

The authors developed a network approach that simplifies the identification of natural variation in complex temporal systems. Using this approach, they identified significant linkages between natural variation in the circadian clock and plant metabolism, including a newly described capacity for structural variation in plant secondary metabolism to influence the periodicity of the circadian clock.
Abstract
Modern systems biology permits the study of complex networks, such as circadian clocks, and the use of complex methodologies, such as quantitative genetics. However, it is difficult to combine these approaches due to factorial expansion in experiments when networks are examined using complex methods. We developed a genomic quantitative genetic approach to overcome this problem, allowing us to examine the function(s) of the plant circadian clock in different populations derived from natural accessions. Using existing microarray data, we defined 24 circadian time phase groups (i.e., groups of genes with peak phases of expression at particular times of day). These groups were used to examine natural variation in circadian clock function using existing single time point microarray experiments from a recombinant inbred line population. We identified naturally variable loci that altered circadian clock outputs and linked these circadian quantitative trait loci to preexisting metabolomics quantitative trait loci, thereby identifying possible links between clock function and metabolism. Using single-gene isogenic lines, we found that circadian clock output was altered by natural variation in Arabidopsis thaliana secondary metabolism. Specifically, genetic manipulation of a secondary metabolic enzyme led to altered free-running rhythms. This represents a unique and valuable approach to the study of complex networks using quantitative genetics.
INTRODUCTION
Phenotypes within species are not fixed and instead have significant levels of natural genetic variation that distinguishes individuals. This includes traits ranging from development and metabolism to pathogen resistance, with selection often maintaining the underlying genetic variation (Hopper, 1999; Veening et al., 2008). Understanding the molecular and genetic basis of complex quantitative traits is an important goal in genetics with wide-ranging ramifications across the scientific community. Unfortunately, this effort is complicated by the fact that most phenotypic variation is quantitative and polygenic with at least binary interactions with the environment, development, and second site genetic variation. This variation is further complicated by higher-order interaction among these factors (Falconer and Mackay, 1996; Lynch and Walsh, 1998; Wentzell and Kliebenstein, 2008). Thus, there is a desire to begin identifying the molecular systems controlling these interactions, likely requiring systems biology approaches.
One network that is known to be naturally variable while also having global ramifications for organisms across metabolism and development is the circadian clock. The clock attempts to coordinate metabolism and development within an organism to optimally coincide with the local day/night cycle (Harmer, 2009). Interestingly, in spite of its central importance, there is natural variation in plant circadian clocks that may help promote optimization to the local environment (Swarup et al., 1999; Michael et al., 2003; Darrah et al., 2006). This variation also affects interactions between environmental factors and the clock (Edwards et al., 2005, 2006). Thus, natural variation in the circadian clock could lead to complex genotype-by-environment interactions, perhaps causing large effects on transcript or metabolite levels.
One barrier to analyzing natural variation within the circadian clock is that the majority of existing methods require the development of new experimental tools, such as the introduction of promoter-reporter constructs into each genotype to be tested (Darrah et al., 2006). Alternatively, individual phenotypes such as leaf movement can be measured across time to estimate clock output for that phenotype; however, this still requires a large number of samples to generate a time course for each genotype (Swarup et al., 1999; Michael et al., 2003; Edwards et al., 2005, 2006). Even after this significant effort, only a few parameters are derived that measure clock function in a given population, limiting the ability to systematically identify variation in the circadian system. Thus, there is interest in developing tools that would allow the identification of natural variation in circadian clock function without having to invest significant new resources.
Recently, large populations of natural genotypes have been analyzed for expression quantitative trait loci (eQTL) via microarrays (Brem et al., 2002; Schadt et al., 2003; Keurentjes et al., 2007; West et al., 2007; Potokina et al., 2008). eQTL analysis typically identifies interesting loci by querying the data set on a gene-by-gene basis, but in addition generates residual databases of expression data that can be used in a systems approach to analyze complex phenotypes. One strategy uses a priori defined gene networks to conduct a network statistics analysis of the microarray data (Ueda et al., 2004; Kliebenstein et al., 2006; Bussemaker et al., 2007; Christley et al., 2009; Kliebenstein, 2009a, 2009b). These a priori networks can be defined from a number of sources, including biochemical pathways (Kliebenstein et al., 2006), coexpression data sets (Saito et al., 2008), genetics (Keurentjes et al., 2007), and protein–protein interactions (Ito et al., 2001). One approach for conducting a network analysis of circadian clock outputs has been to use preexisting data to assign genes to time bins based upon coincidence of peak expression within a circadian period (Ueda et al., 2004). These bins can then be used in the analysis of microarray data obtained from a sample harvested at a single time point to estimate the individual’s internal circadian time (CT) and identify altered circadian clock function (Ueda et al., 2004). This provides a network approach to analyze altered circadian clock outputs without requiring the development of new experimental materials, such as the introduction of promoter fusions into each genotype.
