

Abstract
Peroxisome, a ubiquitous subcellular organelle, plays an important function in cellular metabolism, and its importance for human health is underscored by the identification of fatal disorders caused by genetic abnormalities. Recent findings indicate that peroxisomal dysfunction is not restricted only to inherited peroxisomal diseases but also to disease processes associated with generation of inflammatory mediators that downregulate cellular peroxisomal homeostasis. Evidence indicates that leukodystrophies (X-linked adrenoleukodystrophy, globoid cell leukodystrophy, periventricular leukomalacia) may share common denominators in the development and progression of the inflammatory process and thus in the dysfunctions of peroxisomes. Dysfunctions of peroxisomes may therefore contribute in part to white matter disease and to the mental and physical disabilities that develop in patients affected by these diseases.
Keywords: cytokines, inflammation, leukodystrophies, myelin, neuroinflammation, peroxisomal disorders
Introduction
Peroxisome is a subcellular organelle widely present in eukaryotic cells that play important metabolic roles in the synthesis of myelin lipid plasmalogens and the catabolism of fatty acids (very-long-chain fatty acids, phytanic acid/branched-chain fatty acids, dicarboxylic acids, eicosanoids derived [prostaglandins, thromboxanes, leukotrienes, hydroxyeicosatetraenoic acids]) by a set of β-oxidation reactions,1,2 and detoxification of oxidase-produced hydrogen peroxide (H2O2) (Figures 1 A-C).3 Based on peroxisomal protein content in the cell and on the many oxidases it contains,4 peroxisomes can metabolize as much as 20% of cellular oxygen,1,5 converting most of it to H2O2. Hence peroxisomal oxygen metabolism contributes significantly to overall cellular oxygen consumption and H2O2 production.1,5 However, the peroxisomal enzyme catalase detoxifies H2O2 to water and molecular oxygen.2 Lack of detoxification of H2O2 in peroxisomes as a result of absence of catalase targeting to peroxisomes (catalase-negative peroxisomes) was described in patients with progressive developmental delay, micronodular cirrhosis, and elevated very-long-chain fatty acids in plasma and skin fibroblasts.6 Inherited abnormalities in peroxisomal β-oxidation such as X-adrenoleukodystrophy (X-ALD) leads to progressive inflammatory demyelinating disease (Figure 1D).7 In addition to catabolic and detoxifying functions, peroxisome participates in the synthesis of important membrane structural components such as plasmalogens, long-chain alcohol, and n-3/n-6 polyunsaturated fatty acids.2 Therefore, peroxisome has the potential to not only regulate the bioavailability of important intermediates of inflammation and oxygen metabolism but also to provide structural components of myelin membranes in the central and peripheral nervous system as well as of others organs’ membranes.
Figure 1.

Peroxisomes, peroxisomal pathways, and white matter disease. Morphology of catalase-stained liver peroxisomes (A); β-oxidative pathway for degradation of very-long-chain fatty acids, hydrogen peroxide generation, and detoxification by catalase (B); and initial reactions of plasmalogens synthesis in peroxisomes (C). Course of progression of demyelination in the occipital region (arrows) of a brain affected with the severe form of adrenoleukodystrophy, as seen by magnetic resonance imaging. Images were obtained at patient age 7 (a), age 8 (b), and age 9 (c).30
The identification of fatal neurological peroxisomal disorders (Table 1)7-9 encompassing dysfunction in the peroxisomal assembly (biogenesis)9 or individual enzyme functions8,10 highlights the importance of peroxisomes for human health. Peroxisomes are also responsible for turnover of a number of proinflammatory mediators such as metabolite of arachidonic acid and thus participate in inflammatory disease10, and abnormality in very-long-chain fatty acid turnover by peroxisomes, such as in adrenoleukodystrophy, upregulates the expression of inflammatory mediators and thus inflammatory disease.7 Therefore, once peroxisomal functions are affected, a self-amplification cycle perpetuates itself by further inhibiting peroxisomal functions, resulting in further amplification of the inflammatory cascade. We summarize the observations that dysfunction of peroxisomes leads to neuroinflammatory disease in childhood adrenoleukodystrophy and that neuroinflammatory disease in lysosomal disorders like globoid cell leukodystrophy (Krabbe disease) and inflammatory disease in cerebral palsy results in peroxisomal dysfunction and dysmyelination. Recent studies on the pathology of these disorders provide evidence that peroxisomal functions are essential for proper brain growth and development.11-13
Table 1.
