

Abstract
Zinc accumulates in the synaptic vesicles of certain glutamatergic forebrain neurons and modulates neuronal excitability and synaptic plasticity by multiple poorly understood mechanisms. Zinc directly inhibits NMDA-sensitive glutamate-gated channels by two separate mechanisms: high-affinity binding to N-terminal domains of GluN2A subunits reduces channel open probability, and low-affinity voltage-dependent binding to pore-lining residues blocks the channel. Insight into the high-affinity allosteric effect has been hampered by the receptor's complex gating; multiple, sometimes coupled, modulatory mechanisms; and practical difficulties in avoiding transient block by residual Mg2+. To sidestep these challenges, we examined how nanomolar zinc concentrations changed the gating kinetics of individual block-resistant receptors. We found that block-insensitive channels had lower intrinsic open probabilities but retained high sensitivity to zinc inhibition. Binding of zinc to these receptors resulted in longer closures and shorter openings within bursts of activity but had no effect on interburst intervals. Based on kinetic modeling of these data, we conclude that zinc-bound receptors have higher energy barriers to opening and less stable open states. We tested this model for its ability to predict zinc-dependent changes in macroscopic responses and to infer the impact of nanomolar zinc concentrations on synaptic currents mediated by 2A-type NMDA receptors.
Introduction
Zinc is an essential micronutrient that accumulates in brain and is required for normal development and function (1). Both deficiency and excess of zinc alter behavior and can cause brain abnormalities and neuropathies, of which epilepsy, ischemia, and Alzheimer's degeneration have been the most studied (2,3). Aside from catalytic and structural functions in many proteins, ionic zinc (Zn2+) may play important roles in neurotransmission (4). Free Zn2+ accumulates in the synaptic vesicles of a specific subset of glutamatergic neurons and is coreleased with glutamate in an activity-dependent manner (5–7). Upon release, free Zn2+ may modulate neurotransmitter receptors and transporters, activate zinc-sensing metabotropic receptors, and/or gain cellular access through Ca2+-permeable channels (8–10). At certain glutamatergic synapses, a primary role for vesicular zinc is to reduce N-methyl-D-aspartate (NMDA) receptor currents (11). A wide range of extracellular Zn2+ concentrations directly and specifically inhibit NMDA receptor responses (12–16), and in the hippocampus, a region highly enriched in vesicular zinc, zinc-positive glutamatergic synapses are also enriched in NMDA receptors (17). The inhibitory effects of Zn2+ on NMDA receptors have received considerable attention due in part to the pivotal role played by these receptors in synaptic transmission and plasticity. Still, the mechanism by which the inhibition occurs is incompletely understood.
NMDA receptors are glutamate-gated channels responsible for the slow component of excitatory postsynaptic currents at central synapses. They assemble as tetramers of two glycine-binding GluN1 subunits and two glutamate-binding GluN2 subunits (18–20). Optional splicing of three exons in GluN1 subunits and differential expression of GluN2 subunits from four separate genes (A–D) result in substantial molecular diversity of NMDA receptors during development, across brain regions and during physiologic and pathologic conditions (21–24). The subunit composition of NMDA receptors determines their biophysical and pharmacological properties, including sensitivity to Zn2+ (25–28). The extracellular portion of each NMDA receptor subunit consists of an N-terminal domain (NTD) that binds allosterically active ligands including Zn2+, and a contiguous ligand-binding domain (LBD), which binds the obligatory coagonists glutamate and glycine. The LBD also connects with the transmembrane domain (TMD), which forms the ion-conducting pore and presumably the gate (Fig. 1 A) (29,30). The recently elucidated 3-D structure of a tetrameric receptor reveals that across subunits homologous modules organize in layers and identifies important intermodule interfaces that may mediate allosteric communication (31). Two types of Zn2+-binding sites are located on separate modules and inhibit NMDA receptor currents through distinct mechanisms.
Figure 1.
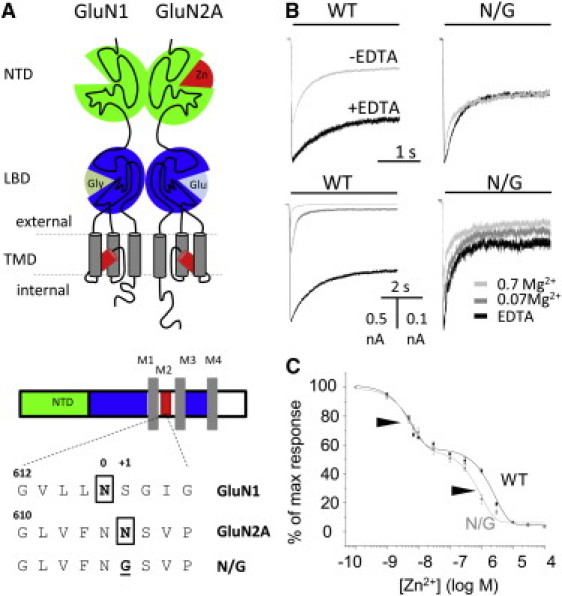
N/G responses have reduced sensitivity to block by extracellular Mg2+. (A, upper) Diagram of a 1/2A dimer highlights the low-affinity Zn2+-binding site within the TMD and the high-affinity Zn2+-binding site within the NTD (red). (A, lower) Topology of GluN subunits with expanded pore sequence illustrates the asparagine residues (boxed) involved in cation coordination and the pore sequence of the block-resistant construct. (B, upper) Whole-cell currents recorded in the presence and absence of EDTA (10 μM) reveal tonic inhibition of WT but not N/G receptors. (B, lower) Traces recorded in several external concentrations of Mg2+ (mM) illustrate the resistance of N/G receptors to block by external Mg2+. (C) Dose response of N/G (gray; 7.3 nM and 1.0 μM) and WT (black; 5.8 nM and 2.5 μM) receptors show similar sensitivities to nanomolar concentrations of Zn2+.
