

Abstract
The G-protein coupled receptor associated protein β-arrestin-1 is crucial for the regulation of numerous biological processes involved in cancer progression, such as intracellular signaling and cell motility. The encoding gene ARRB1 is harbored in the same chromosomal region as the CCND1 gene (11q13). Amplification of CCND1, frequently encountered in breast cancer, often involves coamplification of additional oncogenes, as well as deletion of distal 11q genes. We investigated the clinical relevance of β-arrestin-1 in breast cancer and elucidated a potential link between β-arrestin-1 expression and CCND1 amplification. β-Arrestin-1 protein expression was evaluated in two breast cancer patient cohorts, comprising 179 patients (cohort I) and 500 patients randomized to either tamoxifen or no adjuvant treatment (cohort II). Additionally, migration after β-arrestin-1 overexpression or silencing was monitored in two breast cancer cell lines. Overexpression of β-arrestin-1 reduced the migratory propensity of both cell lines, whereas silencing increased migration. In cohort I, high expression of stromal β-arrestin-1 was linked to reduced patient survival, whereas in cohort II both high and absent stromal expression predicted a poor clinical outcome. Patients exhibiting low or moderate levels of stromal β-arrestin-1 did not benefit from tamoxifen, in contrast to patients exhibiting absent or high expression. Furthermore, CCND1 amplification was inversely correlated with tumor cell expression of β-arrestin-1, indicating ARRB1 gene deletion in CCND1-amplified breast cancers.
β-arrestin-1 belongs to a small family comprised of four members, designated arrestin 1 to 4. Arrestins 1 and 4 are exclusively expressed in retinal photoreceptors, but arrestins 2 and 3 (termed β-arrestin-1 and -2, respectively) are expressed in virtually all tissues.1 After their activation and phosphorylation by G-protein-coupled receptor kinases,2 the versatile β-arrestin adaptor proteins function in regulation of signaling and trafficking of G-protein-coupled receptors. The binding of β-arrestin sterically prevents further G-protein signaling and hence desensitizes the receptor.3 In recent years, the novel role of β-arrestins in signaling has been extensively studied, and their function as scaffold proteins interacting with a number of different signaling molecules has emerged.1,4,5 Of note, as G-protein signaling is turned off, a second set of signals can be initiated. Signaling pathways reported to be modulated by β-arrestins include the TGF-β, IGF-1R, PI3K, and MAPK pathways.2,6
A number of studies have investigated the role of the β-arrestins in cancer. In colorectal cancer, the interaction between β-arrestin-1 and proto-oncogene SRC (alias c-Src) has been shown to be critical for cell migration in vitro and also for metastatic spread to the liver in vivo.7 Zou et al6 demonstrated that murine liver cancer and lymphoma cells inoculated into β-arrestin-1 transgenic mice formed tumors more rapidly than in either β-arrestin-2 transgenic or wild-type mice. Furthermore, migration of the invasive breast cancer cell line MDA-MB-231 has been shown to be reduced after knockdown of the β-arrestins.8,9 β-arrestin-2 has also been implicated in cell death in MCF-7 and MDA-MB-231 cells, by blocking morphine-induced cell death through proapoptotic caspase-8 pathways.10 In a study analyzing expression of the β-arrestins in 48 breast cancer samples, mRNA levels of β-arrestin-2 were elevated in advanced breast cancers.9 To date, however, no previous studies have described the importance of β-arrestin-1 protein expression in a clinical setting and including analysis of a large number of breast cancer cases.
The ARRB1 gene coding for the β-arrestin-1 protein maps to chromosomal locus 11q13,11 a region that is frequently amplified in certain human cancers, including lung, bladder, breast, and ovarian carcinomas.2,7,12 The well-characterized gene CCND1 is also harbored in the 11q13 region, and its amplification has been associated with worse clinical outcome in several cancers.13,14 Jirström et al15 reported that patients with CCND1-amplified breast cancers were subject to an increased risk of disease recurrence after treatment with the selective estrogen receptor modulator tamoxifen, compared with patients with nonamplified tumors. PAK1, another gene located at 11q13, has been reported to be coamplified with CCND1,16 and overexpression of PAK1 protein is associated with impaired tamoxifen response in premenopausal breast cancer patients, despite the presence of estrogen receptor (ERα) expression.17 The ARRB1 gene is located between the CCND1 and the PAK1 genes, suggesting that a coamplification of these two genes may also include ARRB1.18 Moreover, loss of heterozygosity at the distal region of chromosome 11q, including genes such as CHEK1 (alias CHK1) (11q24), has also been associated with 11q13 amplification.19 We have previously reported loss of CHEK1 protein to be a predictor of tamoxifen response in premenopausal breast cancer, abolishing the response seen in patients presenting with tumors that exhibit normal levels of CHEK1.20 Based on these data, we hypothesized that ARRB1 may be a candidate gene affected by the amplification/deletion event occurring at chromosome 11q. Furthermore, β-arrestin-1 may also be a relevant predictor of response to tamoxifen treatment, given the involvement of cyclin D1, PAK1, and CHEK1 in breast cancer.
Recently, a comprehensive atlas of human protein expression patterns was generated through the Human Protein Atlas program (http://www.proteinatlas.org; last accessed March 13, 2010).21,22 At present, >3000 antibodies (corresponding to >2600 different human proteins) have been screened in TMAs, representing 48 types of normal tissues, the 20 most common forms of human cancer, and 47 cell lines, using an antibody-based proteomics strategy.23 Notably, the Human Protein Atlas can also be used as a platform for in silico discovery of new cancer biomarkers.24–26 In this fashion, we ventured to perform a systematic screening of 11q13 gene products and found that β-arrestin-1, although sparsely expressed in normal breast tissue, exhibited a differential expression ranging from negative to high among breast cancers. Notably, no other forms of cancer displayed a high expression of this protein.
To assess the importance of β-arrestin-1 in breast cancer, TMAs with tumor samples from two independent breast cancer cohorts were analyzed and, based on the initial evaluation, the relevance for both tumor and stromal cell protein expression was investigated. A possible link between ARRB1 and CCND1 amplification was also elucidated, by studying β-arrestin-1 protein expression in relation to amplification status of CCND1, analyzed by fluorescence in situ hybridization. Of note, stromal β-arrestin-1, irrespective of tumor cell expression, was shown to be a critical prognostic marker in both cohorts. Furthermore, patients exhibiting low or moderate stromal β-arrestin-1 expression did not benefit from treatment with tamoxifen, whereas those showing negative or high stromal expression responded well. Finally, a link between β-arrestin-1 protein expression and CCND1 amplification was observed in the larger cohort. To our knowledge, this is the first study demonstrating the potential importance of β-arrestin-1 as a prognostic and treatment predictive marker in breast cancer.
Materials and Methods
Cell Lines, Western Blot, and Immunocytochemistry
The human breast cancer cell lines MDA-MB-468 and MDA-MB-231 (ATCC, Manassas, VA) were used to verify the reactivity of the β-arrestin-1 antibody [rabbit monoclonal against human β-arrestin-1 (1:200, E246; Epitomics, Burlingame, CA)], by immunocytochemistry. For detailed description of culturing conditions, immunocytochemistry, and Western blot, we refer to a previous report.27 For detection of β-arrestin-1 overexpression, rabbit polyclonal anti-GFP antibody (1:1000, sc-8334; Santa Cruz, Biotechnology, Santa Cruz, CA) was used. To monitor cell proliferation, rabbit polyclonal anti-human cyclin A (1:500, H-432, sc-751, Santa Cruz, Biotechnology, Santa Cruz, CA) was used; for apoptosis detection, rabbit polyclonal anti-human caspase-3 antibody (1:500, P42574; Cell Signaling Technology, Danvers, MA) was used.
Transfection
For transient expression of wild-type β-arrestin-1, we used the pcDNA3 expression plasmid encoding EGFP-β-arrestin-1,28 kindly provided by Dr. Vsevolod V. Gurevich (Vanderbilt University, Nashville, TN). For transfection in six-well plates, MDA-MB-468 and MDA-MB-231 were transiently transfected with β-arrestin-1 vector using Lipofectamine 2000 according to the manufacturer's recommendations (Invitrogen Life Technologies, Carlsbad, CA). Two micrograms DNA was used per well of a six-well plate. For β-arrestin-1 knockdown, MDA-MB-468 and MDA-MB-231 cells were transfected with 50 nmol/L control small interfering RNA (siRNA) or siRNA against β-arrestin-1 (ON-TARGETplus siRNA, SMARTpool) using Dharmafect (both from Dharmacon, Lafayette, CO). Cells were allowed to grow for 48 hours after transfection before being harvested for migration assay. Cell pellets were made from control and β-arrestin-1 transfected cells 48 hours after transfection, and an immunocytochemistry array was constructed and stained with β-arrestin-1 antibody.