Peroxisomal Disorders
| Group 1: Peroxisome Biogenesis Disorders |
| Zellweger spectrum |
| Zellweger syndrome (severe) |
| Neonatal adrenoleukodystrophy (mild) |
| Infantile Refsum disease (mild) |
| Classical rhizomelic chondrodysplasia punctate (RCDP type 1) |
| Group 2: Single Enzyme Deficiencies |
| Disorders of fatty acid 3-oxidation |
| X-linked adrenoleukodystrophy |
| Acyl-CoA oxidase deficiency |
| D-bifunctional protein deficiency |
| Thiolase deficiency |
| 2-methylacyl-CoA racemase deficiency |
| Disorders of ether-phospholipids (plasmalogens) biosynthesis |
| Dihydroxyacetonephosphate acyltransferase (DHAPAT) deficiency (RCDP type 2) |
| Alkyldihydroxyacetonephosphate synthase (akyl DHAP synthase) deficiency (RCDP type 3) |
| Disorder of fatty acid α-oxidation |
| Phytanoyl-CoA hydroxylase deficiency (Refsum disease) |
| Disorder of the isoprenoid biosynthesis |
| Mevalonate kinase deficiency |
| Disorder of L-lysine metabolism |
| L-pipecolate oxidase deficiency |
| Disorder of glutaryl-CoA metabolism |
| Glutaryl-CoA oxidase deficiency |
| Disorder of the hydrogen peroxide metabolism |
| Catalase deficiency |
| Disorder of glyoxylate detoxification: |
| Alanine:glyoxylate aminotransferase deficiency (hyperoxaluria type I) |
| Others |
| TRIM37 (RING-B-box-coiled-coil protein) deficiency (Mulibrey nanism) |
| DLP1 (dynamin-like GTPase) deficiency (affecting mitochondria and peroxisomes) |
Childhood White Matter Diseases with Peroxisomal Dysfunction
X-Adrenoleukodystrophy (X-ALD)
The most common peroxisomal disorder, X-ALD, is an inherited metabolic disease that affects the nervous system of boys during early childhood (cerebral form, cALD), the severe form of the disease, or early adulthood (adrenomyeloneuropathy, AMN), the mild form of the disease.1,7,8 The affected gene encodes a peroxisomal membrane protein14 that is a member of the ATP-binding cassette family of transporters (ABCD1 or adrenoleukodystrophy protein [ALDP])15 whose function still is unclear. Consequence of the ABCD1 gene defect is the accumulation of straight, saturated very-long-chain fatty acids (those containing more than 22 carbons) in nervous and peripheral tissues and organs.1 Biochemical analysis using cultured skin fibroblasts derived from X-ALD patients demonstrated alteration in the degradation of those fatty acids by peroxisomal β-oxidation16 and an enhanced elongation of fatty acids, an endoplasmic reticulum function.17 Although the precise function of ABCD1 is not understood at present, the fact that transfection of ABCD1 into adrenoleukodystrophy fibroblasts normalizes the fatty acids levels suggests that ABCD1 is involved in the metabolism of very-long-chain fatty acids.7,8 However, lack of correlation between genotype and phenotype as identical mutations can induce the mild or severe form of the disease, indicates that an additional genetic factor (modifier gene) may play a role in the progression of the disease.18,19 In addition to ABCD1, 3 other peroxisomal membrane transporters with sequence homology to ABCD1 (ie, ABCD2,20 ABCD3,21 and ABCD422) were also found in the peroxisomal membrane. Although these transporters share the ability to compensate/overlap functionality of ABCD1 deficiency in vitro under pharmacologic or genetic manipulations,23-25 their in vivo compensation is an open question due to their differential developmental expression26 as well as differential tissue expression.27 Recently, a genetic study of sequence and segregation of the ABCD2 gene in a large adrenoleukodystrophy kindred has excluded this gene as a possible modifier locus for the observed clinical diversity in X-ALD.28
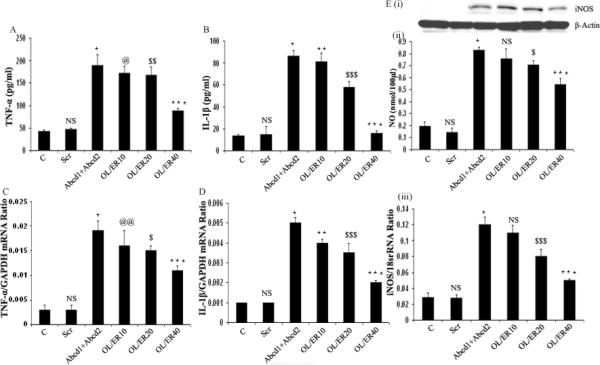
The disease development in X-ALD is associated with the cytotoxic effect of the accumulation of very-long-chain fatty acids (metabolic disease)29 and the subsequent triggering of an inflammatory cascade (inflammatory disease)1,7,8 that ultimately leads to demyelination and loss of oligodendrocytes (Figure 1D).8,30 The observation of increased accumulation of very-long-chain fatty acids in inflammatory areas than in histological normal areas of X-ALD brain (3-folds) suggests that inflammatory mediators in turn downregulate peroxisomes/peroxisomal functions, thus creating a perpetual inflammatory cascade.31 These conclusions are consistent with excessive accumulations of very-long-chain fatty acids in C6 glial cells after treatment with proinflammatory cytokines (Table 2).32 In addition, the observed loss of plasmalogens in inflammatory areas of cALD brain strongly indicates loss of peroxisomal function in those areas.33 This data suggests a relationship between the expression of inflammation mediators, impairment of peroxisomal function, and accumulation of very-long-chain fatty acids in cALD brain.7 Although the expression of mediators of inflammatory disease in cALD have been known since the 1990s,8 the role of very-long-chain fatty acids in the induction of inflammatory disease eluded very many efforts. The absence of an inflammatory neurological disease in the Aldp (Abcd1) knockout mouse,34 