Several divalent cations including Mg2+, Ca2+, and Zn2+ bind with voltage-dependent affinities to residues in the TMD and transiently block current flow through open channels. Asparagine residues in the second transmembrane helix, N616 in GluN1-1a and N615 in GluN2A, contribute to the narrowest portion of the permeation pathway and are critical for voltage-dependent block (32–36). Glycine substitutions at these critical positions, GluN1-1a(N0G) or GluN2A(N+1G), increase pore diameter and reduce channel sensitivity to voltage-dependent block by orders of magnitude (32,35,37) (Fig. 1, A and B). In addition, zinc ions also produce voltage-independent inhibition of NMDA receptor currents. This effect is mediated by NTD of GluN2 subunits and has a purely allosteric mechanism: Zn2+ binding changes the receptor's gating kinetics but not its conductance (28,38–40). The NMDA receptor sensitivity to this type of inhibition is remarkably isoform-specific, with 2A receptors having high sensitivity (IC50 = 5–30 nM) and 2B receptors having low sensitivity (∼1 μM) (25,27,28,41). The mechanism, although incompletely understood, is believed to be similar for both isoforms, with the large difference in sensitivity reflecting a much slower Zn2+ dissociation rate from 2A receptors (28).
Within the NTD of GluN2 subunits, Zn2+ binds to polar residues that line the cleft between two hinged lobes and stabilizes closed-cleft conformations (42–46). Glutamate binding to residues in the adjacent LBD facilitates this change in NTD structure, which in turn lowers the channel's open probability (Po) (47,48). Because glutamate binding is required for channel opening and at the same time increases the NTD's affinity for Zn2+, when Zn2+ concentrations are subsaturating, the initial glutamate-elicited current declines relatively quickly as more receptors bind Zn2+, a phenomenon known as Zn2+-dependent desensitization (48,49). However, in saturating concentrations of Zn2+, when NTDs of resting receptors are fully occupied, this phenomenon is absent and Zn2+ inhibition reflects solely the altered gating kinetics of Zn2+-bound receptors (39). Why Zn2+-bound receptors gate with lower Po than Zn2+-free receptors is unknown.
To focus specifically on this question, we examined the gating kinetics of block-resistant Zn2+-bound NMDA receptors over the entire range of experimentally resolvable gating transitions. We found that when compared to Zn2+-free receptors, Zn2+-bound receptors had higher energy barriers to opening and populated open states that were less stable. These changes caused Zn2+-bound receptors to dwell longer in pre-open states, to remain open for shorter durations, and to accumulate in desensitized states, although the desensitized states themselves were not altered. This redistribution of state occupancies provided a quantitative account for the observed lower Po of Zn2+-bound NMDA receptors.
Materials and Methods
Rat GluN1-1a (NR1-1a, U08261) and GluN2A (NR2A, M91561) or GluN2A (N615G) were expressed together with green fluorescent protein in HEK293 cells. Single-channel currents were recorded with the cell-attached patch-clamp technique in extracellular solution containing (in mM) 150 NaCl, 2.5 KCl, 10 HEPBS (pKa, 8.3), 1 Glu, and 0.1 Gly, adjusted to pH 8. EDTA or tricine were added as specified. Free Zn2+ concentrations in 10 mM tricine-buffered solutions were calculated using Maxchelator software (www.stanford.edu/∼cpatton/maxc.html) using Kd = 10−5 M, as previously reported (28). For processing and analyses, we selected only one-channel records that required minimal processing, as described in detail in a previous work (50). Idealization was done with the segmental-k-means (SKM) algorithm in QuB on 12 kHz filtered data, with 0.15 ms dead time. Modeling was done with the maximum-interval-likelihood (MIL) algorithm. Whole-cell currents (clamped at −70 mV) and excised patch currents (clamped at −100 mV) were recorded with intracellular solution consisting of (in mM) 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 10 HEPES, and 11 EGTA, pH 7.4. Clamped cells were perfused with extracellular solutions containing (in mM) 150 NaCl, 2.5 KCl, 0.5 CaCl2, 0.1 Gly, and 10 HEPBS, pH 8. Glutamate, chelators and tricine-buffered Zn2+ were included as specified. Differences were evaluated with two-tailed Student's t-tests assuming equal variance and were considered significant for P-values <0.05. (See Supporting Material.)
Results
To determine how Zn2+ binding to the NTD affects the gating sequence of GluN1-1a/GluN2A receptors (1/2A) it is necessary to evaluate quantitatively and compare all the kinetic species populated by Zn2+-free and Zn2+-bound receptors. This is experimentally challenging for several reasons: NMDA receptors gate with multistate kinetic mechanisms; several soluble ligands modulate channel kinetics and also have reciprocal effects on each other's potency (39,48); and divalent cations such as Mg2+, Ca2+, and Zn2+, which are experimentally difficult to control simultaneously, block the channel in a voltage-dependent manner, further complicating the single-channel signal.
We isolated allosteric effects from voltage-dependent effects by examining block-insensitive receptors, 1/2A(N615G), with stationary single-channel recordings from cell-attached one-channel patches clamped at low membrane potentials (+40 mV in the recording pipette) (35,37). To minimize the effects of the various ligands on each other's affinity, we used maximally effective concentrations of glutamate (Glu, 1 mM), glycine (Gly, 0.1 mM), and free Zn2+ (67 nM, 10 mM tricine with 200 μM added Zn2+), and minimally effective proton concentrations (10 nM, pH 8).
N/G receptors are resistant to Mg2+-block but retain high-affinity Zn2+ inhibition
First, to test whether the N-to-G substitution in the pore-lining region of GluN2A subunits eliminates block by trace divalent cations, we examined whole-cell responses from HEK293 cells expressing wild-type 1/2A (WT) or 1/2A(N615G) receptors (N/G). Adding a strong metal chelator (EDTA, 0.01 mM) to the extracellular solution potentiated WT responses but not N/G responses, indicating that, as expected, N/G receptors were rendered resistant to trace divalent cations (Fig. 1 B). Consistent with this interpretation, WT responses but not N/G responses, were reduced substantially by adding Mg2+ (0.07 or 0.7 mM) to the extracellular solution (Fig. 1 B). Importantly for our purpose, N/G receptors maintained high-affinity Zn2+ inhibition. Successive applications of increasing concentrations of nanomolar-range tricine-buffered Zn2+ reduced to the same extent the equilibrium WT and N/G whole-cell responses (IC50, 5.8 nM and 7.3 nM, respectively) (Fig. 1 C). Based on these results, we conclude that N/G receptors are well suited for investigations into the mechanism of high-affinity Zn2+ inhibition.
