

Abstract
Recombinant lentivector immunization has been demonstrated to induce potent CD8 T cell responses in vivo. In this study, we investigated whether lentivector delivering a self/tumor Ag, tyrosinase related protein 1 (TRP1), could stimulate effective antitumor T cell responses. We found that immunization with lentivector expressing mutated TRP1 Ag elicited potent CD8 T cell responses against multiple TRP1 epitopes. Importantly, the activated CD8 T cells effectively recognize wild-type TRP1 epitopes. At peak times, as many as 10% of CD8 T cells were effector cells against TRP1 Ag. These cells killed wild-type TRP1 peptide-pulsed target cells in vivo and produced IFN-γ after ex vivo stimulation. The CD8 T cell responses were long-lasting (3– 4 wk). Immunized mice were protected from B16 tumor cell challenge. In a therapeutic setting, lentivector immunization induced potent CD8 T cell responses in tumor bearing mice. The number of infiltrating T cells and the ratio of CD8/CD4 were dramatically increased in the tumors of immunized mice. The tumor-infiltrating CD8 T cells were functional and produced IFN-γ. The potent CD8 T cell responses stimulated by lentivector immunization eliminated small 3-day s.c. B16 tumors and strongly inhibited the growth of more established 5-day tumors. These studies demonstrate that genetic immunization with lentivector expressing mutated self/tumor Ag can generate potent CD8 T cell immune responses and antitumor immunity that prevent and inhibit B16 tumor growth, suggesting that lentivector immunization has the potential for tumor immunotherapy and immune prevention.
T cell-mediated immunity plays an important role in controlling tumor growth (1). Elimination of tumor cells by the immune system is mainly dependent on the activation of tumor-specific T cells, and especially CD8+ T cells, to recognize and kill tumor cells. It has been demonstrated that the antitumor efficacy of tumor vaccines correlates with the magnitude of vaccine-induced CD8 T cell responses (2). Thus, the goal of active immunization is to elicit potent and durable effector and memory T cell responses to eliminate existing tumors and recognize and destroy evolving tumor cells as they arise. However, despite intensive effort, the antitumor efficacy of current active tumor immunotherapy is limited (3). Multiple factors, including the immunosuppressive condition in the tumor, the intrinsic weak immunogenicity of tumor Ags, and suboptimal Ag delivery, may together contribute to the limited success of tumor immunotherapy.
One major obstacle to successful active tumor immunotherapy is that most tumor Ags are shared by the tissues from which the tumors developed and thus they are shared self/tumor Ags (4, 5). Induction of T cell immune responses against such Ags is actively controlled by mechanisms of central and peripheral tolerance to prevent unwanted immune responses against self. However, such mechanisms also prevent generation of antitumor immune responses in the case of tumor immunotherapy (6). To break this immune tolerance to the shared self/tumor Ag and induce antitumor immune responses, altered peptide ligands, xenoantigens, or mutated Ags have been used (7–10). In a recent study, Guevara-Patino et al. introduced a series of amino acid mutations into mouse tyrosinase related protein 1 (TRP1)3 to generate epitope with higher MHC class I (MHC I) binding affinity. Additionally, amino acid changes were also brought into TRP1 to reduce protein glysosylation. Such modifications destabilized protein via enhanced protein degradation and thereby increased Ag processing to generate antigenic epitopes that strongly bound to MHC I molecules for better presentation (8). However, even with such altered strong Ag, four consecutive gene gun deliveries were required to generate CD8 T cell responses. Thus, altered peptides have been successful in stimulating cross-reactive T cell responses against self Ags. However, the problem remains as to how to most effectively deliver them to create effective T cell responses in vivo.
Recombinant viral vectors are effective vehicles for delivering Ag to the immune system and offer a promising method for eliciting potent antitumor immunity (11). Recently, we and others have shown that recombinant lentivector effectively transduced dendritic cells (DCs) in vitro and in vivo and stimulated more potent and longer lasting Ag-specific CD8 T cell immune responses than other viral vectors, as well as other genetic immunization approaches such as naked plasmid DNA (12–17). The potency and durability of lentivector-induced immune responses may be related to the vector’s capacity to efficiently transduce nondividing skin DC subsets without compromising their Ag processing and presenting function in vivo (12, 18, 19). The absence of preexisting antilentivector immunity in the host and the focus of resulting immune responses on intended Ag because of the absence of viral genes in the vector are other merits of using lentivector to stimulate adaptive immune responses. Indeed, lentivector has been demonstrated to stimulate T cell responses against melanoma NY-ESO Ag (20, 21), tyrosinase related protein 2 (TRP2) (22), Melan-A (15), combined polyepitopes melanoma Ag (23), and tumor-associated OVA foreign Ag (12–14, 16). However, most studies to date have been in vitro investigations of T cell responses against human tumor Ag, or the in vivo T cell responses in HLA-A2/Kb mice; these models could not examine the biological antitumor effect in relevant tumor therapy models in vivo. In the present study, we hypothesize that delivery of mutated self/tumor Ag by lentivector can break immune tolerance to self/tumor Ag and stimulate potent CD8 T cell responses that have preventive and therapeutic antitumor efficacy. To test this hypothesis, we used lentivector to deliver mutated TRP1 gene. We found that potent and long-lasting CD8 T cell responses against multiple epitopes of TRP1 Ag were induced, resulting in dramatic CD8 T cell infiltration into tumor lesions, which eliminated small early stage B16 tumors and strongly inhibited the growth of well-established B16 tumors.
Materials and Methods
Cell lines and mice
The B16F0 melanoma and 293T cells were purchased from the American Type Culture Collection. C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD) and housed under specific pathogen-free condition in Laboratory Animal Services (LAS) of the Medical College of Georgia. OT-I TCR transgenic mice were kindly provided by Dr. Andrew Mellor of the Medical College of Georgia. Animal care protocols were approved by the Institutional Animal Care and Use Committee of the Medical College of Georgia.