and observed similar levels of very-long-chain fatty acids in cALD that express neuroinflammatory disease and adult form of X-ALD (AMN) that does not express neuroinflammatory clinical disease,8 even questioned the relationship of very-long-chain fatty acid accumulation to neuroinflammatory disease of X-ALD. However, recently we reported that silencing the peroxisomal transporters Abcd1 and Abcd2 expression in mouse primary cultures of astrocytes result in expression of inflammatory mediators by these cells.35 We demonstrated that silencing of both genes induced oxidative imbalance (reactive oxygen species production), expression of enzymes of the eicosanoid pathway (5-lipoxoygenase, cycloxygenase-2), and induction of inflammatory mediators such as cytokines (tumor necrosis factor alpha, TNFα; interleukin-1beta, IL-1β) and inducible nitric oxide synthase (iNOS) and production of nitric oxide (Figure 2) as well as alteration of peroxisomal metabolism.35 All these changes were downregulated by reducing the very-long-chain fatty acid load by treatment with a mixture of oleic acid (C18:1, n-9) and erucic acid (C22:1, n-9), known as Lorenzo's oil.8,35 This is the first study that clearly established a relationship between deletion of Abcd1 expression, excessive accumulation of very-long-chain fatty acids, and induction of neuroinflammatory disease. Moreover, silencing of Abcd3 also induced the expression of inflammatory mediators in C6 cells.36 These observations support the role of very-long-chain fatty acids in the observed Th1 response by peripheral blood mononuclear cells.37,38 Taken together, these studies indicate that cells exposed to or induced to accumulate very-long-chain fatty acids become prone to higher expression and release of inflammatory intermediates.
Table 2.
Levels of Saturated Very-Long-Chain Fatty Acids in X-ALD-Affected Brains
Figure 2.

Silencing of Abcd1/Abcd2 genes in primary astrocytes induce protein levels (A, B, Ei) and expression (C, D, Eiii) of the proinflammatory mediators tumor necrosis factor-α (A, C), interleukine-1β (B, D), and inducible nitric oxide synthase (Ei, Eiii) as well as of nitric oxide levels (Eii). Those changes were downregulated by “Lorenzo's oil” treatment (mixture of oleic acid (OL) and erucic acid [ER]).35
X-Adrenoleukodystrophy mice (Aldp-deficient)34 do not express a clear phenotype of neurological disease process as observed in cALD39; however, they present a mild oxidative imbalance in spinal cord40 and a mild phenotype of the disease later in life.41 Understanding the difference between the human and animal phenotype may hold the key to understand the cause of the transition from metabolic to inflammatory disease in X-ALD.
Globoid Cell Leukodystrophy (Krabbe Disease)
Recent studies described the loss of peroxisomes/ peroxisomal functions secondary to dysfunction of lysosomes in globoid cell leukodystrophy (GCL),42,43 a genetic disorder caused by loss of activity of the lysosomal enzyme galactosylceramidase,44 leading to accumulation of galactosylsphingosine (psychosine) in the nervous tissue44,45 and peripheral organs.46 Psychosine-accumulation becomes cytotoxic especially for oligodendrocytes, resulting in early demyelination.44 Studies using an animal model of GCL (twitcher mouse)47 and patients’ postmortem tissues indicate that psychosine accumulation triggers an inflammatory process, inducing proinflammatory cytokines,48 iNOS,49 and an oxidative imbalance43 in the central nervous system. Those intermediates ultimately exert a cytotoxic effect, inducing apoptotic cell death in oligodendrocyte,50,51 glial activation,52 and demyelination/dysmyelination53 as a mechanism of progressive neurodegeneration in this disease. Progression of inflammatory disease may be accelerated by impairment of peroxisomal function as a consequence of the action of proinflammatory mediators. Experimental evidence suggests that peroxisomal dysfunction during inflammation can be the consequence of 2 different mechanisms: Inhibition of peroxisomal enzymes,54,55 mediated by inflammatory intermediates such as nitric oxide and free radicals56,57; and inhibition of peroxisomal biogenesis,55,58,59 mediated by downregulation of the transcription factor peroxisome proliferator-activated receptor-alpha (PPARα) activity.42,60. PPARα plays a role in inflammation control,61 but how the progression of inflammation can affect its activity is unknown. Because psychosine accumulation induces an inflammatory process in nervous tissue,48,49 this event in turn likely leads to impaired peroxisomal structure and function during disease progression. Indeed, during postnatal development we observed an increase in inflammatory mediator TNFα parallel to the decrease in PPARα and PPAR-dependent peroxisomal protein expression (acyl-CoA oxidase-1 and alkyl-DHAP synthase; Figure 3) and thus peroxisomal functions.42,43 Therefore, psychosine-mediated induction of inflammatory disease downregulates peroxisomes, and the loss of peroxisomes, in turn, upregulates inflammatory disease. Hence, these events perpetuate inflammatory disease.