Using existing microarray data from detailed time courses on the Arabidopsis thaliana accession Columbia (Col-0; Covington et al., 2008), we developed 24 CT groups, hereafter called CT phase groups, that allowed us to query natural variation in circadian clock outputs using existing microarray experiments performed on the Bayreuth-0 (Bay-0) × Shahdara (Sha; syn: Shakdara) recombinant inbred line (RIL) population (Kliebenstein et al., 2006; West et al., 2007). This allowed us to rapidly identify naturally variable loci that alter circadian clock outputs. We then compared the CT phase group eQTLs to preexisting metabolomics QTL from this same population (Wentzell et al., 2007; Rowe et al., 2008) and found possible links between alterations in clock function and metabolism. Using single-gene isogenic lines, we were able to show that multiple circadian clock outputs, including CT expression networks, circadian regulation of the photosynthetic machinery, and daylength–dependent changes in flowering time, were altered by natural variation in a secondary metabolic enzyme. Together, these data show it is possible to use the CT phase group approach to query network alterations in circadian clock outputs and highlight the crosstalk between the circadian clock and plant metabolism.
RESULTS
CT Phase Group Expression Analysis for Measuring Natural Variation of Circadian Clock Output
Numerous studies have shown natural variation of various aspects of the circadian clock, such as periodicity of leaf movement or promoter activity of an individual gene (Swarup et al., 1999; Edwards et al., 2005, 2006; Darrah et al., 2006; Loudet et al., 2008). However, these studies required significant investments in time and revealed how natural variation may influence a single clock output. To both provide a more global view of natural variation in circadian outputs and to take advantage of existing data, we employed a network analysis of circadian-regulated genes to test for natural variation in clock outputs using a previously established method (Ueda et al., 2004). In this analysis, the authors showed that a single time point network-based measure of the circadian clock can rapidly identify perturbations within the circadian clock caused by genotypic alterations (Ueda et al., 2004). This approach relies upon the use of a carefully curated list of genes known to peak at different times of the day to generate a set of a priori defined phase groups that can then be used to estimate the overall state of the circadian system based upon a single time point, rather than relying upon multiple time points for a single gene (Figure 1) (Ueda et al., 2004; Kliebenstein et al., 2006; Kliebenstein, 2009b).
Figure 1.

Estimating a Circadian Pattern from a Single Time Point.
Shown is a three-dimensional standing wave plot of all circadian-regulated genes in a theoretical system. There are genes exhibiting all different times of peak expression, or phase. Differences in shading represent different levels of expression.
(A) If the expression level of a single gene is measured across time, the usual circadian expression pattern for that gene is obtained.
(B) Alternatively, if an experiment (i.e., microarray) measures expression of multiple genes with known different times of peak expression at a single time point (CT6 in this example), a similar estimate of the circadian expression pattern across all these genes can be obtained. This single time point snapshot thus allows the circadian status of the sample to be assessed.
We used a previously published list of 3975 genes to define 24 circadian output gene bins, here defined as CT phase groups, with coordinate peak time of expression between CT0 and CT23 (Covington et al., 2008). To test if this network approach would be able to capture the standing wave of circadian oscillations across a CT course, we used the network definitions to estimate the average CT phase group expression across a 4-d time course (Covington et al., 2008). This showed that each CT phase group had a single peak of expression per day and that it was possible to image the standing wave of oscillations (see Supplemental Figures 1 and 2 online). Individual genes within a CT phase group displayed some variation, but the confidence limits were very tight around the average across the time course (see Supplemental Figure 3 online). This demonstrates that analysis of the average expression patterns of the different CT phase groups allows for estimation of the global behavior of clock-regulated genes.
To test the utility of this network approach for analyzing clock outputs in other samples, we examined the difference in expression of clock output genes between the Arabidopsis accessions Bay-0 and Sha, the parents of a large RIL population for which several microarray experiments have been conducted. We used the average log2 expression of all genes within a CT phase group to estimate that group’s expression value. There were significant differences between Bay-0 and Sha across the CT phase groups, with expression of predusk-phased genes higher in Bay-0 and expression of night-phased genes higher in Sha (Figure 2, Table 1). To independently validate the slight difference in expression patterns of clock-regulated genes, we transformed the Bay-0 and Sha accessions with the luciferase reporter gene driven by the CCR2 (At2g21660) promoter (CCR2:luc), which is a member of the CT12 expression phase group (Strayer et al., 2000). Using three independent T2 lines per genotype, the free-running period of luminescence activity was found to be significantly different between Bay-0-CCR2:luc and Sha-CCR2:luc (Figure 3; see Supplemental Figure 4 online). Thus, the luciferase reporter assay supports the CT phase group approach for identifying genetic differences underlying alterations in outputs from the circadian clock.