We did not anticipate that the N-to-G pore substitution would affect receptor gating. However, the faster and deeper desensitization of N/G whole-cell responses was a strong indication of intrinsic changes in the activation of mutant receptors, which forced us to examine the kinetic mechanism of N/G receptors (Fig. 1 B). We recorded stationary currents from one-channel patches of cells transfected with WT or N/G receptors in the presence of saturating concentrations of agonists, with or without EDTA (1 mM), and +100 mV in the recording pipette. Consistent with the macroscopic measurements, WT receptors in the absence of EDTA displayed low single-channel activity (Po = 0.10 ± 0.02, n = 8) and EDTA increased the observed activity (0.65 ± 0.04, n = 12) (Fig. 2, A and B, and Table S1 in the Supporting Material). This large increase in Po reflected two major differences in the single-channel record, both attributable to relief from low-affinity voltage-dependent block. First, in the absence of EDTA, WT openings were short (mean open time (MOT) = 1.3 ± 0.1 ms) and of uniform duration (only one time component). The addition of EDTA revealed ∼10-fold longer openings (11.7 ± 0.6 ms) and complex open interval distributions. Second, when EDTA was omitted, bursts included frequent brief gaps responsible for the additional, sixth component in the closed-event distributions (τblock = 1.2 ± 0.1 ms, ablock = 35 ± 1%). This single-channel structure indicated, most likely, that in the absence of EDTA, currents through long-lived open receptors were interrupted often by trace Mg2+-binding to pore residues. The apparent binding (1/MOT = ∼800 s−1) and dissociation rates (1/τblock = ∼1000 s−1) were within the range reported previously for this receptor isoform, assuming ∼1 μM contaminant Mg2+ (51). As expected, the frequent 1-ms gaps observed for WT receptors in the absence of EDTA, which most likely represented Mg2+-bound open receptors, were absent in N/G records, whether EDTA was present or not (Fig. 2, C and D, and Table S1).
Figure 2.
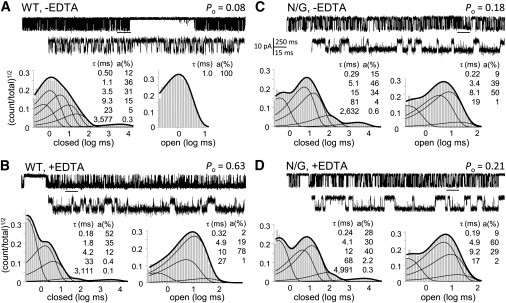
N/G receptors are resistant to block by contaminant Mg2+ and have altered kinetics. Cell-attached traces (open is down) displayed at two time resolutions. Event histograms below each trace represent interval distributions for one record obtained with (A) WT with no added EDTA (104,352 events), (B) WT with 1 mM EDTA (509,043 events), (C) N/G with no added EDTA (188,412 events), or (D) N/G with 1 mM EDTA (416,360 events) (+100 mV in cell-attached pipette, pH 8).
Despite lacking Mg2+ block, N/G channels displayed lower activity than WT, indicative of mutation-induced changes in gating. EDTA slightly but significantly increased single-channel Po of N/G channels, from 0.22 ± 0.05 (n = 7) to 0.36 ± 0.04 (n = 10) (P < 0.05), an indication that EDTA-chelatable allosteric inhibitors, most probably Zn2+, may also contaminate our recording solutions (49). However, comparing N/G and WT records obtained in the presence of EDTA, we were able to attribute the observed 45% reduction in Po specifically to the N-to-G pore substitution. It is important to point out that although the N/G mutation results in measurable differences in pore diameter, as reported previously (35), it had no effect on the channel conductance for monovalent cations. In our recordings, where Na+ was the principal charge carrier, both the WT and N/G receptors had statistically indistinguishable single-channel amplitudes: 8.5 ± 0.4 and 9.6 ± 0.7 pA, respectively (P > 0.5).
Like WT, N/G records had five closed components (E1–E5) and two to four open components (τf and at least one of τL, τM, and τH), an indication that the mutation did not change the fundamental gating mechanism of NMDA receptors (49,52–55). The lower Po measured in N/G records was fully accounted for by 2.5-fold longer closures and 30% shorter openings (Fig. 3 A). Except for the principal desensitized component τE5, which was unaffected by the mutation, all other detected closures were around twofold longer (Fig. 3 B and Table S2). The slight decrease in MOT reflected a fivefold decrease in the area of the longest open component (aH), with no change in the individual open-component durations (Table S3). Based on this observation and the knowledge that within the NMDA receptor gating reaction open states are coupled, we opted to interpret the data with a simplified kinetic model, 5C1O, in which all open states are represented as a single aggregate state. Fitting this model to individual one-channel records within the two data sets, showed that the kinetic differences between N/G and WT receptors can be explained by slower rate constants for the three activation transitions: C3→C2, C2→C1, and C1→O (Fig. 3 C). Based on these results, we suggest that by removing the asparagine side chain, which is hypothesized to face the channel lumen of GluN2A subunits, the N/G substitution increases the receptor's activation energy barriers (Fig. 3 D).
Figure 3.