Figure 3.

Reduction of protein levels of the peroxisomal enzyme alkyldihydroxyacetone phosphate synthase (alkyl-DHAP synthase) (B, C) parallels the induction of the inflammatory cytokine tumor necrosis factor-α (TNF-α)(A) in twitcher mouse brain, an animal model of globoid cell leukodystrophy.42
Periventricular Leukomalacia (Cerebral White Matter Injury)
Periventricular leukomalacia is the predominant form of brain injury and the leading known cause of neurodevelopmental impairments such as cerebral palsy and cognitive deficits in premature infants.62,63 The neuropathologic hallmarks of this disease are diffuse microglial activation64 and focal and diffuse periventricular depletion of premyelinating oligodendroglia.62,65 Premyelinating oligodendroglia are highly vulnerable to death caused by glutamate,66 free radicals,67 and proinflammatory cytokines;65,68 some common factors described also in the development and progression of adrenoleukodystrophy and globoid cell leukodystrophy disease.
We have induced periventricular white matter lesion in the forebrain of immature experimental animals by activating the innate immune system by administration of bacterial lipopolysaccharide to pregnant rats.69,70 Under these conditions, we have reported a decrease in numbers of developing oligodendrocytes (determined by the cellular markers O4+ and O1+;Table 3) and in the number of platelet-derived growth factor-alpha receptor (PDGF-αR) and NG2 chondroitin sulphate proteoglycan (NG2) immunoreactive oligodendrocyte precursor cells as well as in total content of PDGF-αR during brain early development (embryonic days 18-20).69 Concomitant with the loss of oligodendrocytes, we observed alteration in myelination, as decreased expression of myelin proteins levels (myelin basic protein) in the injured embryonic brains (Figure 4).70 The recovery of cell number, PDGF-αR mRNA, and myelin basic protein levels in the central nervous system of animals pretreated with the antioxidant and glutathione precursor N-acetylcysteine indicates the role of oxidative imbalance in the depletions of oligodendrocyte precursor cells from embryonic brains (Figure 4).67,69,70 In addition, the presence of malondialdehyde, 4-hydroxynonenal and reduction of reduced glutathione levels in injured fetal brains that were corrected by N-acetylcysteine pretreatment supports previous reports that suggested induction of oxidative imbalance as a mechanism for the observed oligodendrocyte depletion.70
Table 3.
Rat Brain Glial Cell Survival to Mothers’ Systemic Exposure to Bacterial LPS and Protection by NAC Pretreatment
| Groups | Primary Cell Culture Developing Oligodendrocytes | Astrocytes (GFAP+) | Neurons (ENOLASE+) | |
|---|---|---|---|---|
| CTL | 17.54 ± 0.02 *** | 21.76 ± 0.02 *** | 39.72 ± 0.23 | 13.71 ± 0.12 |
| LPS | 6.73 ± 0.01 | 4.06 ± 0.01 | 46.47 ± 0.46 | 12.57 ± 0.001 |
| NAC + LPS | 18.14 ± 0.02 *** | 21.38 ± 0.01 *** | 34.60 ± 0.26 | 14.93 ± 0.23 |
P < .001 versus LPS group. Rat brain glial cell survival in the fetal brain at embryonic day 20, 48 hours postbacterial LPS systemic exposure to pregnant mothers (embryonic day 18) and protection by NAC pretreatment (2 hours prior to LPS administration).
Abbreviations: CTL, control; GFAP+, glial fibrillary acidic protein (cellular marker for astrocytes); LPS, lipopolysaccharide; NAC, N-acetylcysteine; O4+; O1+, cellular markers for oligodendrocytes precursor cells. Modified from reference 70.
Figure 4.