Migration Assay
Migration assays were performed in Transwell migration chambers with 8-μm pore membranes (Corning, Corning, NY). Cells (105) were resuspended in upper Transwell chambers in serum-free media and were allowed to migrate toward a serum gradient (10%) in the lower chamber for 3 hours (MDA-MB-231) or 6 hours (MDA-MB-468). Membranes were then excised, and cells on the upper side were removed before fixation in 4% paraformaldehyde and staining with DAPI prior to mounting. The number of migrating cells was counted in three randomly chosen fields on each membrane, photographed at ×10 magnification. Values reported are the averages of three experiments performed in triplicate.
Patients
Cohort I
Designed as a first-line screening cohort for Human Protein Atlas antibodies with potential relevance in breast cancer, cohort I comprises 179 patients diagnosed with primary invasive breast cancer between 2000 and 2002 at the Department of Pathology, Malmö University Hospital. Median age at diagnosis was 65 years (range, 35 to 97 years), and median follow-up time was 69 months. All patients in cohort I had been treated after surgery. For detailed description of clinicopathological features of the tumor samples, we refer to previous studies.29,30 Ethical permission for the study was obtained from the Ethics Committee at Lund University (ref 445/2007), whereby informed consent was deemed not to be required other than by the opt-out method.
Cohort II
Cohort II was established from a previous clinical trial population. Between 1986 and 1991, 564 premenopausal breast cancer patients with invasive stage II disease were enrolled in a Swedish trial (SBII:2a) in which they were randomized to receive either 2 years of adjuvant tamoxifen (n = 276) or no adjuvant treatment (control, n = 288). All patients were followed for recurrence-free and overall survival. Recurrence was defined as local, regional, or distant recurrence or breast cancer-specific death; contralateral breast cancer was excluded. Each patient received surgery (either modified radical mastectomy or breast-conserving surgery), followed by radiotherapy and in few cases (<2%) by adjuvant polychemotherapy. After surgery, the median follow-up time without breast cancer event was 13.9 years. Detailed description of the SBII:2a study design is available in a previous report.31 Informed consent was obtained from the patients participating in the randomized study, and the ethical committees at Lund and Linköping Universities approved the study and the reanalysis of the tumor material.
Tissue Specimens and Immunohistochemistry
FFPE tumor material was available from 500 of the 564 patients in cohort II. Areas representative of invasive cancer were selected and assembled in a TMA. Two tissue cores (cohort I, 1.0 mm; cohort II, 0.6 mm) from each donor block were placed in recipient paraffin block by using an automated tissue arrayer (Beecher Instruments Microarray Technology, Woodland, MD). Sections (4 μm) from this block were mounted onto slides before being deparaffinized, rehydrated. and microwave-treated in target retrieval solution pH 9.9 (Dako, Glostrup, Denmark) and then processed in an automated immunostainer (Techmate 500; Dako, Copenhagen, Denmark), using Envision software version 3.0 (Dako, Glostrup). The β-arrestin-1 antibody was diluted 1:200 also for IHC. Because β-arrestin-1 was expressed in various intensities both in the cytoplasm of the tumor cells and in the surrounding stromal cells, staining intensity of both compartments was evaluated and was scored as follows: 0 = negative, 1 = low, 2 = moderate, or 3 = high. Evaluation was performed independently by two different observers (K.L. and G.L.). Conflicting observations were low (<5%) for all evaluations made. All immunohistochemical evaluations were performed without knowledge of tumor characteristics. In cases of no evaluation, the tumor cores were either nonrepresentative (ie, contained no invasive tumor cells) or missing.
Expression of carbonic anhydrase 9 (CAIX) and vimentin had previously been assessed in cohort I (unpublished data). CAIX staining was assessed as negative or positive and stromal staining intensity for vimentin was graded from 0 to 3. In cohort II, data for expression of ERα and progesterone receptor (PR),32 as well as amplification status of ERBB2 (alias HER2),33 were available from previous studies.32,33 When performing survival analyses in which treatment response between different subgroups was compared, only ERα-positive tumors were analyzed (cutoff at 10% positively stained nuclei according to Swedish clinical protocols). We selected ERα-positive patients for the analyses, because this subgroup of patients can be regarded as potential responders to ER-targeting therapy, such as tamoxifen. Staining of the proliferation marker Ki-6732 (not available for cohort I) and HIF-1α34 in cohort II had also been performed previously. Data regarding CCND1 gene amplification status (assessed by fluorescence in situ hybridization analysis)15 and protein expression data for CHEK120 and stromal α-smooth muscle actin (α-SMA) (scored 0 to 3, negative to high) were also available.15,20
Statistical Methods
Statistical analyses were performed using SPSS software version 15.0 (SPSS, Chicago, IL). For examination of the statistical significance of associations between β-arrestin-1 expression and other categorical variables, Spearman's rank-order correlation coefficient and Wilcoxon-Mann-Whitney U-tests were used. To study recurrence-free survival (RFS), the Kaplan-Meier method was used; to compare RFS among different treatment groups, the log-rank test was used. A Cox proportional hazards regression model was used for relative risk estimation in multivariate analysis. All hazard ratios have been adjusted for age (continuous), Nottingham histological grade (NHG; I/II versus III), lymph node status (negative versus positive), and for cohort II also for Ki-67 nuclear fraction (0% to1%, 2% to 10%, 11% to 25%, 26% to 50%, and 51% to 100%); data for Ki-67 were not available for cohort I. We evaluated whether the effect of tamoxifen treatment was modified by stromal β-arrestin-1 by adding an interaction term to the Cox regression model. All P values corresponded to two-sided tests and a P value of <0.05 was considered statistically significant. This study was conducted according to the guidelines provided by the REMARK study.35
Results
β-Arrestin-1 Expression Affects Breast Cancer Cell Migration
β-Arrestin-1 antibody reactivity was first tested by siRNA silencing of β-arrestin-1 in MDA-MB-468 and MDA-MB-231 breast cancer cells. Significantly decreased levels of the protein were detected by immunocytochemistry for β-arrestin-1 siRNA transfected cells, compared with control, 48 hours after transfection (Figure 1B). In parallel with the immunohistochemical validation of the antibody, we tested the migratory capacity of MDA-MB-468 and MDA-MB-231 cells after overexpression and silencing of β-arrestin-1. Overexpression was detected with anti-GFP antibody using Western blot analysis (Figure 1A). Of note, β-arrestin-1 overexpression significantly reduced the migratory propensity of both cell lines 48 hours after transfection (Figure 1C), whereas silencing resulted in prominently increased migration (Figure 1D). To exclude the possibility that decreased migration was an effect of reduced cell viability due to vector transfection, we used Western blot analysis to monitor proliferation and apoptosis in both cell lines 48 hours after vector transfection. No change in proliferation or increased apoptosis was observed in cells overexpressing β-arrestin-1, compared with control transfected cells, indicating that the decreased migration observed was a true effect of β-arrestin-1 overexpression (see Supplemental Figure S1 at http://jmd.amjpathol.org).
Figure 1.
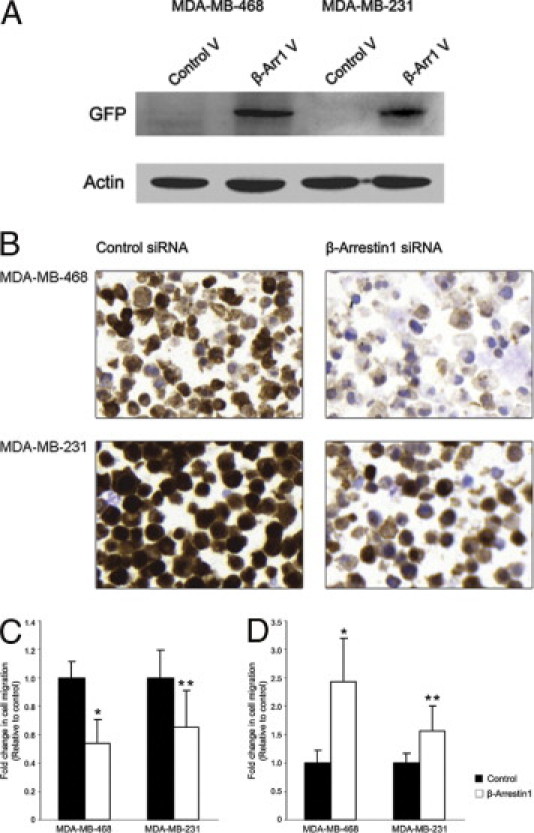
β-Arrestin-1 antibody validation and migratory propensity of breast cancer cells (MDA-MB-468 and MDA-MB-231 cell lines) after β-arrestin-1 overexpression and silencing. A: β-Arrestin-1 overexpression was detected with anti-GFP antibody on Western blot, 48 hours after transfection. V, vector. B: Protein expression was significantly decreased 48 hours after transfection with siRNA against β-arrestin-1 in both cell lines, analyzed by immunocytochemistry. Magnification ×40. C: Overexpression of β-arrestin-1 reduced migration of both cell lines, compared with control, 48 hours after transfection. *P < 0.001; **P < 0.001. D: Knockdown of β-arrestin-1 resulted in an increased migratory propensity of both cell lines, compared with control, 48 hours after transfection. *P < 0.001; **P < 0.001.
