

Abstract
Although next-generation sequencing (NGS) has been the domain of large genome centers, it is quickly becoming more accessible to general pathology laboratories. In addition to finding single-base changes, NGS allows for the detection of larger structural variants, including insertions/deletions, translocations, and viral insertions. We describe the use of targeted NGS on DNA extracted from formalin-fixed, paraffin-embedded (FFPE) tissue, and show that the short read lengths of NGS are ideally suited to fragmented DNA obtained from FFPE tissue. Further, we describe a novel method for performing hybrid-capture target enrichment using PCR-generated capture probes. As a model, we captured the 5.3-kb Merkel cell polyomavirus (MCPyV) genome in FFPE cases of Merkel cell carcinoma using inexpensive, PCR-derived capture probes, and achieved up to 37,000-fold coverage of the MCPyV genome without prior virus-specific PCR amplification. This depth of coverage made it possible to reproducibly detect viral genome deletions and insertion sites anywhere within the human genome. Out of four cases sequenced, we identified the 5′ insertion sites in four of four cases and the 3′ sites in three of four cases. These findings demonstrate the potential for an inexpensive gene targeting and NGS method that can be easily adapted for use with FFPE tissue to identify large structural rearrangements, opening up the possibility for further discovery from archival tissue.
High-throughput, massively paralleled DNA sequencing methods, colloquially grouped as next-generation sequencing (NGS), have revolutionized many aspects of human disease study by quickly providing massive amounts of sequencing data for relatively little cost.1,2 Several recent, high-profile studies have examined the tumor genomes/exomes of acute myeloid leukemia, prostate cancer, and breast cancer, resulting in novel findings that have changed our understanding of these disease processes.3–5 As the cost of sequencing continues to fall to <$20,000/genome [National Human Genome Research Institute (NHGRI), http://www.genome.gov/27540667, last accessed January 30, 2011], compared to $2.7 billion for the initial human genome (NHGRI, http://www.genome.gov/11006943, last accessed September 2010), it is clear that the application of NGS to patient specimens will increase.
Although NGS methodologies were initially applied for whole-genome analysis, recent technical advances have made it possible to target defined regions of the genome. Such methods include the Roche NimbleGen Capture Array (Roche NimbleGen Inc, Madison, WI), Agilent SureSelect (Agilent Technologies, Santa Clara, CA), RainDance Technologies emulsion PCR (RainDance Technologies, Lexington, MA), and standard PCR.6,7 The utility of these so-called genome-partitioning approaches is well established and has made it possible to perform deep-sequence analysis of the exome or of groups of genes correlated with specific diseases.8,9 Nonetheless, clinical application of NGS in the routine pathology laboratory has been limited by the fact that virtually all existing approaches were developed and optimized for high-quality DNA extracted from fresh tissue, and require a high initial investment (often >$10,000) for custom targeting, depending on the number of genes/regions to be evaluated. Although fresh tissue is available for laboratory testing in many inherited diseases, hematolymphoid malignancies, and some solid tumors, in routine clinical practice, the most common substrate is formalin-fixed, paraffin-embedded (FFPE) tissue specimens. Furthermore, banked frozen tissue for many rare tumors is scarce, making it difficult to perform retrospective studies of these malignancies.
In this study, we report a simple, inexpensive, laboratory-generated hybrid-capture enrichment method that is optimized for DNA extracted from FFPE tissue and NGS on the Illumina GAIIx genome analyzer (San Diego, CA) (summarized in Figure 1). As a model, we analyzed the 5.3-kb genome of Merkel cell polyomavirus (MCPyV) in cases of Merkel cell carcinoma (MCC), to evaluate the ability of NGS to detect viral insertion sites. Originally identified in 2008, MCPyV is a small DNA virus in the same family as the BK, JC, WU, and KI viruses, and has been identified in up to 80% of cases of MCC, a rare and often fatal cutaneous tumor of the elderly and immunocompromised.10–13 Because MCC is rare, the lack of fresh tissue and concomitant high-quality DNA has significantly hampered efforts to study both the viral genomic structure and the organization of human genomic insertion sites.14 Further difficulty arises in the study of MCC because MCPyV shows frequent genomic deletions and sequence mutations that make it difficult to amplify the virus from cases of MCC by PCR.15 Finally, the identification of polyomavirus insertion sites within the human genome is technically challenging, as the 5.3-kb circular genome does not contain a defined linearization sequence. Since the viral genome can linearize at any location during integration into the host genome, analysis by traditional methods such as RACE (rapid amplification of cDNA ends) PCR, ligation-mediated PCR, or inverse PCR, is tedious with fresh tissue and impossible with low-quality FFPE tissue–derived DNA.
Figure 1.