Systemic injection of bacterial lipopolysaccharide into pregnant rats at embryonic date 18 induces demyelination in the brains of animals at postnatal days 23 and 30, determined by expression (A) and proteins levels of myelin basic protein (MBP)(B-D) in 2 different regions of the brains, corpus callosum (C) and cingulum (D). Pretreatment with the antioxidant drug N-acetylcysteine provide protection against lipopolysaccharide insult.70
Similar to the reported effects of inflammation on peroxisomal functions,42,54,55,58,59 the injured fetal brain from lipopolysaccharide-exposed mothers had significantly lower expression of peroxisomes measured as reduced level of peroxisomal enzymes for the synthesis of plasmalogens in peroxisomes (dihydroxyacetone phosphate acyltransferase),70 peroxisomal membrane transporter Abcd3 (Pmp70),21,70 and expression of transcription factor PPARα.70,71 These changes in peroxisomes correlated with the low expression of myelin basic protein in the injured fetal brain (Figure 5).70 The mRNA expression and protein levels of all the indicated proteins were restored by N-acetylcysteine pretreatment (Figure 5). Furthermore, cell culture studies demonstrated that lipopolysaccharide-stimulated glial cell (microglia/astrocyte)-released factors induce negative effect(s) on proliferation of oligodendrocytes progenitors (BrdU uptake by NG2+), thus the development of pro-oligodendrocytes (O4+).72 These altered functions were restored by N-acetylcysteine pretreatment.72 Again, mRNA expression and protein levels of peroxisomal protein Pmp70 was downregulated in the oligodendrocyte precursors (NG2+) as well as in developing oligodendrocytes (RIP+),72 and, as expected, N-acetylcysteine pretreatment restored the expression and level of these proteins.72 These observations indicate that loss of peroxisomes may interfere with maturation of oligodendrocytes (ie, brain myelination),11 thus suggesting a role of peroxisomal biology in maturation of oligodendrocytes for white matter myelination and repair.
Figure 5.
Systemic injection of bacterial lipopolysaccharide into pregnant rats at embryonic date 18 reduces protein levels (A-C, F, G) and expression (D, E) of the peroxisomal membrane protein of 70kDa (PMP70) (A-D) and of the transcription factor peroxisome proliferator-activated receptor-α (PPAR-α)(E), in parallel to decrease of myelin basic protein (MBP)(F, G), at the indicated postnatal days (PND). Pretreatment with N-acetylcysteine restored the expression and proteins levels of those proteins.70
Summary
The causes of white matter disease vary and include inherited disorders of lysosomes and peroxisomes, hypoxia, and infection-mediated toxicities (Figure 6). Inflammation seems to be common to many white matter disorders. In this review, we described the role of peroxisomes in the biology of oligodendrocyte progenitors and their differentiation into myelin-producing oligodendrocytes and, thus, myelination. Therefore, dysfunctions of peroxisomal biology observed in the described disease conditions may contribute, at least in part, to white matter disease.
Figure 6.

Peroxisomal dysfunction as an intermediate of dysmyelination/demyelination in white matter disease. Peroxisomal and lysosomal disorders and maternal infection converging in inflammatory disease, with further derangement of peroxisomal functions during development and/or progression of white matter disease.
Acknowledgment
Presented at the Neurobiology of Disease in Children Conference: Symposium on Injury to the Preterm Brain and Cerebral Palsy, in conjunction with the 37th Annual Meeting of the Child Neurology Society, Santa Clara, California, November 5, 2008. Supported by grants from the National Institutes of Health (5R13NS040925-09, NS-22576, NS-34741, NS-37766, NS-40810, 5R13NS040925-09), the Cerebral Palsy International Research Foundation, the Kennedy Krieger Institute, and the Child Neurology Society.