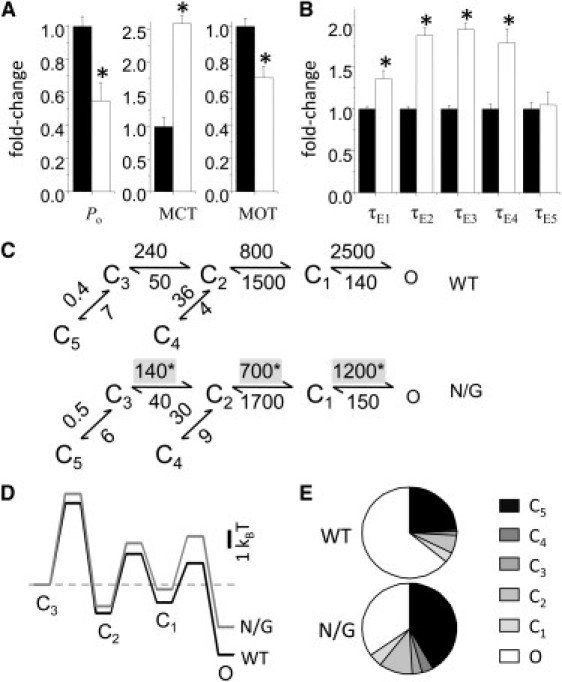
Reaction mechanism of N/G receptors. (A) Single-channel kinetic parameters for N/G (white; n = 10, 8 × 105 events) relative to WT (black; n = 12, 3.3 × 106 events). (B) Closed-component time constants (τEn) for N/G (white) relative to those for WT (black). (C) Reaction mechanisms estimated from fits of 5C1O models to individual one-channel records obtained from WT (upper) and N/G (lower). Rounded means of rate constants for each condition are in s−1. (D) Relative free-energy profiles calculated from the mechanisms illustrated in C displayed relative to the initial liganded state C3; off-pathway transitions into the long-lived C4 and C5 states are omitted. (E) Predicted fractional occupancies for WT and N/G receptors. Bars and whiskers represent means and standard errors; ∗P < 0.05 in a Student's t-test.
Notably, these results demonstrate that the pore mutation had no observable effect on the kinetics of microscopic desensitization (Fig. 3 C), although the N/G macroscopic traces we measured desensitized around twofold faster (τD, 0.5 ± 0.1 s vs. 1.2 ± 0.1 s) and ∼1.3-fold deeper (Iss/Ipk, 0.47 ± 0.05 vs. 0.60 ± 0.02) (Fig. 1 B). To investigate this possible discrepancy, we simulated macroscopic traces with the models in Fig. 3 C assuming equal glutamate association rates for N/G and WT receptors (1.7 × 106 M−1 s−1) (56). Despite the differences in experimental conditions for the single-channel (cell-attached) and macroscopic (whole-cell) experiments, the models reproduced well the measured change in time course, predicting that N/G receptors will generate currents that desensitize 1.9-fold faster (1.2 s vs. 2.1 s) and 1.3-fold deeper (59% vs. 79%) (Fig. 1 B). Based on this result, we conclude that the measured increase in macroscopic desensitization produced by the N/G substitution is not indicative of changes in microscopic desensitization. Rather, it illustrates that delayed access into open conformations due to higher activation barriers causes the accumulation of N/G receptors in desensitized states (Fig. 3, D and E).
Zn2+ effects on the gating kinetics of N/G receptors
Having established that N/G receptors retain the fundamental gating mechanism of WT receptors and their high sensitivity to Zn2+ inhibition, and are resistant to Mg2+ block, we proceeded to examine how nanomolar concentrations of zinc change this receptor's gating reaction. We recorded, analyzed, and compared cell-attached one-channel traces obtained from Zn2+-free (10 mM tricine, n = 6) and Zn2+-bound N/G receptors (67 nM free Zn2+, n = 9) in the presence of high concentrations of glutamate (1 mM) and glycine (0.1 mM) and in low proton concentrations (pH 8) (Fig. 4, A and B). As an added precaution against voltage-dependent block, these recordings were done at lower membrane potentials, by applying only +40 mV in the recording pipette as compared to the +100 mV applied in the measurements described above. Zn2+ binding had no significant effect on single-channel current amplitudes elicited from N/G receptors (5.0 ± 0.4 pA vs. 5.5 ± 0.4 pA, P > 0.3), but Zn2+-bound receptors gated with threefold-lower Po (0.10 ± 0.02 vs. 0.31 ± 0.07, P < 0.003) and had shorter openings (4.9 ± 0.4 ms vs. 7.8 ± 0.6 ms, P < 0.002) (Fig. 4 C and Table S4). These results are in agreement with conclusions reached previously based on single-channel measurements done at physiologic pH, which showed that: the high-affinity Zn2+ inhibition of 1/2A receptors occurs through an entirely allosteric mechanism, is incomplete, and results, at least in part, from shorter channel openings (39,57,58). In addition, because all our recordings originated from one-channel patches, we were able to determine that Zn2+-bound channels also had longer mean closed durations (63 ± 12 ms vs. 21 ± 4 ms, P < 0.02) (Fig. 4 C and Table S4).
Figure 4.
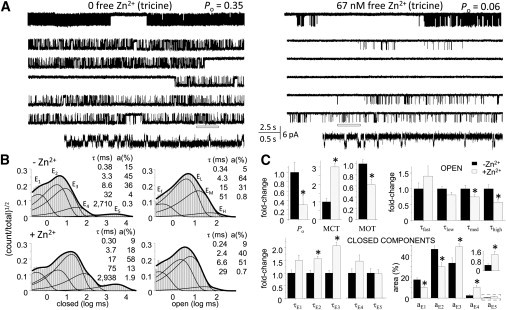
Allosteric effects of extracellular Zn2+ on individual N/G receptors. (A) Cell-attached recordings of N/G currents in the absence (left, 10 mM tricine) and presence of Zn2+ (right, 67 nM free Zn2+). Top traces illustrate a 25-s period that is expanded in the five traces beneath it, with the underlined segment shown at higher resolution at the bottom. (B) Histograms for the records illustrated in A (24 min, 122,562 events; and 27 min, 39,961 events). (Insets) Component time constants (τ) and areas (a). (C) Summary of kinetic parameters estimated for added zinc (gray, N/G with 67 nM free Zn2+, n = 9) relative to control (black, N/G with zero zinc, n = 6). Control open components (in ms): τf, 0.44; τL, 4.3; τM, 12; τH, 41. Control closed components (in ms): τE1, 0.41; τE2, 3.5; τE3, 9.8; τE4, 53; τE5, 3112. Bars and whiskers represent means and standard errors; ∗P < 0.05 in a Student's t-test.
