

Abstract
Relating specific genetic alterations to prognosis may help improve prognostication in melanoma, may identify key oncogenic drivers in cancer, and may assist in developing targeted therapies. Characteristic genetic alterations in melanoma include chromosomal copy number aberrations. We evaluated 97 melanomas (55 metastasizing and 42 nonmetastasizing) after a minimum 5-year follow-up in a case-control study using fluorescence in situ hybridization, targeting commonly altered chromosomal loci in melanoma. Eight probes arranged in two panels were used, and 11 parameters were evaluated. Parameters showing a statistically significant difference between the metastasizing and nonmetastasizing groups were evaluated with multivariate logistic regression analysis to compare their prognostic potential with other traditional prognostic markers used by the American Joint Committee on Cancer. Four of 11 parameters evaluated, including CCND1 (alias Bcl-1) gain, CCND1 r-gain, MYC (alias c-myc) gain, and MYC r-gain, had a statistically significant difference in the metastasizing versus nonmetastasizing group. All four parameters maintained statistical significance when evaluated in separate multivariate logistic regression analyses that included the seven currently used American Joint Commission on Cancer prognosticators in melanoma. In multivariate analyses, these four parameters were second only to ulceration in their prognostic potential. Copy number changes at 11q13 and 8q34 harboring CCND1 and MYC, respectively, are highly associated with prognosis. Fluorescence in situ hybridization targeting these loci may be a useful standardized prognostic marker in melanoma skin cancer.
The incidence and mortality rates of melanoma have been increasing during the past few decades.1 The American Cancer Society estimates that the lifetime risk of developing melanoma is approximately 1 in 50 for whites, 1 in 1000 for blacks, and 1 in 200 for Hispanics. Overall, melanoma is the sixth most common cancer in men and the seventh most common cancer in women; in 2009, 68,720 new cases of invasive melanoma and 8650 deaths were reported in the United States (http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2009, last accessed October 10, 2010). Although the number of melanoma cancer–related deaths continues to increase and treatment of advanced melanoma continues to show dismal results, there have been several breakthroughs in the past decade. This includes stratification of melanoma into molecular subtypes that correlate to prognosis2 and targeted therapy that can be tailored to the specific activated oncogenic pathway.3,4 For example, specific targeted inhibitors, such as v-raf murine sarcoma viral oncogene homolog B1 (BRAF) inhibitors or v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) inhibitors (eg, imatinib mesylate), have been successfully used to treat patients with advanced melanoma.3,5 Therefore, identifying specific oncogenic pathways in melanoma can help stratify patients with melanoma prognostically and predict therapeutic results.
In addition to somatic mutations, copy number aberrations through gain of specific chromosomal segments containing relevant oncogenes or loss of chromosomal segments harboring critical tumor suppressor genes are highly characteristic of melanoma. In fact, studies with comparative genomic hybridization show that 95% of melanomas have chromosomal copy number aberrations.6 Frequent chromosomal copy number losses include deletions at 9p (82%), 10q (63%), 6q (28%), and 8p (22%). Frequent copy number gains may occur at 7q (50%), 8q (34%), 6p (28%), and 1q (25%), among others.6 By using comparative genomic hybridization data, we have recently been involved in the development of a targeted fluorescence in situ hybridization (FISH) assay to assist in the diagnosis of melanoma when the histopathological characteristics show ambiguous or conflicting features.7 This assay looks for copy number gains in 11q13 (CCND1; alias Bcl-1) and imbalances in chromosome 6 with gains in the short arm at 6p25 (RREB1) and/or loss of the long arm at 6q23 (MYB). In the development of this assay, we previously published several original observations8,9 linking high-level gains or amplification of CCND1 at 11q13 with poor prognosis in melanoma. Thus, in our extensive experience using FISH to evaluate chromosomal copy number aberrations in melanoma, we observed the following: i) melanomas are heterogeneous at the molecular level and may vary in the specific chromosomal aberrations present, and ii) specific copy number aberrations may have significant prognostic implications.
Copy number gains of specific oncogenes have been linked to prognosis in several cancers. For example, amplification of v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2) has been associated with poor prognosis in breast cancers,10 whereas elevated copy numbers of the epidermal growth factor receptor (EGFR) gene are highly associated with likely response and survival benefit of non–small-cell lung cancer when treated with EGFR tyrosine kinase inhibitors.11 The goal of this study was to evaluate the prognostic significance of the most common chromosomal copy number aberrations in melanomas in a case-control study looking at melanomas with and without metastasis after prolonged follow-up.