We thank A. Contreras for reading the manuscript.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Singh I. Biochemistry of peroxisomes in health and disease. Mol Cell Biochem. 1997;167:1–29. doi: 10.1023/a:1006883229684. [DOI] [PubMed] [Google Scholar]
- 2.Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 3.Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Baudhuin P, Beaufay H, Rahman-Li Y, et al. Tissue fractionation studies. 17. Intracellular distribution of monoamine oxidase, aspartate aminotransferase, alanine aminotransferase, D-amino acid oxidase and catalase in rat-liver tissue. Biochem J. 1964;92:179–184. doi: 10.1042/bj0920179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh I, Voigt RG, Sheikh FG, et al. Biochemical features of a patient with Zellweger-like syndrome with normal PTS-1 and PTS-2 peroxisomal protein import systems: a new peroxisomal disease. Biochem Mol Med. 1997;61:198–207. doi: 10.1006/bmme.1997.2593. [DOI] [PubMed] [Google Scholar]
- 7.Contreras M, Singh I. Adrenoleukodystrophy: Molecular, Metabolic, Pathologic, and Therapeutic Aspects. In: Lajtha A, editor. Handbook of Neurochemistry and Molecular Neurobiology. Springer; New York: 2009. pp. 13–42. [Google Scholar]
- 8.Moser HW, Smith KD, Watkins PA, et al. X-Linked Adrenoleukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3257–3301. [Google Scholar]
- 9.Gould SJ, Raymond GV, Valle D. The peroxisome biogenesis disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3181–3217. [Google Scholar]
- 10.Wanders RJ, Barth PG, Heymans HS. Single peroxisomal enzyme deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3219–3256. [Google Scholar]
- 11.Lazo O, Singh AK, Singh I. Postnatal development and isolation of peroxisomes from brain. J Neurochem. 1991;56:1343–1353. doi: 10.1111/j.1471-4159.1991.tb11431.x. [DOI] [PubMed] [Google Scholar]
- 12.Kassmann CM, Lappe-Siefke C, Baes M, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- 13.Krysko O, Hulshagen L, Janssen A, et al. Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J Neurosci Res. 2007;85:58–72. doi: 10.1002/jnr.21097. [DOI] [PubMed] [Google Scholar]
- 14.Contreras M, Mosser J, Mandel JL, et al. The protein coded by the X-adrenoleukodystrophy gene is a peroxisomal integral membrane protein. FEBS Lett. 1994;344:211–215. doi: 10.1016/0014-5793(94)00400-5. [DOI] [PubMed] [Google Scholar]
- 15.Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730. doi: 10.1038/361726a0. [DOI] [PubMed] [Google Scholar]
- 16.Lazo O, Contreras M, Hashmi M, et al. Peroxisomal lignoceroyl-CoA ligase deficiency in childhood adrenoleukodystrophy and adrenomyeloneuropathy. Proc Natl Acad Sci U S A. 1988;85:7647–7651. doi: 10.1073/pnas.85.20.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuji S, Sano T, Ariga T, Miyatake T. Increased synthesis of hexacosanoic acid (C23:0) by cultured skin fibroblasts from patients with adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN). J Biochem. (Tokyo) 1981;90:1233–1236. doi: 10.1093/oxfordjournals.jbchem.a133578. [DOI] [PubMed] [Google Scholar]
- 18.Maestri NE, Beaty TH. Predictions of a 2-locus model for disease heterogeneity: application to adrenoleukodystrophy. Am J Med Genet. 1992;44:576–582. doi: 10.1002/ajmg.1320440509. [DOI] [PubMed] [Google Scholar]
- 19.Moser HW, Moser AB, Smith KD, et al. Adrenoleukodystrophy: phenotypic variability and implications for therapy. J Inherit Metab Dis. 1992;15:645–664. doi: 10.1007/BF01799621. [DOI] [PubMed] [Google Scholar]
- 20.Lombard-Platet G, Savary S, Sarde CO, et al. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc Natl Acad Sci U S A. 1996;93:1265–1269. doi: 10.1073/pnas.93.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamijo K, Taketani S, Yokota S, et al. The 70-kDa peroxisomal membrane protein is a member of the Mdr (P-glycoprotein)-related ATP-binding protein superfamily. J Biol Chem. 1990;265:4534–4540. [PubMed] [Google Scholar]
- 22.Shani N, Jimenez-Sanchez G, Steel G, et al. Identification of a fourth half ABC transporter in the human peroxisomal membrane. Hum Mol Genet. 1997;6:1925–1931. doi: 10.1093/hmg/6.11.1925. [DOI] [PubMed] [Google Scholar]
- 23.Kemp S, Wei HM, Lu JF, et al. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261–1268. doi: 10.1038/3242. [DOI] [PubMed] [Google Scholar]
- 24.Gondcaille C, Depreter M, Fourcade S, et al. Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. J Cell Biol. 2005;169:93–104. doi: 10.1083/jcb.200501036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pujol A, Ferrer I, Camps C, et al. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet. 2004;13:2997–3006. doi: 10.1093/hmg/ddh323. [DOI] [PubMed] [Google Scholar]