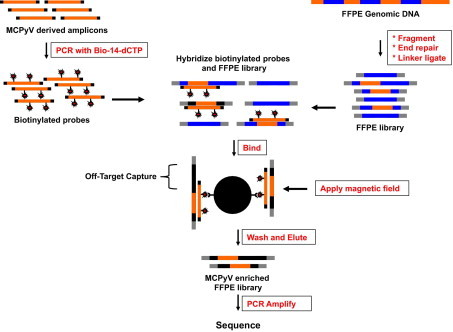
Pictorial representation of Washington University Capture. Washington University Capture (WUCap) enables solution-phase hybridization between double-stranded DNA PCR “bait” and whole-genome shotgun libraries. The solution-phase method we have developed for hybrid capture is robust and involves only a few basic steps. The bait used for targeting is dictated by primer-specific amplification of genomic targets generated during the PCR. Subsequently, the amplicons are used as a template in a second PCR incorporating biotin-14-dCTP. Genomic DNA is prepared from each of the samples to be sequenced, sheared to an average fragment size of 300 bp, enzymatically repaired to blunt the ends, and ligated to Illumina adapter sequences (at both ends). Five hundred nanograms of genomic DNA library is denatured, combined with 100 ng of the biotinylated “bait,” and hybridized for 48 hours. Mixing this hybridization reaction with streptavidin-coated superparamagnetic beads allows binding of biotinylated bait–target hybrids and selective removal from solution by applying a magnet field. The remaining supernatant is removed, and the beads are washed, removing nonspecific DNA. The enriched target sequences are released from the bead-bound bait sequences by denaturation (0.125 N NaOH), neutralized, amplified in the PCR to generate double-stranded Illumina libraries, and then sequenced.
Our data demonstrate that a simple, novel hybrid-capture methodology coupled with Illumina GAIIx sequencing can be applied to DNA derived from FFPE tissue, with a resulting capture efficiency comparable to that reported when fresh tissue–derived DNA is used for analysis.16 Through exploitation of the “off-target” or “shoulder” coverage inherent in capture-based enrichment (but not present in PCR-based enrichment), we demonstrate that this method allows for the efficient detection of large structural DNA variation including viral insertion sites from FFPE tissue. Furthermore, the ability to generate hybrid-capture probes by simple PCR rather than relying on commercial synthesis greatly reduces the cost and time required to perform targeted sequencing. These results establish that FFPE tissue is a suitable substrate for targeted NGS analysis, a result that immediately opens clinical and archival specimens to high-throughput sequencing approaches for analysis of the full spectrum of DNA mutations, ranging from single base pair changes to larger structural changes such as translocations, viral insertion sites, and indels.
Materials and Methods
Case Selection
We selected four cases of MCPyV-positive, surgically resected MCC from the files of the Lauren V. Ackerman Laboratory of Surgical Pathology at Washington University Medical Center for which adequate FFPE tissue was available for testing. A paired primary tumor (sample 12) and subsequent local metastasis (sample 23) occurring approximately 1 year later were included. The remaining two cases, samples 15 and 27, were from a 10-cm perigastric metastasis and a cutaneous recurrence, respectively, from two different patients.
Generation of Capture Probes
Biotinylated capture probes were generated by first designing a series of 23 overlapping PCR products that tiled across the MCPyV genome (gi: 165973999). Amplicons were spaced such that on average a 50-bp overlap was achieved; however, variable GC content prohibited this degree of overlap in some amplicons. The PCR products had an average size of 275 bp (range, 222 to 353 bp) to permit amplification from FFPE tissue–derived template DNA. A case of MCC with an intact copy of MCPyV was used as a template for amplification (sample 25, unpublished data). PCR was performed using 50 ng of template DNA in a 50-μL reaction with Phusion Taq Polymerase (New England Biolabs, Ipswich, MA) under the following conditions: 35 cycles of template denaturation at 94°C for 30 seconds, annealing for 30 seconds at 55°C, and extension at 68°C for 1 minute. Biotin-14-dCTP (Invitrogen, Carlsbad, CA) was added to the PCR reaction at a 1:5 molar ratio with unlabeled dCTP. PCR products of the appropriate size were then gel purified, and an aliquot of each (250 to 500 ng) was subjected to a “bind and boil” assay to determine the extent of biotin incorporation. Briefly, the capture probes were assayed for biotin incorporation by binding 500 ng of biotinylated PCR products to 200 μg of streptavidin-coated paramagnetic beads (cat. #S1420S; New England Biolabs, Worcester, MA). An external magnetic field was then applied and the supernatant retained. The beads were then boiled in 0.1% SDS for 5 minutes to release the biotinylated capture probes from the streptavidin-coated paramagnetic beads, and the resulting supernatant was collected. A second boil in 0.1% SDS was performed and the supernatant collected. Aliquots from the binding supernatant and both boils were run on an Agilent Bioanalyzer 2100 (Agilent Technologies) using DNA 7500 cartridges (Figure 2).
Figure 2.
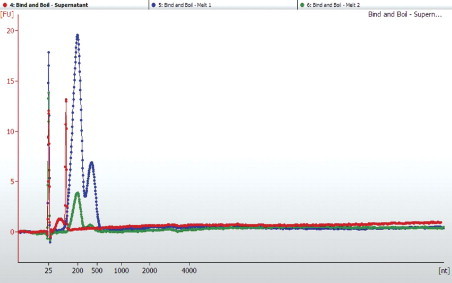
Validating biotin-14-dCTP incorporation by a bind and boil method. The biotin–streptavidin dissociation constant (kD) is on the order of 4 × 10−14 mol/L. However, boiling the biotin-steptavidin complexes in the presence of SDS releases these noncovalent complexes. To validate the incorporation of biotin-14-dCTP during the PCR, we assayed the supernatant of our PCR “bait” solution after mixing with Dynal M-280 beads. If biotin-14-dCTP did not incorporate during the PCR, we would expect the supernatant to contain PCR-amplified DNA. To assay the supernatant, we performed a buffer exchange using AmpureXP beads, eluted in water, and evaluated the supernatant of PCR on the Agilent BioAnalyzer 2100 High Sensitivity DNA chip (red dotted line). The Dynal M-280 beads were boiled in 0.1% SDS, and this supernatant was assayed for PCR products (dotted blue line). A second boil treatment was performed, and this supernatant was assayed for the presence of PCR products (dotted green line). FU, fluorescent units; nt, nucleotides.