Materials and Methods
After approval from the Northwestern University Robert H. Lurie Comprehensive Cancer Center, Chicago, IL, and the Northwestern University Internal Review Board, we searched the Northwestern University dermatology and surgical pathology archives for cases of melanoma with documented metastasis. We identified 55 melanoma cases from January 1, 1992, to December 31, 2007, in which patients had documented metastasis and the slides and tissue blocks were available. This included patients with metastasis limited to the lymph nodes (n = 15), in-transit disease (n = 8), and distant metastasis (n = 32). Among the 15 patients with lymph node–limited disease, 8 had palpable lymph node disease and 7 had the metastasis identified on sentinel node biopsy. Twenty-seven of these patients were deceased as a result of their disease. With the exception of one patient, all patients from the metastatic group had a Breslow depth of at least 0.55 mm. We then searched for patients with melanoma from the same time frame with a Breslow depth of at least 0.55 mm or greater, a minimum follow-up of 5 years, and clear documentation of no disease recurrence/metastasis. Forty-two such patients were identified, for whom slides and tissue blocks were available. Only patients for whom the slides, tissue blocks, and clinical course were all available were included in the study. The histopathological characteristics of all patients were verified by a dermatopathologist (P.G.). Along with the clinical course, for each patient, Breslow depth, age, sex, site of disease, presence or absence of ulceration, mitotic count, and Clark's level was recorded. Table 1 summarizes the characteristics of the metastasizing and nonmetastasizing groups.
Table 1.
Comparison of Metastatic and Nonmetastatic Samples on Traditional Markers, Including All Prognosticators Used by the AJCC for Melanoma
| Variable | Metastatic sample (n = 54) | Nonmetastatic sample (n = 42) | P value |
|---|---|---|---|
| Age (years) | |||
| Mean ± SEM | 66.0 ± 2.1 | 59.9 ± 2.5 | 0.03 |
| Median (range) | 67.0 (33–96) | 62.5 (13–84) | 0.001 |
| Breslow tumor thickness (mm) | |||
| Mean ± SEM | 2.28 ± 0.42 | 2.16 ± 0.35 | 0.06 |
| Median (range) | 2.15 (0.15–16.00) | 1.43 (0.55–12.50) | 0.11 |
| Mitoses (No./mm2) | |||
| Mean ± SEM | 4.50 ± 0.71 | 2.33 ± 0.36 | 0.01 |
| Median (range) | 3.0 (0–20) | 1.5 (0–10) | 0.12 |
| Sex⁎ | |||
| Female | 20 (36.4) | 12 (28.6) | 0.51 |
| Male | 35 (63.6) | 30 (71.4) | |
| Total | 54 (100.0) | 42 (100.0) | |
| Clark's level⁎ | |||
| II or III | 7 (13.2) | 13 (31.0) | 0.044 |
| IV or V | 46 (86.8) | 29 (69.0) | |
| Total | 53 (100.0) | 42 (100.0) | |
| Ulceration⁎ | |||
| Yes | 26 (48.1) | 3 (7.1) | <0.0001 |
| No | 28 (51.9) | 39 (92.9) | |
| Total | 54 (100.0) | 42 (100.0) | |
| Site⁎ | |||
| Trunk | 21 (38.2) | 13 (31.0) | 0.30 |
| Head and neck | 14 (25.4) | 10 (23.8) | 0.99 |
| Extremities | 20 (36.4) | 19 (45.2) | 0.41 |
| Total | 55 (100.0) | 42 (100.0) |
AJCC, American Joint Committee on Cancer.
Data are given as number (percentage) of each sample unless otherwise indicated.
Probe Selection
We evaluated all 97 melanomas with probes targeting eight distinct chromosomal loci frequently altered in melanoma. This included RREB1 at 6p25, the chromosome 6 centromere (cen 6), v-myb myeloblastosis viral oncogene homolog (MYB) at 6q23, and CCND1 at 11q13, which are part of the melanoma diagnostic test used (Abbott Molecular Laboratories, Des Plaines, IL). We used four additional probes targeting cyclin-dependent kinase inhibitor 2A (CDKN2A) at 9p21, the chromosome 9 centromere (cen 9), v-myc myelocytomatosis viral oncogene homolog (MYC; alias c-myc) at 8q34, and zinc finger protein 217 at 20q13. The latter four probes were identified in a previous study as also being frequently altered in melanoma and, in a report in progress, we identified these probes as highly complementary to the initial set of four probes. In each panel, the FISH probes were labeled with Spectrum Aqua, Spectrum Green, Spectrum Gold, and Spectrum Red for simultaneous hybridization and enumeration. All FISH probes were obtained from Abbott Molecular Laboratories and included both Vysis LSI and CEP commercial and developmental probes.