- 26.Berger J, Albet S, Bentejac M, et al. The four murine peroxisomal ABC-transporter genes differ in constitutive, inducible and developmental expression. Eur J Biochem. 1999;265:719–727. doi: 10.1046/j.1432-1327.1999.00772.x. [DOI] [PubMed] [Google Scholar]
- 27.Troffer-Charlier N, Doerflinger N, Metzger E, et al. Mirror expression of adrenoleukodystrophy and adrenoleukodystrophy related genes in mouse tissues and human cell lines. Eur J Cell Biol. 1998;75:254–264. doi: 10.1016/S0171-9335(98)80121-0. [DOI] [PubMed] [Google Scholar]
- 28.Maier EM, Mayerhofer PU, Asheuer M, et al. X-linked adrenoleukodystrophy phenotype is independent of ABCD2 genotype. Biochem Biophys Res Commun. 2008;377:176–180. doi: 10.1016/j.bbrc.2008.09.092. [DOI] [PubMed] [Google Scholar]
- 29.Hein S, Schonfeld P, Kahlert S, Reiser G. Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet. 2008;17:1750–1761. doi: 10.1093/hmg/ddn066. [DOI] [PubMed] [Google Scholar]
- 30.Aubourg P. X-linked adrenoleukodystrophy. In: Vinken PJ, Bruyn GW, Moser HW, editors. Handbook of Clinical Neurology: Neurodystrophies and Neurolipidoses. Elsevier; Amsterdam: 1996. pp. 447–483. [Google Scholar]
- 31.Paintlia AS, Gilg AG, Khan M, et al. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425–439. doi: 10.1016/j.nbd.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Khan M, Pahan K, Singh AK, Singh I. Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem. 1998;71:78–87. doi: 10.1046/j.1471-4159.1998.71010078.x. [DOI] [PubMed] [Google Scholar]
- 33.Khan M, Singh J, Singh I. Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J Neurochem. 2008;106:1766–1779. doi: 10.1111/j.1471-4159.2008.05513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu JF, Lawler AM, Watkins PA, et al. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci U S A. 1997;94:9366–9371. doi: 10.1073/pnas.94.17.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh J, Khan M, Singh I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X-adrenoleukodystrophy. J Lipid Res. 2009;50:135–147. doi: 10.1194/jlr.M800321-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Benedetto R, Denti MA, Salvati S, et al. PMP70 knock-down generates oxidative stress and pro-inflammatory cytokine production in C6 glial cells. Neurochem Int. 2009;54:37–42. doi: 10.1016/j.neuint.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Di Biase A, Merendino N, Avellino C, et al. Th 1 cytokine production by peripheral blood mononuclear cells in X-linked adrenoleukodystrophy. J Neurol Sci. 2001;182:161–165. doi: 10.1016/s0022-510x(00)00469-x. [DOI] [PubMed] [Google Scholar]
- 38.Lannuzel A, Aubourg P, Tardieu M. Excessive production of tumour necrosis factor alpha by peripheral blood mononuclear cells in X-linked adrenoleukodystrophy. Eur J Paediatr Neurol. 1998;2:27–32. doi: 10.1016/1090-3798(98)01002-7. [DOI] [PubMed] [Google Scholar]
- 39.Powers JM, Pei Z, Heinzer AK, et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64:1067–1079. doi: 10.1097/01.jnen.0000190064.28559.a4. [DOI] [PubMed] [Google Scholar]
- 40.Fourcade S, Lopez-Erauskin J, Galino J, et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet. 2008;17:1762–1773. doi: 10.1093/hmg/ddn085. [DOI] [PubMed] [Google Scholar]
- 41.Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002;11:499–505. doi: 10.1093/hmg/11.5.499. [DOI] [PubMed] [Google Scholar]
- 42.Haq E, Contreras MA, Giri S, Singh I, Singh AK. Dysfunction of peroxisomes in twitcher mice brain: a possible mechanism of psychosine-induced disease. Biochem Biophys Res Commun. 2006;343:229–238. doi: 10.1016/j.bbrc.2006.02.131. [DOI] [PubMed] [Google Scholar]
- 43.Khan M, Haq E, Giri S, et al. Peroxisomal participation in psychosine-mediated toxicity: Implications for Krabbe's disease. J Neurosci Res. 2005;80:845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- 44.Wenger DA, Suzuki K, Suzuki Y, Suzuki K. Galactosylceramide Lipidosis: Globoid cell Leukodystrophy (Krabbe Disease). In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3669–3694. [Google Scholar]
- 45.Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi T, Shinoda H, Goto I, et al. Globoid cell leukodystrophy is a generalized galactosylsphingosine (psychosine) storage disease. Biochem Biophys Res Commun. 1987;144:41–46. doi: 10.1016/s0006-291x(87)80472-2. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki K. The twitcher mouse. A model of human globoid cell leukodystrophy (krabbe's disease). Am J Pathol. 1983;111:394–397. [PMC free article] [PubMed] [Google Scholar]
- 48.LeVine SM, Brown DC. IL-6 and TNFalpha expression in brains of twitcher, quaking and normal mice. J Neuroimmunol. 1997;73:47–56. doi: 10.1016/s0165-5728(96)00166-x. [DOI] [PubMed] [Google Scholar]