N/G receptors maintained the complex structure of open interval distributions characteristic of NMDA receptors even in the absence of strong metal chelators, an indication of their immunity to contaminant Mg2+. In some records, we could clearly separate four open components, a clear indication that Zn2+-bound receptors retain modal gating (56). Further, we were able to determine that the overall shorter openings reflected shorter openings in each mode (Fig. 4 C) and that zinc did not significantly change the relative areas of open components (P > 0.2). In contrast, the cause of longer closures could be traced specifically to longer intraburst closures, specifically τE2 (5.4 ± 0.5 ms vs. 3.5 ± 0.4 ms, P < 0.02) and τE3 (20 ± 3 vs. 10 ± 1, P < 0.02) (Fig. 4 C). It is important to note that we were able to determine that Zn2+ did not change the durations of desensitized intervals τE4 and τE5 but increased their relative areas from 3 ± 1% to 10 ± 2% (P < 0.01) and from 0.5 ± 1% to 1.3 ± 0.3% (P < 0.02), respectively (Fig. 4 C).
The observation that Zn2+ did not affect the number of components in closed- and open-interval distributions represents a strong argument that Zn2+-bound receptors activate with a basic mechanism similar to that of Zn2+-free receptors. For this reason, we chose to fit these two data sets by the same 5C1O model used to describe WT and N/G activity (Fig. 3 C). This analysis showed that Zn2+ binding further decreased the activation rate constants C3→C2, C2→C1, and C1→O that were already slowed by the N/G mutation (Fig. 5 A). In addition, the closing rate constant O→C1 was significantly faster, an indication that the open states of Zn2+-bound receptors were overall less stable. Notably, the rates at which receptors entered the desensitized states C4 and C5 were not significantly changed by Zn2+. Instead, the observed increase in the areas of long closed components was explained solely on the basis of longer dwells in states from which desensitized states can be accessed. Thus, the model postulates that although Zn2+-bound receptors have intact microscopic desensitization rates, at equilibrium, more receptors will accumulate in desensitized states, because by dwelling longer in preopen conformations they have more opportunities to transition into desensitized states. These analyses explain the allosteric effect of Zn2+ at NMDA receptors as an increase in the activation energy barriers to opening and a destabilization of open states (Fig. 5 B). They also predict a substantially increased occupancy of desensitized states (∼35–55%) at the expense of open states (approximately fourfold lower occupancy) (Fig. 5 C).
Figure 5.
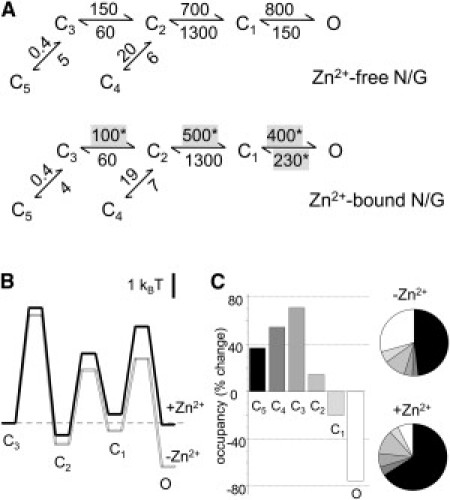
Kinetic mechanism of NMDA receptor inhibition by Zn2+. (A) Reaction mechanisms estimated for N/G receptors with 10 mM tricine (n = 6, 5.7 × 105 events) (upper) or with tricine-buffered zinc (free Zn2+, 67 nM) (n = 9, 7.5 × 105 events) (lower). Rounded mean rate constants for each condition are in s−1; ∗P < 0.05 in a Student's t-test. (B) Free-energy profiles calculated from the kinetic models in A. The desensitized states C4 and C5 are omitted. (C) Changes in state equilibrium occupancies calculated from the models in A (bar graph) and predicted fractional occupancies for Zn2+-free and Zn2+-bound receptors (pie chart).
These conclusions necessarily rely on the assumption that the 5C1O model we used to evaluate single-channel activity is accurate, at least with regard to the salient features of Zn2+-induced changes in receptor gating. As a way of testing this assumption, we asked whether the mechanism we derived from single-channel data can match the known effects of Zn2+ on macroscopic currents, such as dose response of inhibition and kinetics of onset and recovery. We were successful in reproducing the dose response relationship illustrated in Fig. 1 C with the tiered model illustrated in Fig. 6 A, which has the following features. First, the upper arm represents the activation reaction of zinc-free N/G receptors: the reaction is initiated by the sequential binding of two glutamate molecules to resting (CU) and monoliganded (CM) receptor states, respectively (k+ = 1.7 × 107 M−1 s−1, k− = 60 s−1, Kd = 3 μM) (59). Further, fully liganded receptors transition through: several preopen states (C3-C2-C1), a collection of open states (C1-O), and two separate desensitized states (C3-C5 and C2-C4) (50). Second, the lower arm of the tiered model represents the activation for Zn2+-bound receptors and postulates identical transitions that occur along a slightly altered energy landscape, as determined in this study (Fig. 5 B). Third, the model incorporates explicit Zn2+-binding and dissociation steps for glutamate-bound, but not for resting, receptors. This feature represents the higher affinity for Zn2+ of glutamate-bound receptors, as shown previously by elegant experiments from the Traynelis group (48).
Figure 6.
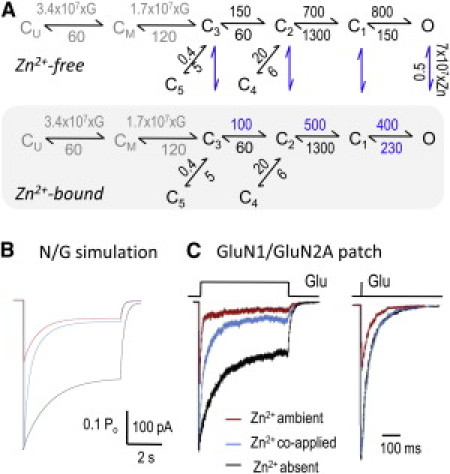
Zn2+ effects on nonstationary NMDA receptor responses. (A) Tiered model represents glutamate binding and the gating reaction of Zn2+-free receptors (upper), the binding of Zn2+ (vertical arrows, blue), and the gating reaction of Zn2+-bound receptors (lower, gray-shaded). Except for binding reactions, which are in M−1 s−1, all other rate constants are in s−1. [Glu] was set to 1 mM (G) and [Zn2+] to 67 nM (Zn). (B) Simulated responses with the N/G model in A during a 5-s pulse of 1 mM Glu in the absence (black), coapplication (blue), or ambient presence (red) of 67 nM Zn2+. (C) Responses from WT receptors measured in excised patches after fast application of 1 mM Glu for 5 s (left) or 1 ms (right). Conditions are as in B.