Preparation of Sample DNA
FFPE tissue blocks were first aligned to corresponding hematoxylin and eosin–stained slides; areas containing nonnecrotic tumor with minimal stroma were identified and cored with 2-mm sterile punches (Miltex, Tuttlingen, Germany). The tissue was then deparaffinized in xylene, and DNA extracted using the Ambion Recoverall Kit (Applied Biosystems, Foster City, CA). One microgram of FFPE-extracted genomic DNA was fragmented to between 200 and 400 bp using the Covaris S2 Sonolab (Covaris, Woburn, MA). Fragmentation conditions used a frequency sweeping mode with two successive acoustic treatments of: i) duty cycle = 20%, intensity = 5, cycle/bursts = 500 for 60 seconds, and ii) duty cycle = 5%, intensity = 9, cycles/burst = 100 for 60 seconds. Samples were fragmented while incubating at 4°C. The fragments were end repaired in 1× end-repair buffer in the presence of an end-repair enzyme cocktail (Lucigen Corp. Madison, WI) at 25°C for 15 minutes. Blunt-end fragments were tailed with deoxyadenosine triphosphate in the presence of 3 U Klenow exo− (New England Biolabs, Worcester, MA) at 37° for 30 minutes. Illumina adapters were ligated to A-tailed DNA fragments per the manufacturer's protocol in the presence of 1× Quick Ligase Buffer and 15 U Quick Ligase (New England Biolabs) at 25°C for 15 minutes. Small fragments <100 bp and unligated adapters were removed from the mix by AMPure purification (Agencourt Bioscience, Beverly, MA).
Hybridization/Capture
The biotinylated DNA probes were first pooled to form an equimolar solution. Next, 100 ng of pooled capture products in 5 μL of water were mixed with 0.5 μg of sequencing-prepared genomic DNA in 4 μL of water and 1 μL of human Cot-1DNA (Invitrogen, Carlsbad, CA). These components were then added to 10 μL of MWG hybridization buffer (Eurofins MWG Operon, Ebersberg, Germany), denatured at 95°C for 5 minutes, and allowed to hybridize at 71°C for 48 hours. A total of 50 μL of Dynal M-280 streptavidin-coated magnetic beads (Invitrogen) were then washed three times in 200 μL of the supplied bead-binding buffer, and finally resuspended in 80 μL of bead-binding buffer. The beads were then added to the 20-μL hybridization mixture and allowed to bind for 30 minutes at room temperature on a lab shaker. An external magnetic field was then applied and the supernatant containing unbound DNA discarded. The beads were then subjected to one wash with 1000 μL of 1× SSC/0.1% SDS for 5 minutes at 71°C, followed by three 5-minute washes with 1000 μL of 0.1× SSC/0.1% SDS at 71°C, and a final wash with 1000 μL 0.2× SSC for 5 minutes at room temperature. Captured DNA was then eluted from the streptavidin-coated beads by adding 10 μL of 0.125 N NaOH and allowing the mixture to incubate at room temperature for 4 minutes. An external magnetic field was then applied and the supernatant (now containing the captured DNA sequences) collected and placed in a new tube with 20 μL of 1 mol/L Tris-HCL (pH 7.5). The entire elution process was then repeated resulting in 40 μL of enriched ssDNA. Finally, a buffer exchange by dialysis filtration (Millipore V series filter; Millipore Inc, Billerica, MA) was performed for 1 hour to both remove any residual NaOH and reduce the sample volume. The sample was then resuspended in 30 μL of water. As an alternative to dialysis filtration, an equal volume of neutralization solution [1 mol/L Tris-HCl (pH 8.8)] may be added to the eluted/captured DNA and buffer exchange/volume reduction accomplished by Solid Phase Reversible Immobilization (SPRI) technology in the form of AmpureXP paramagnetic particles (Beckman Coulter, Morrisville, NC).
Post-Capture Amplification
To ensure sufficient DNA for cluster generation, the captured DNA was then subjected to low-cycle PCR using 7.5 μL of capture material, 160 nmol/L of the Illumina paired-end oligonucleotides 1.0 (PE1.0: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) and 2.0 (PE2.0: 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′) in 1× Phusion High Fidelity PCR Master Mix with HF Buffer (New England Biolabs). To ensure no preferential amplification occurred in this capture-amplified Illumina library that could lead to biased sequencing coverage, the library was sampled at various intermediate cycles (12, 15, 18, 21, 24, and 27), and the products were visualized on 2.2% Lonza FlashGel System (Lonza Rockland, Rockland ME) (data not shown). Based on this cycle optimization, three reactions per sample were prepared using 7.5 μL of adapter-ligated DNA in 42.5 μL of PCR amplification master mix and PCR amplified with an initial thermal denaturing step of 98°C for 30 seconds followed by 24 rounds of amplification: 98°C, 15 seconds; 60°C, 30 seconds; and 72°C, 30 seconds. After PCR amplification, reactions were pooled and concentrated in 30 μL of elution buffer [10 mmol/L Tris-HCl (pH 8.0)] using the Qiagen MinElute Reaction Cleanup columns (Qiagen, Valencia, CA).
