of the molecular control of growth plate chondrocytes.
The Sox trio is still required for the development of proliferative and prehypertrophic chondrocytes (Fig. 4B). It continues to generate expression of early cartilage markers in these cells, and it also activates expression of Matn1 (Smits et al., 2001; Rentsendorj et al., 2005). Its role in chondrocyte proliferation is unclear, as chondrocyte proliferation is blocked in the absence of Sox5/6 or Sox9, but also slowed down when Sox9 is overexpressed (Smits et al., 2001 and 2004; Akiyama et al., 2002 and 2004). Sox5/6 are needed to delay chondrocyte prehypertrophy, i.e., Ppr and Ihh expression, and to allow the cells to activate Col10a1 and to undergo hypertrophy instead of immediately undergoing terminal maturation (Smits et al., 2004). Sox9 may delay chondrocyte prehypertrophy downstream of Pthrp signaling (Huang et al. 2001), but in vivo validation of this property is still missing.
Chondrocyte maturation is driven from prehypertrophy to the terminal stage by the Runt-domain transcription factors Runx2 and Runx3 (also known as Cbfa1 and Cbfa3, respectively) and by the MADS-box transcription factors Mef2c and Mef2d (myocyte enhancer factor 2c and 2d, respectively). The activity of these factors is inhibited by the histone deacetylase Hdac4. Runx2 and Runx3 were shown in knockout mice to be needed for chondrocyte maturation in a largely redundant manner (Inada et al., 1999; Kim et al., 1999; Yoshida et al., 2004), and gain-of-function experiments in transgenic mice demonstrated that Runx2 is sufficient to cause ectopic and precocious maturation of chondrocytes (Takeda et al., 2001). Runx2 directly binds and activates Ihh, Col10a1, and Mmp13 (Zheng et al., 2003; Selvamurugan et al., 2004; Yoshida et al., 2004). Mef2c and Mef2d were originally identified as essential regulators of muscle and cardiovascular development. Deletion of Mef2c and Mef2d in the mouse results in severe impairment of chondrocyte maturation, including downregulation of Runx2 expression, while forced expression of a superactivating form of MEF2C causes precocious maturation (Arnold et al., 2007). Mef2c directly binds and activates Col10a1, but it has not been shown whether it cooperates with Runx2. Mice lacking the Hdac4 histone deacetylase precociously mature chondrocytes, whereas mice overexpressing Hdac4 in proliferating chondrocytes exhibit a delay of chondrocyte maturation (Vega et al., 2004). Hdac4 can physically interact with Runx2 and Mef2c and thereby block Col10a1 transactivation (Vega et al., 2004; Arnold et al., 2007; Kozhemyakina et al., 2009). Runx2/3 and Mef2c/d are thus master switches for chondrocyte maturation, with Mef2c/d acting upstream and possibly together with Runx2, and with Hdac4 blocking the activity of both types of factors.
Many signaling pathways interact with each other to control chondrocyte proliferation and maturation in the growth plate. Their roles are so critical that mutations in any one of them cause severe skeleton malformation diseases (reviewed by Rimoin et al., 2007). Indian Hedgehog (Ihh) signaling has a pivotal role. Secreted by prehypertrophic chondrocytes, Ihh stimulates chondrocyte proliferation on one hand, and chondrocyte maturation on the other hand (St-Jacques et al., 1999). Furthermore, it activates the gene for the parathyroid hormone-related peptide (Pthrp) in subarticular chondrocytes. Pthrp signals through its Ppr receptor expressed at a low but functional level in proliferating chondrocytes and at a high level in prehypertrophic chondrocytes. It does not affect chondrocyte proliferation directly, but establishes a negative feedback loop with Ihh to delay prehypertrophy. Counteracting Ihh signaling, FGF signaling inhibits chondrocyte proliferation and maturation. The Fgf18 and Fgf9 ligands produced by the perichondrium bind to the FGF receptor-3 (Ffgr3) abundantly expressed on proliferating chondrocytes (reviewed by Ornitz, 2005). They activate the MAPK pathway to inhibit chondrocyte maturation and the Stat1 transcription factor to inhibit cell proliferation (Murakami et al., 2004). Several Wnt ligands and pathways control growth plate development. Best known is the canonical pathway mediated by beta-catenin. Its main action appears to lead hypertrophic chondrocytes to the terminal stage (Guo et al., 2009). Other pathways with key roles in the growth plate during gestation and postnatally are initiated by bone morphogenetic proteins (BMPs), TGF-beta, growth hormone (GH), insulin-like growth factors (IGFs), thyroid hormone, retinoic acid, connective tissue growth factor (CTGF), C-type natriuretic peptide (CNP), thyroid hormone (TH) and estrogen (ER). We refer the readers to recent reviews for details on their actions (Pogue and Lyons, 2006; Mackie et al., 2008; Olney, 2009). Cartilage growth plates are thus essential drivers of skeleton growth and the multi-step maturation program of chondrocytes in these plates is tightly controlled by numerous signaling pathways and transcription factors. While this aspect of skeletogenesis appears to be one of the best known, additional studies are still warranted to identify additional regulatory players and fully understand how all pathways and factors interact with each other.