Lentivector preparation and immunization
Plasmid DNA containing the mutated TRP1 (muTRP1) or wild-type TRP1 (wtTRP1) gene was previously described (8). Wild-type and mutated TRP1 DNA was subcloned into lentivector transfer plasmid pLenti-TRIP-enhanced GFP by replacing the enhanced GFP gene to generate plasmid pLenti-muTRP1 or pLenti-wtTRP1-lv. Recombinant lentivectors expressing wtTRP1 (wtTRP1-lv) and muTRP1 (muTRP1-lv) were prepared by transient cotransfection method as described (13, 24). Approximately 2.5 × 107 transducing units (TU) of muTRP1-lv or 2.5 × 106 TU of OVA-lv was utilized for immunization via footpad in 50 μl in all experiments. Compared with OVA-lv immunization, 5–10-fold more muTRP1-lv was needed to achieve similar levels of CD8 T cell activation.
Abs and intracellular staining
Abs used in this study were purchased from BD Biosciences or eBioscience. Intracellular staining was performed as we described previously (13).
In vivo killing assay
To measure the cytotoxic function of activated T cells in vivo, we performed an in vivo killing assay as described previously (13, 25). Briefly, peptide-pulsed (targets) and nonpulsed (control) mouse splenocytes were labeled with 5 or 0.5 μM CFSE, respectively, and injected into mice. After 12 h, splenocytes were collected from mice and the specific lysis of target cells was examined and calculated.
In vivo Ag presentation assay
The duration of in vivo Ag presentation was determined as we previously described (12). Thy1.1 congenic OT-I TCR transgenic cells were labeled with 10 μM CFSE. At different time points after immunization, 2 × 106 of CFSE labeled OT-I cells were injected into naive or immunized mice. Three days later, draining popliteal lymph node was collected and stained for Thy1.1. Thy1.1+ cells were gated and the OT-I cell proliferation was analyzed by the progressive banding of CFSE intensity.
In vivo tumor study
B16F0 cells were inoculated s.c. into the flank of C57BL/6 mice. For preventive tumor challenge experiments, 2 × 105 cells were utilized. For all other experiments, 1 × 105 cells were used. Tumor growth was monitored by measuring the perpendicular diameters of the tumor and recorded as tumor area. Mouse survival was recorded daily.
Tumor-infiltrating lymphocytes (TILs)
TILs were analyzed as reported previously (26). The whole B16F0 tumor was collected from mice and weighted. Approximately 20 –100 mg of tumors was cut into small pieces and incubated at 37°C for 1 h in RPMI 1640 containing 1 mg/ml collagenase, 1 mg of hyaluronidase, and 100 U of DNase I. Single-cell suspensions were prepared and stained for CD90.2, CD4, and CD8. Another part of cells was pulsed with or without exogenous TRP1 peptides in the presence of GolgiStop for 3 h and intracellularly stained for IFN-γ.
Immunofluorescent staining
Tumor tissues were immediately frozen in dry ice-ethanol after collecting from mice. Tumor tissues were sectioned at 5 μm and stained for CD8 and CD4 T cells. Slides were first blocked with 10% normal donkey serum (Jackson ImmunoResearch Laboratories) for 30 min at room temperature in a humidified chamber and stained with primary anti-CD4 or anti-CD8 Ab (BD Biosciences) for 1 h. After rinses, slides were then incubated for 30 – 60 min with Cy3-labeled donkey anti-Rat Ab (Jackson Immuno Research Laboratories). Slides were then counterstained with Hoechst 33258 (Sigma-Aldrich).
JAM assay
In vitro CTL assay was performed as JAM test described previously (27). Briefly, B16 tumor cells treated with IFN-γ (20 ng/ml) for 48 h were labeled with [3H]thymidine overnight in 96-well plates. Splenocytes from immunized mice were stimulated in vitro with 1 μg/ml wtTRP1 peptide 455 (wt455) overnight and then added to the B16 tumor cells at indicated ratios. Cells were cocultured for 7 h before harvesting onto fiberglass mat and measuring the counts per minute in each well. Specific lysis was calculated as: % lysis = [(cpm target without CTL − cpm target with CTL)/cpm target without CTL] × 100.
Results
Immunization with recombinant lentivector induces potent CD8 T cell responses against multiple epitopes of wtTRP1 tumor Ag
We previously demonstrated that lentivector was very effective in stimulating CD8 T cell responses against OVA as a model non-self Ag (12, 13). To examine whether immunization with lentivector could stimulate potent CD8 T cell responses against a shared self/tumor protein, TRP1 Ag, we generated recombinant lentivectors expressing the murine wtTRP1 (wtTRP1-lv) or muTRP1 (muTRP1-lv) (Fig. 1A). C57BL6 mice were immunized with lentivector wtTRP1-lv or muTRP1-lv. Nine days after immunization, in vivo killing assays were performed to determine whether Ag-specific CTL activity was induced against wtTRP1 epitopes in vivo. Immunization with lentivector encoding shared self/tumor wtTRP1 Ag did not stimulate measurable in vivo killing activity against wild-type epitopes (Fig. 1B). In contrast, a single immunization with the muTRP1-lv stimulated potent in vivo CTL activity that eliminated nearly all wt455-pulsed target cells (as much as 96% killing of 1 × 107 target cells) within 12 h in vivo. In this experiment, we further showed that lentivector immunization stimulated much stronger CD8 T cell responses than those induced by naked DNA expressing the same mutated Ag, which induced only ~5% killing (Fig. 1C).