Figure 2.

Circadian Phase Group Expression Difference in Arabidopsis Accessions.
Genes showing circadian expression in the Arabidopsis accession, Col-0, were grouped into 24 phase groups differing by their hour of maximal expression (i.e., the CT0 phase group contains all the genes with peak expression between CT0 and CT1). These 24 phase groups were then used to measure the average phase group expression in a single sampling time-point (CT8) from the Arabidopsis Bay-0 (blue) and Sha (red) accessions.
(A) The average log2 phase group expression in Bay-0 and Sha.
(B) The average Z scaled phase group expression in Bay-0 and Sha. Asterisks show statistically significant differences between Bay-0 and Sha as determined using ANOVA and a P < 0.05; significances were corrected post hoc using Tukey’s HSD comparisons.
Table 1.
ANOVA Comparison of CT Phase Groups in Bay-0 and Sha Parents
| Source | DF | SS | F | P |
| Model | 191 | 33.611 | 3.18 | <0.001 |
| Error | 768 | 42.516 | ||
| Total | 959 | 76.127 | ||
| Class | DF | Type III SS | F | P |
| Genotype | 1 | 0.088 | 1.59 | 0.208 |
| CT phase group | 23 | 0.073 | 0.06 | 1.000 |
| Treatment | 1 | 0.110 | 2.00 | 0.158 |
| Replicate | 1 | 1.416 | 25.58 | <0.001 |
| Genotype × replicate | 1 | 0.000 | 0.00 | 0.949 |
| Genotype × treatment | 1 | 0.011 | 0.20 | 0.653 |
| CT × genotype | 23 | 6.213 | 4.88 | <0.001 |
| CT × genotype × replicate | 46 | 2.177 | 0.85 | 0.742 |
| CT × genotype × treatment | 46 | 20.855 | 8.19 | <0.001 |
| CT × genotype × replicate × treatment | 48 | 2.632 | 0.99 | 0.494 |
DF is the degrees of freedom for a term within the model. Type III SS is the Type III sums of squares. SS is normal sums of squares. F indicates the F value, and P indicates the statistical significance for a given term within the model. Genotype is a comparison of Bay-0 versus Sha. CT phase group is the term incorporating the 24 CT phase groups. Treatment is control versus salicylate-treated plants. Replicate is experiment 1 versus 2. X indicates an interaction test within the ANOVA. There were 10 replicates per genotype and five per experiment.
Figure 3.

Circadian Period Length Differences between Bay-0 and Sha.
Transgenic Bay-0 and Sha accessions carrying the CCR2:luc reporter were entrained for 7 d in 12-h white light/dark cycles. Plants were released into constant red light, and luminescence was measured every 2 h. Free-running periods of individual plants were determined using the program fast Fourier transformed nonlinear least squares (Plautz et al., 1997). Data shown combine measurements from six independent T2 families assayed in two independent experiments. Data points represent average ± se of plants from both experiments combined. The colored bars represent the one standard deviation range with open circles representing outlier data points. Period length was significantly different between Bay-0 and Sha as shown by the asterisk (ANOVA, P < 0.05).
eQTL Mapping Using CT Phase Groups
The difference in CT phase group expression between Bay-0 and Sha suggested that there should be QTLs underlying these distinct patterns in the Bay-0 × Sha RIL population. We used an existing database of replicated single time point microarrays on 211 of the Bay-0 × Sha RILs to map eQTL controlling the CT phase groups (Kliebenstein et al., 2006; West et al., 2007; Kliebenstein, 2009b). We were able to identify a number of genomic regions that contained eQTL controlling CT phase group expression. The three largest CT phase group eQTL were on chromosome II and chromosome V (Figure 4) in the same positions as the largest trans-eQTL hotspots for this population (West et al., 2007). A similar analysis performed on these RILs after treatment with salicylic acid revealed eQTLs in all of the same locations, as well as an additional eQTL at the top of chromosome V and two eQTLs on chromosome III (see Supplemental Figure 5 online). The similar results obtained with independent biological replicates serves to confirm the location of these eQTLs.
Figure 4.

Additive QTL Estimates across the Genome.
Shown is the range of predicted additive effects for all marker × phase group combinations in Z scale, with the direction and magnitude of the effect indicated by the color. Only the additive effects for marker × phase group positions that showed a significant QTL are presented, with all nonsignificant positions given a value of 0 to improve clarity. The x axis indicates the 24 different CT phase groups, while the y axis shows the genetic position on the five different Arabidopsis chromosomes (I to V). Labels along the right side of the y axis show the genomic position of metabolomic QTLs identified previously (Rowe et al., 2008).