BONE DEVELOPMENT
Bones form through two processes: endochondral and intramembranous ossification. The former consists in replacing growth plate cartilage by bone (Fig. 5A and B). As soon as terminal chondrocytes die at the end of growth plates, their lacunae are invaded by a team of bone-forming cells: osteoclasts help remove the cartilage matrix; osteoblasts lay down bone matrix; endothelial cells vascularize the newly formed tissue; and hematopoietic and stromal cells generate bone marrow. Chondrocytes actively contribute to this process. At prehypertrophy, their production of Ihh induces osteoblast differentiation in the perichondrium, then renamed periosteum or bone collar (St-Jacques et al., 1999). At hypertrophy, they produce the vascular endothelial growth factor (Vegf) to trigger blood vessel formation in developing bone, and the matrix metalloproteinases Mmp13 and Mmp14 to help osteoclast-produced Mmp9 degrade the cartilage matrix (Zelzer et al., 2004; Stickens et al., 2004). The exact mechanisms that cause chondrocyte death remain unknown, but it does not occur if bone formation is impaired, further demonstrating that close interactions occur between cartilage and bone cells during endochondral ossification. The bone tissue forming in the diaphysis of fetal long bones is called the primary ossification center. Secondary centers similarly develop in the epiphyses postnatally, and the two types of centers fuse when growth plates close in early adulthood.
Figure 5. Osteoblast differentiation and intramembranous and endochondral ossification.

A. Sections through an endochondral bone in a newborn mouse show the replacement of cartilage by bone. The left section is stained with Alcian blue and the right one with the von Kossa reagent, which leaves a brown precipitate on the mineralized bone matrix.
B. Schematic showing how growth plate chondrocytes and bone-forming cells interact with each other to achieve endochondral ossification.
C. Coronal sections of a newborn mouse head. In the suture linking the two frontal bones (top panel), osteoblast precursors are surrounded by an abundant collagenous matrix. Further away (bottom panel), osteoblasts mature and deposit a mineralized bone matrix. This matrix is stained with the von Kossa reagent.
D. Schematic of the molecular control of osteoblast differentiation.
Intramembranous ossification occurs when skeletogenic cells condense into compact nodules in the sites of future skull bones and in the perichondrium of growth plates at the level of prehypertrophic chondrocytes (reviewed by Opperman, 2000) (Fig. 5C). While some cells develop capillaries, most become osteoblastic. They first produce an organic matrix and later calcify it through radial production of bony spicules from an ossification center located in the middle of each future bone. Developing skull bones are connected by fibrous, elastic tissues called sutures and whose primary role is not to serve as joints, but as bone growth centers. They maintain a pool of undifferentiated, osteogenic cells and simultaneously produce new bone cells that are recruited into ossification fronts. They disappear at about two years of age in humans, resulting in fusion of cranial bones. Insufficient bone growth results in suture agenesis, whereas too much bone growth results in craniosynostosis, i.e., precocious obliteration of sutures. Both types of defects result in severe skull malformations.
Both endochondral and intramembranous bones are built by osteoblasts, while osteoclasts degrade the tissue and have a key role in bone homeostasis postnatally. We will restrict our review of bone cells to osteoblasts, and will refer readers interested in osteoclast development to several recent reviews (reviewed by Yavropoulou and Yovos, 2008; Karsenty et al., 2009). Osteoblasts differentiate from skeletogenic cells in two main steps (reviewed by Karsenty et al., 2009; Hartmann, 2009; Jensen et al., 2010) (Fig. 5D). As mentioned earlier, they first build a organic non-mineralized (osteoid) matrix. They strongly upregulate collagen-1 expression and express alkaline phosphatase, an important enzyme in matrix mineralization. As they mature, they produce bone-specific proteins, such as osteocalcin (Bgp) and bone sialoprotein (Bsp), and mineralize the osteoid matrix. These differentiation steps are governed by three specific transcription factors: Runx2, Osx, and ATF4.
Runx2 is a master osteogenic factor. While both Runx2 and Runx3 are expressed in growth plate chondrocytes, Runx2 but not Runx3 is expressed in skeletogenic cells and it remains expressed in differentiating osteoblasts until maturation. The importance of Runx2 in this lineage was uncovered in 1997 when it was found that RUNX2 haploinsufficiency causes cleidocranial dysplasia (CCD), a human disorder characterized by clavicle hypoplasia or aplasia (cleido-) and persistently open skull sutures (-cranial) (Mundlos et al., 1997). At the same time, inactivation of Runx2 in mice was shown to result in a block of osteoblast differentiation at the skeletogenic stage and thus in complete absence of bone (Komori et al., 1997; Otto et al., 1997). Runx2 directly binds and activates Col1a1 (collagen-1), Bsp and Bgp, and is sufficient to activate these genes in mesenchymal cells in vitro (Ducy et al., 1997). Interestingly, however, forced expression of Runx2 in chondrocytes in transgenic mice results in precocious and ectopic maturation of the cells, but not in their osteoblastic transformation (Takeda et al., 2004). Runx2 is thus needed, but may not be sufficient for osteoblast differentiation.