β-Arrestin-1 Is Expressed in both Tumor Cells and Stroma of Primary Breast Tumors
Based on the discovery that overexpression of β-arrestin-1 expression was restricted exclusively to breast cancers in the Human Protein Atlas, we further investigated its protein expression in two independent cohorts of breast cancer cases. β-Arrestin-1 protein expression was first evaluated in cohort I, consisting of a TMA of 179 breast tumors. Notably, apart from the expected cytoplasmic tumor cell expression, β-arrestin-1 was also detectable in stromal cells, including fibroblasts, endothelial cells, and various immune cells. To validate the staining of the TMAs, normal breast tissue was also stained for IHC. Expression of β-arrestin-1 was observed exclusively in endothelial and luminal epithelial cells in normal breast tissue; no staining of the stromal compartment was detected. Both tumor cell and stromal staining of the breast cancer samples varied greatly, from no staining at all to very intense staining. From this point onwards, stromal staining intensity was assessed separately from the tumor cell staining intensity (Figure 2). In cohort I, tumor cell staining of β-arrestin-1 was assessable in 131/179 tumors and stromal staining in 130/179 tumors. Next, we validated our findings in cohort II (the larger cohort, consisting of 500 tumors); and 359/500 tumors were assessable for both tumor cell and stromal staining.
Figure 2.
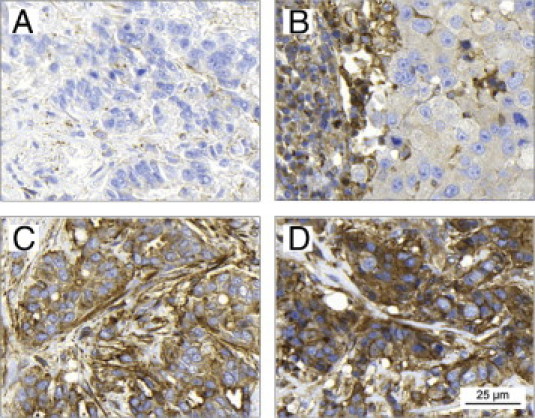
Immunohistochemical staining of β-arrestin-1 in primary breast tumors. Cytoplasmic tumor cell β-arrestin-1 expression was evaluated as negative (A), low (B), moderate (C), or high (D). Stromal expression (Neg-High) was observed to be high (B) in some tumors in which tumor cell expression was quite low, and the opposite was also observed: low (D), when tumor cell expression was high. Scale bar applies to all images.
Tumor Cell and Stromal β-Arrestin-1 Expression Is Associated with Different Tumor Characteristics
First, we wanted to elucidate possible associations between the expression of β-arrestin-1 and clinicopathological parameters, as well as other markers for tumor behavior. In cohort I, tumor cell expression of β-arrestin-1 was inversely correlated with ERα expression (P = 0.028) and positively correlated with ERBB2 amplification status (P = 0.001) and age (P = 0.006), whereas stromal expression was inversely correlated with ERα expression (P = 0.001) and positively correlated with ERBB2 status (P = 0.042), NHG (P = 0.017), tumor size (P = 0.029), lymph node status (P = 0.014), distant metastases (P = 0.004), and expression of the hypoxia marker CAIX (P = 0.027) (Table 1). A positive correlation between tumor cell and stromal expression of β-arrestin-1 was also observed (P < 0.001). These contrasting associations between relevant clinicopathological variables and tumor cell versus stromal β-arrestin-1 expression imply distinct roles for this protein in different cellular compartments of a tumor.
Table 1.
Distribution of β-Arrestin-1 Staining Category According to Clinicopathological Parameters and Tumor Markers in Cohort I
| Variable | β-Arrestin-1 |
P⁎ | β-Arrestin-1 |
P⁎ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor cell staining intensity |
Stromal staining intensity |
|||||||||
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | |||
| Sample size | n = 56 | n = 58 | n = 14 | n = 3 | n = 13 | n = 57 | n = 54 | n = 6 | ||
| Sample % of total | 42.7 | 44.3 | 10.7 | 2.3 | 10.0 | 43.8 | 41.5 | 4.6 | ||
| ERα positive (%) | 0.028† | 0.001† | ||||||||
| <10 | 3 | 5 | 5 | 0 | 0 | 2 | 8 | 3 | ||
| ≥10 | 52 | 52 | 9 | 3 | 13 | 53 | 46 | 3 | ||
| Missing cases: 48/49 | ||||||||||
| PR positive (%) | 0.325† | 0.430† | ||||||||
| <10 | 14 | 19 | 6 | 0 | 4 | 15 | 15 | 4 | ||
| ≥10 | 42 | 39 | 8 | 3 | 9 | 42 | 39 | 2 | ||
| Missing cases: 48/49 | ||||||||||
| ERBB2 status | 0.001 | 0.042 | ||||||||
| Negative | 43 | 31 | 5 | 2 | 10 | 36 | 33 | 1 | ||
| 1+ | 9 | 14 | 3 | 1 | 1 | 16 | 8 | 2 | ||
| 2+ | 2 | 9 | 2 | 0 | 1 | 3 | 8 | 1 | ||
| 3+ | 2 | 4 | 4 | 0 | 1 | 2 | 5 | 2 | ||
| Missing cases: 48/49 | ||||||||||
| NHG | 0.879 | 0.017 | ||||||||
| I | 9 | 8 | 4 | 2 | 2 | 14 | 7 | 0 | ||
| II | 26 | 28 | 2 | 1 | 6 | 27 | 24 | 0 | ||
| III | 21 | 22 | 8 | 0 | 5 | 16 | 23 | 6 | ||
| Missing cases: 48/49 | ||||||||||
| Tumor size | 0.485† | 0.029† | ||||||||
| ≤20 | 28 | 34 | 6 | 3 | 10 | 34 | 24 | 3 | ||
| >20 | 28 | 24 | 8 | 0 | 3 | 23 | 30 | 3 | ||
| Missing cases: 48/49 | ||||||||||
| Lymph node status | 0.952 | 0.014 | ||||||||
| N0 | 31 | 27 | 8 | 2 | 12 | 28 | 26 | 1 | ||
| N1–3 | 15 | 15 | 2 | 0 | 0 | 16 | 15 | 1 | ||
| N≥4 | 7 | 8 | 3 | 0 | 1 | 6 | 8 | 3 | ||
| Missing cases: 61/62 | ||||||||||
| Distant metastases | 0.970† | 0.004† | ||||||||
| No | 51 | 51 | 13 | 3 | 13 | 54 | 47 | 3 | ||
| Yes | 5 | 7 | 1 | 0 | 0 | 3 | 7 | 3 | ||
| Missing cases: 48/49 | ||||||||||
| CAIX | 0.072† | 0.027† | ||||||||
| Negative | 45 | 39 | 6 | 1 | 10 | 36 | 41 | 3 | ||
| Positive | 3 | 5 | 3 | 0 | 0 | 3 | 5 | 3 | ||
| Missing cases: 77/78 | ||||||||||
| Age at surgery (yr) | 0.006 | 0.126 | ||||||||
| Median | 61 | 72 | 72 | 66 | 63 | 65 | 65 | 78 | ||
| Range | 40–89 | 35–97 | 42–81 | 64–83 | 47–82 | 35–92 | 35–97 | 40–81 | ||
| Missing cases: 48/49 | ||||||||||
CAIX, carbonic anhydrase IX; ER, estrogen receptor; ERBB2, epidermal growth factor receptor; NHG, Nottingham histologic grade; PR, progesterone receptor.
Correlations were calculated using Spearman's ρ unless otherwise specified.
Wilcoxon/Mann-Whitney test (two-sided).
The associations of β-arrestin-1 to prognostic markers found in cohort I were analyzed in cohort II with consistent results. In cohort II, comprising 500 premenopausal breast cancer cases, the expression of tumor cell β-arrestin-1 was inversely correlated with ERα expression (P = 0.015) and PR expression (P = 0.002) (Table 2). Notably, in contrast to cohort I, tumor cell β-arrestin-1 expression was inversely correlated with age (P = 0.003). In addition, tumor cell expression was positively correlated with ERBB2 status (P = 0.003) and NHG (P = 0.003) (Table 2). Stromal expression of β-arrestin-1 was inversely correlated with ERα (P = 0.006) and PR expression (P = 0.001) and was positively correlated with NHG (P < 0.001), tumor size (P = 0.038), proliferation (Ki-67) (P < 0.001), and hypoxia inducible factor-1α (HIF-1α) expression (P = 0.024) (Table 2). As for cohort I, a positive correlation was observed between tumor cell and stromal β-arrestin-1 expression in cohort II (P < 0.001).
Table 2.
