

Abstract
The current need for high-throughput genotyping platforms for targeted validation of disease-associated single nucleotide polymorphisms (SNPs) motivated us to evaluate a novel nanofluidics platform for genotyping DNA extracted from peripheral blood and buccal wash samples. SNP genotyping was performed using a Fluidigm 48.48 Dynamic Array biochip on the BioMark polymerase chain reaction platform and results were compared against standard TaqMan assays and DNA sequencing. Pilot runs using these dynamic arrays on 90 samples against 20 SNP assays had an average call rate of 99.7%, with 100% call rates for 16 of the assays. Manual TaqMan genotyping of these samples against three SNPs demonstrated 100% correlation between the two platforms. To understand the influence of DNA template variability, three sources of blood samples (CH-1, n = 20; CH-2, n = 47; KK, n = 47) and buccal washes (n = 37) were genotyped for 24 SNPs. Although both CH-1 and CH-2 batches showed good base calling (≥98.8%), the KK batch and buccal wash samples exhibited lower call rates (82.1% and 94.0%). Importantly, repurification of the KK and buccal wash samples resulted in significant improvements in their call rates (to ≥97.9%). Scale-up for genotyping 1698 cases and controls for 24 SNPs had overall call rates of 97.6% for KK and 99.2% for CH samples. The Dynamic Array approach demonstrated accuracy similar to that of TaqMan genotyping, while offering significant savings in DNA, effort, time, and costs.
The HapMap and genome-wide association (GWA) projects have led to the identification of hundreds of single nucleotide polymorphisms (SNPs) that are associated with more than 80 disease states and traits.1–8 Although most of these SNPs have revealed novel risk loci or genes for particular diseases, they are mostly of moderate to low penetrance. Thus, the next stage of research will likely involve the validation and replication of smaller, relevant subsets of SNPs against targeted populations of interest; that is, smaller subsets of interesting SNPs will need to be genotyped against a large set of individuals.
Classical genotyping assays using gel electrophoresis-based procedures, restriction fragment length polymorphism (RFLP) analysis, and allele-specific PCR analyses are labor intensive and unsuitable for high-throughput approaches, whereas genotyping platforms such as the GeneChip (Affymetrix, Santa Clara, CA) and Hap300 (Illumina, San Diego, CA) are more appropriate for GWA studies involving hundreds to thousands of SNPs.9,10 Clearly, there is an immediate need for scalable and cost-effective platform technologies capable of analyzing flexible sample sizes on a specific set of SNPs and suitable for validation of SNPs identified from disease association studies. Two technologies, the SNPlex platform (Applied Biosystems, Foster City, CA) and the iPLEX platform (Sequenom, San Diego, CA)11,12 meet these criteria, but neither is based on the TaqMan assay,13 which is generally accepted as the gold standard for genotyping.
Recent advances in nanofluidics technology have made possible the use of integrated fluidic circuits (IFCs) for high-throughput real-time PCR.14 Nanoliter-scale quantities of samples and reagents are channeled into thousands of nanoliter-scale chambers in which distinct real-time PCRs can be run. Nanofluidic arrays have been successfully used in quantifying the absolute amounts of circulating fetal DNA in a background of maternal DNA by exploiting the compartmentalization feature of nanofluidic chips.15 Furthermore, single-molecule detection of epidermal growth receptor mutations in plasma has been achieved by partitioning DNA molecules in the nanofluidic chip,16 thereby allowing the fraction of two common epidermal growth receptor mutant alleles in plasma to be accurately quantified. Other applications of nanofluidic arrays include studies on copy number variation17 and accurate calibration of input DNA for next-generation sequencing.18
Recently, Fluidigm (South San Francisco, CA) has introduced a nanofluidics chip, the Dynamic Array chip, that is compatible with existing TaqMan genotyping assays.19 Potentially, this technology allows up to 9216 individual TaqMan reactions to be run in a single experiment, with the promise of considerable reagent and time savings achievable from using nanofluidics arrays, compared with standard TaqMan genotyping. Of note, the Dynamic Array has been successfully used by U.S. federal and Alaskan fishery organizations to genotype salmon samples for the purpose of fisheries management.20,21 Although one group has demonstrated the proof-of-concept of genotyping human samples from the PLCO Screening Trial22 and HapMap samples from cell lines19 using the Dynamic Array, their reports focused mainly on the description of chip design, and details of the DNA extraction method and its effects on data quality were not included.
Population genotyping typically uses genomic DNA from different resources, extracted using a variety of methods, including automated platforms, and often from limited amounts of clinical specimens (eg, peripheral blood, saliva, and buccal swabs or washes). Thus, it is imperative to understand the influence of various DNA extraction methods on the quality of the results, and also to establish procedures that allow for small amounts of DNA to be used. In the present study, we evaluated the performance of the Dynamic Array nanofluidics platform for SNP genotyping against standard TaqMan genotyping assays, by comparing overall call rates and concordance. The effect of DNA extraction methods, types of clinical specimens, and the use of archival frozen whole blood were also assessed.