Sequencing
Cluster generation and sequencing were performed on an Illumina cluster station and a GAIIx analyzer with paired-end module, respectively. Each sample was run in duplicate. Paired-end clusters were generated in flow cells with 8 pmol/L capture-amplified Illumina library DNA according to the manufacturer's instructions. Three samples (12, 15, and 23) were run using 75-bp paired-end reads with two Illumina version 4 sequencing kits, whereas one sample (sample 27) was run with 50-bp paired-end reads. Base calls were provided by the included Illumina software.
Data Analysis
The resulting FASTQ files from the Illumina GAIIx were first aligned to a reference MCPyV genome (gi: 165973999) using quality-weighted alignments within the freely available MAQ Software package (http://maq.sourceforge.net) allowing for an assessment of capture efficiency and consensus sequence generation.17 Further analysis to identify viral integration sites and deletions was performed using both the BreakDancer and SLOPE software packages on MAQ-aligned data.18,19 Briefly, BreakDancer identifies paired-end reads in which one end aligns to the MCPyV genome and the other to the human genome, whereas SLOPE identifies actual chimeric virus–human reads within the single ends. Both methods produced similar results and were used in tandem to reduce the possibility of false-positive detection of putative insertion events.
Insertion Site Verification
To validate the NGS findings, primer pairs were designed using the computationally derived contigs that spanned the viral insertion sites. Primers were designed using the Vector NTI suite (Invitrogen) and had an average melting temperature of 60°C. PCR was performed for 35 cycles with an annealing temperature of 55°C using Platinum TaqHF DNA polymerase (Invitrogen). PCR products of the predicted size were visualized by ethidium bromide staining following agarose gel electrophoresis. The products were then excised from the gel, purified (Qiagen gel extraction kit, Qiagen, Valencia, CA), and subcloned into vector pCR2.1 using the TA Cloning kit (Invitrogen); DNA obtained by plasmid purification (PureYield Plasmid Miniprep Promega, Madison, WI) was then sequenced using standard M13 primers, ABI BigDye v3.1 terminator cycle sequencing kit (Applied Biosystems, Foster City, CA), and a fluorescent DNA sequencer (3730XL; Applied Biosystems).
Regulatory Approval
This study was approved by the Human Studies Committee of Washington University School of Medicine.
Results
Deep Sequencing of MCPyV Genome from FFPE MCC Cases
We performed Illumina GAII deep sequencing in duplicate on MCPyV DNA extracted from FFPE tissue in four cases of MCC, including one paired primary tumor and subsequent metastasis. Average MCPyV coverage ranged from 4700- to 37,000-fold, corresponding to a sequence enrichment of up to 100,000-fold, with up to 18% of the total sequence mapping back to the MCPyV genome (Table 1). Those sequences that did not align to the MCPyV genome represented nonenriched human genomic DNA. Sequence coverage varied minimally across each viral genome, with small differences in coverage likely representing differences in capture probe melting temperature, partial hybridization between probes and off-target fragments, or nonuniform biotin representation within probes (Figure 3). Although capture probe size varied between 222 bp and 353 bp, there was no clear association between size and hybridization efficiency. Further, optimal lengths of the capture probes were not empirically derived, and probe size differences were an artifact of PCR optimization and probe melting-temperature balancing. All cases showed viral genomic deletions (areas with zero coverage), consistent with previous PCR and Sanger sequencing findings (data not shown). The paired primary and metastasis (samples 12 and 23, respectively) showed identical deletion patterns that were conserved at the single-base level.
Table 1.
Next-Generation Sequencing Statistics
| Coverage of hybrid capture samples | ||||||
|---|---|---|---|---|---|---|
| Read length | Total sequence | Number of reads | Reads mapped to viral geome | Fold enrichment | Viral coverage (fold) | |
| Sample 12 | 75 bp ×2 | 2.2 Gbp | 30 × 106 | 1.5 × 106 | 28,000 | 9953 |
| Sample 15 | 75 bp ×2 | 2.6 Gbp | 28 × 106 | 5.5 × 106 | 107,000 | 36,961 |
| Sample 23 | 75 bp ×2 | 1.9 Gbp | 26 × 106 | 3.2 × 106 | 62,000 | 19,860 |
| Sample 27 | 50 bp ×2 | 0.75 Gbp | 15 × 106 | 1.1 × 106 | 40,000 | 4800 |
Data represent averages of duplicate runs. “On-target” matches to the MCPyV target were determined by paired-end alignment of all raw sequence data to the MCPyV genome using the MAQ software package with default paired-end parameters. ‘Fold enrichment’ was calculated by comparing the expected frequency of MCPyV sequences compared to the human genome. ‘Fold coverage’ represents the average number of times each MCPyV nucleotide was sequenced.
Figure 3.