Distribution of β-Arrestin-1 Staining Category According to Clinicopathological Parameters and Tumor Markers in Cohort II
| Variable | β-Arrestin-1 |
P⁎ | β-Arrestin-1 |
P⁎ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor cell staining intensity |
Stromal staining intensity |
|||||||||
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | |||
| Sample size | n = 27 | n = 202 | n = 110 | n = 20 | n = 30 | n = 168 | n = 140 | n = 21 | ||
| Sample % of total | 7.5 | 56.3 | 30.6 | 5.6 | 8.4 | 46.8 | 39.0 | 5.8 | ||
| ERα positive (%) | 0.015† | 0.006† | ||||||||
| <10 | 6 | 55 | 41 | 8 | 7 | 42 | 47 | 14 | ||
| ≥10 | 21 | 140 | 63 | 11 | 19 | 117 | 92 | 7 | ||
| Missing cases: 155 | ||||||||||
| PR positive (%) | 0.002† | 0.001† | ||||||||
| <10 | 5 | 51 | 43 | 9 | 5 | 40 | 51 | 12 | ||
| ≥10 | 20 | 135 | 62 | 9 | 19 | 115 | 85 | 7 | ||
| Missing cases: 166 | ||||||||||
| ERBB2 status | 0.003† | 0.370† | ||||||||
| Nonamplified | 18 | 137 | 69 | 11 | 17 | 107 | 96 | 15 | ||
| Amplified | 1 | 13 | 13 | 6 | 0 | 16 | 14 | 3 | ||
| Missing cases: 232 | ||||||||||
| NHG | 0.003 | <0.001 | ||||||||
| I | 2 | 28 | 6 | 3 | 3 | 25 | 10 | 1 | ||
| II | 13 | 96 | 42 | 2 | 16 | 79 | 53 | 5 | ||
| III | 12 | 73 | 54 | 15 | 11 | 58 | 71 | 14 | ||
| Missing cases: 154 | ||||||||||
| Tumor size, mm | 0.988† | 0.038† | ||||||||
| ≤20 | 10 | 75 | 41 | 7 | 15 | 67 | 44 | 7 | ||
| >20 | 17 | 127 | 68 | 13 | 15 | 100 | 96 | 14 | ||
| Missing cases: 142 | ||||||||||
| Lymph node status | ||||||||||
| N0 | 7 | 49 | 35 | 6 | 0.703 | 6 | 42 | 40 | 9 | 0.188 |
| N1–3 | 18 | 108 | 47 | 12 | 18 | 86 | 76 | 5 | ||
| N≥4 | 2 | 43 | 28 | 2 | 6 | 39 | 23 | 7 | ||
| Missing cases: 141 | ||||||||||
| Distant metastases | 0.632† | 0.496† | ||||||||
| No | 12 | 118 | 57 | 10 | 14 | 92 | 80 | 11 | ||
| Yes | 15 | 84 | 53 | 10 | 16 | 76 | 60 | 10 | ||
| Missing cases: 141 | ||||||||||
| Ki67 positive (%) | 0.097 | <0.001 | ||||||||
| 0–10 | 14 | 84 | 39 | 7 | 17 | 80 | 43 | 4 | ||
| 11–25 | 7 | 45 | 30 | 3 | 4 | 40 | 36 | 5 | ||
| 26–100 | 4 | 49 | 25 | 9 | 2 | 27 | 47 | 11 | ||
| Missing cases: 184 | ||||||||||
| CCND1 status | 0.041† | 0.429† | ||||||||
| Nonamplified | 12 | 98 | 55 | 13 | 12 | 87 | 66 | 13 | ||
| Amplified | 5 | 20 | 8 | 0 | 4 | 15 | 14 | 0 | ||
| Missing cases: 289 | ||||||||||
| CHEK1 positive nuclei (%) | 0.030 | <0.001 | ||||||||
| 0–5 | 6 | 52 | 23 | 4 | 9 | 47 | 26 | 3 | ||
| 6–50 | 14 | 74 | 48 | 6 | 9 | 56 | 67 | 10 | ||
| 51–100 | 0 | 15 | 12 | 6 | 0 | 10 | 18 | 5 | ||
| Missing cases: 240 | ||||||||||
| HIF-1α positive (%) | 0.388 | 0.024 | ||||||||
| 0–1 | 17 | 116 | 61 | 13 | 18 | 100 | 77 | 12 | ||
| 2–10 | 2 | 23 | 16 | 4 | 1 | 15 | 26 | 3 | ||
| 11–100 | 3 | 12 | 9 | 1 | 2 | 9 | 14 | 0 | ||
| Missing cases: 223 | ||||||||||
| Age at surgery (yr) | 0.003 | 0.903 | ||||||||
| Median | 45 | 46 | 43 | 45 | 44 | 45 | 45 | 42 | ||
| Range | 35–50 | 29–57 | 26–55 | 37–5 | 30–51 | 29–57 | 26–57 | 39–5 | ||
| Missing cases: 141 | ||||||||||
CHEK1, checkpoint kinase 1; ER, estrogen receptor; ERBB2, epidermal growth factor receptor; HIF, hypoxia inducible factor; NHG, Nottingham histologic grade; PR, progesterone receptor.
Correlations were calculated using Spearman's ρ unless otherwise specified.
Wilcoxon/Mann-Whitney test (two-sided).
β-Arrestin-1 Is A Prognostic and Treatment Predictive Marker in Breast Cancer
To investigate the influence of β-arrestin-1 expression on disease outcome, we studied patient survival according to tumor cell or stromal β-arrestin-1 expression. Although tumor cell expression did not affect RFS in cohort I (Figure 3A), patients with tumors exhibiting high levels of stromal β-arrestin-1 were subject to a shorter RFS than were those with tumors of lower expression levels (P < 0.001) (Figure 3A). Notably, multivariate Cox regression analysis revealed an independent prognostic value for stromal β-arrestin-1 expression in cohort I (hazard ratio HR = 2.72, 95% confidence interval CI = 1.06 to 6.98; P = 0.038) (Table 3). To exclude the possibility that stromal β-arrestin-1 was associated with worse prognosis because its expression was dependent on the amount of stromal tissue in the tumor, we analyzed the link to the mesenchymal cell marker protein vimentin, abundantly expressed by fibroblasts. A positive correlation between stromal β-arrestin-1 and vimentin was observed in cohort I, but vimentin did not reveal any independent prognostic information, implying a specific role for β-arrestin-1, irrespective of tumor stroma, for clinical outcome in breast cancer (data not shown).
Figure 3.
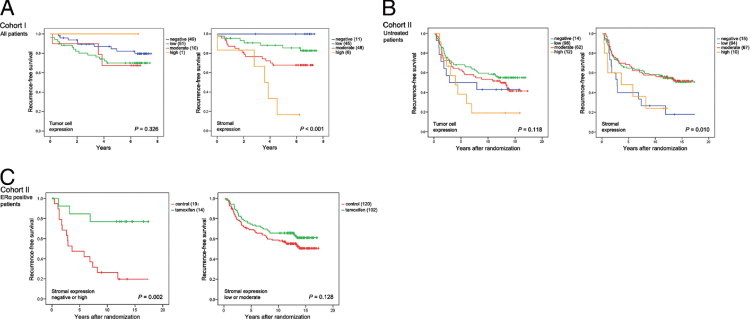
Kaplan-Meier curves showing the effect of β-arrestin-1 on RFS. A: In cohort I, only stromal expression (right panel) had an effect on overall survival. B: In the subgroup of untreated control patients of cohort II, negative or high expression of stromal β-arrestin-1 was associated with a shorter RFS, compared with low or moderate expression (right panel). There was a trend indicating that also high tumor cell expression was linked to a reduced RFS in cohort II (left panel). C: ERα-positive patients with tumors of low or moderate stromal β-arrestin-1 expression (right panel) did not benefit from tamoxifen treatment, in contrast to patients with tumors exhibiting negative or high expression (left panel).
Table 3.
Recurrence-Free Survival with Cox Multivariate Analysis for Patients in Cohort I
| Variable | Multivariate HR (95% CI) | P |
|---|---|---|
| β-arrestin-1 stroma | 0.038 | |
| Low, 0–1 | 1.00 | |
| High, 2–3 | 2.72 (1.06–6.98) | |
| NHG | 0.019 | |
| I–II | 1.00 | |
| III | 2.81 (1.18–6.70) | |
| Lymph node status | <0.001 | |
| N0 | 1.00 | |
| N+ | 1.18 (1.09–1.29) | |
| Age | 0.110 | |
| Continuous (per year) | 1.03 (0.99–1.06) |
CI, Confidence interval; HR, hazard ratio; NHG, Nottingham histologic grade.
In cohort II, RFS in the subgroup of untreated control patients was analyzed first, to avoid the potential bias of treatment influencing the results. Only a trend indicating a shorter RFS in patients with tumors of high tumor cell β-arrestin-1 expression was observed (Figure 3B). In contrast to cohort I, both negative and high stromal β-arrestin-1 expression were associated with a reduced RFS, compared with low or moderate expression (P = 0.010) (Figure 3B). Because the curves for patients with tumors of negative and high stromal β-arrestin-1 expression overlapped and were associated with a shorter RFS compared with patients with tumors of low or moderate expression, patients were divided into two subgroups (negative or high versus low or moderate) for the multivariate Cox regression analysis. Notably, β-arrestin-1 expression was an independent prognostic factor (HR = 0.44, 95% CI = 0.25 to 0.79; P = 0.006) in cohort II as well (Table 4). Expression of stromal β-arrestin-1 was positively correlated with stromal α-SMA, a marker for carcinoma-associated fibroblasts (P = 0.009); however, expression of α-SMA was not associated with recurrence rate.