Materials and Methods
Sample Collection
Archived frozen peripheral blood samples were obtained from the Singapore SingHealth Tissue Repository. Before DNA extraction, these blood samples had been stored at −80°C for up to 12 years. The CH-1 batch of samples comprised 111 blood samples from the SingHealth Tissue Repository, and the extracted DNA had been archived at −20°C for approximately 7 years. The CH-2 batch of blood samples (n = 179) were also obtained from the SingHealth Tissue Repository, and DNA from these samples was extracted within the last year. In addition, the CH-3 samples (peripheral blood, n = 134; buccal wash, n = 37) were collected from patients attending outpatient clinics at the National Cancer Centre; DNA from these samples was extracted within the last year. Written informed consent was obtained from all contributing volunteers, and ethics approval was obtained from the Centralised Institutional Review Board of SingHealth. The CH-1, CH-2, and CH-3 samples are collectively referred to as CH samples. The KK batch of samples (n = 1237) comprised purified DNA obtained from the DNA Diagnostic and Research Lab, KK Women's and Children's Hospital, Singapore.
DNA Extraction
The CH DNA samples were extracted using an optimized in-house method. Red blood cell lysis was performed by adding 9 volumes of buffer A (0.32 mol/L sucrose, 10 mmol/L Tris-HCl pH 7.5, 5 mmol/L MgCl2, 1% Triton X-100) to 5 mL of frozen or freshly drawn peripheral blood. The mixture was mixed well and centrifuged at 2095 × g for 20 minutes, after which the supernatant was discarded and the lysis step was repeated once. Buccal wash samples were directly centrifuged to pellet the buccal cells. Cell pellets containing lymphocytes or buccal cells were resuspended in 5 mL Buffer B (25 mmol/L EDTA, 75 mmol/L NaCl) and lysed with the addition of 250 μL of 10% SDS. Proteinase treatment was performed using 20 μL proteinase K (20 mg/mL), followed by incubation at 56°C for 24–48 hours with shaking. The DNA was precipitated using ethanol and sodium acetate and was washed with 70% ethanol. The dried DNA was dissolved in a TE buffer with reduced EDTA (10 mmol/L Tris, 1 mmol/L EDTA). Buccal wash samples were subsequently further purified by organic extraction using an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) (Sigma-Aldrich, St. Louis, MO) and were reprecipitated as described above.
The KK DNA samples were extracted from 300 μL of blood using the MagNA Pure compact system (Roche, Basel, Switzerland),23 a robotic system based on magnetic-bead technology, and were dissolved in a proprietary elution buffer. These DNA samples were subsequently purified using a column method (QIAamp blood mini kit; Qiagen, Hilden, Germany) according to a modification of the manufacturer's protocol. Briefly, DNA samples were reconstituted to 200 μL with PBS buffer, after which 200 μL of buffer AL (a proprietary buffer from the QIAamp kit) and 100% ethanol were added. The solution was then loaded onto the mini spin columns and was processed according to the manufacturer's protocol. The DNA was eluted with 30 μL of autoclaved reverse osmosis water.
Genotyping Using the TaqMan Real-Time PCR Assay
Real-time PCR with the TaqMan assay (Applied Biosystems) was performed according to the manufacturer's instructions. Briefly, 5-μL reactions were run, comprising 2.5 μL of TaqMan universal genotyping master mix, 0.25 μL of TaqMan 20× SNP assay, 1.125 μL autoclaved reverse osmosis water, and 1.125 μL DNA (5 ng/μL) per reaction. Each run included non-template controls (NTC). The real-time PCR reactions were run using a 7500 Fast Real-Time PCR system (Applied Biosystems). The SNPs that were genotyped to validate the Dynamic Array results were rs2981582 (SNP1; Assay ID: C_2917302_10), rs3803662 (SNP2; Assay ID: C_25968567_10), and rs3817198 (SNP10; Assay ID: C_27493923_10).
Genotyping Using the 48.48 Dynamic Array
The 48.48 Dynamic Array used in the present study is able to analyze 48 samples with 48 assays on the BioMark platform (Fluidigm). The array is mounted on a plastic interface containing 48 sample and 48 assay inlets on the left and right of the array. In the present study, each array was loaded with 47 samples and one non-template control. Twenty-four SNP assays were loaded in duplicate into the 48 assay inlets. The array contains a network of fluid lines (integrated fluidic circuit, IFC) and chambers that are controlled by elastomeric valves. These valves deflect under pressure to create a tight seal, thereby regulating the flow of liquids into the IFC. Before reagents are loaded, the array is primed using the IFC Controller MX, which pressurizes the control lines and closes the interface valves.