We next examined the effects of individual eQTL on expression of the different CT phase groups. The two CT phase group eQTL on chromosome II showed differential effects on night and day networks, while the chromosome V locus caused all CT phase groups to have elevated expression albeit with variable magnitudes (Figures 4 and 5). This suggests that these three trans-hotspots may contain polymorphisms that alter outputs of the Arabidopsis circadian clock. The vertical striping in the eQTL analysis (Figure 4; see Supplemental Figure 5 online) is likely due to certain CT phase groups having less statistical power, possibly due to inherent imprecision in the phase group definition. In accord with this, these lower significance CT phase groups showed similar additive effect trends; however, the magnitude of these changes was lower than for the CT phase groups that showed statistical significance (see Supplemental Figure 6 online).
Figure 5.

Individual QTL Effects across CT Phase Groups.
Shown are the percentage of the Sha allele additive effects for all CT phase groups for the three most significant QTL. Differences are shown in log2 for more direct interpretations, but identical results were obtained using Z scale values. The Arabidopsis Genome Initiative numbers indicate the gene from which the genetic marker closest to the LOD peak was obtained and represent the marker used for statistical analysis. At2g05630 (A), At2g32150 (B), and At5g45110 (C).
[See online article for color version of this figure.]
Since we have only one time point, it is not possible to determine whether the CT phase group eQTLs are due to changes in the amplitude, phase, or periodicity of the clock. However, the different free-running periodicities of Bay-0 and Sha (Figure 3) suggest that changes in clock pace likely play a role. We thereby modeled the impact of all three changes upon a CT phase group analysis and plotted out the predicted CT phase group eQTL results (see Supplemental Figure 7 online). The modeling showed that amplitude shifts of 20% or periodicity shifts of 1 h would generate a similar spectrum of effect sizes on differential expression as observed for the CT phase group eQTL (see Supplemental Figure 7 online). Although the large effect eQTL on chromosome V does not correspond to any of the circadian differences that we modeled, a visual comparison suggested that the two chromosome II CT phase group eQTL might be due to changes in either periodicity or amplitude of circadian clock outputs (Figure 5; see Supplemental Figure 7 online). Both period and amplitude have been found to vary in other studies of natural variation in Arabidopsis clock outputs (Swarup et al., 1999; Edwards et al., 2005, 2006; Darrah et al., 2006; Loudet et al., 2008). Extensive time-course analyses would be required to differentiate between these two possibilities.
Metabolomic QTL to CT Phase Group eQTL Relationship
After identification of the CT phase group eQTLs in control and salicylic acid–treated plants, we noticed that 11 of the 13 loci were also identified as major metabolomic QTL hotspots in a previous analysis of this same population (Figure 4; see Supplemental Figure 5 online) (Rowe et al., 2008). These metabolic QTL had previously been shown to describe an epistatic network whereby there are five core loci as defined from the metabolomics QTL analysis (Figure 6, red nodes) surrounded by two secondary metabolite loci (Figure 6, yellow nodes) and four interrelated peripheral loci (Figure 6, white nodes) (Rowe et al., 2008) regulating the abundance of glucosinolates. To test if these same loci also form an epistatic network altering the circadian clock output, we measured all pairwise interactions between these loci as previously described (Rowe et al., 2008). The circadian eQTLs also formed an epistatic network controlling CT phase group output within this population. Interestingly, all of the loci are involved in an epistatic network controlling both the metabolomic and CT phase group QTLs. This suggests that these 11 loci affect both circadian clock output and metabolism. This suggests that it is possible that these phenotypes are causally related, as it has been previously reported that clock function can affect metabolism and that metabolism can affect the clock (Fukushima et al., 2009). Given the number of RILs used for eQTL and metabolite QTL mapping, it is not possible to fully query the structure of this network because there are more genotypic combinations possible (2048) than there are lines available (211).
Figure 6.

Comparison of Epistatic Networks Generated from Metabolomic and Circadian Phase Group QTLs.
Pairwise epistatic interaction networks were determined between all 11 shared QTLs using the previously published metabolomic data (Rowe et al., 2008) and circadian phase group expression data and plotted using Pajek. Thickness of the lines represents the fraction of total epistatic interactions within the respective data set that were significant for that pair. For both plots, the five proposed core metabolomic QTLs are colored in red, the two major glucosinolate QTL are in yellow, and the more peripheral yet interconnected metabolomic QTLs are in white.
(A) Epistatic metabolomics QTL network.
(B) Epistatic circadian eQTL network (shown at half scale relative to the metabolomics network).