Table 4.
Recurrence-Free Survival with Cox Multivariate Analysis for Patients in Cohort II
| Variable | Multivariate HR (95% CI) | P |
|---|---|---|
| β-arrestin-1 stroma | 0.006 | |
| Negative or high | 1.00 | |
| Low or moderate | 0.44 (0.25−0.79) | |
| NHG | 0.038 | |
| I–II | 1.00 | |
| III | 1.62 (1.03–2.55) | |
| Lymph node status | 0.030 | |
| N0 | 1.00 | |
| N+ | 1.88 (1.06–3.34) | |
| Age | 0.165 | |
| Continuous (per year) | 0.97 (0.94–1.01) | |
| Ki67 positive (%) | 0.923 | |
| ≤25 | 1.00 | |
| >25 | 1.03 (0.61–1.73) |
CI, Confidence interval; HR, hazard ratio; NHG, Nottingham histologic grade.
Because several gene products of chromosomal region 11q have been reported to be associated with impaired tamoxifen response, we wanted to further delineate the effect of β-arrestin-1 expression on response to this selective estrogen receptor modulator. Cohort II is based on a large clinical trial including breast cancer patients randomly assigned to either 2 years of tamoxifen treatment or to no adjuvant treatment. Tumor cell expression of β-arrestin-1 had no effect on tamoxifen response in the subgroup of ER-positive patients selected for these analyses (data not shown). Furthermore, no effect on treatment response was observed for stromal β-arrestin-1 expression when patients were not subcategorized (data not shown). However, when analyzing the difference in tamoxifen response according to stromal expression subcategorized as negative or high versus low or moderate, based on previous survival analyses, an intriguing feature was observed: RFS among patients who presented with tumors of negative or high β-arrestin-1 expression was improved with tamoxifen (P = 0.002), in contrast to patients exhibiting low or moderate expression (P = 0.128) (Figure 3C). A multivariate Cox proportional hazard regression model for stromal β-arrestin-1 and treatment interaction revealed a statistically significant difference between the two subgroups defined as negative or high versus low or moderate (HR = 5.31, 95% CI = 1.34 to 20.96; P = 0.017) (Table 5). These results indicate that patients exhibiting low or moderate stromal β-arrestin-1 expression are less likely to respond to tamoxifen, compared with patients exhibiting negative or high expression.
Table 5.
Recurrence-Free Survival with Cox Multivariate Analysis According to β-Arrestin-1 Expression and Treatment Interaction for Patients in Cohort II
| ERα positive (n = 384)⁎ | Multivariate HR (95% CI) | P |
|---|---|---|
| β-arrestin-1 stroma | <0.001 | |
| Negative or high | 1.00 | |
| Low or moderate | 0.27 (0.13−0.55)† | |
| Treatment | 0.001 | |
| Control | 1.00 | |
| Tamoxifen | 0.12 (0.03−0.44)‡ | |
| Interaction variable§ | 0.017 | |
| Value | 5.31 (1.34–20.96) |
CI, Confidence interval; HR, hazard ratio; NHG, Nottingham histologic grade.
Other factors included in the multivariate analysis: age (continuous), tumor grade (NHG I+II versus III), proliferation (Ki67 0–1, 2–10, 11–25, 26–50, 51–100%), and nodal status (negative versus positive).
Stromal β-arrestin-1 hazard ratio for the control group.
Treatment hazard ratio for the stromal β-arrestin-1 subgroup of negative or high expression.
Interaction variable states whether there is a difference in the treatment response in relation to stromal β-arrestin-1 status.
β-Arrestin-1 Expression Is Inversely Correlated with Amplification of CCND1 and Positively Correlated with CHEK1 Expression
Given that both CCND1 and the ARRB1 gene map to the chromosomal amplification region 11q13, our next objective was to investigate a possible link between β-arrestin-1 expression and amplification of CCND1. Unfortunately, no data for ARRB1 gene expression were available in our tumor material. Surprisingly, tumor cell β-arrestin-1 expression was inversely correlated with amplification of CCND1 (P = 0.041) (Table 2) in cohort II (no data for CCND1 amplification were available for cohort I). Furthermore, in cohort II both tumor cell and stromal expression of β-arrestin-1 was positively correlated with expression of CHEK1 (alias CHK1), a marker for 11q deletion (tumor cell, P = 0.030; stroma, P < 0.001) (Table 2). We have previously shown that loss of CHEK1 protein expression in tumor cells was inversely correlated with CCND1 amplification in the same patient cohort,20 implying that the CHEK1 gene is deleted concurrently with amplification of 11q13. These observations suggest an inverse relationship between the CCND1 and the ARRB1 genes, and thus ARRB1 might be deleted instead of being coamplified with CCND1, despite the close proximity of the two genes.
Discussion
The role of β-arrestin-1 in breast cancer has not been extensively studied, and in particular its clinical relevance has not previously been addressed. By analyzing two different cohorts, one including a subset of both pre- and postmenopausal breast cancer patients and another including premenopausal patients randomized to either 2 years of adjuvant tamoxifen or no adjuvant treatment, we had the opportunity to validate the significance of β-arrestin-1 protein expression in relation to patient outcome. Partial agreement between the observations made in the different cohorts suggests an important role for β-arrestin-1 protein in the context of tumor behavior. The positive correlations between stromal β-arrestin-1 expression and clinicopathological markers such as NHG, tumor size, node status (cohort I), and proliferation (cohort II) suggests that β-arrestin-1 is a negative predictor of outcome. However, in cohort II, both high expression and absent expression of β-arrestin-1 was associated with a poor clinical outcome. The present findings demonstrate that both loss and elevated expression of β-arrestin-1 in the stromal compartment of breast tumors are associated with poor prognosis, independent of standard clinicopathological risk factors.
In contrast to previous reports for MDA-MB-231 breast cancer cells,8,9 migration of both MDA-MB-468 and MDA-MB-231 cells was increased when β-arrestin-1 was silenced. In accordance with these results, overexpression of the protein decreased migration, implying that β-arrestin-1 is a negative regulator of migration in these breast cancer cell lines. In previous studies, however, migration was induced by either protease-activated receptor-2 or lysophosphatidic acid receptor stimulation,8,9 whereas we monitored migration toward serum without additional stimulating agents in uncoated wells, which might explain the conflicting results. Notably, tumor cell expression of β-arrestin-1 was not the crucial factor determining tumor aggressiveness in our patient cohorts. Instead, lymph node status and distant metastases (two features that define the capability of cells to migrate) were related to the expression of stromal β-arrestin-1.
In recent years, a wealth of data regarding the influence of the tumor microenvironment in breast cancer has stimulated new ideas about the mechanism for development and progression of the disease.36–38 Communication between tumor cells and the surrounding cells is central to these hypotheses.39 The present study addressed β-arrestin-1, a protein prominent for the communication between different cell types, via G-protein-coupled receptors, and whose expression levels have here been shown to have important effects on tumor progression. The actual role of β-arrestin-1 expression by stromal cells is unclear, and the specific cell types expressing this protein have not yet been identified. Nonetheless, in our tumor materials, fibroblasts, endothelial cells and immune cells all displayed β-arrestin-1 staining of varying intensities. Our results suggest that stromal expression is indicative of the clinical outcome independent of tumor cell expression. Notably, in staining of normal breast tissue the appearance of β-arrestin-1 was strikingly different, compared with tumor tissue, with only weak staining of endothelial cells and normal luminal epithelial cells and without staining of the stroma. Studies are needed to investigate the effect of β-arrestin-1 in stromal cells in vitro, and also to elucidate the effect of altered protein expression in fibroblasts on breast cancer cells (to clarify its importance for paracrine signaling). Unfortunately, such experiments were beyond the scope of this clinically oriented study.