The same genotyping assay reagents and enzyme master mixes used for conventional genotyping were used for the nanofluidics array. Each assay (5 μL) comprised 2.5 μL DA assay loading reagent (2×) (Fluidigm), 1.25 μL SNP genotyping assay mix (40×) (Applied Biosystems), 0.25 μL ROX (50×) (Invitrogen, Carlsbad, CA), and 1 μL autoclaved reverse osmosis water. Each sample (5 μL) comprised 2.5 μL TaqMan genotyping master mix (2×), 0.05 μL AmpliTaq Gold DNA polymerase, 0.25 μL GT sample loading reagent (20×) (Fluidigm), 0.1 μL autoclaved reverse osmosis water, and 2.1 μL genomic DNA. Of note, during the course of experimentation we found it imperative to set up assays fresh, because freezing and thawing premixed assays appeared to have a detrimental effect on the quality of the results (data not shown). Each of the assays (4 μL) and samples (5 μL) was pipetted into separate inlets on the frame of the chip according to the manufacturer's instructions. The assays and samples were loaded into the reaction chambers and mixed using the IFC Controller MX. Arrays should be run immediately after assays, with samples pipetted into wells. The arrays were processed using the BioMark system (Fluidigm), which performs the thermal cycling and fluorescent image acquisition.
The data were analyzed using the BioMark SNP Genotyping Analysis software version 2.1.1, to obtain genotype calls. Briefly, the software calculates the FAM and VIC relative fluorescence intensities (relative to ROX background), and then automatically classifies samples into three genotypes and NTCs using a k-means clustering algorithm. In a typical computer-generated image of the data obtained, each row represents data from one DNA sample loaded from each sample inlet, with data assigned using four color codes (one for each genotype and black for NTC).
DNA Sequencing
To confirm the genotyping results from both the standard TaqMan assay and the 48.48 Dynamic Array, PCR followed by DNA sequencing was performed on 10% of the samples used in the pilot run of two chips (90 samples). DNA sequence confirmation was performed on 9 of the 90 samples against 3 SNPs (ie, 27 out of 270 data points). The targeted loci were PCR-amplified from DNA samples and were sequenced using BigDye chemistry (Applied Biosystems) on a 3130xl DNA analyzer (Applied Biosystems) according to manufacturer's instructions, as described previously.24
Results
Comparison of SNP Genotyping by Dynamic Array, TaqMan Real-Time PCR, and DNA Sequencing
Genotyping of 90 samples (from the CH-2 batch) for 20 SNPs was performed on two 48.48 Dynamic Array chips, with each SNP assay run in duplicate. Thus, a total of 3600 data points were collected (90 × 20 × 2), corresponding to 1800 genotyping calls. These runs resulted in an average call rate of 99.7%, with call rates of 100% in most of the assays (16 of 20), 99.4% in 3 of 20 assays, and 97.2% in 1 of 20 assays (Figure 1A).
Figure 1.
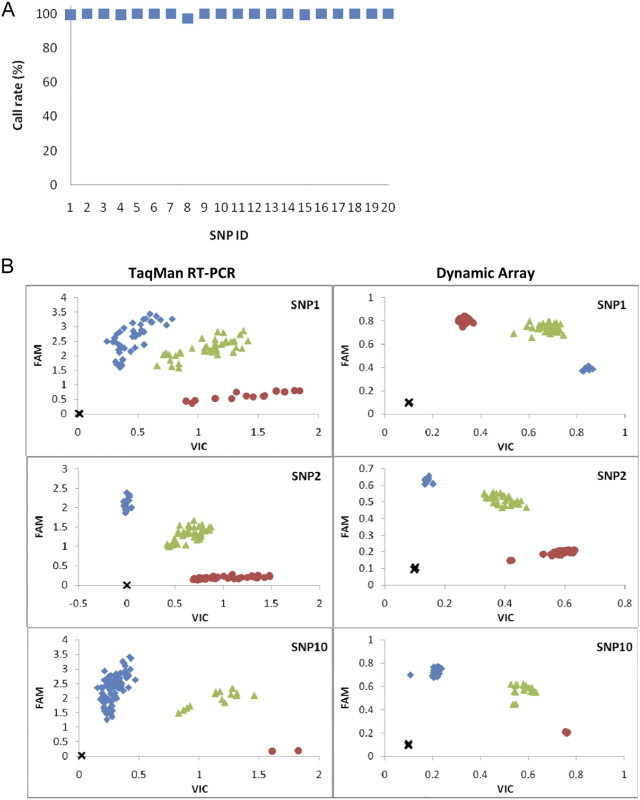
A: Plot of call rates (average of duplicate wells from two Dynamic Array chips) for 90 samples (from CH-2) against 20 SNP assays. B: Comparison of the allelic discrimination plots between the standard TaqMan Assay and the Dynamic Array for one run of CH-2 samples (47 samples in duplicate).
The same set of 90 samples used on the arrays was also genotyped manually using TaqMan real-time PCR for three SNPs. The three SNPs used were SNPs 1, 2, and 10, which had 100% call rates on the Dynamic Arrays. The results between the two genotyping approaches were 100% consistent, demonstrating a high degree of accuracy for the genotyping approach established for the Dynamic Arrays. Both experimental systems showed similar clustering patterns in allelic discrimination plots generated by the respective customized analysis software package (Figure 1B).
To further confirm the accuracy of the genotyping data, PCR followed by DNA sequencing was performed on 9 of the same 90 samples against SNPs 1, 2, and 10. As expected, the DNA sequencing results were also 100% consistent with the TaqMan and Dynamic Array results, confirming the accuracy of the genotyping approaches.