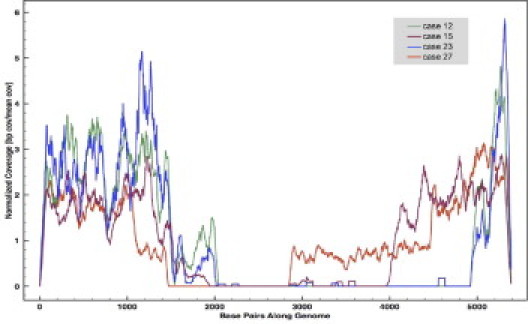
MCPyV sequence coverage in four cases of MCC. To account for variability of sequence depth, the plot displays normalized coverage where “1” represents the average coverage in each case. The differences in sequence coverage across each viral genome vary minimally and likely reflect differences in melting temperatures of the capture probes and sequence heterogeneity of the captured DNA. All cases showed evidence of viral deletions (areas of zero coverage) that were previously confirmed by PCR and Sanger sequencing. Note that cases 12 and 23 (the paired primary and metastasis samples, respectively) show identical deletion patterns, which were appreciated at the single-base level.
As part of initial validation experiments, we performed the hybrid-capture protocol with and without Cot-1 DNA on sample 27 using 36-bp paired-end reads. We found that adding 1 μg of Cot-1 DNA to the hybridization mixture effectively increased the capture efficiency by approximately 2.5-fold (Table 2), resulting in up to 6.8% of the captured DNA mapping back to the viral sequence. Later experiments using longer (75 bp) paired-end reads resulted in even greater capture efficacy, approaching 20%, likely representing the increased specificity of longer reads.
Table 2.
The Use of 1 μg of Cot-1 DNA Added to the Hybridization Reaction Blocks Repetitive Sequences and Increases Hybridization Efficiency Approximately 2.5-Fold
| Hybridization with Cot-1 | |
| Read 1 | |
| Total reads | 7603,264 |
| Capture-specific reads | 520,304 |
| % Match to virus | 6.8% |
| Read 2 | |
| Total reads | 7603,264 |
| Capture-specific reads | 495,973 |
| % Match to virus | 6.5% |
| Hybridization without Cot-1 | |
| Read 1 | |
| Total reads | 2313,487 |
| Capture-specific reads | 57,967 |
| % Match to virus | 2.5% |
| Read 2 | |
| Total reads | 7603,264 |
| Capture-specific reads | 54,897 |
| % Match to virus | 2.4% |
“On-target” matches to the MCPyV target were determined by paired-end alignment of raw 50-bp reads to the MCPyV genome.
Identification of Viral Insertion Sites from Next-Generation Sequence Data
We relied on off-target coverage flanking the capture regions to find viral integration sites (Figure 4A). Unlike PCR-based enrichment methods where both the 3′ and 5′ ends of targeted DNA must be specified through the use of template-specific primers, hybrid capture allows for the enrichment of sequences containing only partial homology to the viral insertion site. These off-target areas consist of chimeric viral/human DNA sequences presumably representing the virus–human insertion site boundaries, and were identified by two computational methods. Using the SLOPE software package, specifically written to quickly identify large DNA structural variation in targeted NGS data, we mapped viral insertion sites by performing partial alignments to the MCPyV genome within the single-end reads (Figure 4B).18 Briefly, SLOPE looks for paired-end reads where one end matches the target sequence while the mate does not. SLOPE then performs weighted partial alignments within these regions to find chimeric reads spanning the viral insertion site. To further exclude the possibility of false-positive discovery, we used the BreakDancer software package to identify viral insertion sites by a second, paired-end dependent methodology in which sequences with one end aligned to the viral genome and the corresponding mate pair aligned to the human genome were identified (Figure 4C).19 Both methods produced similar findings, although SLOPE offered the advantage of locating the actual insertion site with single-base accuracy due to its use of single-end partial alignments.
Figure 4.
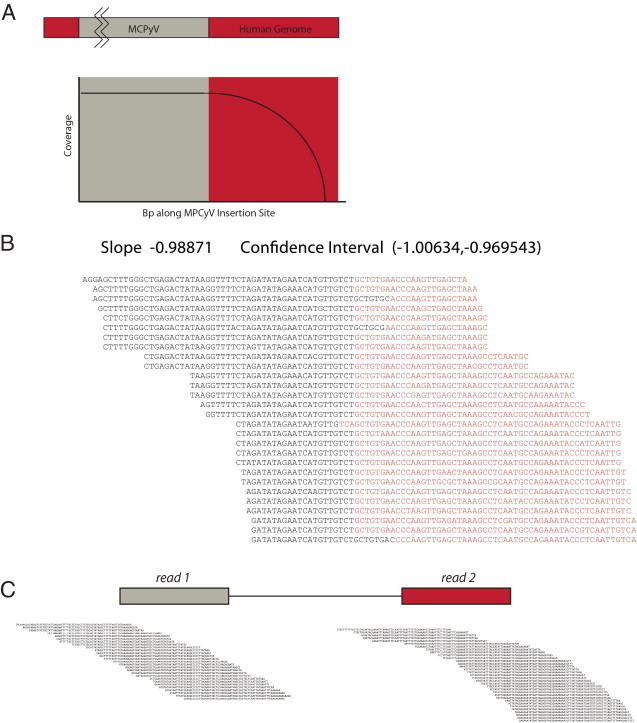
A: Diagrammatic representation of the off-target coverage model used to identify viral integration sites. Genomic DNA fragments with partial homology to the MCPyV capture probes are captured with decreasing efficiency as the amount of targeted sequence is reduced. These chimeric reads represent viral insertion sites. B: Chimeric sequences were identified using the SLOPE software package. Human sequences are in black, whereas viral sequences are in red; duplicate reads were removed. Sequences were aligned to form clusters that tile across the insertion boundary, and a score was assigned to each cluster based on the slope of a linear regression line drawn through the insertion site. C: As an alternative methodology, paired-end identification was also performed using the BreakDancer software package. Paired-end reads in which one end mapped to the viral genome and the other end to the human genome were identified and clustered to demonstrate the integration site.