Glucosinolates and Circadian Clock Output
To investigate whether there is a causal relationship between changes in clock function and changes in metabolites, we made use of previously isolated mutants. Two of the minor CT phase group eQTLs mapped to the previously cloned AOP and ELONG loci, which contain multiple genes (Figure 4; see Supplemental Figure 5 online). Natural alleles lead to loss of AOP and ELONG function and result in natural variation in levels of glucosinolate secondary metabolites (Kliebenstein et al., 2001; Kroymann et al., 2001; Benderoth et al., 2006; Wentzell et al., 2007). Bay-0 has a natural knockout of AOP2, whereas Sha expresses the functional enzyme due to a local inversion that alters the promoter–open reading frame relationship (Chan et al., 2010). To test whether alterations in the glucosinolate pathway might affect regulation of the CT phase groups, we used previously published microarray data on single-gene isogenic lines that recreate the Bay-0 (nonfunctional) versus Sha (functional) polymorphism at AOP2 and compared expression of the CT phase group genes (West et al., 2007). We found that the introduction of a single functional AOP2 back into a naturally null Col-0 background caused a CT phase group shift, with predusk genes being lowered and night genes being elevated (Figure 7B). This shift is remarkably similar to that seen in Sha (functional AOP2) relative to Bay-0 (nonfunctional AOP2) (cf. Figures 7A and 7B). Thus, our quantitative complementation test in a completely different accession indicates that natural variation in a gene encoding a metabolic enzyme can cause changes in circadian regulation of gene expression and strongly suggests that AOP2 is at least partly responsible for one of our CT phase group eQTLs.
Figure 7.

Glucosinolate Genes Regulate Expression of CT Phase Groups.
The percentage of the Sha allele additive effects are shown for all CT phase groups. Only three of the significant QTL are shown for clarity. Asterisks indicate significant differences between Bay-0 and Sha in (A) and between single gene variants versus Col-0 for (B) to (E). The percentage of expression differences are shown for more intuitive interpretation. Because identical results were seen using the Z scale values for statistical analysis, the differences in log2 are shown to be more directly interpretable.
(A) AOP QTL within the Bay-0 and Sha population.
(B) AOP2 transgene in Col-0 versus wild-type Col-0 (natural knockout of AOP2).
(C) myb28-1 knockout versus wild-type Col-0.
(D) myb29-1 knockout versus wild-type Col-0.
(E) myb28-1/myb29-1 double knockout versus wild-type Col-0.
(F) Flowering time effects of glucosinolate single gene variants as measured by time to bolting. The difference with respect to Col-0 is presented with the glucosinolate single gene variants as follows: AOP2, circle; myb28, triangle; myb29, diamond; and myb28/myb29 double knockout, square. All genotypes are represented by at least 12 individual plants per condition. Error bars are not provided for each circle as they represent the comparison to the wild type. Instead, symbols show if the comparison was statistically significant. Open symbols show phenotypes with no difference from Col-0, while closed symbols are statistically different from Col-0 within that light cycle at P < 0.05.
[See online article for color version of this figure.]
Since the AOP2 QTL interacts epistatically with two MYB QTL, MYB28 and MYB29, in the regulation of glucosinolates (Sønderby et al., 2007; Wentzell et al., 2007), we hypothesized that alterations in MYB28 and MYB29 expression would also alter CT phase group expression. To test this hypothesis, we examined previous microarray data from plants mutant for these two transcription factors. Mutation of either of these Mybs individually recreates the polymorphism at two separate natural QTLs (Gigolashvili et al., 2007, 2008; Hirai et al., 2007; Sønderby et al., 2007, 2010; Wentzell et al., 2007). Interestingly, we found that mutation of either Myb gene by a T-DNA insertion in the Col-0 background caused a significant shift in the output of the circadian clock when compared with the wild type, as measured by the CT phase group analysis (Figures 7C and 7D) (Sønderby et al., 2007). Since MYB28, MYB29, and AOP2 coordinately function within a complex regulatory network, these findings further support the conclusion that AOP2 is a causal gene controlling natural variation in CT phase group expression and that glucosinolates are involved in this process.
The genetic relationship between MYB28 and MYB29 is complex, as they are thought to function in an incoherent feed-forward loop in the regulation of some genes (Sønderby et al., 2010). In accordance with this complex relationship, the double MYB28/MYB29 knockout line altered CT phase group output in a manner different from the single knockouts (Figure 7E). Since the wild-type and MYB mutant microarrays were all done on plants in the same growth chamber and all samples were harvested within 15 min of each other, technical errors are not a likely explanation for the striking similarities and differences between expression of the CT phase groups in these different genotypes (Sønderby et al., 2010). Unfortunately, the independent MYB/wild type and AOP2/wild type microarray experiments were harvested at different times of day, precluding a direct comparison between the effects of AOP2 and the MYB genes on clock outputs.