As already noted, cancer cells have the ability to alter their surroundings to create a permissive environment (the reactive tumor stroma) that supports tumor progression.40 The stromal compartment is made up of a number of different secreted growth factors that disrupt normal tissue homeostasis,41 which in turn activates distinct subpopulations of stromal cells. An important feature of the reactive tumor stroma is the carcinoma-associated fibroblasts (also known as activated fibroblasts). These cells are mesenchymal-like and share characteristics with smooth-muscle cells and fibroblasts.40 Carcinoma-associated fibroblasts express markers such as α-SMA, vimentin, desmin, and fibroblast activation protein. Carcinoma-associated fibroblasts have been observed in the stroma of the majority of invasive breast cancers42,43 and have been shown to stimulate tumor progression by secreting a wide range of different growth factors, hormones, and cytokines targeting the tumor cells.44 We speculate that a fraction of the β-arrestin-1 expressing cells of the stroma are carcinoma-associated fibroblasts, which might be important for the clinical outcome of these breast cancers. A positive correlation between β-arrestin-1 and stromal α-SMA implies that this actually is the case, further supporting the emerging theory describing the stroma as a determinant of tumor aggressiveness. Altered expression of β-arrestin-1 in the stroma might be regulated by a paracrine signaling with the tumor cells and so in turn emanate in a release of the growth suppressing properties of the stroma on the tumor cells, resulting in tumor cell proliferation, migration, and local invasion. Moreover, a number of studies have reported genetic alterations in the stroma to be associated with clinicopathological parameters defining a more aggressive tumor phenotype.38,45
Two other factors determining tumor aggressiveness are the hypoxia markers HIF-1α and CAIX, which were associated with high stromal expression of β-arrestin-1 in cohort II (HIF-1α) and in cohort I (CAIX). Hypoxic tumors often have a worse clinical outcome, compared with their well-vascularized counterparts.46 Furthermore, both high tumor cell and stromal expression of β-arrestin-1 was associated with ERα (both cohorts) and PR (cohort II) negativity, and with ERBB2 amplification (only tumor cell expression in cohort II). ERα and PR negativity and ERBB2 amplification have been associated with the more aggressive subtypes of breast cancer,47 and these patients are also less likely to respond to antihormonal therapy.
The almost identical correspondence of the curves in the survival analysis of cohort II patients exhibiting negative and high stromal β-arrestin-1 expression is somewhat surprising (Figure 3B), but might be due to differences between the cohorts. In cohort I, all patients included in the analysis received adjuvant therapy, whereas the survival analysis of cohort II included untreated patients only, which might be a factor explaining the differing results observed for stromal expression. When including all patients or when analyzing treated patients exclusively in cohort II, no significant difference was observed between patients showing different expression levels of β-arrestin-1 (data not shown). The analysis criteria differed somewhat between the two cohorts; however, because all patients in cohort I received adjuvant treatment, an analysis unbiased by treatment could not be performed for these patients. Furthermore, cohort I included both pre- and postmenopausal patients, whereas cohort II included only premenopausal patients, possibly contributing to the inconsistent results. Nonetheless, it is difficult to draw any conclusions about the underlying cause of the differential effect of β-arrestin-1 in pre- and postmenopausal breast cancers.
Of note, patients with tumors displaying negative or high stromal β-arrestin-1 expression were the ones who benefited from adjuvant treatment with tamoxifen, in contrast to those with low or moderately expressing tumors. In the absence of tamoxifen, patients exhibiting low or moderate β-arrestin-1 expression did well, but in the presence of tamoxifen these were the patients with the highest risk of recurrence. No previous reports investigating the role of β-arrestin-1 as a treatment predictive marker in breast cancer exist, and further studies including randomized patient cohorts would be required to confirm our results. Moreover, functional studies are needed to elucidate the mechanism by which expression of this protein interferes with the action of tamoxifen and confers resistance. Hattar et al48 reported that mammary fibroblasts isolated from tamoxifen-treated rats exhibited features of quiescent mammary microenvironment, with decreased levels of fibronectin and reduced extracellular matrix turnover. Thus, the stroma was functionally altered by tamoxifen to inhibit tumor cell progression. Although the underlying mechanism is poorly understood, a number of experimental studies in breast cancer cells have shown that the therapeutic effects of antiestrogens are mediated not only by targeting the epithelial cell compartment, but also by targeting the stroma.49,50 Our results further indicate that fibroblasts might be direct targets of tamoxifen treatment, and might also be a source of therapy resistance.
In recent years, the emerging role of the insulin-like growth factor-1 receptor 1 (IGF-1R) in cancer progression has been recognized. IGF-1R is a receptor tyrosine kinase, and signaling through this receptor is a central mechanism in carcinogenesis, promoting tumor cell proliferation and cell motility, as well as resistance to apoptosis.51 β-Arrestin-1 has been implicated in IGF-1R signaling in vitro, as a mediator of ubiquitination and proteasomal degradation of the receptor.52 Furthermore, silencing of β-arrestin-1 in human melanoma cells results in ablation of ERK stimulation by IGF-1R and prolonged G1-phase of the cell cycle.53 These results imply a role for β-arrestin-1 in IGF-1R signaling mediated by ERK and regulation of cell cycle progression.
We speculate that expression of β-arrestin-1 might be associated with the extent of IGF-1R signaling in the patient cohorts; that is, patients with tumors of low β-arrestin-1 expression would exhibit an increased IGF-1R signaling, because of impaired receptor degradation. This might explain the link between absence of stromal β-arrestin-1 and reduced RFS observed for patients in cohort II. However, patients who presented with tumors showing high expression of stromal β-arrestin-1 were in addition subject to a worse clinical outcome in both cohorts. The involvement of β-arrestin-1 in IGF-1R-mediated ERK signaling implies a link between high β-arrestin-1 expression and more aggressive disease, because reduced protein levels results in decreased proliferation. Moreover, the mechanisms promoting tumor progression are likely to differ between tumor cells and cells of the reactive tumor stroma. Hypothetically, altered IGF-1R signaling in stromal cells is more critical in promoting tumor aggressiveness than aberrant signaling via IGF-1R in tumor cells in breast cancer. However, no data for IGF-1R expression were available for our breast cancer cohorts, and the mechanism underlying the observation that both negative and high stromal β-arrestin-1 expression was associated with shorter RFS in cohort II is still elusive. β-Arrestin-1 is involved in various signaling pathways, likely to be differentially regulated by this protein depending on the context. Notably, the results should be interpreted with caution because these two subgroups (negative and high expression) included fewer patients than the remaining two subgroups of patients (low and moderate expression). Nonetheless, the independent prognostic value of stromal β-arrestin-1 observed in both cohorts implies that this is a potential novel biomarker in breast cancer.
Loss of expression of caveolin-1 (Cav-1), a protein involved in receptor-independent endocytosis exclusively in the tumor-associated fibroblast compartment of breast tumors, has been linked to early recurrence, lymph node metastasis, and tamoxifen resistance.54,55 Caveolin-1 has been reported to enhance the interaction between β-arrestin-2 and the neurokinin-1 receptor in clathrin-mediated endocytosis and signaling,56 suggesting that regulation of Cav-1 might also affect the expression of β-arrestin-1. Altered protein expression of β-arrestin-1 in breast cancer stroma may be a result of lost Cav-1 expression in stromal fibroblasts, resulting in worse prognosis and tamoxifen insensitivity. Furthermore, high protein levels of stromal but not tumor cell platelet-derived growth factor receptor β (PDGF-R-β) has been associated with unfavorable clinical characteristics and reduced survival, particularly prominent in premenopausal breast cancer patients, a finding that further highlights the importance of stromal markers in breast cancer prognosis.57 PDGF-R-β associates with G-protein-coupled receptors in mammalian cells, and it is thus likely that a relationship between this receptor and the β-arrestins exists, potentially highlighting another link between stromal β-arrestin-1 expression and clinical outcome in breast cancer.
Expression of tumor cell β-arrestin-1 was inversely correlated with amplification of CCND1, a novel and intriguing discovery. The β-arrestin-1 gene ARRB1 maps to chromosomal locus 11q13 (11q13.4), the same region that harbors CCND1. Amplification of CCND1 in cancer has been reported to include several other genes located in the 11q13 chromosomal region, including PAK116 and cortactin (CTTN).58 In a previous study, we reported that protein expression of PAK1 and cortactin was positively correlated with CCND1 amplification in breast cancer, suggesting a link between CCND1 amplification and amplification of PAK1 or CTTN.20 We speculate that a correlation between CCND1 amplification and β-arrestin-1 protein expression would be positive, considering the close proximity of the two genes. However, a deletion of parts of the 11q chromosome has been reported, concurrent with the amplification event occurring at 11q13.19 In general, reports describe deletion of distal 11q, but deletions in the 11q13 region have also been reported.59 Accordingly, ARRB1 might be one of the genes deleted in the amplification event frequently occurring at 11q13. Supporting this finding is the positive correlation with the CHEK1 protein, which is encoded by CHEK1 at 11q24 (ie, distal 11q). We previously showed that protein expression of CHEK1 was inversely correlated with CCND1 amplification,20 indicating a loss of distal 11q concurrent with the amplification of 11q13. The ARRB1 gene is located between CCND1 and PAK1, demonstrating that two amplicons of the 11q13 might be amplified without the intervening chromosomal segment, as has been described previously.18 Our present results indicate that, instead of being coamplified with CCND1, the ARRB1 gene may be deleted. No relationship between CCND1 gene amplification and stromal β-arrestin-1 expression was found, implying that the tamoxifen resistance observed for patients with tumors of low or moderate expression was not due to CCND1 amplification, which we have previously reported to be associated with an agonistic effect of tamoxifen.15
The present study demonstrates a previously unrecognized finding, that the expression pattern of β-arrestin-1 in tumor stroma exclusively predicts both clinical outcome and response to tamoxifen in breast cancer. The results highlight the emerging significance of stromal characteristics as prognostic and predictive markers in cancer. Nevertheless, much remains to be clarified in the field of breast cancer and the tumor microenvironment, and the role of the β-arrestins in communication between the cells that make up a solid tumor remains unknown.