We successfully identified both 5′ and 3′ viral insertion sites in all three cases sequenced with 75-bp paired-end reads, and identified only the 5′ insertion site in case 27, sequenced with 50-bp paired-end reads. Neither the BreakDancer nor the SLOPE method was able to identify a 3′ insertion site; we speculate that this site occurred in a repetitive region of human genomic DNA, resulting in nonspecific alignment of sequences, a situation that may be avoided by using longer read lengths or mate-pair libraries. All cases showed unique viral insertion sites (Table 3), with the exception of the paired primary tumor and metastasis samples, which showed identical sites. Duplicate sequencing runs using the same libraries further confirmed the findings in each case (data not shown).
Table 3.
MCC Case Clinical Characteristics, PCR-Verified Viral Insertion Sites, and Viral Deletions
| Sample | Type | Age of block (years) | Sex | Site | 3′ insertion site | 5′ insertion site | Viral genomic deletion size |
|---|---|---|---|---|---|---|---|
| 12 | Primary | 6 | M | Buttocks | ch8: 65568962 | ch8: 65566806 | 3.0 kb |
| 23 | Metastasis | 5 | M | Back | ch8: 65568962 | ch8: 65566806 | 3.0 kb |
| 27 | Metastasis | 2 | F | Right arm | ch9: 121417276 | undetermined | 2.4 kb |
| 15 | Metastasis | 6 | M | Small bowel | ch6: 19684666 | ch6: 19684859 | 1.3 kb |
Confirmation of Results by Sanger Sequence Analysis
To evaluate the validity of the viral insertion sites identified by NGS, we performed PCR and Sanger sequencing using primers specific for the insertion sites demonstrated by NGS. Primers were designed based on the computationally constructed contigs that spanned both human and viral sequences at the insertion sites. Amplification by PCR-generated bands of the predicted size (∼250 bp to ensure amplification from FFPE material). Sanger sequence analysis of the PCR products demonstrated viral insertion sites identical to those identified within the NGS data at all seven insertion site boundaries, confirming the validity of our methodology (data not shown).
Discussion
Here, we demonstrate a simple and inexpensive method for targeted NGS from FFPE tissue. This method can be used to achieve high enrichment of kilobase-long target regions (in our model, the approach permitted almost 38,000-fold sequence coverage of the 5.3-kb MCPyV genome) and can be used to detect both small single base pair changes and larger rearrangements including translocations/insertions.
Our data make several important points. First, FFPE tissue provides DNA that is an acceptable substrate for use with NGS approaches, specifically targeted sequence analysis via a hybrid capture coupled with Illumina GAIIx sequencing. Second, our laboratory-derived enrichment method using DNA extracted from FFPE tissue offers only slightly less efficient capture than more expensive commercial methods using DNA extracted from high-quality fresh tissue. For example, in a recent paper, Hoppman-Chaney et al20 demonstrate that the capture efficiency for 22 genes (272 kb total) obtained by a custom-made Nimblegen 385K array and Illumina GAII sequencing was between 21% and 58%, compared with 5% to 19.6% with our methodology (5.3-kb capture region). It is important to note, however, that capture efficiencies are likely influenced by the size of the capture region as reported by Hodges et al, with the size of the capture region being inversely proportional to its efficiency.21 Third, from a technical viewpoint, targeted NGS results can be achieved with the use of capture probes produced in-house, eliminating the cost and delays associated with a reliance on commercial vendors. Taken together, our data show that NGS performed on DNA extracted from FFPE tissue can produce targeted sequence results on par with more expensive capture methods using DNA from fresh tissue.
Our data also illustrate the power of hybrid-capture–based enrichment approaches over traditional PCR-based methods. For example, while a priori PCR produces higher efficiencies in terms of “on-target” sequence, PCR is unable to identify events involving large genomic changes that result in sufficient sequence changes to prevent primer annealing, such as translocations, inversions, viral insertion events, and so on. Furthermore, PCR enrichment of large stretches of fragmented FFPE tissue–derived genomic DNA is tedious and requires placement of primers approximately every 300 bp to ensure amplification.22,23 For intron/exon coverage of large genes, including those of clinical interest such as BRCA1/2, EGFR, c-kit, or KRAS, PCR-based amplification from FFPE tissue would require hundreds of individual PCR reactions, greatly increasing test complexity and cost. Methods such as RainDance emulsion PCR could potentially reduce this complexity by performing numerous parallel PCR reactions in micelles; however, such methods remain unproven for FFPE tissue, and the associated overhead cost is prohibitive.24 The use of laboratory-derived hybrid capture offers the potential to overcome these limitations with minimal cost since large quantities of capture probes can be generated using a high-quality DNA template, although the lower on-target efficiencies of such methods could result in low coverage of larger capture regions. Furthermore, it is likely possible to generate larger capture probes (on the order of 10 kb) by long-range PCR from high-quality templates that can then be sheared to obtain an optimal 200- to 500-bp size. Such methods could greatly reduce the number of individual PCR reactions needed to make smaller 270-bp capture probes, as was necessary in the current study.