Effect of DNA Extraction Method, Type of Sample, and Long-Term Storage of DNA and Blood on Genotyping Call Rate
We next sought to investigate the influence of different DNA resources on the outcome of the established nanofluidics genotyping approach. SNP genotyping of CH-1 samples (n = 20), comprising DNA samples that had been stored for approximately 7 years, and CH-2 (n = 47) samples, which had been stored for ≤1 year, showed good base calling results (average call rates of 99.8% and 98.8%, respectively, for 24 SNPs), but the success rates for the KK samples (n = 47) and buccal wash samples (n = 37) were significantly lower (average call rates of 82.1% and 94.0%, respectively) (Figure 2), suggesting that the KK and buccal wash samples cannot be used directly, but require further purification.
Figure 2.
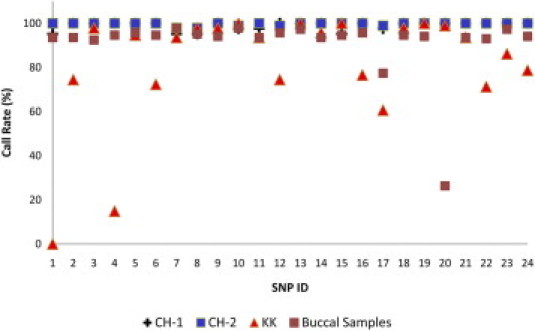
Plot of call rates for samples from CH-1 (n = 20), CH-2 (n = 47), and KK (n = 47) against 24 SNP assays.
To understand the cause of lower success rates associated with the KK DNA samples, which had been extracted from peripheral blood using the automated MagNA Pure compact system, these samples were purified by column purification and compared against those before column purification, for 24 SNPs run in duplicate. In the resultant call map (Figure 3), the columns correspond to 24 SNP assays loaded in duplicate, and the rows correspond to DNA samples and one NTC. In sample groups A, B, and C, unpurified KK samples tested at 100, 50, and 25 ng/μL demonstrated low call rates (47.5% to 77.5%). In contrast, group E, corresponding to column-purified samples, had average call rates of ≥97.9%, showing significant improvement with column purification.
Figure 3.
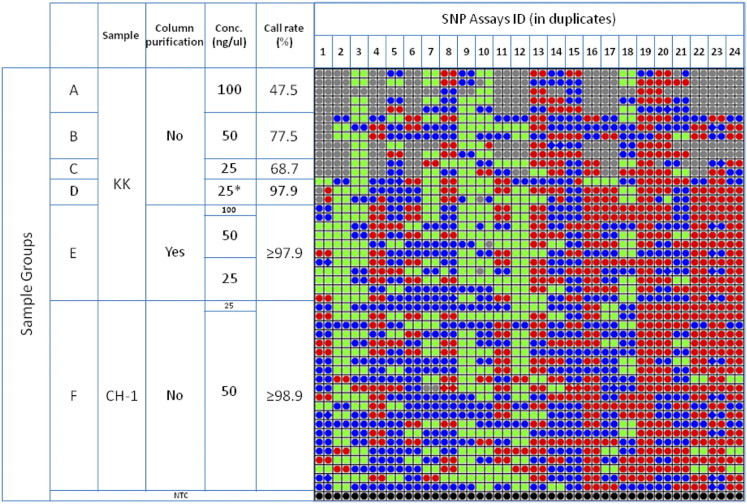
Call map from a 48.48 Dynamic Array using non-column-purified KK samples, column-purified KK samples, and CH-1 samples, run at working concentrations as indicated. The columns correspond to 24 SNP assays loaded in duplicate and the rows correspond to DNA samples (47 samples) and one non-template control (NTC). The red, blue, green, and gray dots correspond to FAM, FAM+VIC (ie, heterozygote), VIC, and no call, respectively. The concentration for sample group D, marked with an asterisk, was derived by dilution from non-column-purified DNA samples at 100 ng/μL.
Furthermore, a fourfold dilution of non-column-purified samples to 25 ng/μL (group D) showed significantly higher call rate (97.9%), compared with 68.7% for samples that were originally at 25 ng/μL (group C). This suggests that the low call rate for group C was not due to insufficient template, but more likely was caused by inhibitory compounds present in the unpurified samples. For comparison purposes, CH-1 samples (group F) were assayed on the same chip. Twenty of these samples had a standardized final concentration of 50 ng/μL, and one sample was used at 25 μg/μL, for confirming the minimum usable concentration. As expected, group F showed high overall call rates (≥98.9%). These observations suggested that inhibitory compounds might be present in the KK samples, which could be removed either by column repurification or by dilution. It should be noted that there were several discordant duplicates in Group D, suggesting that column purification should be the method of choice for processing the KK samples.
The success rates for buccal wash samples could be improved significantly, from 94.0% to 98.7%, by the inclusion of a phenol-chloroform extraction step, followed by ethanol reprecipitation of DNA. Phenol-chloroform extraction was chosen as the approach for repurification, instead of using spin-columns, because of the presence of particulate impurities that could clog the spin columns.