FIGURE 1.

Lentivector immunization induces potent CD8 T cell response against multiple epitopes of TRP1. C57BL/6 mice were immunized with 2.5 × 107 TU of lentivector wtTRP1-lv, muTRP1-lv (A), or plasmid pLenti-TRP1 DNA (100 μg) expressing mutated TRP1 (muTRP1 DNA). After 9 days, in vivo CTL activity was measured by 12 h in vivo killing assay. A representative experiment is presented for each group and the calculated percentage of specific killing is summarized in bar graphs (B and C). On day 10 after immunization, splenocytes from immunized mice were collected and intracellularly stained for IFN-γ after 3 h of ex vivo stimulation with three different peptides derived from wtTRP1 (D). Splenocytes from naive mice, DNA-immunized mice, or wtTRP1-lv-immunized mice were unable to produce IFN-γ (data not depicted). The summarized data from three mice of each group are presented in the right panels. E, Splenocytes from muTRP1-lv-immunized mice were also compared for IFN-γ production after restimulation with either wt455 peptides or IFN-γ-treated B16 tumor cells (center panels). IFN-γ treatment increased the MHC level on B16 tumor surface (left panel). Splenocytes stimulated with wt455 peptides in vitro were then examined for their capability of killing B16 tumor cells (right panel). Numbers in B and C represent the percentage of each population. Numbers in the upper right quadrant of D and E indicate the percentage of IFN-γ+ cells of total CD8 T cells. Experiments in B–D were repeated at least five times with similar results. Experiments in E were repeated three times with similar results.
The ability of lentivector-mediated immunization to break tolerance to self Ag was additionally examined by intracellular staining of the effector cytokine IFN-γ in the T cells. Ten days after immunization with wtTRP1-lv or muTRP1-lv, mouse splenocytes were collected and stimulated in vitro with different epitopes of wtTRP1. Consistent with the in vivo killing assay, after one intramuscular injection of naked plasmid DNA, pLenti-TRP1, essentially no IFN-γ-producing CD8 T cells were detected (bar graph of Fig. 1D). Similarly, immunization with lentivector expressing wtTRP1, wtTRP1-lv, did not stimulate CD8 T cells to produce IFN-γ (bar graph of Fig. 1D), indicating that the mice were extremely tolerant to native self Ag. In contrast, after one immunization with lentivector expressing muTRP1, muTRP1-lv, CD8 T cell responses against multiple epitopes of wtTRP1 Ag were induced. The percentage of CD8 T cells secreting IFN-γ was between 2% and 6% for each epitope (Fig. 1D).
Next, we investigated if lentivector-stimulated CD8 T cells could recognize B16 tumors, on which the MHC molecule and peptide complex are much lower than splencoytes pulsed with TRP1 peptides. First, we examined if B16 tumor cell surface possessed sufficient MHC-Ag complex that could stimulate CD8 T cells from immunized mice. B16 tumor cells have very low MHC I molecules, which can be increased by IFN-γ treatment (28, 29) (Fig. 1E, right panel). As expected, CD8 T cells from immunized mice responded very well to wt455 peptide stimulation, and higher number of the CD8 T cells produced IFN-γ, but there was only a nominal level of responses from B16 tumor stimulation (Fig. 1E, center panels). As a negative control, no IFN-γ production by CD8 T cells was detected from irrelevant OVA SIINFEKL peptide stimulation. Thus, B16 tumor cells did not stimulate CD8 T cells for IFN-γ production by FACS staining. However, this is relatively insensitive readout. We therefore asked if activated T cells from immunized mice could recognize and kill B16 tumor cells in vitro. Splenocytes were stimulated with wt455 peptides overnight and then used as CTLs and added to radiolabeled B16 tumor cells. Killing of B16 tumor cells was readily detected when B16 tumor cells were pretreated with IFN-γ (Fig. 1E, right panel), with ~30% of IFN-γ-treated B16 tumor cells lysed in 7 h. Even without IFN-γ pretreatment, there was detectable target cell killing, although it was less efficient.
Collectively, our data showed that CD8 T cells activated by muTRP1 Ag could effectively recognize and lyse wtTRP1 peptide-pulsed target cells in vivo and could respond to wtTRP1 peptide stimulation in vitro. Importantly, activated T cells could also recognize and kill target B16 tumor cells. Thus, all of the subsequent experiments were conducted with muTRP1-lv immunization and the immune responses were monitored by wt-TRP1 peptides.
CD8 T cell responses following muTRP1-lv immunization are multifunctional
Recent emerging data suggest that CD8 T cells capable of producing multiple cytokines are critical for controlling viral infections (30 –33). To investigate if the CD8 T cells elicited by lentivector immunization could produce multiple cytokines, production of three effector function cytokines by splenocytes from immunized mice, IFN-γ, TNF-α, and IL2, were measured by intracellular staining following wt455 peptide stimulation in vitro. As demonstrated in Fig. 2A, approximately one-third of the IFN-γ-producing CD8 T cells were also TNF-α +. However, only approximately 3% of IFN-γ + CD8 T cells were IL-2+, suggesting that two cytokines (IFN-γ and TNF-α) producer were abundant, but that CD8 T cells producing IL-2 were rare.
FIGURE 2.