We used this model to simulate the effects of several Zn2+ concentrations (0–300 nM) on Zn2+-free resting receptors. All 100 receptors invoked initially occupied the CU state of the upper tier arm, were stimulated with a prolonged pulse of 1 mM Glu, and had intermittent pulses of Zn2+ superimposed on the Glu pulse. We obtained the best match between simulated traces and measured whole-cell responses by assuming Zn2+ rate constants of: k+ = 7 × 107 M−1 s−1 and k− = 0.5 s−1. These corresponded to our experimental value of Kd = 7 nM (N/G receptors, pH 8) and were very close to values reported previously for WT receptors at pH 7.4 by two groups: k+ = 108 M−1 s−1; k− = 0.6 s−1 (28) and k+ = 3.4 × 107 M−1 s−1; k− = 1.4 s−1 (49). These results illustrate that the relatively simple operational model we derived from single channels residing in cell-attached patches reproduced well salient behaviors of macroscopic whole-cell responses.
Next, we asked how Zn2+ may affect NMDA receptors in physiological situations that at present are technically off-limits to direct observation, namely, how synaptic NMDA receptors may be affected by nanomolar concentrations of free Zn2+ when 1), the modulator is coreleased with the neurotransmitter glutamate, or 2), the modulator predates synaptic release, as, for example, during high-frequency trains. We set out to mimic these situations with two stimulation paradigms: 1), brief (1 ms) pulses of glutamate (1 mM) (60,61), and 2), seconds-long glutamate applications. For each, the following conditions were compared: 1), 1 mM Glu in zero Zn2+ (using only the upper arm of the model); 2), 1 mM Glu and 67 nM coapplied Zn2+ (tiered model with all receptors starting in the Zn2+-free resting state); and 3), 1 mM Glu applied onto receptors preequilibrated with Zn2+ (using only the lower arm of the model). Results showed that for brief and long pulses alike, the peak N/G response was not affected by coapplied Zn2+, but was dramatically reduced (>50%) when Zn2+ predated the Glu pulse (Fig. 6, B and C). In addition, as observed after long pulses (5 s), coapplied Zn2+ increased dramatically the rate and degree of macroscopic desensitization. This result represents additional validation for the model, because it demonstrates the model's ability to reproduce a prominent and well-known feature of Zn2+ effects on macroscopic NMDA receptor responses: the fast zinc-dependent component of macroscopic desensitization (25,28,48).
Zn2+ effects on synaptic-like NMDA receptor responses
Finally, to test whether the predictions made based on experimental results obtained from block-resistant N/G receptors also held true for WT receptors, we measured currents elicited by 5-s or 1-ms fast applications of 1 mM Glu onto excised patches of HEK293 cells expressing WT GluN1/GluN2A receptors. Responses were recorded successively from the same patch in the absence of Zn2+ (10 mM tricine); in the ambient presence of 67 nM free Zn2+ (preequilibrated); and when 67 nM free Zn2+ was coapplied with Glu (Fig. 6 C). Results showed that coapplied Zn2+ had no effect on the peak or time course of NMDA receptor responses to brief stimulation, as may be the case during normal transmission. However, when Zn2+ predated the Glu pulse, responses had drastically lower peaks, desensitized faster, and equilibrated to lower activity levels. Traces elicited with 5-s coapplications of Zn2+ and Glu revealed the same effects on WT currents as predicted from the N/G model: faster and deeper desensitization. The model explains this result by postulating a combined effect of nanomolar concentrations of co-applied Zn2+ on NMDA receptor responses to fast applications of Glu: Glu-elicited increase in Zn2+-affinity and altered activation kinetics of Zn2+-bound receptors.
Discussion
In this article, we examined how zinc binding to the N-terminal domains of NMDA receptors affected the receptors' gating kinetics. We compared current traces obtained from individual block-resistant receptors in the absence and presence of nanomolar concentrations of extracellular zinc and found that zinc shortened all openings, increased the duration of specific closures within bursts, and had no effect on intervals residing outside bursts. Further, based on these data, we derived a gating model for zinc-bound receptors that explained the inhibitory effect of zinc as a combination of increased energy barriers to opening and less stable open states. The deduced mechanism predicted correctly the known effects of zinc on macroscopic responses, including dose response and macroscopic kinetics. Last, we used the model to ask how nanomolar zinc concentrations may influence NMDA receptor responses to physiologically relevant stimulation protocols. Responses recorded from WT receptors residing in excised patches exposed to fast pulses of glutamate confirmed that zinc had no effect on peak currents when coapplied with glutamate, but it strongly decreased receptor-mediated charge transfer when zinc predated the glutamate pulse.
Together, these results make a strong case for the mechanism of allosteric zinc inhibition proposed here. However, a good fit to experimental data was possible only after making a number of experimental simplifications and theoretical assumptions. Keeping these in mind helps to place results in perspective and identifies current obstacles toward the goal of understanding in quantitative detail the mechanism of synaptic transmission at glutamatergic synapses. Among the experimental limitations faced, we note that NMDA receptor kinetics vary considerably, though not fundamentally, between preparations. Perhaps recordings from cell-attached patches of intact cells are closest to native responses, given the observation that recordings from whole cells and excised patches both illustrate changes in kinetics related to the time of membrane rupture (62–65). Thus, without knowledge about the mechanisms responsible for these changes in kinetics, quantitative fits cannot be expected. Despite these limitations, the operational model developed in this study reproduced with useful accuracy the behaviors of individual receptor molecules and allowed important, testable predictions about Zn2+ effects on NMDA receptors.