Osterix (Osx or Sp7) is a Krüppel-like zinc finger domain-containing transcription factor expressed exclusively in osteoblasts (Nakashima et al., 2002). It was identified through potent induction of its expression by Bmp2 in mesenchymal cells in vitro. Its inactivation in the mouse revealed that it is required for the differentiation of Runx2-expressing skeletogenic cells into osteoblasts in both endochondral and intramembranous bones, including expression of Col1a1, Bgp and Bsp. Osx is not only needed, but also sufficient to activate Col1a1, Bgp and Bsp in mesenchymal cells in vitro and evidence exists that it directly activates these genes (Nakashima et al., 2002; Sinha et al., 2010). It is unknown, however, whether Osx cooperates with Runx2 in this function. Interestingly, Osx-null osteogenic cells exhibit a prehypertrophic chondrocyte phenotype in endochondral and intramembranous bone primordia (Nakashima et al., 2002). Osx is thus needed to repress the chondrocytic fate of skeletogenic cells and it is directly needed to ensure osteoblast differentiation of Runx2-positive osteogenic cells.
Atf4 (activating transcription factor-4, or cyclic AMP-responsive element-binding protein 2, Creb2) controls osteoblast maturation. Its gene is widely expressed, but the protein is detected almost exclusively in osteoblasts. Atf4 deficiency results in delayed bone formation during mouse embryonic development (Yang et al., 2004). Atf4 is needed for amino acid import and thereby for synthesis of collagen, the main protein in bone. It is also directly involved in activating terminal markers in a Runx2-dependent manner. Each step of osteoblast differentiation is thus under the control of a specific master transcription factor.
Besides Runx2, Osx, and Atf4, many widely expressed transcription factors also have important roles in osteoblast development. They control cell proliferation or modulate the expression and activity of the masters. They include Twist1/2, already mentioned earlier, AP1 family members (reviewed by Wagner, 2002), and the forkhead factor FoxO1. The latter was very recently shown to stimulate osteoblast proliferation through interaction with Atf4, as well as through regulation of a stress-dependent pathway influencing p53 signaling (Rached et al., 2010).
Many signaling pathways control osteoblastogenesis. We have already mentioned the key role of Ihh in the induction of osteoblast differentiation (Day and Yang, 2008). Downstream of Ihh signaling, the Wnt/beta-catenin pathway plays an important role in the decision of skeletogenic cells to undergo osteogenesis rather than chondrogenesis, and in boosting osteoblast differentiation (Rodda and McMahon, 2006; reviewed by Karsenty et al., 2009). It is believed to work by downregulating Sox9 expression and upregulating Runx2 expression. FGF signaling plays important roles in endochondral and intramembranous ossification, as demonstrated for instance by the fact that gain-of-function mutations in the human FGFR1, FGFR2 and FGFR3 receptors cause craniosynostosis (reviewed by Ornitz, 2005; Su et al., 2008). FGF signaling influences various stages of osteoblast development. FGFR1 promotes the differentiation of early osteoblasts, but prevents their maturation, whereas FGFR2 and FGFR3 promote osteoblast maturation. While Fgf2, 4 and 8 are essential in skeletogenic mesenchyme, Fgf18 promotes osteoblast progenitor proliferation as well as osteoblast maturation, mainly through Fgfr2. Bmps were the first molecules identified as possessing bone-inducing properties in vivo (reviewed by Rosen, 2006). Their osteogenic activities in vitro have been profusely documented. For instance, Bmp2 was shown to upregulate Runx2 and Osx expression in osteogenic cells, and in turn, Bmp2 expression was shown to regulate Runx2 expression. However, as of today, our knowledge of the specific roles of Bmps in osteoblasts in vivo remains scanty. Like many other cell developmental pathways, osteoblast generation thus involves a few cell type-specific master transcription factors that are controlled by and act together with many other widely expressed factors and signaling pathways. As for the chondrocyte lineage, most osteogenic regulatory factors were identified in the last two decades, and important progress remains to be done to elucidate the precise actions and mutual interactions of many known factors and likely to identify additional key factors.
SYNOVIAL JOINT FORMATION
As mentioned earlier, several types of joints link skeletal elements together. Their composition and roles are very different from one type to another. Sutures are fibrous joints allowing minimal movement between skull bones and ensuring bone growth. Intervertebral and other cartilaginous joints allow more movement than sutures, but much less movement than synovial joints. We will focus here on the latter because they have key roles and are subject to highly prevalent of degeneration diseases in humans, but also because our knowledge of the mechanisms underlying their development remains very modest compared that of other skeletogenic processes. We thus owe to draw attention to this process and stimulate research efforts in this area.
Synovial joints develop concomitantly with the skeletal elements that they articulate (reviewed by Pacifici et al., 2005; Khan et al., 2007; Pitsillides and Ashhurst, 2008). They do so in two main steps. In the first step, called joint specification, skeletogenic cells are directed to the articular fate (Fig. 6A and B). In the second step, called joint morphogenesis, articular cells differentiate and develop the various joint structures.
Figure 6. Synovial joint development.

A. Sections through the mouse knee joint at various stages of development. At E12, the presumptive joint region (arrow) is not distinguishable from the femur (F) and tibia (T) precartilaginous condensations. At E13.5, this region becomes distinguishable as surrounding cartilage primordia are overtly developing. At E16.5, joint morphogenesis is well advanced. The joint cavity has formed between the patella (P) and femur. Cruciate ligaments (CL) and fat pad (FP), lined with synovial tissue, are developed. At the postnatal day 19, the knee joint is mature. The articular cartilage (AC) is separated from the epiphyseal growth plate (GP) by a secondary center of ossification. The sections are stained with Alcian blue and nuclear fast red.