FISH Analysis
All 97 cases, including 55 with metastasis and 42 without metastasis, were analyzed by FISH with two probe sets. The first probe set included the index probe set for melanoma, targeting 6p25, 6q23, cen 6, and 11q13. The second set included 9p21, cen 9, 8q34, and 20q13. A reviewer blinded to the status of the case as metastasizing or nonmetastasizing enumerated all specimens. Sufficient tissue with high-quality hybridizations for enumeration was obtained in 97 of 97 patients with the first index probe set and in 91 of 97 patients with the second probe set.
In Situ Hybridization and Enumeration of FISH Signals
The hybridization procedure was performed as previously described.7 The slides were analyzed with an epifluorescence microscope equipped with single band-pass filter sets (Abbott Molecular Inc.). The analyses were performed by a trained technician and a dermatopathologist. All analyses were performed blinded to the specimens' diagnoses. Tumor-bearing areas were localized using the DAPI filter at low magnification. The tumor area was then thoroughly inspected for the presence of nuclei harboring abnormal copy numbers of any probe. Areas with the most significant copy number changes were selected for enumeration. Wherever possible, three abnormal areas were selected; within each area, 10 random nuclei were analyzed under high power (×60 objective). To qualify, nuclei had to be nonoverlapping and had to harbor sufficiently bright signals. Nuclei that showed no signals for more than one probe were not analyzed. Thirty cells were enumerated in each specimen.
Statistical Analysis
Eleven parameters from the two probe sets were subjected to statistical analysis: 8q34 gain (percentage of cells with gains in 8q34 gain), 8q34 r-gain (average 8q34 value per cell), 11q13 gain, 11q13 r-gain, 6p25 gain, 6p25 r-gain, 6q23/cen6% loss (percentage of cells with a loss of 6q23 relative to cen 6), 6p25/cen6% gain (percentage of cells with a gain in 6p25 relative to cen 6), 9p21/cen9% loss (percentage of cells with a loss in 9p21 relative to cen 9), 20q13 gain, and 20q13 r-gain.
For each specimen (n = 97), we calculated the following 11 parameters that quantitate the level of gains in the targeted chromosomal loci, including the targeted oncogenes and the level of loss in targeted tumor suppressor genes: percentage of cells with 8q34 gain, 8q34 r-gain, 11q13 gain, 11q13 r-gain, 6p25 gain, 6p25 r-gain, 6q23/cen6% loss, 6p25/cen6% gain, 9p21/cen9% loss, 20q13 gain, and 20q13 r-gain. The values for each of these parameters were compared between the metastasizing and nonmetastasizing groups by the Student's t-test for the mean, the Wilcoxon rank sum test for the median, and logistic regression. Categorical variables were compared between groups using Fisher's exact test. The area under the receiver operating characteristic curve was calculated as the c-statistic in the logistic regression. There were 11 markers that were investigated. To account for multiple testing, a Bonferroni adjustment to the significance criterion was made so that any marker with at least one of the three P values (t-test, rank sum test, or logistic regression) <0.0045 (0.05/11) was analyzed in a multivariate analysis. In the multivariate analysis, the statistical significance of the selected marker was tested in the presence of the following traditional melanoma risk factors: age (in years), sex (female or male), site of disease (head and neck, trunk, or extremities), Clark's level, Breslow depth (mm), mitoses (in mm2), and presence of ulceration (yes or no). For each marker, a pair of logistic regression models was estimated: one using the marker plus all risk factors and one stepwise model that selected only variables (marker or risk factor) significant at P < 0.05. No variables were forced into the stepwise model. A separate pair of multivariate analyses was performed for each marker, and the marker remained statistically significant if that marker's P value in the multivariate analysis was <0.05.
Sample sizes of 55 metastatic samples and 42 nonmetastatic samples have 80% power to detect an absolute difference in proportions of 23% or a mean difference of 0.58 SDs, assuming a two-tailed test and a type I error rate of 5%.
Results
Table 1 compares the metastatic and nonmetastatic samples on traditional markers, including all those used by the American Joint Committee on Cancer (AJCC) for melanoma. The metastatic group was significantly older (mean age, 66 years) compared with the nonmetastatic group (mean age, 60 years). There was also a greater frequency for Clark's level IV or V in the metastatic group versus the nonmetastatic group (87% versus 69%). Not surprisingly, the metastatic group also had more frequent ulceration and a higher mean level of mitoses. The two samples were comparable on Breslow thickness, median level of mitoses, sex, and site of disease.