Large-Scale SNP Genotyping Using the Dynamic Array
Scale-up genotyping of 1698 subjects was performed with 461 CH samples (CH-1, CH-2, and CH-3 DNA samples) and 1237 samples from the KK batch (Table 1). On scale-up, one SNP for CH samples (SNP 20) and two for KK samples (SNPs 17 and 20) had failure rates of >19% and were excluded from analysis. Exclusion of a small fraction of SNP assays is unavoidable, given that all assays were amplified together, and optimization of thermal cycling conditions for individual SNP assays was not possible. The total sample size of 1698 was derived after the exclusion of 10 samples showing >58% failure across all 24 SNPs. The call rates obtained for both KK samples (97.6 ± 1.93%) and CH samples (99.2 ± 0.60%) were largely similar to those calculated for the validation phase. Although KK samples had been repurified for the Dynamic Array, on scale-up the KK samples still demonstrated slightly lower call rates (97.6 ± 1.93% versus 99.2 ± 0.60%) and slightly lower reproducibility between replicates (96.3 ± 1.90% versus 99.4 ± 0.77%), compared with the CH samples. The call and reproducibility rates indicated are expressed as means ± SD. This difference was statistically significant, with P values of <0.005 for both call and reproducibility rates obtained from standard two-tailed Student's t-tests (paired). Within the CH samples, there was no observable difference in data quality among the CH-1, CH-2 or CH-3 samples, or between buccal wash and peripheral blood samples.
Table 1.
Call Rates and Reproducibility of the Dynamic Array for Scale-up Genotyping of 1698 Samples against 24 SNPs
| SNP | CH samples (n = 461) |
KK samples (n = 1237) |
||
|---|---|---|---|---|
| Call rate (%) | Reproducibility (%) | Call rate (%) | Reproducibility (%) | |
| Avg | 99.2 | 99.4 | 97.6 | 96.3 |
| 1 | 100 | 99.8 | 99.7 | 96.2 |
| 2 | 99.7 | 99.1 | 99.2 | 94.3 |
| 3 | 98 | 98.5 | 89.5 | 95.3 |
| 4 | 99.8 | 100 | 96.4 | 97.3 |
| 5 | 99.1 | 98.7 | 99.4 | 98.5 |
| 6 | 100 | 100 | 99.3 | 95.5 |
| 7 | 100 | 100 | 99.3 | 97.6 |
| 8 | 99.9 | 98.9 | 95.8 | 95.6 |
| 9 | 99.7 | 99.3 | 97.9 | 94.7 |
| 10 | 100 | 100 | 99.6 | 98.1 |
| 11 | 99 | 99.8 | 98.6 | 94 |
| 12 | 99.6 | 99.1 | 97.9 | 91.5 |
| 13 | 98.2 | 99.3 | 99.2 | 96 |
| 14 | 99.7 | 99.6 | 99.4 | 97.5 |
| 15 | 99.9 | 99.8 | 95.3 | 97.6 |
| 16 | 99.7 | 99.6 | 99.5 | 95.7 |
| 17 | 94 | 98.9 | (76.7)⁎ | (93.8)⁎ |
| 18 | 99.7 | 99.3 | 99.7 | 98.4 |
| 19 | 98.9 | 99.6 | 94.7 | 97.7 |
| 20 | (31.8)⁎ | (97.6)⁎ | (6.2)⁎ | (94.1)⁎ |
| 21 | 99.9 | 99.8 | 99.2 | 96.8 |
| 22 | 98 | 99.3 | 93.5 | 97 |
| 23 | 99.8 | 97.6 | 97.3 | 96.7 |
| 24 | 98 | 99.6 | 96.8 | 97.1 |
The performance indicators used to evaluate the genotyping platforms were defined according to the following formulae: i) Call rate (%) = (No. of successful base calls)/(Total no. of assays performed) × 100 and ii) Reproducibility between replicates (%) = {1 − [(No. of assays inconsistent between replicates)/(Total no. of assays)]} × 100.
Values in parentheses were excluded from the overall rate computations as they had failure rates of >19%.
To further study the concordance between the Dynamic Array and TaqMan real-time PCR platforms on scale-up, a representative number of samples (n = 261 for CH samples, comprising of 111 CH-1 and 150 CH-2 samples; and n = 292 for KK samples) were genotyped on the TaqMan real-time PCR platform for SNPs 1, 2, and 10. The concordance between the two platforms were calculated as the percentage of assays with consistent allelic discrimination divided by the total number of samples tested. The concordance between the Dynamic Array and TaqMan real-time PCR platforms was 99.8% for CH samples and 97.7% for KK samples. Examination of the individual data points on both platforms suggested that these discordant calls were high-quality calls on both platforms. The precise explanation for this difference in concordance between the CH and KK samples is as yet unclear and may require further evaluation.
Discussion
For the evaluation of disease-associated SNPs identified from GWA studies, there is a need for SNP genotyping methodologies that are capable of medium- to high-throughput genotyping of hundreds to thousands of patient samples. We therefore assessed the performance of a nanofluidics approach using the Dynamic Array, compared with TaqMan real-time PCR analysis. We also determined whether DNA extracted using automated or manual methods, or DNA from different sources (fresh versus frozen blood samples, or blood versus buccal wash samples), was amenable to SNP genotyping using the Dynamic Array.