Acknowledgment
We thank Ms. Elise Nilsson (Malmö University Hospital, Malmö, Sweden) for excellent technical implementation.
Footnotes
Supported by grants from the Swedish Cancer Society, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, Malmö University Hospital Research and Cancer Funds, Lund University Research Funds, South Swedish and South-East Swedish Breast Cancer groups, and Breakthrough Breast Cancer Unit, Manchester, UK.
CME Disclosure: The authors did not disclose any relevant financial relationships.
Supplemental material for this article can be found at http://jmd.amjpathol.org and at doi: 10.1016/j.jmoldx.2011.01.009.
Supplementary data
Supplemental Figure S1.
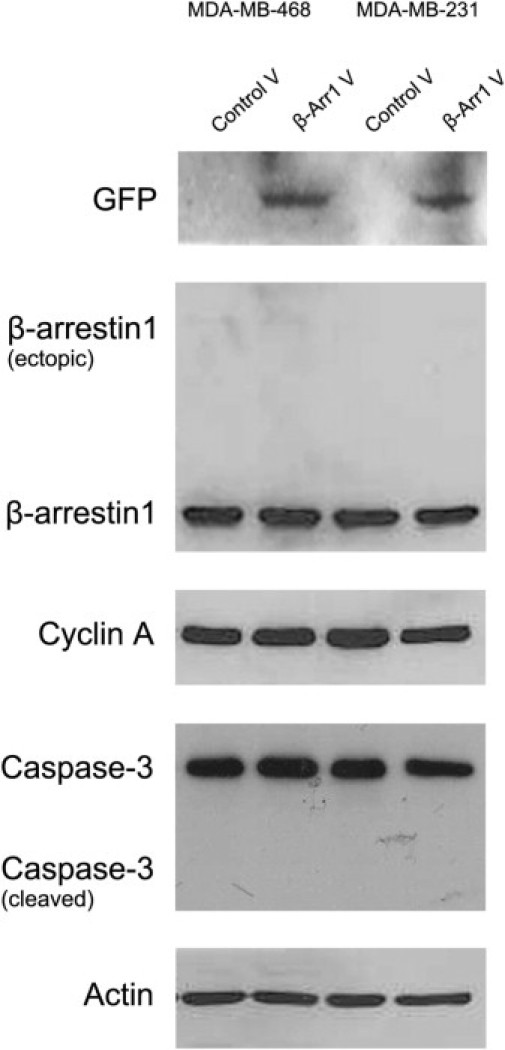
Western blot monitoring proliferation and apoptosis in MDA-MD-468 and MDA-MD-231 breast cancer cells overexpressing β-arrestin-1, 48 hours after transfection. Overexpression of GFP tagged β-arrestin-1 was detected by anti-GFP antibody (75 kDa). Ectopic expression of the protein was not recognized by the β-arrestin-1 antibody, possibly because of interference of the tag with the antibody-specific epitope. Proliferation detected by cyclin A antibody was unchanged in samples overexpressing β-arrestin-1, compared with control. Cells overexpressing β-arrestin-1 did not display increased apoptosis, detected by caspase-3 antibody. No cleaved caspase-3 was detected, indicating that cells were not apoptotic. V, vector.
References
- 1.Lefkowitz R.J., Whalen E.J. beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Shenoy S.K., Lefkowitz R.J. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J., Shi Y., Xiang B., Qu B., Su W., Zhu M., Zhang M., Bao G., Wang F., Zhang X., Yang R., Fan F., Chen X., Pei G., Ma L. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [Erratum appeared in Cell 2006, 124:645] [DOI] [PubMed] [Google Scholar]
- 4.Ma L., Pei G. Beta-arrestin signaling and regulation of transcription. J Cell Sci. 2007;120:213–218. doi: 10.1242/jcs.03338. [DOI] [PubMed] [Google Scholar]
- 5.Lefkowitz R.J., Shenoy S.K. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 6.Zou L., Yang R., Chai J., Pei G. Rapid xenograft tumor progression in beta-arrestin1 transgenic mice due to enhanced tumor angiogenesis. FASEB J. 2008;22:355–364. doi: 10.1096/fj.07-9046com. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan F.G., Gorden D.L., Matta P., Shi Q., Matrisian L.M., DuBois R.N. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–1497. doi: 10.1073/pnas.0510562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge L., Shenoy S.K., Lefkowitz R.J., DeFea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J Biol Chem. 2004;279:55419–55424. doi: 10.1074/jbc.M410312200. [DOI] [PubMed] [Google Scholar]
- 9.Li T.T., Alemayehu M., Aziziyeh A.I., Pape C., Pampillo M., Postovit L.M., Mills G.B., Babwah A.V., Bhattacharya M. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res. 2009;7:1064–1077. doi: 10.1158/1541-7786.MCR-08-0578. [DOI] [PubMed] [Google Scholar]
- 10.Zhao M., Zhou G., Zhang Y., Chen T., Sun X., Stuart C., Hanley G., Li J., Zhang J., Yin D. Beta-arrestin2 inhibits opioid-induced breast cancer cell death through Akt and caspase-8 pathways. Neoplasma. 2009;56:108–113. doi: 10.4149/neo_2009_02_108. [DOI] [PubMed] [Google Scholar]
- 11.Calabrese G., Sallese M., Stornaiuolo A., Morizio E., Palka G., De Blasi A. Assignment of the beta-arrestin 1 gene (ARRB1) to human chromosome 11q13. Genomics. 1994;24:169–171. doi: 10.1006/geno.1994.1594. [DOI] [PubMed] [Google Scholar]
- 12.Luttrell L.M., Ferguson S.S., Daaka Y., Miller W.E., Maudsley S., Della Rocca G.J., Lin F., Kawakatsu H., Owada K., Luttrell D.K., Caron M.G., Lefkowitz R.J. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 13.Bièche I., Olivi M., Noguès C., Vidaud M., Lidereau R. Prognostic value of CCND1 gene status in sporadic breast tumours, as determined by real-time quantitative PCR assays. Br J Cancer. 2002;86:580–586. doi: 10.1038/sj.bjc.6600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenny F.S., Hui R., Musgrove E.A., Gee J.M., Blamey R.W., Nicholson R.I., Sutherland R.L., Robertson J.F. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999;5:2069–2076. [PubMed] [Google Scholar]
- 15.Jirström K., Stendahl M., Rydén L., Kronblad A., Bendahl P.O., Stål O., Landberg G. Adverse effect of adjuvant tamoxifen in premenopausal breast cancer with cyclin D1 gene amplification. Cancer Res. 2005;65:8009–8016. doi: 10.1158/0008-5472.CAN-05-0746. [DOI] [PubMed] [Google Scholar]
- 16.Schraml P., Schwerdtfeger G., Burkhalter F., Raggi A., Schmidt D., Ruffalo T., King W., Wilber K., Mihatsch M.J., Moch H. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am J Pathol. 2003;163:985–992. doi: 10.1016/S0002-9440(10)63458-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holm C., Rayala S., Jirström K., Stål O., Kumar R., Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst. 2006;98:671–680. doi: 10.1093/jnci/djj185. [DOI] [PubMed] [Google Scholar]
- 18.Ormandy C.J., Musgrove E.A., Hui R., Daly R.J., Sutherland R.L. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003;78:323–335. doi: 10.1023/a:1023033708204. [DOI] [PubMed] [Google Scholar]
- 19.Shuster M.I., Han L., Le Beau M.M., Davis E., Sawicki M., Lese C.M., Park N.H., Colicelli J., Gollin S.M. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chromosomes Cancer. 2000;28:153–163. [PubMed] [Google Scholar]
- 20.Lundgren K., Holm K., Nordenskjöld B., Borg A., Landberg G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008;10:R81. doi: 10.1186/bcr2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson P., Paavilainen L., Larsson K., Odling J., Sundberg M., Andersson A.C., Kampf C., Persson A., Al-Khalili Szigyarto C., Ottosson J., Björling E., Hober S., Wernérus H., Wester K., Pontén F., Uhlén M. Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics. 2005;5:4327–4337. doi: 10.1002/pmic.200500072. [DOI] [PubMed] [Google Scholar]
- 22.Uhlén M., Björling E., Agaton C., Szigyarto C.A., Amini B., Andersen E. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Andersson A.C., Strömberg S., Bäckvall H., Kampf C., Uhlén M., Wester K., Pontén F. Analysis of protein expression in cell microarrays: a tool for antibody-based proteomics. J Histochem Cytochem. 2006;54:1413–1423. doi: 10.1369/jhc.6A7001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pontén F., Jirström K., Uhlén M. The Human Protein Atlas–a tool for pathology. J Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- 25.Jögi A., Brennan D.J., Rydén L., Magnusson K., Fernö M., Stål O., Borgquist S., Uhlén M., Landberg G., Påhlman S., Pontén F., Jirström K. Nuclear expression of the RNA-binding protein RBM3 is associated with an improved clinical outcome in breast cancer. Mod Pathol. 2009;22:1564–1574. doi: 10.1038/modpathol.2009.124. [DOI] [PubMed] [Google Scholar]
- 26.Björling E., Lindskog C., Oksvold P., Linné J., Kampf C., Hober S., Uhlén M., Pontén F. A Web-based tool for in silico biomarker discovery based on tissue-specific protein profiles in normal and cancer tissues. Mol Cell Proteomics. 2008;7:825–844. doi: 10.1074/mcp.M700411-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Lundgren K., Nordenskjöld B., Landberg G. Hypoxia, Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br J Cancer. 2009;101:1769–1781. doi: 10.1038/sj.bjc.6605369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song X., Raman D., Gurevich E.V., Vishnivetskiy S.A., Gurevich V.V. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rexhepaj E., Brennan D.J., Holloway P., Kay E.W., McCann A.H., Landberg G., Duffy M.J., Jirström K., Gallagher W.M. Novel image analysis approach for quantifying expression of nuclear proteins assessed by immunohistochemistry: application to measurement of oestrogen and progesterone receptor levels in breast cancer. Breast Cancer Res. 2008;10:R89. doi: 10.1186/bcr2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helczynska K., Larsson A.M., Holmquist Mengelbier L., Bridges E., Fredlund E., Borgquist S., Landberg G., Påhlman S., Jirström K. Hypoxia-inducible factor-2alpha correlates to distant recurrence and poor outcome in invasive breast cancer. Cancer Res. 2008;68:9212–9220. doi: 10.1158/0008-5472.CAN-08-1135. [DOI] [PubMed] [Google Scholar]