Table 2 compares the metastatic and nonmetastatic samples on the 11 investigational parameters. By using the criterion of any test for an individual marker having to be <0.0045, four of the 11 markers are significantly different between the two samples. The 8q34 (gain and r-gain) and 11q13 (gain and r-gain) values are all significantly higher in the metastatic sample compared with the nonmetastatic sample. Table 3, Table 4, Table 5, and Table 6 provide the multivariate analyses for these four markers. In all analyses, all four markers remained statistically significant (P < 0.05) after adjusting for the traditional markers of age, sex, site of disease, Clark's level, Breslow depth, presence of mitoses, and presence of ulceration. In fact, in all four multivariate analyses, the markers were second only to ulceration in their prognostic power in our data set, with 8q34 (MYC) gain having a P value of 0.0108 and, when performed stepwise, a P value of 0.0014. In the multivariate analysis, the 11q13 (CCND1) gain had a P value of 0.0045 and, when performed stepwise, a P value of 0.0029, again second only to ulceration in its prognostic power.
Table 2.
Comparison of Metastatic and Nonmetastatic Samples on Investigational Parameters
| Investigational parameters | Metastatic sample | Nonmetastatic sample | P value |
|---|---|---|---|
| 8q34 gain⁎ | |||
| Mean ± SEM | 40.0 ± 2.6 | 25.6 ± 3.0 | 0.0005 |
| Median (range) | 40.0 (6.7–96.7) | 26.7 (0.0–66.7) | 0.0008 |
| Logistic regression (c = 0.705): 8q34 r-gain⁎ | 0.0011 | ||
| Mean ± SEM | 2.41 ± 0.06 | 2.13 ± 0.06 | 0.0023 |
| Median (range) | 2.33 (1.8–4.6) | 2.20 (1.2–2.9) | 0.0023 |
| Logistic regression (c = 0.686): 11q13 gain† | 0.0043 | ||
| Mean ± SEM | 43.6 ± 3.2 | 27.0 ± 3.1 | 0.0004 |
| Median (range) | 43.3 (3.3–96.7) | 25.0 (0.0–76.7) | 0.0005 |
| Logistic regression (c = 0.707): 11q13 r-gain† | 0.0011 | ||
| Mean ± SEM | 2.81 ± 0.17 | 2.16 ± 0.08 | 0.0009 |
| Median (range) | 2.47 (1.57–7.27) | 2.10 (1.2–3.5) | 0.0017 |
| Logistic regression (c = 0.686): 6p25 gain† | 0.0070 | ||
| Mean ± SEM | 46.8 ± 2.7 | 48.1 ± 3.3 | 0.76 |
| Median (range) | 46.7 (6.7–96.7) | 45.0 (6.7–96.7) | 0.85 |
| Logistic regression (c = 0.511): 6p25 r-gain† | 0.76 | ||
| Mean ± SEM | 2.59 ± 0.07 | 2.58 ± 0.08 | 0.97 |
| Median (range) | 2.47 (1.7–3.9) | 2.49 (1.4–4.1) | 0.95 |
| Logistic regression (c = 0.512): 6q23/cen6% loss† | 0.96 | ||
| Mean ± SEM | 29.0 ± 1.9 | 28.0 ± 2.9 | 0.79 |
| Median (range) | 26.7 (6.7–67.9) | 23.3 (0.0–93.3) | 0.48 |
| Logistic regression (c = 0.541): 6p25/cen6% gain† | 0.78 | ||
| Mean ± SEM | 57.4 ± 2.3 | 57.3 ± 2.9 | 0.96 |
| Median (range) | 53.3 (16.7–100.0) | 57.2 (16.7–90.0) | 0.86 |
| Logistic regression (c = 0.497): 9p21/cen9% loss‡ | 0.96 | ||
| Mean ± SEM | 51.7 ± 2.4 | 49.6 ± 3.4 | 0.61 |
| Median (range) | 50.0 (20.0–96.7) | 46.7 (3.3–100.0) | 0.50 |
| Logistic regression (c = 0.541): 20q13 gain⁎ | 0.60 | ||
| Mean ± SEM | 41.7 ± 2.5 | 44.8 ± 3.4 | 0.46 |
| Median (range) | 40.0 (13.3–100.0) | 43.3 (2.0–93.3) | 0.53 |
| Logistic regression (c = 0.538): 20q13 r-gain‡ | 0.45 | ||
| Mean ± SEM | 2.59 ± 0.17 | 2.56 ± 0.09 | 0.88 |
| Median (range) | 2.37 (1.6–10.0) | 2.43 (1.5–4.2) | 0.38 |
n = 50 for the metastatic sample, and n = 41 for the nonmetastatic sample.
n = 55 for the metastatic sample, and n = 42 for the nonmetastatic sample.
n = 49 for the metastatic sample, and n = 41 for the nonmetastatic sample.
Table 3.