- 49.Giri S, Jatana M, Rattan R, et al. Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implications for Krabbe disease. Faseb J. 2002;16:661–672. doi: 10.1096/fj.01-0798com. [DOI] [PubMed] [Google Scholar]
- 50.Haq E, Giri S, Singh I, Singh AK. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86:1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- 51.Taniike M, Mohri I, Eguchi N, et al. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J Neuropathol Exp Neurol. 1999;58:644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Itoh M, Hayashi M, Fujioka Y, et al. Immunohistological study of globoid cell leukodystrophy. Brain Dev. 2002;24:284–290. doi: 10.1016/s0387-7604(02)00057-8. [DOI] [PubMed] [Google Scholar]
- 53.Ohno M, Komiyama A, Martin PM, Suzuki K. MHC class II antigen expression and T-cell infiltration in the demyelinating CNS and PNS of the twitcher mouse. Brain Res. 1993;625:186–196. doi: 10.1016/0006-8993(93)91058-z. [DOI] [PubMed] [Google Scholar]
- 54.Yasmineh WG, Parkin JL, Caspers JI, Theologides A. Tumor necrosis factor/cachectin decreases catalase activity of rat liver. Cancer Res. 1991;51:3990–3995. [PubMed] [Google Scholar]
- 55.Singh I, Paintlia AS, Khan M, et al. Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res. 2004;1022:1–11. doi: 10.1016/j.brainres.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 56.Dobashi K, Pahan K, Chahal A, Singh I. Modulation of endogenous antioxidant enzymes by nitric oxide in rat C6 glial cells. J Neurochem. 1997;68:1896–1903. doi: 10.1046/j.1471-4159.1997.68051896.x. [DOI] [PubMed] [Google Scholar]
- 57.Keng T, Privalle CT, Gilkeson GS, Weinberg JB. Peroxynitrite formation and decreased catalase activity in autoimmune MRL-lpr/lpr mice. Mol Med. 2000;6:779–792. [PMC free article] [PubMed] [Google Scholar]
- 58.Beier K, Volkl A, Fahimi HD. Suppression of peroxisomal lipid beta-oxidation enzymes of TNF-alpha. FEBS Lett. 1992;310:273–276. doi: 10.1016/0014-5793(92)81347-o. [DOI] [PubMed] [Google Scholar]
- 59.Contreras MA, Khan M, Smith BT, et al. Endotoxin induces structure-function alterations of rat liver peroxisomes: Kupffer cells released factors as possible modulators. Hepatology. 2000;31:446–455. doi: 10.1002/hep.510310226. [DOI] [PubMed] [Google Scholar]
- 60.Beier K, Volkl A, Fahimi HD. TNF-alpha downregulates the peroxisome proliferator activated receptor-alpha and the mRNAs encoding peroxisomal proteins in rat liver. FEBS Lett. 1997;412:385–387. doi: 10.1016/s0014-5793(97)00805-3. [DOI] [PubMed] [Google Scholar]
- 61.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 62.Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008;93:F153–F161. doi: 10.1136/adc.2006.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng W, Pleasure J, Pleasure D. Progress in periventricular leukomalacia. Arch Neurol. 2008;65:1291–1295. doi: 10.1001/archneur.65.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang Y, Cai Z, Rhodes PG. Effects of lipopolysaccharide on oligodendrocyte progenitor cells are mediated by astrocytes and microglia. J Neurosci Res. 2000;62:510–520. doi: 10.1002/1097-4547(20001115)62:4<510::AID-JNR5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 65.Bell MJ, Hallenbeck JM. Effects of intrauterine inflammation on developing rat brain. J Neurosci Res. 2002;70:570–579. doi: 10.1002/jnr.10423. [DOI] [PubMed] [Google Scholar]
- 66.Deng W, Wang H, Rosenberg PA, et al. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci U S A. 2004;101:7751–7756. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haynes RL, Folkerth RD, Keefe RJ, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 68.Li J, Ramenaden ER, Peng J, et al. Tumor necrosis factor alpha mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. J Neurosci. 2008;28:5321–5330. doi: 10.1523/JNEUROSCI.3995-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paintlia MK, Paintlia AS, Barbosa E, et al. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- 70.Paintlia MK, Paintlia AS, Contreras MA, et al. Lipopolysaccharide-induced peroxisomal dysfunction exacerbates cerebral white matter injury: attenuation by N-acetyl cysteine. Exp Neurol. 2008;210:560–576. doi: 10.1016/j.expneurol.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 72.Paintlia MK, Paintlia AS, Khan M, et al. Modulation of peroxisome proliferator-activated receptor-alpha activity by N-acetyl cysteine attenuates inhibition of oligodendrocyte development in lipopolysaccharide stimulated mixed glial cultures. J Neurochem. 2008;105:956–970. doi: 10.1111/j.1471-4159.2007.05199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]