Although our data demonstrated that altered AOP2 function caused changes in expression of clock output genes (Figures 7A and 7B), it did not reveal whether this was due to alteration of a subset of outputs or changes in the entire circadian system. We therefore compared the expression levels of central clock genes in Col-0, Col-0 expressing the AOP2 transgene, and the myb28 and myb 29 mutants (Table 2) (Sønderby et al., 2007, 2010; Wentzell et al., 2007; Harmer, 2009). We found that the introduction of AOP2 into a natural null background significantly decreased the expression of PRR7, CCA1, and PRR3 at the time point examined (Table 2). Furthermore, the knockouts in MYB28 and MYB29 led to significantly increased expression of GI and PRR7 at the sampled time (Table 2). Thus, genetic perturbations in the glucosinolate pathway can influence transcript abundance for the core oscillator genes (Harmer, 2009), providing an example of how changes in metabolism affect clock function.
Table 2.
Altered Expression of Clock-Related Genes in Glucosinolate Mutants
|
AOP2 |
myb28 |
myb29 |
|||||
| Gene | AGI | P | Chg | P | Chg | P | Chg |
| CCA1 | At2g46830 | <0.001 | −85% | 0.133 | 17% | 0.941 | −28% |
| GI | At1g22770 | 0.168 | −9% | <0.001 | 57% | 0.001 | 54% |
| LHY | At1g01060 | 0.046 | 10% | 0.017 | 19% | 0.313 | 21% |
| PRR3 | At5g60100 | 0.012 | −71% | 0.990 | 3% | 0.561 | 4% |
| PRR7 | At5g02810 | 0.043 | −183% | <0.001 | 58% | <0.001 | 57% |
| PRR9 | At2g46790 | 0.011 | −10% | 0.172 | −16% | 0.221 | 7% |
| TOC1 | At5g61380 | 0.104 | −15% | 0.079 | 29% | 0.053 | 30% |
| ZTL | At5g57360 | 0.109 | 5% | 0.012 | 21% | 0.433 | 11% |
P is the false discovery rate adjusted P value for the ANOVA comparing the mutant to wild-type Col-0 array-based expression value for the given core circadian clock genes. Chg is the percentage of change of the mutant in relation to the experiment-specific wild-type expression average as calculated by (Mut – wild type)/((wild type + Mut)/2). All data were obtained from the previous microarrays described in Methods. AGI, Arabidopsis Genome Initiative.
Glucosinolates and Circadian Clock Period
To independently validate the effect of the above glucosinolate mutants on the circadian clock, we assayed their free running circadian periods using delayed fluorescence (DF), an assay that determines the photochemical state of photosystem II (Gould et al., 2009). The DF assay for circadian period is a physiological measurement that is completely independent of the CT phase group approach. Using three independent biological replications, both AOP2 and the myb28/myb29 double knockout show a significantly shorter circadian period than Col-0 (Table 3; see Supplemental Figures 8 and 9 online). If the circadian period data are reanalyzed without the third trial, which has elevated variance, the period difference between Col-0 and myb28 also becomes significant (see Supplemental Figure 9 online). Interestingly, AOP2 and the myb28/myb29 double knockout have opposite effects on the total level of glucosinolates, with AOP2 leading to higher glucosinolate accumulation in comparison to the Col-0 genotype, whereas the myb28/myb29 double knockout has no aliphatic glucosinolates (Sønderby et al., 2007, 2010; Wentzell et al., 2007; Beekwilder et al., 2008). However, both genotypes have a shorter circadian period, suggesting that the influence is not simply due to altered carbon/nitrogen/sulfur flux into glucosinolates. These independent data confirm that AOP2 natural variation leads to a shorter circadian period in at least chlorophyll fluorescence under these growth conditions. This also shows that the single time point CT phase group approach can identify candidate genes that influence circadian clock outputs.
Table 3.
Measurement of Circadian Period using Delayed Fluorescence
| Genotype | Period (h) | Period Diff. versus Col-0 | se | P | n |
| Col-0 | 23.95 | NA | 0.39 | 29 | |
| AOP2 | 23.00 | −0.94 | 0.28 | 0.001 | 27 |
| myb28 | 23.51 | −0.44 | 0.28 | 0.119 | 29 |
| myb28/29 | 23.27 | −0.68 | 0.29 | 0.02 | 26 |
| myb29 | 23.67 | −0.28 | 0.28 | 0.322 | 28 |
Period shows the measured circadian period of DF oscillation in hours. P is the P value for the comparison of the specific genotypes period to the wild-type Col-0 period using ANOVA. n is the number of independent groups of plants measured to determine DF period.
Glucosinolates and Complex Physiological Outputs
We next wished to determine whether alterations in the glucosinolate pathway would affect a physiological process strongly linked to the circadian clock: the daylength-dependent regulation of flowering time. When grown in constant light, all three single glucosinolate pathway variants (AOP2, myb28, and myb29) flowered significantly earlier than the wild type (Figure 7F). However, they all showed differential regulation of flowering time when grown in long days (16 h light/8 h dark) versus short days (10 h light/14 h dark). Introduction of AOP2 caused delayed flowering in long days but had no effect in short days. Loss of myb28 or myb29 accelerated flowering in short days relative to the wild type but had no significant effect in long days. By contrast, loss of both myb28 and myb29 caused a strong delay of flowering in short days and a more modest delay in long days (Figure 7F). This latter finding is consistent with the opposite effects of the myb28 and myb29 single and double mutants on regulation of expression of CT phase group genes in comparison to the myb28/29 double knockout (Figures 7C to 7E). Thus, all four glucosinolate variants lead to alteration of flowering time in a daylength-dependent manner.