Multivariate Logistic Regression Analysis of the Effect of 8q34 (MYC) Gain on Predicting Metastases in the Presence of Various Traditional Melanoma Prognostic Factors
| Factors | χ2 Value | Odds ratio⁎ | 95% Confidence interval | P value |
|---|---|---|---|---|
| Prognostic | ||||
| Ulceration (present versus absent) | 10.71 | 20.41 | 3.36–125.0 | 0.0011 |
| 8q34 (MYC) gain | 6.50 | 1.042† | 1.010–1.075 | 0.0108 |
| Sex (female versus male) | 3.97 | 4.15 | 1.02–16.87 | 0.046 |
| Clark's level (IV to V versus II to III) | 3.47 | 3.89 | 0.93–16.13 | 0.062 |
| Site | ||||
| Extremities versus trunk | 2.91 | 2.00 | 0.4–8.74 | 0.088 |
| Head and neck versus trunk | 2.45 | 0.47 | 0.12–1.89 | 0.13 |
| Breslow depth | 1.78 | 1.19† | 0.92–1.54 | 0.28 |
| Mitoses | 0.23 | 0.96† | 0.81–1.14 | 0.63 |
| Age | 0.18 | 1.007† | 0.974–1.042 | 0.67 |
| Stepwise | ||||
| Ulceration (present versus absent) | 11.02 | 10.3 | 2.6–40.0 | 0.0009 |
| 8q34 (MYC) gain | 6.03 | 1.035† | 1.007–1.065 | 0.0014 |
| Clark's level (IV to V versus II to III) | 4.01 | 3.66 | 1.03–12.99 | 0.045 |
n = 91.
Odds of metastases in category of interest/odds of metastases in reference category.
Fold change in odds because of a one-unit change in prognostic factor.
Table 4.
Multivariate Logistic Regression Analysis of the Effect of 8q34 (MYC) R-Gain on Predicting Metastases in the Presence of Various Traditional Melanoma Prognostic Factors
| Factors | χ2 Value | Odds ratio⁎ | 95% Confidence interval | P value |
|---|---|---|---|---|
| Prognostic | ||||
| Ulceration (present versus absent) | 10.82 | 21.60 | 3.46–134.70 | 0.0010 |
| 8q34 (MYC) r-gain | 4.97 | 7.96† | 1.28–49.38 | 0.026 |
| Clark's level (IV to V versus II to III) | 3.53 | 3.92 | 0.94–16.39 | 0.060 |
| Sex (female versus male) | 3.26 | 3.52 | 0.90–13.77 | 0.071 |
| Site | ||||
| Extremities versus trunk | 2.18 | 0.53 | 0.14–2.04 | 0.14 |
| Head and neck versus trunk | 1.72 | 1.77 | 0.40–7.75 | 0.19 |
| Breslow depth | 0.93 | 1.13† | 0.88–1.45 | 0.33 |
| Age | 0.43 | 1.011† | 0.978–1.046 | 0.51 |
| Mitoses | 0.28 | 0.95† | 0.80–1.14 | 0.60 |
| Stepwise | ||||
| Ulceration (present versus absent) | 12.17 | 11.5 | 2.9–45.5 | 0.0005 |
| 8q34 (MYC) r-gain | 5.80 | 7.73† | 1.46–40.86 | 0.016 |
| Clark's level (IV to V versus II to III) | 3.88 | 3.61 | 1.01–12.99 | 0.049 |
n = 91.
Odds of metastases in category of interest/odds of metastases in reference category.
Fold change in odds because of a one-unit change in prognostic factor.
Table 5.
Multivariate Logistic Regression Analysis of the Effect of 11q13 (CCND1) Gain on Predicting Metastases in the Presence of Various Traditional Melanoma Prognostic Factors
| Factors | χ2 Value | Odds ratio⁎ | 95% Confidence interval | P value |
|---|---|---|---|---|
| Prognostic | ||||
| Ulceration (present versus absent) | 11.34 | 17.90 | 3.34–95.96 | 0.0008 |
| 11q13 (CCND1) gain | 8.06 | 1.04† | 1.01–1.07 | 0.0045 |
| Clark's level (IV to V versus II to III) | 4.02 | 4.35 | 1.03–18.18 | 0.045 |
| Sex (female versus male) | 2.21 | 2.64 | 0.73–9.52 | 0.14 |
| Site | ||||
| Head and neck versus trunk | 1.46 | 1.79 | 0.43–7.54 | 0.23 |
| Extremities versus trunk | 1.41 | 0.66 | 0.18–2.41 | 0.23 |
| Breslow depth | 0.76 | 1.11† | 0.88–1.39 | 0.38 |
| Age | 0.71 | 1.015† | 0.981–1.050 | 0.40 |
| Mitoses | 0.08 | 0.98† | 0.85–1.13 | 0.77 |
| Stepwise | ||||
| Ulceration (present versus absent) | 12.44 | 13.7 | 3.2–58.9 | 0.0004 |
| 11q13 (CCND1) gain | 8.85 | 1.040† | 1.013–1.066 | 0.0029 |
| Clark's level (IV to V versus II to III) | 4.78 | 4.5 | 1.16–14.93 | 0.029 |
n = 97.