A, CD8 T cells activated by muTRP1-lv immunization can produce multiple cytokines. CD8 T cells from muTRP1-lv-immunized mice were restimulated ex vivo with TRP1 wt455 peptides or control OVA SIINFEKL peptides and then stained for IFN-γ, TNF-α, and IL-2. Cells shown were gated on CD8 T cells. B, TRP1-specific CD8 T cell response induced by lentivector immunization is long-lasting. C57BL/6 mice were immunized with 2.5 × 107 TU of lentivector muTRP1-lv. At different time points, peripheral blood cells were stimulated with pooled peptides of 455, 481, and 522, whereas the splenocytes were stimulated with each individual wtTRP1 peptides for 3 h ex vivo. Cells were then intracellularly stained for IFN-γ. The percentages of IFN-γ-producing CD8 T cells of total CD8 T cells were calculated and are presented as means plus SE. This experiment was repeated twice. C, Ag presentation in vivo after lentivector immunization is protracted. C57BL/6 mice were immunized with 2.5 × 106 TU of lentivector OVA-lv. At indicated time points, mice were injected with purified CFSE-labeled OT-I cells. Three days after OT-I injection, OT-I T cell proliferation in the draining lymph nodes was analyzed by progressive banding of CFSE intensity. Numbers in each figure indicated the percentage of nondividing injected CFSE+ cells (right) and dividing CFSE-diluted cells (left). The in vivo Ag presentation assay was repeated twice.
CD8 T cell responses against TRP1 Ag following lentivector immunization is long-lasting
Following T cell expansion after immunization or microbial infection, there is a dramatic contraction phase in which 90 –95% of CD8 T cells die, leaving 5–10% of activated CD8 T cells to become memory T cells (34). Using the non-self OVA model Ag, we previously found that the contraction of T cell responses following lentivector immunization was slower and less dramatic than with vaccinia vector immunization (12). In the following experiment, we examined the kinetics of CD8 T cell responses against three epitopes of wtTRP1 Ag after muTRP1-lv immunization. As shown in Fig. 2B, following lentivector immunization, TRP1-specific CD8 T cell responses could be detected within 1 wk, peaked at 2 wk, and slowly declined but were maintained at a fairly high level for at least 5 wk. A similar pattern of CD8 T cell responses was found both in the spleen and in peripheral blood. CD8 T cells responding to all three different epitopes followed similar kinetics, with epitopes 455 and 481 being dominant. The data showed that after lentivector immunization, the contraction phase of CD8 T cell responses to a shared self/tumor Ag was gradual, and the CD8 T cell response was maintained for a prolonged period. This sustained CD8 T cell response may be related to the prolonged in vivo Ag presentation following lentivector immunization (12). To determine the duration of Ag presentation in vivo, TCR transgenic T cells that can respond to Ag presentation are needed. Since there is no TCR transgenic T cell clone specifically recognizing TRP1 epitopes, we utilized the OT-I TCR transgenic cells that proliferate in response to OVA Ag presentation. As shown in Fig. 2C, a significant number of OT-I cells were undergoing proliferation in OVA lentivector-immunized mice even after 1.5 mo.
Lentivector immunization with muTRP1-lv protects mice against B16 tumor challenge
To examine if potent and long-lasting CD8 T cell immune responses against multiple epitopes of TRP1 Ag induced by lentivector could generate effective antitumor immunity, we investigated the prophylactic effect of lentivector immunization in the mouse B16F0 tumor model. C57BL/6 mice were immunized with lentivector muTRP1-lv. Fifteen and 40 days after immunization, immunized and naive mice were s.c. challenged with a high dose (2 × 105) of B16F0 tumor cells. All mice were protected from B16F0 tumor challenge 15 days after immunization (Fig. 3A). Even 40 days after immunization, 50 – 60% of mice were protected by a single immunization (Fig. 3B). This tumor prevention effect correlated with the kinetics of CD8 T cell responses, which peaked at 2 wk when maximal protection from tumor challenge was observed.
FIGURE 3.

Lentivector immunization protects mice against B16 tumor challenge. C57BL/6 mice (10 in each group) were immunized with 2.5 × 107 TU of muTRP1-lv and then s.c. challenged with high does (2 × 105) of B16F0 tumor cells on day 15 (A) or day 40 (B) after immunization. Tumor growth was recorded. By the end of 2 mo, mice showed no tumor growth and were designated as tumor free. Experiments in this figure were repeated twice and similar results were observed.
Lentivector immunization stimulates potent CD8 T cell responses in tumor-bearing mice
Therapeutic immunization is normally conducted in settings where tumors are already established. To investigate if potent CD8 T cell responses could be induced by lentivector immunization when tumor was present, B16 tumors were established in C57BL/6 mice. Five days after tumor inoculation, mice were immunized with muTRP1-lv. CD8 T cell responses were measured by IFN-γ staining. As shown in Fig. 4A, CD8 T cells were activated to express IFN-γ in tumor-bearing mice. However, fewer IFN-γ + CD8 T cells were consistently observed in tumor-bearing mice than in tumor-free control animals even though the differences were not statistically significant at the day 14 time point (Fig. 4B). However, IFN-γ+ CD8 T cells in tumor-bearing mice is significantly lower than that in tumor-free mice at the time points after day 14 (Fig. 4C, p = 0.0117), suggesting that the CD8 responses contracted at a faster rate in tumor-bearing mice.
FIGURE 4.

Lentivector immunization stimulates potent CD8 T cell responses in B16 tumor-bearing mice. C57BL/6 mice (five in each group) were inoculated with 1 × 105 B16F0 tumor cells. Five days later, tumor-bearing mice and tumor-free mice were immunized with 2.5 × 107 TU of muTRP1-lv. Peripheral blood lymphocytes were collected from immunized mice (tumor-bearing and tumor-free) and naive nonimmunized control mice and stimulated ex vivo with pooled wtTRP1 peptides at indicated time points before intracellular staining of IFN-γ was conducted. A, Typical intracellular staining of PBL from tumor-free and tumor-bearing mice on day 14 after immunization. Numbers in the parentheses of upper left quadrants represent the percentage of CD8 T cells that secrete IFN-γ. B, Summary of five mice at day 14 time points. C, Kinetics of CD8 T cell responses in tumor-free mice and tumor-bearing mice. This experiment was repeated three times with similar results.