B. Schematic of the molecular control of synovial joint cell differentiation.
As mentioned earlier, articular progenitor cells arise in the first step of joint formation from the same pool of skeletogenic cells as chondrocytes and osteoblasts (Rountree et al., 2004; Koyama et al., 2008). They express Sox5/6/9 as do adjacent chondrogenic cells, but specifically express the genes for the Tgf-beta receptor 2 (Tgfbr2), Wnt canonical ligands (Wnt4, Wnt9a/14, and Wnt16), and the growth differentiation factor-5 (Gdf5) (Hartmann and Tabin, 2001; Guo et al., 2004; Seo and Serra, 2007; Spagnoli et al., 2007; Dy et al., 2010). A Tgfbr2/Wnt/Gdf5 signaling cascade is required and sufficient to specify progenitors to the articular fate. Its action results in the formation of presumptive joint regions, called interzones. These zones feature highly condensed cells, among which the precursors of non-cartilaginous tissues downregulate expression of Sox5/6/9.
An important aspect of synovial joint morphogenesis is the formation of an internal cavity, a process referred to as joint cavitation. Cell death was originally thought to trigger this process, but this notion is now contested by evidence that cell death is detected in presumptive phalangeal joints, but not in other sites (Ito and Kida, 2000). Instead, hyaluronan secretion and shifts in extracellular matrix composition are now believed to promote joint cavitation (Matsumoto et al., 2009), and skeletal movement and cartilage primordia development were recently demonstrated to be required for this event (Kahn et al., 2009; Dy et al., 2010). The underlying mechanisms, however, remain unknown.
Another important aspect of joint morphogenesis is the differentiation of articular chondrocytes. These cells overtly differentiate in the mouse only postnatally and, in contrast to growth plate chondrocytes, they do never terminally mature. Instead, they maintain a chondrocyte early phenotype throughout life. Not unexpectedly, their differentiation requires expression of Sox5/6 (Dy et al., 2010). It is likely that it requires Sox9 expression as well, but this has not been demonstrated yet. The Ets-domain transcription factor Erg has been proposed to control the specific fate of articular chondrocytes based on the fact that Erg was reported to be highly expressed in articular progenitors, but to be absent in growth plate chondrocytes, and that its forced expression in mouse cartilage primordia appeared to delay growth plate development (Iwamoto et al., 2007). However, recent evidence that Erg is actually highly expressed in both growth plate and articular chondrocyte progenitors and that it is downregulated upon differentiation of both chondrocyte types, strongly suggests that Erg might help maintain chondrogenic cells at a precursor stage rather than help them acquire an articular fate (Dy et al., 2010). There is presently no evidence that transcription factors other than the Sox trio control articular chondrocyte differentiation. It is thus possible that the maintenance of an early phenotype in articular chondrocytes depends on continued expression of the Sox trio rather than on expression of additional lineage-specific transcription factors. Knowledge of the roles of signaling pathways that control articular chondrocytes remains scanty. Notch signaling is receiving increasing attention, as it is believed to have key roles in the maintenance of articular progenitors (reviewed by Karlsson and Lindhal, 2009). Pthrp, Ihh, Bmp and Tgf-beta signaling are required to maintain healthy articular cartilage postnatally, and Wnt/beta-catenin signaling may have a key role in preventing chondrocyte differentiation of articular progenitors not destined to form articular cartilage (Serra et al., 1997; Yang et al., 2001; Rountree et al., 2004; Chen et al., 2008; Zhu et al., 2008; Yuasa et al., 2009). The direct involvement of these pathways in the development of articular cartilage remains, however, unclear. This is mostly due to the lack of suitable mouse models to assess their roles in articular chondrocytes independently of their roles in growth plate chondrocytes and other cell types.
Finally, synovial joint morphogenesis is also characterized by the development of synovial fibroblasts, fat pad cells, and tenocytes that form the non-cartilaginous structures of synovial joints. The mechanisms underlying the differentiation of these cells remain virtually unknown. This brief review thus highlights that our current understanding of synovial joint development lags way behind that of other skeletogenic processes. While several pathways have been identified that are required for joint specification, major gaps in knowledge remain regarding the transcription factors and signaling pathways involved in the differentiation of all types of articular cells and in the coordination of their development into the multiple joint tissues.
SKELETON VARIATION
The skeleton, probably more than any other system, exhibits an astonishingly great variability. Variability exists across species in the number of skeletal elements making up the skeleton, and variability also exists across and within species in the size and shape of each element. A fascinating field of research for skeleton biologists, geneticists, and evolutionary developmental biologists is to elucidate the mechanisms underlying this variability. New mechanisms were recently proposed and will be discussed here.