Odds of metastases in category of interest/odds of metastases in reference category.
Fold change in odds because of a one-unit change in prognostic factor.
Table 6.
Multivariate Logistic Regression Analysis of the Effect of 11q13 (CCND1) R-Gain on Predicting Metastases in the Presence of Various Traditional Melanoma Prognostic Factors
| Factors | χ2 Value | Odds ratio⁎ | 95% Confidence interval | P value |
|---|---|---|---|---|
| Prognostic | ||||
| Ulceration (present versus absent) | 11.12 | 18.51 | 3.33–102.85 | 0.0009 |
| 11q13 (CCND1) r-gain | 5.82 | 3.33† | 1.25–8.83 | 0.016 |
| Clark's level (IV to V versus II to III) | 4.09 | 4.37 | 1.05–18.18 | 0.043 |
| Sex (female versus male) | 2.10 | 2.55 | 0.72–9.04 | 0.15 |
| Site | ||||
| Extremities versus trunk | 1.92 | 0.58 | 0.16–2.12 | 0.17 |
| Head and neck versus trunk | 1.67 | 1.75 | 0.43–7.20 | 0.20 |
| Breslow depth | 0.59 | 1.09† | 0.88–1.36 | 0.44 |
| Age | 0.36 | 1.010† | 0.977–1.045 | 0.55 |
| Mitoses | 0.06 | 0.98† | 0.85–1.14 | 0.81 |
| Stepwise | ||||
| Ulceration (present versus absent) | 12.17 | 13.52 | 3.13–58.44 | 0.0005 |
| 11q13 (CCND1) r-gain | 6.82 | 3.54† | 1.37–9.16 | 0.009 |
| Clark's level (IV to V versus II to III) | 4.99 | 4.39 | 1.20–15.87 | 0.026 |
n = 97.
Odds of metastases in category of interest/odds of metastases in reference category.
Fold change in odds because of a one-unit change in prognostic factor.
Discussion
Several different oncogenic mutations and chromosomal copy number aberrations may be present within melanoma skin cancer.12,13 In the era of targeted molecular therapy for cancer, it is critical to identify key oncogenic pathways acting as drivers of the cancer. These critical activated pathways are the basis for new drug development and future targeted therapies. This has been demonstrated in several other cancers, such as non–small-cell lung cancer, in which EGFR copy number aberrations have been tightly associated with response to EGFR tyrosine kinase inhibitors.11
Further improving prognostication in itself is significant in melanoma skin cancer. For example, the outcome for patients with early- to intermediate-stage (ie, stage IB to IIIB) melanoma may be highly variable. Patients with stage IIB disease have a 10-year survival rate between 50% and 60%.1 Therefore, these patients have an approximately equal chance of being dead of disease or alive at 10 years after their diagnosis. The development of standard tests that can be used to improve staging in such patients would be of significant benefit as far as keeping physicians and their patients better informed and more capable of personalized medicine and optimal management decisions. This would also allow better stratification of patients for clinical trials. FISH is an ideal platform for the development of a standardized prognostic test. FISH can be used with good interobserver reliability in melanoma.7 Further FISH identification of copy number aberrations of critical oncogenic pathways has already been developed as a standard prognostic test in other cancers, such as breast cancer, in which copy number gains/amplifications of Her-2/neu are (ERBB2) highly prognostic.10,14
In this study, we evaluated 97 melanomas, of which 55 resulted in metastasis and 42 did not result in metastasis, with FISH targeting several key chromosomal loci frequently altered in melanoma. In this evaluation, copy number gains in the chromosomal loci harboring CCND1 (11q13) and MYC (8q34) emerged as most highly linked to metastasis. In the multivariate analysis, copy number gains at 11q13 (CCND1) and 8q34 (MYC), whether measured by the percentage of cells with gains at these loci or the average copy number per cell at these loci, showed a highly statistically significant association with the metastatic group. When compared with all of the traditional prognosticators used by the AJCC in the multivariate analysis, the strength of the association between copy number gains at these loci and metastasis was second only to ulceration. Although the copy number changes at 11q13 and 8q34 characteristically seen in melanoma may include broad segments of the chromosomes, including multiple other genes in addition to CCND1 and MYC, the linkage of these genes with metastasis along with their well-recognized role in cell cycle regulations suggests further study is necessary to evaluate whether these genes may, in fact, be key oncogenic drivers of melanoma. The highly significant P values confirm the nonrandom association of copy number changes in the chromosomal loci containing these genes with metastasis. More important, 11q13 gain, 11q13 r-gain, 8q34 gain, and 8q34 r-gain maintained their significance in the multivariate analysis when compared with other traditional markers used by the AJCC. This indicates that these markers that can easily be evaluated by a standardized FISH assay (Figure 1, A and B) may allow for further stratification of patients into prognostically significant subgroups. Our data support the findings of other investigators6 that copy number gains/amplifications in 11q13 and 8q34 are frequently present in melanoma. Furthermore, to our knowledge, this is the first study highly linking specific copy number gains at 11q13 and 8q34 to poor prognosis in cutaneous melanoma. Further research is necessary to elucidate the association of elevated chromosomal copy numbers from these loci and prognosis. One possibility is that CCND1 and MYC are, in fact, oncogenic drivers in melanoma. However, an alternative explanation may be that alterations in the chromosomal loci carrying these genes are an indicator of severe genetic instability.