Lentivector immunization markedly increases T cell infiltration into tumor lesions and enhances the ratio of CD8/CD4
To exert an antitumor effect, activated CD8 T cells must infiltrate into a tumor mass and remain functional. In this study, we enumerated the T cell numbers in tumor lesions and the ratios of CD8/CD4 T cells. Lentivector immunization dramatically increased the number of Thy1+ T cells in the tumor (Fig. 5A). Average total T cells (defined as Thy1.2+) increased from 3 × 105 to 4.5 × 106 cells per gram of tumor. Most of the TILs were CD8 T cells, as the average ratio of CD8/CD4 in the tumors increased from 0.5 to 3. In contrast, without immunization, most TILs were CD4 T cells (Fig. 5, A and B). The flow cytometry data were further substantiated by immunofluorescent staining of tumor tissues. While no T cells were detected in the tumor from nonimmunized mice, lentivector immunization considerably increased both CD4 and CD8 T cell infiltration (Fig. 5C).
FIGURE 5.
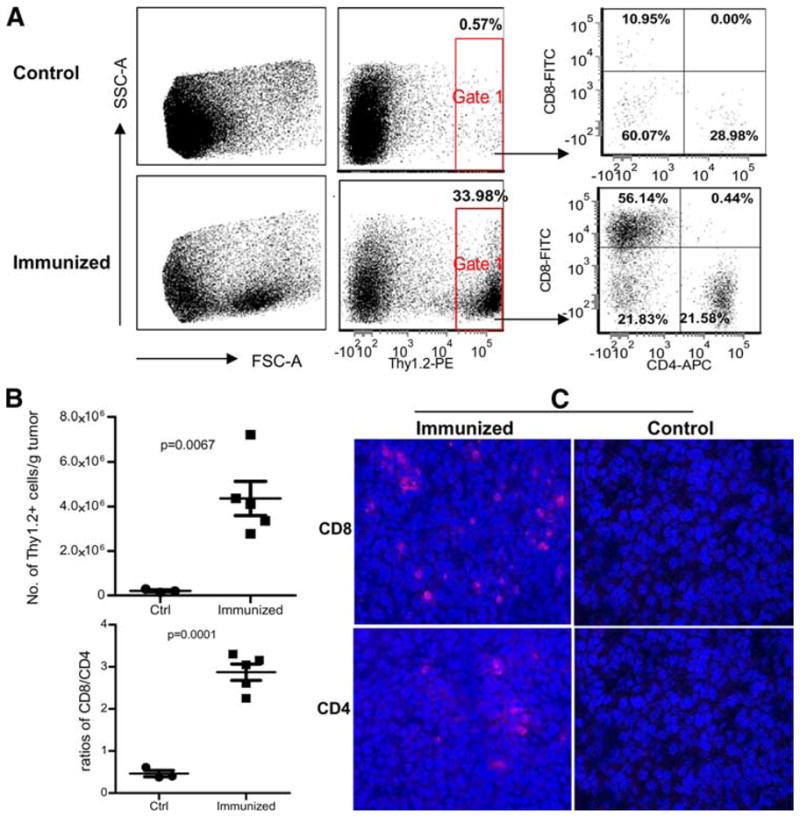
Lentivector immunization dramatically increases T cell infiltration into established B16F0 tumors. C57BL/6 mice were inoculated with 1 × 105 B16F0 cells. Five days later, mice were immunized with 2.5 × 107 TU of muTRP1-lv or left alone (five mice in the treatment group and three mice in control group). Fifteen days after immunization (20 days after tumor inoculation), tumors were collected. A, Half portion of each tumor was examined for TIL by flow cytometry. Flow cytometry gates were set on the live cells based on the size (forward scatter). Thy1.2 was used to stain the T cells, which were then further analyzed for CD8 and CD4 markers. B, Summary of data from all the mice is shown, including the normalized total T cells per gram of tumor and ratio changes of CD8 vs CD4 T cells. C, The other half of tumor was used for immunofluorescent staining of CD8 and CD4 T cells.
Tumor-infiltrating CD8 T cells are functional and produce IFN-γ
To examine if the TIL cells were functional, we cultured single-cell preparations from disintegrated tumors (including tumor cells and TILs) for 3 h ex vivo in the presence of monensin to measure cytokine production. Intracellular staining demonstrated that CD8 T cells from spleen, peripheral blood, and tumors of immunized mice were all able to produce IFN-γ after ex vivo stimulation with wtTRP1 peptides, even though it seems that more CD8 T cells in the TILs produced IFN-γ (Fig. 6, A and B, right panels). However, when the mean fluorescence intensity (MFI) was compared, it was found that CD8 T cells in the TIL produced less IFN-γ per cell than did CD8 T cells in the blood and spleen (Fig. 6, A and B, right panels). In striking contrast, a considerable number (1.5–3.2%) of CD8 T cells from TILs produced IFN-γ in the absence of exogenous peptides stimulation, while no CD8 T cells from spleen and blood were able to do so (Fig. 6, A and B, left panels). Addition of exogenous wtTRP1 peptides could further increase the number of IFN-γ-producing cells (3–5% of CD8 T cells in the TILs) and enhance the amount (MFI from an average of 750 to 850) of IFN-γ production of CD8 T cells of TILs (Fig. 6, A and C).
FIGURE 6.