- 31.Rydén L., Jönsson P.E., Chebil G., Dufmats M., Fernö M., Jirström K., Källström A.C., Landberg G., Stål O., Thorstenson S., Nordenskjöld B., South Swedish Breast Cancer Group; South-East Swedish Breast Cancer Group Two years of adjuvant tamoxifen in premenopausal patients with breast cancer: a randomised, controlled trial with long-term follow-up. Eur J Cancer. 2005;41:256–264. doi: 10.1016/j.ejca.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 32.Jirström K., Rydén L., Anagnostaki L., Nordenskjöld B., Stål O., Thorstenson S., Chebil G., Jönsson P.E., Fernö M., Landberg G. Pathology parameters and adjuvant tamoxifen response in a randomised premenopausal breast cancer trial. J Clin Pathol. 2005;58:1135–1142. doi: 10.1136/jcp.2005.027185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rydén L., Landberg G., Stål O., Nordenskjöld B., Fernö M., Bendahl P.O. HER2 status in hormone receptor positive premenopausal primary breast cancer adds prognostic, but not tamoxifen treatment predictive, information. Breast Cancer Res Treat. 2008;109:351–357. doi: 10.1007/s10549-007-9660-2. [DOI] [PubMed] [Google Scholar]
- 34.Kronblad A., Jirström K., Rydén L., Nordenskjöld B., Landberg G. Hypoxia inducible factor-1alpha is a prognostic marker in premenopausal patients with intermediate to highly differentiated breast cancer but not a predictive marker for tamoxifen response. Int J Cancer. 2006;118:2609–2616. doi: 10.1002/ijc.21676. [DOI] [PubMed] [Google Scholar]
- 35.McShane L.M., Altman D.G., Sauerbrei W., Taube S.E., Gion M., Clark G.M., Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics Reporting recommendations for tumor marker prognostic studies (REMARK) J Natl Cancer Inst. 2005;97:1180–1184. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 36.Elenbaas B., Weinberg R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 37.Rhodes L.V., Muir S.E., Elliott S., Guillot L.M., Antoon J.W., Penfornis P., Tilghman S.L., Salvo V.A., Fonseca J.P., Lacey M.R., Beckman B.S., McLachlan J.A., Rowan B.G., Pochampally R., Burow M.E. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res Treat. 2010;121:293–300. doi: 10.1007/s10549-009-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finak G., Bertos N., Pepin F., Sadekova S., Souleimanova M., Zhao H., Chen H., Omeroglu G., Meterissian S., Omeroglu A., Hallett M., Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 39.Polyak K., Haviv I., Campbell I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Mueller M.M., Fusenig N.E. Friends or foes–bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 41.Werner S., Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 42.Sappino A.P., Skalli O., Jackson B., Schürch W., Gabbiani G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer. 1988;41:707–712. doi: 10.1002/ijc.2910410512. [DOI] [PubMed] [Google Scholar]
- 43.Chauhan H., Abraham A., Phillips J.R., Pringle J.H., Walker R.A., Jones J.L. There is more than one kind of myofibroblast: analysis of CD34 expression in benign, in situ, and invasive breast lesions. J Clin Pathol. 2003;56:271–276. doi: 10.1136/jcp.56.4.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunha G.R., Hayward S.W., Wang Y.Z., Ricke W.A. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 45.Weber F., Xu Y., Zhang L., Patocs A., Shen L., Platzer P., Eng C. Microenvironmental genomic alterations and clinicopathological behavior in head and neck squamous cell carcinoma. JAMA. 2007;297:187–195. doi: 10.1001/jama.297.2.187. [DOI] [PubMed] [Google Scholar]
- 46.Harris A.L. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 47.Sørlie T., Perou C.M., Tibshirani R., Aas T., Geisler S., Johnsen H., Hastie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Thorsen T., Quist H., Matese J.C., Brown P.O., Botstein D., Eystein Lønning P., Børresen-Dale A.L. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hattar R., Maller O., McDaniel S., Hansen K.C., Hedman K.J., Lyons T.R., Lucia S., Wilson R.S., Jr, Schedin P. Tamoxifen induces pleiotrophic changes in mammary stroma resulting in extracellular matrix that suppresses transformed phenotypes. Breast Cancer Res. 2009;11:R5. doi: 10.1186/bcr2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shekhar M.P., Santner S., Carolin K.A., Tait L. Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. Am J Pathol. 2007;170:1546–1560. doi: 10.2353/ajpath.2007.061004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta P.B., Proia D., Cingoz O., Weremowicz J., Naber S.P., Weinberg R.A., Kuperwasser C. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer Res. 2007;67:2062–2071. doi: 10.1158/0008-5472.CAN-06-3895. [DOI] [PubMed] [Google Scholar]
- 51.Paz K., Hadari Y.R. Targeted therapy of the insulin-like growth factor-1 receptor in cancer. Comb Chem High Throughput Screen. 2008;11:62–69. doi: 10.2174/138620708783398313. [DOI] [PubMed] [Google Scholar]
- 52.Girnita L., Shenoy S.K., Sehat B., Vasilcanu R., Girnita A., Lefkowitz R.J., Larsson O. {beta}-Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. J Biol Chem. 2005;280:24412–24419. doi: 10.1074/jbc.M501129200. [DOI] [PubMed] [Google Scholar]
- 53.Girnita L., Shenoy S.K., Sehat B., Vasilcanu R., Vasilcanu D., Girnita A., Lefkowitz R.J., Larsson O. Beta-arrestin and Mdm2 mediate IGF-1 receptor-stimulated ERK activation and cell cycle progression. J Biol Chem. 2007;282:11329–11338. doi: 10.1074/jbc.M611526200. [DOI] [PubMed] [Google Scholar]
- 54.Sloan E.K., Ciocca D.R., Pouliot N., Natoli A., Restall C., Henderson M.A., Fanelli M.A., Cuello-Carrión F.D., Gago F.E., Anderson R.L. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–2043. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Witkiewicz A.K., Dasgupta A., Sotgia F., Mercier I., Pestell R.G., Sabel M., Kleer C.G., Brody J.R., Lisanti M.P. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–2034. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kubale V., Abramović Z., Pogacnik A., Heding A., Sentjurc M., Vrecl M. Evidence for a role of caveolin-1 in neurokinin-1 receptor plasma-membrane localization, efficient signaling, and interaction with beta-arrestin 2. Cell Tissue Res. 2007;330:231–245. doi: 10.1007/s00441-007-0462-y. [DOI] [PubMed] [Google Scholar]
- 57.Paulsson J., Sjöblom T., Micke P., Pontén F., Landberg G., Heldin C.H., Bergh J., Brennan D.J., Jirström K., Ostman A. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol. 2009;175:334–341. doi: 10.2353/ajpath.2009.081030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hui R., Campbell D.H., Lee C.S., McCaul K., Horsfall D.J., Musgrove E.A., Daly R.J., Seshadri R., Sutherland R.L. EMS1 amplification can occur independently of CCND1 or INT-2 amplification at 11q13 and may identify different phenotypes in primary breast cancer. Oncogene. 1997;15:1617–1623. doi: 10.1038/sj.onc.1201311. [DOI] [PubMed] [Google Scholar]
- 59.Chunder N., Mandal S., Roy A., Roychoudhury S., Panda C.K. Analysis of different deleted regions in chromosome 11 and their interrelations in early- and late-onset breast tumors: association with cyclin D1 amplification and survival. Diagn Mol Pathol. 2004;13:172–182. doi: 10.1097/01.pas.0000124337.49401.0b. [DOI] [PubMed] [Google Scholar]