Variability across species could be due to differences in the number of genes involved in skeletogenesis. The SHOX (short stature homeobox) genes, for instance, are candidates for this mechanism. Two of them exist in humans (SHOX and SHOX2), one in mice (Shox2), and none in invertebrates. Mutations in the X-linked SHOX gene occur at a very high frequency, about 1/1000, in humans. Haploinsufficiency causes short stature and malformation of limb extremities (Ellison et al., 1997; Gahunia et al., 2009). Homozygous mutations cause Langer's mesomelic dysplasia, characterized by extremely short and bowed limbs. The human genes and the mouse gene have similar expression patterns (Clement-Jones et al., 2000) and Shox2 inactivation in the mouse results in a phenotype overlapping with that caused by SHOX mutations in humans (Cobb et al., 2006). The Shox genes thus encode essential patterning factors, whose alterations result in striking skeletal differences between human individuals. It can therefore be hypothesized that the duplication of the SHOX genes in humans compared to other species might contribute to some major skeletal differences existing between them.
It is evident that most of the variability existing between and within species cannot be explained by differences in gene numbers, as all humans have the same gene set and largely share this set with other vertebrates. How do divergent skeletal morphologies then arise? Studies on identical twins and siblings have demonstrated that environmental and hormonal conditions affect skeletal growth, but to an extent that is less than 10% (Macgregor et al., 2006). Heritability must thus account for the rest and could have impacts at several levels. For instance, differences in regulatory DNA sequences can cause differences in gene expression patterns and levels. This was recently demonstrated for a Prx1 enhancer variation that may contribute to the spectacular difference in forelimb length between mice and bats (Cretekos et al., 2007). The replacement of a mouse Prx1 enhancer with an orthologous bat sequence resulted in elevated transcript levels in developing forelimbs and in abnormally long limb skeletal elements. Interestingly, deletion of the mouse Prx1 enhancer resulted in normal forelimb length and Prx1 expression, suggesting regulatory redundancy. These findings suggest that mutations in regulatory sequences can result in morphological differences between and within species and that cis-regulatory redundancy may facilitate accumulation of such mutations.
Protein sequence differences also can contribute to variations. This was recently demonstrated in genome-wide studies that associated 47 loci with height variation in humans (Lettre, 2009). In particular, a variant allele of HMGA2 (high-mobility-group DNA-binding protein A2) was estimated to explain approximately 0.3% of the human population variation in height. The effect of each single nucleotide polymorphism was small, but in aggregate these polymorphisms could correctly assign individuals to the lower or upper tail of the human population height distribution. Interestingly, Hmga2 mutations cause dwarfism in pigmy mice (Zhou et al., 1995) and truncations of human HMGA2 and mouse Hmga2 are associated with extreme overgrowth (Ligon et al., 2005). Hmga2 is expressed in mesenchymal cells and its roles are unknown. Several of the other 47 loci also included genes previously implicated in tall or short stature syndromes (Lettre, 2009). These studies thus indicate that genes mutated in severe syndromes can also harbor common alleles with a weaker effect on stature. They also demonstrate that skeletogenesis is under the control of a few master genes as well as numerous patterning genes whose allele variants determine skeletal variability within and among species.
PERSPECTIVES
We have reviewed here many regulatory factors and pathways that were unknown five, ten, or twenty years ago and that have become the focus of attention of skeletal biologists throughout the world since milestone discoveries were made on their roles in skeletogenesis or skeleton malformation diseases. Despite tremendous progress, it is nevertheless striking that our knowledge of the genes involved in human skeletal diseases and in the control of skeletogenesis remains incomplete. Indeed, we often do not fully understand how factors are regulated and participate in the complex molecular networks that govern skeletogenic programs. It also appears likely in some cases and obvious in others that additional factors remain to be uncovered. Having reviewed our current knowledge and identified gaps in knowledge, it is now time to make long-term and short-term plans for future research efforts.
Identifying mechanisms to prevent and cure skeletal malformation diseases must continue to be our chief ultimate goal. To reach it, we must pursue our searches for disease-causing mutations. Positional cloning and candidate gene approaches remain fully appropriate and although laborious they are becoming easier with each technological improvement and the progressive deciphering of human and other genomes. Once mutations are identified, the most logical strategy is to try and fix these mutations using gene therapy or tissue engineering approaches. Major scientific, technological and even ethical challenges, however, have still to be overcome before such a strategy becomes successful. It is therefore wise to consider alternative strategies, which would ameliorate skeletogenesis by bypassing the impact of gene mutations. The proper design of such strategies requires that we understand the roles of affected genes, their modes of regulation, and the impact of the gene mutations. For these purposes, gene manipulations in cultured cells, vertebrates, and even invertebrates, such as the fly and sea urchin, are instrumental, as are various types of cellular and molecular approaches in vitro. Major technological discoveries made in recent years are opening doors to new and more efficient approaches. For instance, comparative genomics has become a powerful approach to identify important coding and non-coding regulatory elements. Mass spectrometry has become highly proficient to identify protein partners and modifications. DNA, RNA, and protein delivery methods in cells using viruses and other reagents have also become very effective to address myriads of questions in vivo or in vitro. One can thus be very optimistic that the landmark discoveries made in recent years have set the stage for many more chief discoveries such that five, ten, or twenty years from now we will not only understand the molecular basis of most skeletal dysplasias, but we may also be able to provide preventive and therapeutic strategies to alleviate the distress of many affected children.
Acknowledgments
Work in the Lefebvre laboratory described in this review was supported by grants from the National Institutes of Health (AR46249 and AR54153 to V.L.). We apologize to all authors whose important work could not be cited due to space constraints.