These data demonstrate that natural variation in the glucosinolate pathway can lead to complex physiological shifts in the plant that may be related to the circadian clock. However, the observed changes in flowering time do not readily fit into existing models linking the output of the circadian clock to flowering decisions; therefore, we are left with the possibility that the two phenotypes, CT phase group and flowering time, are independent outputs of the altered glucosinolate pathway. In support of this altered physiology, the AOP locus, specifically the tandem AOP3 gene that is in tight linkage disequilibrium with AOP2, has been found to be a major flowering time locus using genome-wide association mapping within Arabidopsis (Atwell et al., 2010; Chan et al., 2010; Li et al., 2010).
DISCUSSION
The above results show that we can apply a CT phase group approach to measure potential changes in circadian clock outputs using carefully controlled single time point microarray data. This suggests that it would be possible to reinterrogate all existing microarray experiments (where controls are appropriately timed with the treatment samples) to begin a systematic analysis of how different genotypic and environmental perturbations not frequently tested for circadian clock influences may in fact alter the clock. Given that Arabidopsis has a vast database of microarray experiments, this could provide unique biological insights into novel connections between the circadian clock and physiology (Grennan, 2006; Jen et al., 2006; Obayashi et al., 2007; Thum et al., 2008). Within our analysis of natural variation and circadian clock output, this approach identified significant links between the plant metabolome and circadian clock outputs. In follow-up studies, we directly validated the link between glucosinolates and the clock by showing that glucosinolate genotypes altered the periodicity of a clock output unrelated to glucosinolates, the photochemical state of photosystem II.
Plant Metabolism and Circadian Clock Outputs
Previous work using a single model genotype of Arabidopsis has indicated that the circadian clock may alter the regulation of plant metabolism (Harmer et al., 2000; Gutierrez et al., 2008; Fukushima et al., 2009). Our results suggest that natural variation in the plant metabolome is also intimately linked to natural variation in circadian clock outputs and that the relationship maybe bidirectional. Similar bidirectional relationships have been found in mammalian and plant systems using induced mutants (Dodd et al., 2007; Duez and Staels, 2008; Kovac et al., 2009).
Recent work has shown that the II.47 QTL found for both the circadian clock and metabolomics (At2g32150, Figure 5) is likely caused by natural variation between the Bay and Sha alleles at EARLY FLOWERING3 (ELF3) (Rowe et al., 2008; Jiménez-Gómez et al., 2010). Quantitative complementation tests showed that these alleles led to different periodicities in the circadian clock and nicely agree with the CT phase group effect measured in the Bay × Sha RIL population (Figure 5) (Jiménez-Gómez et al., 2010). Thus, one of the CT phase group eQTLs we identified corresponds to a causal locus previously identified from the same mapping population. Importantly, our prediction that this CT phase group eQTL alters circadian parameters was verified by Jiménez-Gómez et al. (2010), providing further support for our phase group analysis approach.
In this study, we demonstrated that the secondary metabolite QTL, AOP, is a QTL influencing both plant metabolism and the circadian clock period. Transgenic analyses confirmed that introduction of the AOP2 gene into a knockout background altered the circadian clock output in a manner highly similar to the QTL identified using the CT phase group approach (Figure 7). We also found that a functional AOP2 locus significantly shortened circadian period (Table 3), suggesting that this altered clock parameter might cause the observed effects on CT phase group expression in AOP2 transgenic plants. Although AOP2 altered photoperiodic regulation of flowering time (Figure 7), the observed effects cannot easily be explained by a shortening of free-running period; further analysis would be required to prove whether these two phenotypes are causally related to each other.
How might AOP2 affect clock parameters? AOP2 encodes a biosynthetic enzyme that controls the conversion of one glucosinolate into another (Kliebenstein et al., 2001), and as such, this suggests that this is a link whereby the metabolic network alters circadian clock function. Further support for this possibility was found when two MYB transcription factors regulated by AOP2 (Wentzell et al., 2007) were also shown to alter expression of circadian clock outputs and circadian periodicity. Thus, natural variation in biosynthetic enzymes can feedback to alter circadian clock output, suggesting that the flow of information flow between the clock and metabolism is bidirectional. Similar links between clock function and metabolism have been previously reported (Rutter et al., 2002; Dodd et al., 2007; Duez and Staels, 2008; Kovac et al., 2009; Wood et al., 2010). Interestingly, we found that two completely separate genomics technologies, transcriptomics and metabolomics, that measure separate products, transcripts, and metabolites, independently identify the same genetic network within this Arabidopsis population. This provides extra validation for the CT phase group approach to identify novel physiology.