Concordance rates of 99% to 100% between results from the Dynamic Array and TaqMan real-time PCR have been reported from Fluidigm studies.19 We observed 100% concordance between these two platforms for 90 CH-2 samples, and 99.8% for 261 CH samples. Thus, the concordance reported earlier was achievable in a clinical setting. For the KK batch of samples, however, the discordance between the two platforms was slightly more significant, even though the performance of these samples in terms of call rates and reproducibility between duplicates were acceptable (>95%). This difference in concordance was evident only on scale-up testing on the Dynamic Array, which suggests that concordance may vary widely for DNA from different sources. Careful validation of a new source of DNA therefore remains vital to obtaining accurate, useful data.
Our observations clearly suggested that the type of extraction method, in particular the solution in which the DNA is dissolved, exerts a significant influence on the quality of results from the Dynamic Arrays. Problems with samples extracted using the MagNA Pure compact system were observed. In the case of the KK samples, the elution buffer from the MagNA Pure compact system may contain components that interfere with the TaqMan reactions in the nanofluidic chip. Another possibility is that impurities may not have been completely removed by the automated wash procedures. This observation has significant relevance for clinical researchers, because the MagNA Pure system is one of the most common automated platforms used in hospital laboratories for high-throughput DNA extraction for clinical studies.25–27 We subsequently significantly improved call rates on the Dynamic Array by using column purification. Although repurification of the KK samples improved call rates significantly, on scale-up there was still a slightly lower performance, compared with the CH samples extracted by an in-house method (97.6% versus 99.2%). The presence of some residual inhibitory factors in some samples even after repurification may have led to the slightly lower performance observed for the KK samples. Specific target amplification has been proposed for improving call rates of problematic samples on the Dynamic Array.19 We have not found specific target amplification useful, however, because almost all of these samples can in fact be successfully genotyped using the TaqMan assays (data not shown).
Although the Dynamic Array has been reported to be sensitive to variations in DNA concentration,19 in our hands it worked well with DNA concentrations typically obtained for clinical research (approximately 25 to >100 ng/μL). Call rates of 98% to 99% and reproducibility of 99.7% between duplicates have been estimated previously.19 In contrast, we observed very similar performance for the CH samples (≥99.2% call and reproducibility rates), but statistically significant lower performance for the KK samples (97.6% and 96.3%). Although the previously reported reproducibility was derived only from 130 samples, in the present study duplicates were run for all assays, thereby providing a more realistic estimate. The quality or method of the DNA extraction is an important factor for genotyping experiments using the Dynamic Array. We observed similar call rates among the diverse types of samples used for DNA extraction, suggesting that fresh or frozen archival blood samples, and blood or buccal samples, were invariably suitable for use on the Dynamic Array.
Finally, a comparison of performance indicators and consumption of reagents revealed significant savings in effort, time, and reagent costs for the Dynamic Array, compared with the standard TaqMan assay, while achieving the same accuracy as the TaqMan assay (Table 2). For example, a 48.48 Dynamic Array run from priming of chip to acquisition of data typically takes approximately 4 hours and 96 pipetting steps (48 assays and sample inlets). Thus, a complete analysis of 1400 samples in duplicate (approximated to 30 chip runs) will take 120 hours and 2880 pipetting steps. Using the standard TaqMan assay, however, it would take 720 runs (30 runs per SNP × 24 SNPs) and almost 2900 hours to collect the same amount of data. The fold-savings in reagents achievable were similar with the corresponding values provided by the manufacturer (Fluidigm application note: SNP Genotype Profiling. South San Francisco, CA). This may translate to cost savings of >100,000 U.S. dollars. The iPLEX system from Sequenom was calculated to be four times cheaper than TaqMan technology.28 Notably, the Dynamic Array requires almost five times less DNA, compared with TaqMan real-time PCR, an important consideration when working with limited amounts of clinical material.
Table 2.
Comparison of Performance of the Dynamic Array and Standard TaqMan Assays (in 96-Well Formats) for Genotyping 1400 Samples against 24 SNPs Performed in Duplicate
| Dynamic array | TaqMan | Savings | |
|---|---|---|---|
| Call rates (%) | ≥98% | ≥98% | — |
| Time | 30 runs (120 hours) | 720 runs (2880 hours) | 24-fold |
| Pipetting steps | (48 + 48) × 30 = 2880 | 276 480 | 96-fold |
| DNA per sample | 50 ng | 240 ng | 4.8-fold |
| Master mix | 3.6 mL | 180 mL | 50-fold |
| Assay reagent | 1.8 mL | 9 mL | 5-fold |
Nonetheless, certain limitations of the Dynamic Array exist, in particular the inability to run specific real-time PCR conditions for individual SNPs, and the need for specialized hardware. Nevertheless, the accuracy, efficiency, and cost savings in time, reagents, and DNA for a nanofluidics Dynamic Array make it suitable for medium- to high-throughput genotyping against targeted SNPs, and this approach should offer accuracy similar to that of the gold standard, the TaqMan assay, for disease association29 and other pharmacological studies using patient samples.30
Acknowledgments
We are grateful to the staff of Singapore Institute for Clinical Sciences for the use of their BioMark equipment, and to the SingHealth Tissue Repository for providing blood samples.