Although to date most targeted NGS sequencing studies have focused on finding single-nucleotide substitutions, NGS has a tremendous advantage over standard sequencing for the detection of large structural DNA changes.3,20 However, sophisticated software packages such as BreakDancer, SLOPE, moDIL, Pindel, and VariationHunter are required to identify such events, and each software package has its unique tradeoffs in terms of speed, false-positive discovery rate, and types of events detected.18,19,25–27 The fact that software complexity significantly adds to the time required to analyze NGS data cannot be overemphasized. For example, in addition to the time required to perform sequence alignments, initial evaluation of the aligned data by BreakDancer took several days to complete on each case. The use of specific targeted sequence analysis software such as SLOPE can greatly reduce this time to only several minutes, and in the same manner, alignment of sequence data to targeted regions (rather than the whole genome) provides further marked improvements in speed. Nonetheless, it is likely that additional targeted NGS sequence analysis tools will need to be developed for clinical NGS applications to meet the turnaround time requirements intrinsic to patient care settings.
From the perspective of viral oncobiology, the typical armamentarium of molecular genetic methods for finding viral insertion sites is poorly suited for use with FFPE tissue for study of MCPyV since the virus has no linearization sequence and inserts into the human genome ectopically. Because formalin fixation leads to extensive cross-linking and fragmentation of nucleic acids, extractions from FFPE are typically fragmented into pieces <300 bp long, and so methods such as rapid amplification of cDNA ends (RACE), ligation-mediated PCR (LM-PCR), inverse PCR, and so on, are generally inefficient.28,29 By leveraging the power of NGS using hybrid capture, we were able to determine the full viral structure and human integration sites in FFPE cases of MCC with single-base resolution. To our knowledge, this represents the first successful methodology for identifying viral insertion sites from FFPE and is the first application of NGS to identify viral insertion sites from any substrate.
In summary, we demonstrate the utility of an inexpensive system for hybrid-capture enrichment and NGS of DNA extracted from FFPE tissue that can achieve up to 100,000-fold enrichment and 38,000-fold coverage of target region. The method can be used to identify single base pair changes as well as indels. Further, by exploiting the off-target coverage unique to hybrid-capture methods, we were able to successfully identify both 5′ and 3′ viral insertion sites in our model system studying MCPyV characteristics of MCC. This result, in turn, demonstrates that our method can also be used to identify targeted translocations from FFPE tissue of tumors in which one partner gene is known but the other is unknown, such as translocations involving the IgH locus, MLL gene, or PDGRFB gene. We do note, however, that bioinformatically identifying human translocation or inversion events is far more complicated than identifying viral insertions sites, as these events tend to occur in repeat regions or areas with overlapping sequence homologies. The method we describe, therefore, not only opens the FFPE tissue archive to analysis by NGS, but also provides an approach to apply NGS in clinical settings for which fresh tissue is not available.
Acknowledgments
We thank Elaine Mardis and the Genome Sequencing Center at Washington University for their generous support of this work. We thank Xiaopei Zhu for her technical assistance in DNA extraction, PCR, and subcloning.
Footnotes
Supported by the Washington University Department of Pathology and the Washington University Genome Sequencing Center.
E.J.D. and V.M. contributed equally to this manuscript.
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Mardis E.R. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 2.Mardis E.R., Wilson R.K. Cancer genome sequencing: a review. Hum Mol Genet. 2009;18:R163–R168. doi: 10.1093/hmg/ddp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mardis E.R., Ding L., Dooling D.J., Larson D.E., McLellan M.D., Chen K., Koboldt D.C., Fulton R.S., Delehaunty K.D., McGrath S.D., Fulton L.A., Locke D.P., Magrini V.J., Abbott R.M., Vickery T.L., Reed J.S., Robinson J.S., Wylie T., Smith S.M., Carmichael L., Eldred J.M., Harris C.C., Walker J., Peck J.B., Du F., Dukes A.F., Sanderson G.E., Brummett A.M., Clark E., McMichael J.F., Meyer R.J., Schindler J.K., Pohl C.S., Wallis J.W., Shi X., Lin L., Schmidt H., Tang Y., Haipek C., Wiechert M.E., Ivy J.V., Kalicki J., Elliott G., Ries R.E., Payton J.E., Westervelt P., Tomasson M.H., Watson M.A., Baty J., Heath S., Shannon W.D., Nagarajan R., Link D.C., Walter M.J., Graubert T.A., DiPersio J.F., Wilson R.K., Ley T.J. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding L., Ellis M.J., Li S., Larson D.E., Chen K., Wallis J.W., Harris C.C., McLellan M.D., Fulton R.S., Fulton L.L., Abbott R.M., Hoog J., Dooling D.J., Koboldt D.C., Schmidt H., Kalicki J., Zhang Q., Chen L., Lin L., Wendl M.C., McMichael J.F., Magrini V.J., Cook L., McGrath S.D., Vickery T.L., Appelbaum E., Deschryver K., Davies S., Guintoli T., Crowder R., Tao Y., Snider J.E., Smith S.M., Dukes A.F., Sanderson G.E., Pohl C.S., Delehaunty K.D., Fronick C.C., Pape K.A., Reed J.S., Robinson J.S., Hodges J.S., Schierding W., Dees N.D., Shen D., Locke D.P., Wiechert M.E., Eldred J.M., Peck J.B., Oberkfell B.J., Lolofie J.T., Du F., Hawkins A.E., O'Laughlin M.D., Bernard K.E., Cunningham M., Elliott G., Mason M.D., Thompson D.M., Jr, Ivanovich J.L., Goodfellow P.J., Perou C.M., Weinstock G.M., Aft R., Watson M., Ley T.J., Wilson R.K., Mardis E.R. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teer J.K., Mullikin J.C. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19:R145–R151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiss M.M., Ortoleva-Donnelly L., Beer N.R., Warner J., Bailey C.G., Colston B.W., Rothberg J.M., Link D.R., Leamon J.H. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal Chem. 2008;80:8975–8981. doi: 10.1021/ac801276c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloglu A., Ozen S., Sanjad S., Nelson-Williams C., Farhi A., Mane S., Lifton R.P. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A., Redman M.C., Wiltshire E., Wilson C.J., Altshuler D., Gabriel S.B., Daly M.J., Thorburn D.R., Mootha V.K. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duncavage E.J., Zehnbauer B.A., Pfeifer J.D. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516–521. doi: 10.1038/modpathol.2009.3. [DOI] [PubMed] [Google Scholar]
- 12.Kassem A., Schopflin A., Diaz C., Weyers W., Stickeler E., Werner M., Zur Hausen A. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68:5009–5013. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- 13.Becker J.C., Houben R., Ugurel S., Trefzer U., Pfohler C., Schrama D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol. 2009;129:248–250. doi: 10.1038/jid.2008.198. [DOI] [PubMed] [Google Scholar]
- 14.Sastre-Garau X., Peter M., Avril M.F., Laude H., Couturier J., Rozenberg F., Almeida A., Boitier F., Carlotti A., Couturaud B., Dupin N. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48–56. doi: 10.1002/path.2532. [DOI] [PubMed] [Google Scholar]
- 15.Shuda M., Feng H., Kwun H.J., Rosen S.T., Gjoerup O., Moore P.S., Chang Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasta V., Ng S.B., Turner E.H., Shendure J., Hahn S.H. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009;1:100. doi: 10.1186/gm100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abel H.J., Duncavage E.J., Becker N., Armstrong J.R., Magrini V.J., Pfeifer J.D. SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics. 2010;26:2684–2688. doi: 10.1093/bioinformatics/btq528. [DOI] [PubMed] [Google Scholar]
- 19.Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q., Locke D.P., Shi X., Fulton R.S., Ley T.J., Wilson R.K., Ding L., Mardis E.R. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoppman-Chaney N., Peterson L.M., Klee E.W., Middha S., Courteau L.K., Ferber M.J. Evaluation of oligonucleotide sequence capture arrays and comparison of next-generation sequencing platforms for use in molecular diagnostics. Clin Chem. 2010;56:1297–1306. doi: 10.1373/clinchem.2010.145441. [DOI] [PubMed] [Google Scholar]
- 21.Hodges E., Rooks M., Xuan Z., Bhattacharjee A., Benjamin Gordon D., Brizuela L., Richard McCombie W., Hannon G.J. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat Protoc. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagg A., Braziel R.M., Arber D.A., Bijwaard K.E., Chu A.Y. Immunoglobulin heavy chain gene analysis in lymphomas: a multi-center study demonstrating the heterogeneity of performance of polymerase chain reaction assays. J Mol Diagn. 2002;4:81–89. doi: 10.1016/S1525-1578(10)60685-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavelic J., Gall-Troselj K., Bosnar M.H., Kardum M.M., Pavelic K. PCR amplification of DNA from archival specimens: A methodological approach. Neoplasma. 1996;43:75–81. [PubMed] [Google Scholar]
- 24.Leamon J.H., Link D.R., Egholm M., Rothberg J.M. Overview: methods and applications for droplet compartmentalization of biology. Nat Methods. 2006;3:541–543. doi: 10.1038/nmeth0706-541. [DOI] [PubMed] [Google Scholar]
- 25.Hormozdiari F., Hajirasouliha I., Dao P., Hach F., Yorukoglu D., Alkan C., Eichler E.E., Sahinalp S.C. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2010;26:i350–i357. doi: 10.1093/bioinformatics/btq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S., Hormozdiari F., Alkan C., Brudno M. MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nat Methods. 2009;6:473–474. doi: 10.1038/nmeth.f.256. [DOI] [PubMed] [Google Scholar]
- 27.Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greer C.E., Peterson S.L., Kiviat N.B., Manos M.M. PCR amplification from paraffin-embedded tissues: Effects of fixative and fixation time. Am J Clin Pathol. 1991;95:117–124. doi: 10.1093/ajcp/95.2.117. [DOI] [PubMed] [Google Scholar]
- 29.Karlsen F., Kalantari M., Chitemerere M., Johansson B., Hagmar B. Modifications of human and viral deoxyribonucleic acid by formaldehyde fixation. Lab Invest. 1994;71:604–611. [PubMed] [Google Scholar]