TILs are functional and capable of producing IFN-γ after ex vivo stimulation. TILs in the tumor single-cell suspension was prepared as those in Fig. 5 and stimulated ex vivo for 3 h with tumor cells or together with additional exogenous TRP1 peptides. Additionally, PBLs and splenocytes were also collected and stimulated for staining of IFN-γ. A, Representative flow cytometry data of five mice. For analysis of TILs, gates were set in the forward scatter and side scatter dot plots by using splenocytes. Thus, most tumor cells were gated out in the TIL samples. B, Summary of the comparison of IFN-γ production among CD8 T cells from spleen, peripheral blood, and TILs, which were pulsed with and without additional exogenous TRP1 peptides. Percentages of IFN-γ+ CD8 T cells (top) and MFI of the IFN-γ (bottom) are shown. C, Summary of the IFN-γ production of TIL with or without exogenous wtTRP1 peptides. Percentages of IFN-γ+ CD8 T cells (left) and MFI of the IFN-γ (right) are shown. This experiment was repeated three times with similar results.
Lentivector immunization eliminates small early stage tumors and inhibits growth of established tumors
To determine whether lentivector immunization affected growth of established B16 tumors in mice, we injected B16F0 tumor cells s.c. Mice were then immunized with muTRP1-lv 3 or 5 days after tumor inoculation. Immunization with lentivector eliminated early stage tumors (day 3 tumors) in 40% of mice (Fig. 7A). In the remaining immunized mice, tumor growth was strongly inhibited (Fig. 7A). The survival of muTRP1-lv-treated mice was significantly longer than control tumor-bearing mice (Fig. 7B). Even in mice with more established tumors (5 days), immunization with muTRP1-lv strongly inhibited B16F0 tumor growth and prolonged mouse survival, although no mice were cured in this case (Fig. 7, C and D). It is noteworthy that these highly resistant, more established tumors, which were unaffected by aggressive immunotherapy by using vaccines, anti-CTLA4, and therapeutic depletion of CD25 (35), were strongly suppressed by a single immunization with recombinant lentivector, muTRP1-lv.
FIGURE 7.

Lentivector immunization can eliminate small B16 tumor and significantly inhibit the growth of well-established tumors. C57BL/6 mice were inoculated with 1 × 105 B16F0 cells. Three days (A and B) or 5 days (C and D) later, mice were immunized with 2.5 × 107 TU of muTRP1-lv. Tumor growth (A and C) and mouse survival (B and D) were measured and recorded every other day. The numbers in the parentheses indicate the number of mice that have tumors out of total 10 (A) or 5 (C) mice.
Discussion
In the present study we show that lentivector immunization is an effective strategy to stimulate CD8 T cell responses against multiple epitopes of shared self/tumor Ags TRP1. Lentivector immunization dramatically increases functional T cell infiltration into tumors and generates protective and therapeutic anti-tumor immunity. However, the induction of potent CD8 T cell responses requires a combination of effective Ag delivery by lentivector and the mutated self/tumor Ag. Lentivector expressing native TRP1 protein failed to stimulate TRP1-specific T cell responses (Fig. 1B), and naked plasmid DNA expressing mutated TRP1 only stimulated very weak CD8 T cell responses (Fig. 1C). Data from our previous study showed that with four rounds of gene gun immunization, mutated TRP1 plasmid DNA stimulated ~0.5% of CD8 T cells (8). In contrast, recombinant lentivector expressing mutated TRP1 variant stimulated as many as 10% of CD8 T cells against multiple epitopes of TRP1 Ag (Fig. 1, B and C). Activated CD8 T cells could produce both IFN-γ and TNF-α, although the cells producing IL-2 were rare (Fig. 2A). The IL-2−IFN-γ +TNF-α+ CD8 T cells have been suggested to represent early stage partially exhausted T cells in chronic viral infection models (30). Whether their effector function is also partially damaged in antitumor immunity remains unclear. CD8 T cells effectively lysed target cells in vivo and produced IFN-γ after ex vivo stimulation with wtTRP1 peptides (Fig. 1). When B16 tumor cells were used to stimulate T cells, IFN-γ production was less robust. This lack of IFN-γ stimulation of CD8 T cells by B16 tumor cells may reflect the low MHC-Ag level on the B16 tumor cell surface that does not reach the threshold for stimulating IFN-γ production of primed CD8 T cells (Fig. 1E, center panels). Importantly, however, CD8 T cells elicited by immunization with mutated TRP1 lentivector were capable of recognizing and killing B16 tumor cells after in vitro recall stimulation with peptide (Fig. 1E, right panel), suggesting that in vivo primed and ex vivo restimulated CD8 T cells may not need high levels of MHC-Ag to kill target cells. Thus, our data suggested that lentivector is a highly effective immunization strategy for delivering mutated self/tumor Ag and eliciting potent CD8 T cell responses that cross-react with wild-type self/tumor Ag.
Consistent with previous findings in the OVA Ag system (12), the contraction phase of CD8 T cell response against TRP1 Ag induced by lentivector immunization was gradual and prolonged (Fig. 2B). The reasons for these prolonged T cell responses are not clear but are likely related to the protracted in vivo Ag presentation (Fig. 2C). Several studies, including our own, showed that lentivector transduction has minimal cytopathic effects on transduced cells, which exhibited persistent Ag expression, processing, and presentation (13, 19). The persistent in vivo Ag presentation after lentivector immunization may allow continuous stimulation of newly generated CD8 T cells and repeated stimulation of activated T cells, leading to slower and less drastic contraction. Such characteristics may be beneficial for generating potent and durable immune effectors against tumor cells. Although persistent chronic infection and inflammation could cause T cell exhaustion (31, 36, 37), the effect of prolonged Ag presentation on the effector and memory CD8 responses following lentivector immunization needs further investigation.