Footnotes
Supported in part by a grant from the National Medical Research Council of Singapore (NMRC/1194/2008).
References
- 1.International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 2.Nischwitz S., Cepok S., Kroner A., Wolf C., Knop M., Müller-Sarnowski F., Pfister H., Roeske D., Rieckmann P., Hemmer B., Ising M., Uhr M., Bettecken T., Holsboer F., Müller-Myhsok B., Weber F. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J Neuroimmunol. 2010;227:162–166. doi: 10.1016/j.jneuroim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Dick D.M., Aliev F., Krueger R.F., Edwards A., Agrawal A., Lynskey M., Lin P., Schuckit M., Hesselbrock V., Nurnberger J., Jr, Almasy L., Projesz B., Edenberg H.J., Bucholz K., Kramer J., Kuperman S., Bierut L. Genome-wide association study of conduct disorder symptomatology. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D., Thompson D., Ballinger D.G. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F., Lowe ce, Szeszko J.S., Hafler J.P., Zeitels L., Yang J.H., Vella A., Nutland S., Stevens H.E., Schuilenburg H., Coleman G., Maisuria M., Meadows W., Smink L.J., Healy B., Burren O.S., Lam A.A., Ovington N.R., Allen J., Adlem E., Leung H.T., Wallace C., Howson J.M., Guja C., Ionescu-Tîrgovişte C., Simmonds M.J., Heward J.M., Gough S.C., Dunger D.B., Wicker L.S., Clayton D.G., Genetics of Type 1 Diabetes in Finland. Wellcome Trust Case Control Consortium Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A., Styrkarsdottir U., Gretarsdottir S., Thorlacius S., Jonsdottir I., Jonsdottir T., Olafsdottir E.J., Olafsdottir G.H., Jonsson T., Jonsson F., Borch-Johnsen K., Hansen T., Andersen G., Jorgensen T., Lauritzen T., Aben K.K., Verbeek A.L., Roeleveld N., Kampman E., Yanek L.R., Becker L.C., Tryggvadottir L., Rafnar T., Becker D.M., Gulcher J., Kiemeney L.A., Pedersen O., Kong A., Thorsteinsdottir U., Stefansson K. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 8.Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long J., Cai Q., Shu X.O., Qu S., Li C., Zheng Y., Gu K., Wang W., Xiang Y.B., Cheng J., Chen K., Zhang L., Zheng H., Shen C.Y., Huang C.S., Hou M.F., Shen H., Hu Z., Wang F., Deming S.L., Kelley M.C., Shrubsole M.J., Khoo U.S., Chan K.Y., Chan S.Y., Haiman C.A., Henderson B.E., Le Marchand L., Iwasaki M., Kasuga Y., Tsugane S., Matsuo K., Tajima K., Iwata H., Huang B., Shi J., Li G., Wen W., Gao Y.T., Lu W., Zheng W. Identification of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium. PLoS Genet. 2010;6:e1001002. doi: 10.1371/journal.pgen.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helgadottir A., Thorleifsson G., Manolescu A., Gretarsdottir S., Blondal T., Jonasdottir A., Jonasdottir A., Sigurdsson A., Baker A., Palsson A., Masson G., Gudbjartsson D.F., Magnusson K.P., Andersen K., Levey A.I., Backman V.M., Matthiasdottir S., Jonsdottir T., Palsson S., Einarsdottir H., Gunnarsdottir S., Gylfason A., Vaccarino V., Hooper W.C., Reilly M.P., Granger C.B., Austin H., Rader D.J., Shah S.H., Quyyumi A.A., Gulcher J.R., Thorgeirsson G., Thorsteinsdottir U., Kong A., Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 11.Tobler A.R., Short S., Andersen M.R., Paner T.M., Briggs J.C., Lambert S.M., Wu P.P., Wang Y., Spoonde A.Y., Koehler R.T., Peyret N., Chen C., Broomer A.J., Ridzon D.A., Zhou H., Hoo B.S., Hayashibara K.C., Leong L.N., Ma C.N., Rosenblum B.B., Day J.P., Ziegle J.S., De La Vega F.M., Rhodes M.D., Hennessy K.M., Wenz H.M. The SNPlex genotyping system: a flexible and scalable platform for SNP genotyping. J Biomol Tech. 2005;16:398–406. [PMC free article] [PubMed] [Google Scholar]
- 12.Long J., Shu X.O., Cai Q., Gao Y.T., Zheng Y., Li G., Li C., Gu K., Wen W., Xiang Y.B., Lu W., Zheng W. Evaluation of breast cancer susceptibility loci in Chinese women. Cancer Epidemiol Biomarkers Prev. 2010;19:2357–2365. doi: 10.1158/1055-9965.EPI-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak K.J., Marmaro J., Todd J.A. Towards fully automated genome-wide polymorphism screening. Nat Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- 14.Spurgeon S.L., Jones R.C., Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic Dynamic Array. PLoS One. 2008;3:e1662. doi: 10.1371/journal.pone.0001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lun F.M., Chiu R.W., Chan K.C., Leung T.Y., Lau T.K., Lo Y.M. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–1672. doi: 10.1373/clinchem.2008.111385. [DOI] [PubMed] [Google Scholar]