Figure 1.
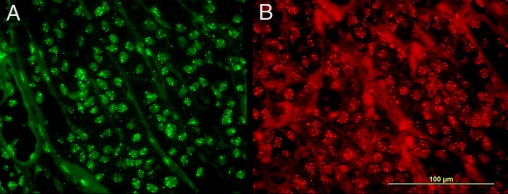
A: FISH image with the spectrum green fluorochrome showing melanoma with prominent gains in 11q13 (CCND1) (original magnification, ×1000). B: FISH image with the spectrum red fluorochrome showing melanoma with prominent gains in 8q34 (MYC) (original magnification, ×1000).
These findings may also have significant theragnostic relevance. Because both CCND1 and MYC are downstream of KIT, BRAF, and NRAS, it is not unlikely that MYC gains may have a similar effect.15 Oncogenic activation of the mitogen-activated protein kinase pathway leads to increased signal transduction of several cytoplasmic kinases, eventually leading to signal transduction into the nucleus, resulting in transcription of MYC and a host of other nuclear transcription factors. The MYC oncoprotein then binds to the DNA causing transcriptional activation of CCND1 and other growth–related genes. The ultimate outcome of activation of these oncogenic pathways is progression through the cell cycle after activation of cyclin–dependent kinases by CCND1 and other cyclins.15 CCND1 directly phosphorylates retinoblastoma protein, allowing cells to move beyond the G1 phase into the S phase. Comparative genomic hybridization data show deletions in cyclin-dependent kinase inhibitor 2A, a direct inhibitor of CCND1, in >60% of melanomas.6 This further emphasizes the importance of unregulated activity of CCND1 in tumor progression.
Currently used targeted therapies for melanoma, including BRAF inhibitors [eg, sorafenib or compound PLX-4032 (Plexxikon Inc., Berkeley, CA)] and KIT inhibitors (eg, imatinib), have shown dramatic responses; however, these responses occur only in a few patients with melanoma, including those with known mutations in the respective oncogene BRAF or KIT.3,5,16,17 This may be related to the presence of other activated downstream oncogenes. Therefore, it is possible that there may be prognostic and theragnostic value to further classify BRAF- or KIT-mutated patients into those with high-level copy number aberrations in CCND1 or MYC and those without either of these aberrations. Further studies are needed to determine whether CCND1 and MYC are, in fact, playing a role in tumor progression in melanomas, with gains at 11q13 and 8q34, and whether copy number gains in either of these two loci influence the response to treatment with BRAF or KIT inhibition.
Footnotes
Supported in part by Abbott Molecular Laboratories; the Innovation, Development, and Progess Foundation; The Dermatology Foundation; and the American Cancer Society, Illinois division (sponsored reference award 160272).
P.G. has served as a consultant to and has received honoraria from Abbott Molecular Laboratories and Neogenomics. S.S.J. and L.M. are employees of Abbott Molecular Laboratories and have stock options. All other authors report no relevant conflicts of interest or funding sources.