To exert a therapeutic antitumor effect, effector T cells must infiltrate into tumor mass to destroy tumor cells. We found a drastic (15-fold) increase of T cell infiltration into tumor lesions following lentivector immunization compared with control tumors (Fig. 5). CD8 T cells were the predominant T cells in the tumors of immunized mice, in striking contrast to the mainly CD4 T cells in untreated tumors. Importantly, the CD8 T cells in the tumors were functional and could immediately produce IFN-γ even in the absence of exogenous TRP1 peptide, suggesting that endogenous tumor Ags released from tumor cells were sufficient to activate CD8 T cells in the tumor environment (Fig. 6). However, compared with the CD8 T cells in the spleen and peripheral blood, CD8 T cells in the tumor did produce less IFN-γ even after exogenous peptide stimulation (lower MFIs of IFN-γ in Fig. 6, A and B), indicating that function of CD8 T cells was compromised to an extent but was not lost. This may be important because a recent report showed a complete loss of IFN-γ production and CTL function after infiltration into tumors (38). Previous reports have shown that both the GM-CSF B16 tumor vaccine (GVAX) and CTLA4 blockade are required to increase CD8 T cell infiltration into small B16 tumor (39); with more established tumor, a combination of vaccines, CTLA4 blockade, and prophylactic depletion of Treg is needed to enhance tumor CD8 T cell infiltration (35, 39). In contrast, we found that a single lentivector immunization was sufficient to induce a dramatic increase of functional CD8 T cell infiltration in tumor lesions. Thus, lentivector immunization is effective at stimulating potent CD8 T cell responses in tumor-bearing mice, which infiltrate into tumor and exert antitumor function.
In the setting of tumor immunotherapy, the time of T cell activation and the potency and longevity of CD8 T cell responses are crucial factors in determining therapeutic outcome. Since adaptive immune responses induced by active immunization need time to develop (usually a week) and have a natural course of expansion and contraction, CTL responses only have a narrow window to eliminate tumor cells before the natural course of contraction ensues and before the growing tumors establish local immune suppression to inhibit the function of effector cells. Thus, immunization strategies that generate rapid, potent, and long-lasting effector responses are essential for effective tumor immunotherapy (especially for the rapidly growing transplanted tumors used in most preclinical studies). We found that lentivector immunization rapidly (1 wk) stimulated ~10% of CD8 T cells to become activated TRP1-specific CD8 T cells against multiple epitopes of TRP1. The strong CD8 response lasted ~3 wk (Figs. 1D, 2B, and 4C). Such potent and durable CD8 T cell response eliminated small B16 tumors established for 3 days in 40% of mice (Fig. 7). Lentivector immunization also inhibited growth of more established B16 tumors (5-day tumors). However, even when 10% of CD8 T cells were effectors, this was not sufficient to eliminate the 5-day tumors. When the CD8 T cell responses were contracted (Fig. 4), B16 tumors grew back (Fig. 7). Unlike the adoptive transfer of activated CD8 T cells, in which T cells can be put into action right away (40), lentivector immunization needs at least 5–7 days to mount a measurable responses against immunized Ag (12). Thus, although we immunized mice on day 5 after tumor inoculation, the immune responses against growing tumor did not begin for 5–7 days, when the B16 tumors had been established for 10 –12 days. At this stage, the large tumor burden and tumor-induced immunosuppressive milieu may have prevented complete elimination of tumors before the T cells contracted. In this scenario, combinatorial approaches including removal of immune suppression with chemotherapy or tumor debulking treatment may be needed for clinical antitumor benefit (41– 43).
In summary, immunization with recombinant lentivector expressing mutated self Ag broke immune tolerance to shared self/tumor Ag and elicited potent and long-lasting effector T cells, which effectively infiltrated into tumor masses to exert their functions, eliminating small B16 tumors. Lentivector immunization offers an attractive approach for stimulating antitumor immunity.
Acknowledgments
We thank Joyce Wilson for her technical help in immunofluorescent staining and Dr. Phil Chandler for helping with the JAM assay. Dr. Janice Sabatine from Avanti Strategies received payment for editing assistance. The authors are grateful for the comments of Dr. Andrew Mellor of the Medical College of Georgia on this work.
Footnotes
Research in this study was supported by National Institutes of Health Grant R01 CA16444 and the Distinguished Investigator Fund from Georgia Research Alliance to Y.H.