In general, kinetic models developed so far for NMDA receptors have been useful in uncovering new aspects of the receptor mechanism precisely because they offer a general agreement between single-channel behaviors and macroscopic recordings (52,54–56,65,66). This advancement is yet to be accomplished for any other ionotropic glutamate receptors; however, even NMDA receptor models offer only coarse simplifications of the receptors' conformational landscapes and are far from representing all functional states known to be present in solution. To enumerate only the most obvious simplifications, the model ignores any influences from glycine and proton binding equilibria. This is an acceptable simplification only when using concentrations outside the physiologic range (in this study, 0.1 mM glycine, pH 8). However, in situ and under nonstationary conditions, these and other modulators will exert changes on the NMDA receptor response that the model does not anticipate. In particular, the mechanism by which zinc and protons collaborate to modify receptor gating and how glutamate dissociation from Zn2+-free receptors modulates the deactivation time course require further investigation.
In summary, we developed a kinetic model for zinc inhibition of NMDA receptor kinetics by observing the effects of zinc on NMDA receptor single-channel and macroscopic responses. The model explains zinc inhibition by postulating a free-energy landscape of zinc-bound receptors that has higher energy barriers to opening and higher-energy open sates. Our results have two important implications. First, they show that zinc-binding to the N-terminal domain has no effect on the receptor's microscopic desensitization kinetics, thus endorsing the view that the molecular determinants of receptor desensitization are separate and independent from conformations controlled by high-affinity zinc-binding. Second, our results suggest that synaptic zinc will strongly inhibit NMDA-receptor-mediated charge transfer only when zinc is present in the cleft before stimulation, perhaps due to slow clearance of zinc released by preceding stimuli. This may represent a mechanism for synaptic short-term depression of the NMDA receptor response and may explain some of the unique features of zinc-containing glutamatergic synapses.
Acknowledgments
S.A.A. recorded all whole-cell responses and most single-channel traces, and analyzed all data. S.E.M. recorded and analyzed patch responses, and T.P.S. contributed single-channel results. S.A.A. and G.K.P. designed the experiments, interpreted the results, and wrote the manuscript. We thank J. M. Myers and Y. Yang for sharing single-channel records and E. M. Kasperek for technical assistance with molecular biology and tissue culture.
This work was supported by NS0052669 (to G.K.P).
Supporting Material
References
- 1.Takeda A. Zinc homeostasis and functions of zinc in the brain. Biometals. 2001;14:343–351. doi: 10.1023/a:1012982123386. [DOI] [PubMed] [Google Scholar]
- 2.Sensi S.L., Paoletti P., Sekler I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009;10:780–791. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- 3.Frederickson C.J., Bush A.I. Synaptically released zinc: physiological functions and pathological effects. Biometals. 2001;14:353–366. doi: 10.1023/a:1012934207456. [DOI] [PubMed] [Google Scholar]
- 4.Smart T.G., Hosie A.M., Miller P.S. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- 5.Assaf S.Y., Chung S.H. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 6.Frederickson C.J., Suh S.W., Thompson R.B. Importance of zinc in the central nervous system: the zinc-containing neuron. J. Nutr. 2000;130(5S, Suppl):1471S–1483S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 7.Howell G.A., Welch M.G., Frederickson C.J. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 8.Paoletti P., Vergnano A.M., Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima A.S., Dyck R.H. Zinc and cortical plasticity. Brain Res. Brain Res. Rev. 2009;59:347–373. doi: 10.1016/j.brainresrev.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Besser L., Chorin E., Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009;29:2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogt K., Mellor J., Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 12.Forsythe I.D., Westbrook G.L., Mayer M.L. Modulation of excitatory synaptic transmission by glycine and zinc in cultures of mouse hippocampal neurons. J. Neurosci. 1988;8:3733–3741. doi: 10.1523/JNEUROSCI.08-10-03733.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters S., Koh J., Choi D.W. Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- 14.Koh J.Y., Choi D.W. Zinc alters excitatory amino acid neurotoxicity on cortical neurons. J. Neurosci. 1988;8:2164–2171. doi: 10.1523/JNEUROSCI.08-06-02164.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westbrook G.L., Mayer M.L. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 16.Mayer M.L., Vyklicky L., Jr. The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J. Physiol. 1989;415:351–365. doi: 10.1113/jphysiol.1989.sp017725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sindreu C.B., Varoqui H., Pérez-Clausell J. Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb. Cortex. 2003;13:823–829. doi: 10.1093/cercor/13.8.823. [DOI] [PubMed] [Google Scholar]
- 18.Laube B., Kuhse J., Betz H. Evidence for a tetrameric structure of recombinant NMDA receptors. J. Neurosci. 1998;18:2954–2961. doi: 10.1523/JNEUROSCI.18-08-02954.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa H., Singh S.K., Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 20.Béhé P., Stern P., Colquhoun D. Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc. Biol. Sci. 1995;262:205–213. doi: 10.1098/rspb.1995.0197. [DOI] [PubMed] [Google Scholar]
- 21.Moriyoshi K., Masu M., Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 22.Durand G.M., Gregor P., Zukin R.S. Cloning of an apparent splice variant of the rat N-methyl-D-aspartate receptor NMDAR1 with altered sensitivity to polyamines and activators of protein kinase C. Proc. Natl. Acad. Sci. USA. 1992;89:9359–9363. doi: 10.1073/pnas.89.19.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutsuwada T., Kashiwabuchi N., Mishina M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 24.Monyer H., Sprengel R., Seeburg P.H. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 25.Chen N., Moshaver A., Raymond L.A. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subtypes to zinc inhibition. Mol. Pharmacol. 1997;51:1015–1023. doi: 10.1124/mol.51.6.1015. [DOI] [PubMed] [Google Scholar]