- 16.Yung T.K., Chan K.C., Mok T.S., Tong J., To K.F., Lo Y.M. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15:2076–2084. doi: 10.1158/1078-0432.CCR-08-2622. [DOI] [PubMed] [Google Scholar]
- 17.Bhat S., Herrmann J., Armishaw P., Corbisier P., Emslie K.R. Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal Bioanal Chem. 2009;394:457–467. doi: 10.1007/s00216-009-2729-5. [DOI] [PubMed] [Google Scholar]
- 18.White R.A., Blainey P.C., Fan H.C., Quake S.R. Digital PCR provides sensitive and absolute calibration for high throughput sequencing. BMC Genomics. 2009;10:116. doi: 10.1186/1471-2164-10-116. [Erratum appeared in BMC Genomics 2009, 10:541] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J., Lin M., Crenshaw A., Hutchinson A., Hicks B., Yeager M., Berndt S., Huang W., Hayes R.B., Chanock S.J., Jones R.C., Ramakrishnan R. High-throughput single nucleotide polymorphism genotyping using nanofluidic Dynamic Arrays. BMC Genomics. 2009;10:561. doi: 10.1186/1471-2164-10-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narum S, Campbell N, Yi Y: High-throughput SNP genotyping in salmon & steelhead (abstract). In Proceedings of Plant and Animal Genomes XVII Conference. 2009January 10-14, San Diego, CA. P578
- 21.Habicht C., Templin W.D., Willette T.M., Fair L.F., Raborn S.W., Seeb L.W. Fishery Manuscript No. 07–07: Anchorage, Alaska Department of Fish and Game. 2007. Post-season stock composition analysis of Upper Cook Inlet sockeye salmon harvest, 2005–2007. [Google Scholar]
- 22.Andriole G.L., Reding D., Hayes R.B., Prorok P.C., Gohagan J.K. The prostate, lung, colon, and ovarian (PLCO) cancer screening trial: status and promise. Urol Oncol. 2004;22:358–361. doi: 10.1016/j.urolonc.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Wittor H., Aschenbrenner A., Thoenes U., Schnittger S., Leying H. Fully automated sample preparation: use for the detection of BCR-ABL fusion transcripts. Biochemica. 2000;3:5–8. [Google Scholar]
- 24.Ong D.C., Ho Y.M., Rudduck C., Chin K., Kuo W.L., Lie D.K., Chua C.L., Tan P.H., Eu K.W., Seow-Choen F., Wong C.Y., Hong G.S., Gray J.W., Lee A.S. LARG at chromosome 11q23 has functional characteristics of a tumor suppressor in human breast and colorectal cancer. Oncogene. 2009;28:4189–4200. doi: 10.1038/onc.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haug K.B., Sharikabad M.N., Kringen M.K., Narum S., Sjaatil S.T., Johansen P.W., Kierulf P., Seljeflot I., Arnesen H., Brørs O. Warfarin dose and INR related to genotypes of CYP2C9 and VKORC1 in patients with myocardial infarction. Thromb J. 2008;6:7. doi: 10.1186/1477-9560-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qutub M.O., Germer J.J., Rebers S.P., Mandrekar J.N., Beld M.G., Yao J.D. Simplified PCR protocols for INNO-LiPA HBV Genotyping and INNO-LiPA HBV PreCore assays. J Clin Virol. 2006;37:218–221. doi: 10.1016/j.jcv.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Bodlaj G., Stöcher M., Hufnagl P., Hubmann R., Biesenbach G., Stekel H., Berg J. Genotyping of the lactase-phlorizin hydrolase −13910 polymorphism by LightCycler PCR and implications for the diagnosis of lactose intolerance. Clin Chem. 2006;52:148–151. doi: 10.1373/clinchem.2005.057240. [DOI] [PubMed] [Google Scholar]
- 28.Schaeffeler E., Zanger U.M., Eichelbaum M., Asante-Poku S., Shin J., Schwab M. Highly multiplexed genotyping of thiopurine S-methyltransferase variants using MALDI-TOF mass spectrometry: reliable genotyping in different ethnic groups. Clin Chem. 2008;54:1637–1647. doi: 10.1373/clinchem.2008.103457. [DOI] [PubMed] [Google Scholar]
- 29.Sun N., Sun X., Chen B., Cheng H., Feng J., Cheng L., Lu Z. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;65:437–446. doi: 10.1007/s00280-009-1046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouguieg K., Picard N., Sauvage F.L., Gaulier J.M., Marquet P. Contribution of the different UDP-glucuronosyltransferase (UGT) isoforms to buprenorphine and norbuprenorphine metabolism and relationship with the main UGT polymorphisms in a bank of human liver microsomes. Drug Metab Dispos. 2010;38:40–45. doi: 10.1124/dmd.109.029546. [DOI] [PubMed] [Google Scholar]