REFERENCE LIST
- Akiyama H. Control of chondrogenesis by the transcription factor Sox9. Mod Rheumatol. 2008;18:213–219. doi: 10.1007/s10165-008-0048-x. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Bhattaram P, Penzo-Méndez A, Sock E, Colmenares C, Kaneko KJ, Vassilev A, DePamphilis ML, Wegner M, Lefebvre V. Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat Commun. 2010;x:x. doi: 10.1038/1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Butterfield NC, McGlinn E, Wicking C. The molecular regulation of vertebrate limb patterning. Curr Top Dev Biol. 2010 doi: 10.1016/S0070-2153(10)90009-4. (submitted) [DOI] [PubMed] [Google Scholar]
- Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strachan T, Lindsay S, Rappold GA. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Molec Genet. 2000;9:695–702. doi: 10.1093/hmg/9.5.695. [DOI] [PubMed] [Google Scholar]
- Chen X, Macica CM, Nasiri A, Broadus AE. Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum. 2008;58:3788–3797. doi: 10.1002/art.23985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb J, Dierich A, Huss-Garcia Y, Duboule D. A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc Nat Acad Sci. 2006;103:4511–4515. doi: 10.1073/pnas.0510544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretekos CJ, Wang Y, Green ED, Martin JF, Rasweiler JJ, IV, Behringer RR NISC Comparative Sequencing Program. Regulatory divergence modifies limb length between mammals. Genes Dev. 2008;22:141–151. doi: 10.1101/gad.1620408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Day TF, Yang Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am. 2008;90(Suppl 1):19–24. doi: 10.2106/JBJS.G.01174. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Dy P, Penzo-Méndez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. The three SoxC proteins--Sox4, Sox11 and Sox12--exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 2008;36:3101–3117. doi: 10.1093/nar/gkn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy P, Smits P, Silvester A, Penzo-Méndez A, Dumitriu B, Han Y, de la Motte CA, Kingsley DM, Lefebvre V. Synovial joint morphogenesis requires the chondrogenic action of Sox5 and Sox6 in growth plate and articular cartilage. Dev Biol. 2010 doi: 10.1016/j.ydbio.2010.02.024. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames BF, Sharpe PT, Helms JA. Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Dev Biol. 2004;274:188–200. doi: 10.1016/j.ydbio.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Ellison JW, Wardak Z, Young MF, Robey PG, Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Molec Genet. 1997;6:1341–1347. doi: 10.1093/hmg/6.8.1341. [DOI] [PubMed] [Google Scholar]
- Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovi_ M, Weissenbach J, Mansour S, Young ID, Goodfellow PN, Brook JD, Schafer AJ. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525–530. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- Gahunia HK, Babyn PS, Kirsch S, Mendoza-Londono R. Imaging of SHOX- associated anomalies. Semin Musculoskelet Radiol. 2009;13:236–254. doi: 10.1055/s-0029-1237691. [DOI] [PubMed] [Google Scholar]
- Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Mak KK, Taketo MM, Yang Y. The Wnt/beta-catenin pathway interacts differentially with PTHrP signaling to control chondrocyte hypertrophy and final maturation. PLoS One. 2009;4:e6067. doi: 10.1371/journal.pone.0006067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–147. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Han Y, Lefebvre V. L-Sox5/Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol Cell Biol. 2008;28:4999–5013. doi: 10.1128/MCB.00695-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C. Transcriptional networks controlling skeletal development. Curr Opin Genet Dev. 2009;19:437–443. doi: 10.1016/j.gde.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Tabin CJ. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell. 2001;104:341–351. doi: 10.1016/s0092-8674(01)00222-7. [DOI] [PubMed] [Google Scholar]
- Hermanns P, Lee B. Transcriptional dysregulation in skeletal malformation syndromes. Am J Med Genet. 2001;106:258–271. [PubMed] [Google Scholar]
- Hill TP, Später D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Huang W, Chung UI, Kronenberg HM, de Crombrugghe B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc Natl Acad Sci U S A. 2001;98:160–165. doi: 10.1073/pnas.011393998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Kamekura S, Mabuchi A, Kou I, Seki S, Takato T, Nakamura K, Kawaguchi H, Ikegawa S, Chung UI. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signal sufficient for induction of permanent cartilage. Arthritis Rheum. 2004;50:3561–3573. doi: 10.1002/art.20611. [DOI] [PubMed] [Google Scholar]
- Ikegawa S. Genetic analysis of skeletal dysplasia: recent advances and perspectives in the post-genome-sequence era. J Hum Genet. 2006;51:581–586. doi: 10.1007/s10038-006-0401-x. [DOI] [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, Komori T. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Tamamura Y, Koyama E, Komori T, Takeshita N, Williams JA, Nakamura T, Enomoto-Iwamoto M, Pacifici M. Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev Biol. 2007;305:40–51. doi: 10.1016/j.ydbio.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ED, Gopalakrishnan R, Westendorf JJ. Regulation of gene expression in osteoblasts. Biofactors. 2010;36:25–32. doi: 10.1002/biof.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C, Lindahl A. Notch signaling in chondrogenesis. Int Rev Cell Mol Biol. 2009;275:65–88. doi: 10.1016/S1937-6448(09)75003-8. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol. 2009;25:629–648. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- Khan IM, Redman SN, Williams R, Dowthwaite GP, Oldfield SF, Archer CW. The development of synovial joints. Curr Top Dev Biol. 2007;79:1–36. doi: 10.1016/S0070-2153(06)79001-9. [DOI] [PubMed] [Google Scholar]
- Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, Rattenbach R, Relaix F, Maire P, Rountree RB, Kingsley DM, Zelzer E. Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell. 2009;16:734–743. doi: 10.1016/j.devcel.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- Kirn-Safran CB, Gomes RR, Brown AJ, Carson DD. Heparan sulfate proteoglycans: coordinators of multiple signaling pathways during chondrogenesis. Birth Defects Res C Embryo Today. 2004;72:69–88. doi: 10.1002/bdrc.20005. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Koyama E, Shibukawa Y, Nagayama M, Sugito H, Young B, Yuasa T, Okabe T, Ochiai T, Kamiya N, Rountree RB, Kingsley DM, Iwamoto M, Enomoto-Iwamoto M, Pacifici M. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev Biol. 2008;316:62–73. doi: 10.1016/j.ydbio.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozhemyakina E, Cohen T, Yao TP, Lassar AB. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol. 2009;29:5751–5762. doi: 10.1128/MCB.00415-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Lassar AB. The transcriptional activity of Sox9 in chondrocytes is regulated by RhoA signaling and actin polymerization. Mol Cell Biol. 2009;29:4262–4273. doi: 10.1128/MCB.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–2346. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Li P, de Crombrugghe B. A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are co-expressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998;17:5718–5733. doi: 10.1093/emboj/17.19.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–212. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- Lettre G. Genetic regulation of adult stature. Curr Opin Pediatr. 2009;21:515–22. doi: 10.1097/MOP.0b013e32832c6dce. [DOI] [PubMed] [Google Scholar]
- Leucht P, Kim JB, Amasha R, James AW, Girod S, Helms JA. Embryonic origin and Hox status determine progenitor cell fate during adult bone regeneration. Development. 2008;135:2845–2854. doi: 10.1242/dev.023788. [DOI] [PubMed] [Google Scholar]
- Li Y, Toole BP, Dealy CN, Kosher RA. Hyaluronan in limb morphogenesis. Dev Biol. 2007;305:411–420. doi: 10.1016/j.ydbio.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon AH, Moore SDP, Parisi MA, Mealiffe ME, Harris DJ, Ferguson HL, Quade BJ, Morton CC. Constitutional rearrangement of the architectural factor HMGA2: a novel human phenotype including overgrowth and lipomas. Am J Hum Genet. 2005;76:340–348. doi: 10.1086/427565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limura T, Denans N, Pourquié O. Establishment of Hox vertebral identities in the embryonic spine precursors. Curr Top Dev Biol. 2009;88:201–234. doi: 10.1016/S0070-2153(09)88007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor S, Cornes BK, Martin NG, Visscher PM. Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum Genet. 2006;120:571–580. doi: 10.1007/s00439-006-0240-z. [DOI] [PubMed] [Google Scholar]
- Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol. 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Li Y, Jakuba C, Sugiyama Y, Sayo T, Okuno M, Dealy CN, Toole BP, Takeda J, Yamaguchi Y, Kosher RA. Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development. 2009;136:2825–2835. doi: 10.1242/dev.038505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre DC, Rakshit S, Yallowitz AR, Loken L, Jeannotte L, Capecchi MR, Wellik DM. Hox patterning of the vertebrate rib cage. Development. 2007;134:2981–2989. doi: 10.1242/dev.007567. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2000;97:1113–1118. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- Ng LJ, Wheatley S, Muscat GE, Conway-Campbell J, Bowles J, Wright E, Bell DM, Tam PP, Cheah KS, Koopman P. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108–121. doi: 10.1006/dbio.1996.8487. [DOI] [PubMed] [Google Scholar]
- Olney RC. Mechanisms of impaired growth: effect of steroids on bone and cartilage. Horm Res. 2009;72(Suppl 1):30–35. doi: 10.1159/000229761. [DOI] [PubMed] [Google Scholar]
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–485. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KG, Kuratani S. Evolutionary origin of bone and cartilage in vertebrates. In: Pourquié O, editor. The skeletal system. Cold Spring Harbor Laboratory Press; 2009. pp. 1–18. [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Pacifici M, Koyama E, Iwamoto M. Mechanisms of synovial joint and articular cartilage formation: recent advances, but many lingering mysteries. Birth Defects Res C Embryo Today. 2005;75:237–248. doi: 10.1002/bdrc.20050. [DOI] [PubMed] [Google Scholar]
- Pitsillides AA, Ashhurst DE. A critical evaluation of specific aspects of joint development. Dev Dyn. 2008;237:2284–2294. doi: 10.1002/dvdy.21654. [DOI] [PubMed] [Google Scholar]
- Pogue R, Lyons K. BMP signaling in the cartilage growth plate. Curr Top Dev Biol. 2006;76:1–48. doi: 10.1016/S0070-2153(06)76001-X. [DOI] [PubMed] [Google Scholar]
- Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, Depinho RA, Kousteni S. FoxO1 Is a Positive Regulator of Bone Formation by Favoring Protein Synthesis and Resistance to Oxidative Stress in Osteoblasts. Cell Metab. 2010;11:147–160. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentsendorj O, Nagy A, Sinkó I, Daraba A, Barta E, Kiss I. Highly conserved proximal promoter element harbouring paired Sox9-binding sites contributes to the tissue- and developmental stage-specific activity of the matrilin-1 gene. Biochem J. 2005;389:705–716. doi: 10.1042/BJ20050214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimoin DL, Cohn D, Krakow D, Wilcox W, Lachman RS, Alanay Y. The skeletal dysplasias: clinical-molecular correlations. Ann N Y Acad Sci. 2007;1117:302–309. doi: 10.1196/annals.1402.072. [DOI] [PubMed] [Google Scholar]
- Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- Rosen V. BMP and BMP inhibitors in bone. Ann N Y Acad Sci. 2006;1068:19–25. doi: 10.1196/annals.1346.005. [DOI] [PubMed] [Google Scholar]
- Rountree RB, Schoor M, Chen H, Marks ME, Harley V, Mishina Y, Kingsley DM. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2:e355. doi: 10.1371/journal.pbio.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Selvamurugan N, Kwok S, Alliston T, Reiss M, Partridge NC. Transforming growth factor-beta 1 regulation of collagenase-3 expression in osteoblastic cells by crosstalk between the Smad and MAPK signaling pathways and their components, Smad2 and Runx2. J Biol Chem. 2004;279:19327–19334. doi: 10.1074/jbc.M314048200. [DOI] [PubMed] [Google Scholar]
- Seo HS, Serra R. Tgfbr2 is required for development of the skull vault. Dev Biol. 2009;334:481–490. doi: 10.1016/j.ydbio.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha KM, Yasuda H, Coombes MM, Dent SY, de Crombrugghe B. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 2010;29:68–79. doi: 10.1038/emboj.2009.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum L, Coleman CM, Hatakeyama Y, Tuan RS. Morphogenesis and dysmorphogenesis of the appendicular skeleton. Birth Defects Res C Embryo Today. 2003;69:102–122. doi: 10.1002/bdrc.10012. [DOI] [PubMed] [Google Scholar]
- Smits P, Li P, Mandel J, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B, Lefebvre V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1:277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- Smits P, Dy P, Mitra S, Lefebvre V. Sox5 and Sox6 are needed to develop and maintain source, columnar and hypertrophic chondrocytes in the cartilage growth plate. J Cell Biol. 2004;164:747–758. doi: 10.1083/jcb.200312045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnoli A, O'Rear L, Chandler RL, Granero-Molto F, Mortlock DP, Gorska AE, Weis JA, Longobardi L, Chytil A, Shimer K, Moses HL. TGF-beta signaling is essential for joint morphogenesis. J Cell Biol. 2007;177:1105–1117. doi: 10.1083/jcb.200611031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger J. Skeletal dysplasias. In: Stevenson RE, Hall JG, editors. Human Malformations and Related Anomalies. 2. Chapter 22 Oxford University Press; Oxford, New York: 2006. [Google Scholar]
- Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Du X, Chen L. FGF signaling: its role in bone development and human skeleton diseases. Front Biosci. 2008;13:2842–2865. doi: 10.2741/2890. [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ, Danielson KG, Hall DJ, Tuan RS. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem. 2003;278:41227–41236. doi: 10.1074/jbc.M305312200. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Wagner EF. Functions of AP1 (Fos/Jun) in bone development. Ann Rheum Dis. 2002;61(Suppl 2):ii40–2. doi: 10.1136/ard.61.suppl_2.ii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, Wolf U, Tommerup N, Schempp W, Scherer G. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene. Cell. 1994;79:1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Wellik DM. Hox genes and vertebrate axial pattern. Curr Top Dev Biol. 2009;88:257–278. doi: 10.1016/S0070-2153(09)88009-5. [DOI] [PubMed] [Google Scholar]
- Yang X, Chen L, Xu X, Li C, Huang C, Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153:35–46. doi: 10.1083/jcb.153.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Karsenty G. ATF4, the osteoblast accumulation of which is determined post-translationally, can induce osteoblast-specific gene expression in non-osteoblastic cells. J Biol Chem. 2004;279:47109–47114. doi: 10.1074/jbc.M410010200. [DOI] [PubMed] [Google Scholar]
- Yavropoulou MP, Yovos JG. Osteoclastogenesis--current knowledge and future perspectives. J Musculoskelet Neuronal Interact. 2008;8:204–216. [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y, Komori T. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952–963. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 2009;175:1993–2003. doi: 10.2353/ajpath.2009.081173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]
- Zeng L, Kempf H, Murtaugh LC, Sato ME, Lassar AB. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16:1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun M, Zhao J, Leyva JA, Zhu H, Yang W, Zeng X, Ao Y, Liu Q, Liu G, Lo WHY, Jabs EW, Amzel LM, Shan X, Zhang X. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet. 2007;80:361–371. doi: 10.1086/511387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995;377:771–774. doi: 10.1038/376771a0. [DOI] [PubMed] [Google Scholar]
- Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O'Keefe RJ, Chen D. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008;58:2053–2064. doi: 10.1002/art.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]