- 26.Paoletti P., Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Williams K. Separating dual effects of zinc at recombinant N-methyl-D-aspartate receptors. Neurosci. Lett. 1996;215:9–12. doi: 10.1016/s0304-3940(96)12924-4. [DOI] [PubMed] [Google Scholar]
- 28.Paoletti P., Ascher P., Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wo Z.G., Oswald R.E. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995;18:161–168. doi: 10.1016/0166-2236(95)93895-5. [DOI] [PubMed] [Google Scholar]
- 30.Oswald R.E., Ahmed A., Loh A.P. Structure of glutamate receptors. Curr. Drug Targets. 2007;8:573–582. doi: 10.2174/138945007780618526. [DOI] [PubMed] [Google Scholar]
- 31.Sobolevsky A.I., Rosconi M.P., Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma G., Stevens C.F. A mutation that alters magnesium block of N-methyl-D-aspartate receptor channels. Proc. Natl. Acad. Sci. USA. 1996;93:9259–9263. doi: 10.1073/pnas.93.17.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burnashev N., Schoepfer R., Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- 34.Sakurada K., Masu M., Nakanishi S. Alteration of Ca2+ permeability and sensitivity to Mg2+ and channel blockers by a single amino acid substitution in the N-methyl-D-aspartate receptor. J. Biol. Chem. 1993;268:410–415. [PubMed] [Google Scholar]
- 35.Wollmuth L.P., Kuner T., Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. J. Physiol. 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawajiri S., Dingledine R. Multiple structural determinants of voltage-dependent magnesium block in recombinant NMDA receptors. Neuropharmacology. 1993;32:1203–1211. doi: 10.1016/0028-3908(93)90014-t. [DOI] [PubMed] [Google Scholar]
- 37.Wollmuth L.P., Kuner T., Sakmann B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+ J. Physiol. 1998;506:13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christine C.W., Choi D.W. Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. J. Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erreger K., Traynelis S.F. Zinc inhibition of rat NR1/NR2A N-methyl-D-aspartate receptors. J. Physiol. 2008;586:763–778. doi: 10.1113/jphysiol.2007.143941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madry C., Mesic I., Laube B. The N-terminal domains of both NR1 and NR2 subunits determine allosteric Zn2+ inhibition and glycine affinity of N-methyl-D-aspartate receptors. Mol. Pharmacol. 2007;72:1535–1544. doi: 10.1124/mol.107.040071. [DOI] [PubMed] [Google Scholar]
- 41.Rachline J., Perin-Dureau F., Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J. Neurosci. 2005;25:308–317. doi: 10.1523/JNEUROSCI.3967-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi Y.B., Lipton S.A. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- 43.Fayyazuddin A., Villarroel A., Neyton J. Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron. 2000;25:683–694. doi: 10.1016/s0896-6273(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 44.Low C.M., Zheng F., Traynelis S.F. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-D-aspartate NR1/NR2A receptors. Proc. Natl. Acad. Sci. USA. 2000;97:11062–11067. doi: 10.1073/pnas.180307497. (In Process Citation) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paoletti P., Perin-Dureau F., Neyton J. Molecular organization of a zinc binding n-terminal modulatory domain in a NMDA receptor subunit. Neuron. 2000;28:911–925. doi: 10.1016/s0896-6273(00)00163-x. [DOI] [PubMed] [Google Scholar]
- 46.Karakas E., Simorowski N., Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gielen M., Siegler Retchless B., Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng F., Erreger K., Traynelis S.F. Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat. Neurosci. 2001;4:894–901. doi: 10.1038/nn0901-894. [DOI] [PubMed] [Google Scholar]
- 49.Erreger K., Traynelis S.F. Allosteric interaction between zinc and glutamate binding domains on NR2A causes desensitization of NMDA receptors. J. Physiol. 2005;569:381–393. doi: 10.1113/jphysiol.2005.095497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kussius C.L., Kaur N., Popescu G.K. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J. Neurosci. 2009;29:6819–6827. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y., Auerbach A. Na+ occupancy and Mg2+ block of the n-methyl-d-aspartate receptor channel. J. Gen. Physiol. 2001;117:275–286. doi: 10.1085/jgp.117.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Popescu G., Auerbach A. The NMDA receptor gating machine: lessons from single channels. Neuroscientist. 2004;10:192–198. doi: 10.1177/1073858404263483. [DOI] [PubMed] [Google Scholar]
- 53.Amico-Ruvio S.A., Popescu G.K. Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys. J. 2010;98:1160–1169. doi: 10.1016/j.bpj.2009.12.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dravid S.M., Prakash A., Traynelis S.F. Activation of recombinant NR1/NR2C NMDA receptors. J. Physiol. 2008;586:4425–4439. doi: 10.1113/jphysiol.2008.158634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banke T.G., Traynelis S.F. Activation of NR1/NR2B NMDA receptors. Nat. Neurosci. 2003;6:144–152. doi: 10.1038/nn1000. [DOI] [PubMed] [Google Scholar]
- 56.Popescu G., Auerbach A. Modal gating of NMDA receptors and the shape of their synaptic response. Nat. Neurosci. 2003;6:476–483. doi: 10.1038/nn1044. [DOI] [PubMed] [Google Scholar]
- 57.Mayer M.L., Westbrook G.L., Vyklický L., Jr. Sites of antagonist action on N-methyl-D-aspartic acid receptors studied using fluctuation analysis and a rapid perfusion technique. J. Neurophysiol. 1988;60:645–663. doi: 10.1152/jn.1988.60.2.645. [DOI] [PubMed] [Google Scholar]
- 58.Legendre P., Westbrook G.L. The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. J. Physiol. 1990;429:429–449. doi: 10.1113/jphysiol.1990.sp018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Popescu G., Robert A., Auerbach A. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature. 2004;430:790–793. doi: 10.1038/nature02775. [DOI] [PubMed] [Google Scholar]
- 60.Clements J.D., Lester R.A., Westbrook G.L. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 61.Clements J.D. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 62.Lester R.A., Clements J.D., Jahr C.E. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- 63.Lester R.A., Jahr C.E. NMDA channel behavior depends on agonist affinity. J. Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Legendre P., Rosenmund C., Westbrook G.L. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J. Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang W., Howe J.R., Popescu G.K. Distinct gating modes determine the biphasic relaxation of NMDA receptor currents. Nat. Neurosci. 2008;11:1373–1375. doi: 10.1038/nn.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erreger K., Dravid S.M., Traynelis S.F. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.