Abbreviations used in this paper: TRP1, tyrosinase related protein 1; MHC I, MHC class I; DC, dendritic cell; muTRP1, mutated tyrosinase related protein 1; wtTRP1, wild-type tyrosinase related protein 1; TU, transducing unit; TIL, tumor-infiltrating T cell; wt455, wtTRP1 peptide 455; MFI, mean fluorescence intensity.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Slingluff CL, Jr, Chianese-Bullock KA, Bullock TN, Grosh WW, Mullins DW, Nichols L, Olson W, Petroni G, Smolkin M, Engelhard VH. Immunity to melanoma antigens: from self-tolerance to immunotherapy. Adv Immunol. 2006;90:243–295. doi: 10.1016/S0065-2776(06)90007-8. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Diez A, Spiess PJ, Restifo NP, Matzinger P, Marincola FM. Intensity of the vaccine-elicited immune response determines tumor clearance. J Immunol. 2002;168:338–347. doi: 10.4049/jimmunol.168.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 6.Sotomayor EM, Borrello I, Levitsky HI. Tolerance and cancer: a critical issue in tumor immunology. Crit Rev Oncog. 1996;7:433–456. doi: 10.1615/critrevoncog.v7.i5-6.30. [DOI] [PubMed] [Google Scholar]
- 7.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guevara-Patino JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J Clin Invest. 2006;116:1382–1390. doi: 10.1172/JCI25591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhorn ME, Guevara-Patino JA, Noffz G, Hooper AT, Lou O, Gold JS, Kappel BJ, Houghton AN. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med. 2006;12:198–206. doi: 10.1038/nm1363. [DOI] [PubMed] [Google Scholar]
- 11.Harrop R, John J, Carroll MW. Recombinant viral vectors: cancer vaccines. Adv Drug Delivery Rev. 2006;58:931–947. doi: 10.1016/j.addr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 12.He Y, Zhang J, Donahue C, Falo LD., Jr Skin-derived dendritic cells induce potent CD8+ T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24:643–656. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y, Zhang J, Mi Z, Robbins P, Falo LD., Jr Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol. 2005;174:3808–3817. doi: 10.4049/jimmunol.174.6.3808. [DOI] [PubMed] [Google Scholar]
- 14.Dullaers M, Van Meirvenne S, Heirman C, Straetman L, Bonehill A, Aerts JL, Thielemans K, Breckpot K. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006;13:630–640. doi: 10.1038/sj.gt.3302697. [DOI] [PubMed] [Google Scholar]
- 15.Esslinger C, Chapatte L, Finke D, Miconnet I, Guillaume P, Levy F, MacDonald HR. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8+ T cell responses. J Clin Invest. 2003;111:1673–1681. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe HM, Lopes L, Ikeda Y, Bailey R, Barde I, Zenke M, Chain BM, Collins MK. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol Ther. 2006;13:310–319. doi: 10.1016/j.ymthe.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 17.He Y, Munn D, Falo LD., Jr Recombinant lentivector as a genetic immunization vehicle for antitumor immunity. Expert Rev Vaccines. 2007;6:913–924. doi: 10.1586/14760584.6.6.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Y, Falo LD., Jr Lentivirus as a potent and mechanistically distinct vector for genetic immunization. Curr Opin Mol Ther. 2007;9:439–446. [PMC free article] [PubMed] [Google Scholar]
- 19.Breckpot K, Dullaers M, Bonehill A, van Meirvenne S, Heirman C, de Greef C, van der Bruggen P, Thielemans K. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med. 2003;5:654–667. doi: 10.1002/jgm.400. [DOI] [PubMed] [Google Scholar]
- 20.Garcia Casado J, Janda J, Wei J, Chapatte L, Colombetti S, Alves P, Ritter G, Ayyoub M, Valmori D, Chen W, Levy F. Lentivector immunization induces tumor antigen-specific B and T cell responses in vivo. Eur J Immunol. 2008;38:1867–1876. doi: 10.1002/eji.200737923. [DOI] [PubMed] [Google Scholar]
- 21.Palmowski MJ, Lopes L, Ikeda Y, Salio M, Cerundolo V, Collins MK. Intravenous injection of a lentiviral vector encoding NY-ESO-1 induces an effective CTL response. J Immunol. 2004;172:1582–1587. doi: 10.4049/jimmunol.172.3.1582. [DOI] [PubMed] [Google Scholar]
- 22.Metharom P, Ellem KA, Schmidt C, Wei MQ. Lentiviral vector-mediated tyrosinase-related protein 2 gene transfer to dendritic cells for the therapy of melanoma. Hum Gene Ther. 2001;12:2203–2213. doi: 10.1089/10430340152710540. [DOI] [PubMed] [Google Scholar]
- 23.Firat H, Zennou V, Garcia-Pons F, Ginhoux F, Cochet M, Danos O, Lemonnier FA, Langlade-Demoyen P, Charneau P. Use of a lentiviral flap vector for induction of CTL immunity against melanoma: perspectives for immunotherapy. J Gene Med. 2002;4:38–45. doi: 10.1002/jgm.243. [DOI] [PubMed] [Google Scholar]
- 24.Follenzi A, Naldini L. Generation of HIV-1 derived lentiviral vectors. Methods Enzymol. 2002;346:454–465. doi: 10.1016/s0076-6879(02)46071-5. [DOI] [PubMed] [Google Scholar]
- 25.Barchet W, Oehen S, Klenerman P, Wodarz D, Bocharov G, Lloyd AL, Nowak MA, Hengartner H, Zinkernagel RM, Ehl S. Direct quantitation of rapid elimination of viral antigen-positive lymphocytes by antiviral CD8+ T cells in vivo. Eur J Immunol. 2000;30:1356–1363. doi: 10.1002/(SICI)1521-4141(200005)30:5<1356::AID-IMMU1356>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki K, Zhao X, Pardee AD, Ueda R, Fujita M, Sehra S, Kaplan MH, Kane LP, Okada H, Storkus WJ. Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol. 2008;181:104–108. doi: 10.4049/jimmunol.181.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Usharauli D, Perez-Diez A, Matzinger P. The JAM test and its daughter P-JAM: simple tests of DNA fragmentation to measure cell death and stasis. Nat Protoc. 2006;1:672–682. doi: 10.1038/nprot.2006.107. [DOI] [PubMed] [Google Scholar]
- 28.Bohm W, Thoma S, Leithauser F, Moller P, Schirmbeck R, Reimann J. T cell-mediated, IFN-γ-facilitated rejection of murine B16 melanomas. J Immunol. 1998;161:897–908. [PubMed] [Google Scholar]
- 29.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 30.Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2008;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwissa M, Amara RR, Robinson HL, Moss B, Alkan S, Jabbar A, Villinger F, Pulendran B. Adjuvanting a DNA vaccine with a TLR9 ligand plus Flt3 ligand results in enhanced cellular immunity against the simian immunodeficiency virus. J Exp Med. 2007;204:2733–2746. doi: 10.1084/jem.20071211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 35.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med. 2008;205:2125–2138. doi: 10.1084/jem.20080099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, Hengartner H, Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res. 2008;68:2993–3000. doi: 10.1158/0008-5472.CAN-07-5008. [DOI] [PubMed] [Google Scholar]
- 39.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 42.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 43.Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]