References
- 1.Balch C.M., Gershenwald J.E., Soong S.J., Thompson J.F., Atkins M.B., Byrd D.R., Buzaid A.C., Cochran A.J., Coit D.G., Ding S., Eggermont A.M., Flaherty K.T., Gimotty P.A., Kirkwood J.M., McMasters K.M., Mihm M.C., Jr, Morton D.L., Ross M.I., Sober A.J., Sondak V.K. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viros A., Fridlyand J., Bauer J., Lasithiotakis K., Garbe C., Pinkel D., Bastian B.C. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 2008;5:e120. doi: 10.1371/journal.pmed.0050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi F.S., Friedlander P., Corless C.L., Heinrich M.C., Mac R.S., Kruse A., Jagannathan J., Van den Abbeele A.D., Velazquez E.F., Demetri G.D., Fisher D.E. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol. 2008;26:2046–2051. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X., Zhou J., Yuen N.K., Corless C.L., Heinrich M.C., Fletcher J.A., Demetri G.D., Widlund H.R., Fisher D.E., Hodi F.S. Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin Cancer Res. 2008;14:7726–7732. doi: 10.1158/1078-0432.CCR-08-1144. [DOI] [PubMed] [Google Scholar]
- 5.Lutzky J., Bauer J., Bastian B.C. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–493. doi: 10.1111/j.1755-148X.2008.00475.x. [DOI] [PubMed] [Google Scholar]
- 6.Bastian B.C., LeBoit P.E., Hamm H., Brocker E.B., Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- 7.Gerami P., Jewell S.S., Morrison L.E., Blondin B., Schulz J., Ruffalo T., Matushek P., 4th, Legator M., Jacobson K., Dalton S.R., Charzan S., Kolaitis N.A., Guitart J., Lertsbarapa T., Boone S., LeBoit P.E., Bastian B.C. Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am J Surg Pathol. 2009;33:1146–1156. doi: 10.1097/PAS.0b013e3181a1ef36. [DOI] [PubMed] [Google Scholar]
- 8.Gerami P., Guitart J., Martini M., Wayne J.D., Kuzel T. Cyclin D1 homogeneous staining regions by fluorescent in situ hybridization: a possible indicator of aggressive behavior in melanomas. Arch Dermatol. 2008;144:1235–1236. doi: 10.1001/archderm.144.9.1235-b. [DOI] [PubMed] [Google Scholar]
- 9.Pouryazdanparast P., Newman M., Mafee M., Guitart J., Gerami P. Malignant melanoma with monster cells showing massive cyclin D1 amplification. Am J Dermatopathol. 2009;31:402–403. doi: 10.1097/DAD.0b013e31819f8316. [DOI] [PubMed] [Google Scholar]
- 10.Tovey S.M., Brown S., Doughty J.C., Mallon E.A., Cooke T.G., Edwards J. Poor survival outcomes in HER2-positive breast cancer patients with low-grade, node-negative tumours. Br J Cancer. 2009;100:680–683. doi: 10.1038/sj.bjc.6604940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahabreh I.J., Linardou H., Siannis F., Kosmidis P., Bafaloukos D., Murray S. Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2010;16:291–303. doi: 10.1158/1078-0432.CCR-09-1660. [DOI] [PubMed] [Google Scholar]
- 12.Pleasance E.D., Cheetham R.K., Stephens P.J., McBride D.J., Humphray S.J., Greenman C.D., Varela I., Lin M.L., Ordóñez G.R., Bignell G.R., Ye K., Alipaz J., Bauer M.J., Beare D., Butler A., Carter R.J., Chen L., Cox A.J., Edkins S., Kokko-Gonzales P.I., Gormley N.A., Grocock R.J., Haudenschild C.D., Hims M.M., James T., Jia M., Kingsbury Z., Leroy C., Marshall J., Menzies A., Mudie L.J., Ning Z., Royce T., Schulz-Trieglaff O.B., Spiridou A., Stebbings L.A., Szajkowski L., Teague J., Williamson D., Chin L., Ross M.T., Campbell P.J., Bentley D.R., Futreal P.A., Stratton M.R. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtin J.A., Fridlyand J., Kageshita T., Patel H.N., Busam K.J., Kutzner H., Cho K.H., Aiba S., Bröcker E.B., LeBoit P.E., Pinkel D., Bastian B.C. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 14.Gokhale S., Gatalica Z., Mohammad A., Rampy A.I., Velagaleti Gopalrao V.N. FISH for HER-2/neu in breast cancer: standardization makes the difference! Indian J Cancer. 2004;41:152–158. [PubMed] [Google Scholar]
- 15.Kumar V., Cotran R., Robbins S. ed 6. Saunders Co; Philadelphia: 1997. Basic Pathology. [Google Scholar]
- 16.Brower V. BRAF inhibitors: research accelerates in wake of positive findings. J Natl Cancer Inst. 2010;102:214–215. doi: 10.1093/jnci/djq037. [DOI] [PubMed] [Google Scholar]
- 17.Smalley K.S., Flaherty K.T. Development of a novel chemical class of BRAF inhibitors offers new hope for melanoma treatment. Future Oncol. 2009;5:775–778. doi: 10.2217/fon.09.56. [DOI] [PubMed] [Google Scholar]