Secondary Metabolism and Pleiotropy
It is striking that natural variation in a secondary metabolism gene can alter circadian clock parameters. There are several possible scenarios that may explain this. The first is that secondary metabolites might be energetically expensive to produce and the functional AOP2 allele leads to increased glucosinolate content. Thus, there may have been evolutionary pressure to alter the circadian clock output to shift some C/N/S from growth, which in Arabidopsis occurs largely in the morning (Nozue et al., 2007; Loudet et al., 2008), toward secondary metabolism, which for glucosinolates is typically induced after dusk (Harmer et al., 2000; Covington et al., 2008). Two factors argue against this. First is that the MYB knockout mutants, which led to similar alterations in flowering time and CT phase groups as AOP2, accumulate less glucosinolates than the wild type; by contrast, AOP2 induces higher glucosinolate levels. Thus, at least the effect on flowering time is not merely a consequence of C/N/S flux into glucosinolates. Second, if there is an energetic cost of glucosinolate production, it is very small, and most measured costs are more likely ecological and not energetic, since these are defensive compounds (Mauricio and Rausher, 1997; Lankau, 2007; Lankau and Strauss, 2007, 2008; Lankau and Kliebenstein, 2009; Paul-Victor et al., 2010). These data suggest that the effects of glucosinolates on clock outputs are not likely caused by altered flux of metabolites or energy.
The second possibility has to do with the central importance of secondary metabolites in plant defense against biotic pests, which likely use their own circadian clocks to optimally time their attacks on plants. Thus, there may be a competition between plant and host that would lead the plant to subtly alter clock outputs in response to an attack so as to prevent the other organism from optimizing its ability to attack (Walley et al., 2007). Our data suggest that there may be a reciprocal flow of information whereby the clock can gate responses to defense responses (Martínez et al., 2004; Griebel and Zeier, 2008; Roden and Ingle, 2009), and some of the defense outputs (i.e., glucosinolates) can send information back to the circadian clock or its outputs (Figure 6, Table 3). Future research on the impact of clock mutants upon plant defense under different day/night cycles as well as how other plant defense pathways interact with circadian clock outputs are required to test how frequently this occurs.
Secondary Metabolism, Selection, and Environmental Correlates
Using the CT phase group approach, we generated evidence that natural variation at four polymorphic glucosinolate loci can influence flowering time and the circadian clock. Previous evidence had strongly suggested that the AOP and ELONG loci are under strong natural selection that was presumed to be primarily due to insect herbivores (Giamoustaris and Mithen, 1995; Wright et al., 2002; Kroymann et al., 2003; Kroymann and Mitchell-Olds, 2005; Bidart-Bouzat and Kliebenstein, 2008). Given that it does not appear that the glucosinolate to circadian clock or flowering time links revolve around simple flux/energy balance, it raises the question of what selective pressure could have driven the formation of these links. One possibility comes from the observations that insect populations within the wild vary greatly from year to year but in a manner that is partially dependent upon the environment. Thus, there could be environmental correlations between abiotic stresses and biotic pest occurrence that would potentially generate pressure to highly integrate signals within a plant between what in the laboratory would be considered unlinked phenomena. For instance, slug and snail herbivory is likely more important in the spring when daylength is shorter, while lepidopterans would begin to predominate later in the season as the days lengthen. Similar links could exist across all clines of abiotic environmental parameters leading to highly complex selective pressures caused by the correlational structure across these clines. More research is required to test how often abiotic correlations can be linked to biotic pest resistance diversity within plants.
CT Phase Group Provides a Broad View of the Circadian Clock Output
An interesting aspect of the CT phase group approach of querying circadian clock output is that it provides the ability to measure numerous clock outputs simultaneously (since there are multiple CT phase groups). This is in contrast with single promoter luciferase reporters or individual phenotype outputs that are limited to the part of the clock controlling that phenotype (Swarup et al., 1999; Edwards et al., 2005, 2006; Darrah et al., 2006; Loudet et al., 2008). For instance, the Met.I.80 QTL would not have been found with a circadian clock reporter or phenotype that peaked during the light hours. This single time point analysis, however, limits the CT phase group approach to the identification of experimental factors that alter circadian clock output but does not readily lend itself to determining whether these are due to changes in phase, periodicity, amplitude, or a subset of clock outputs. Thus, the initial use of the CT phase group approach to rapidly survey numerous experimental factors followed by more intensive time course experiments may provide a powerful approach to characterize the roles of the identified factors. It should be noted that the CT phase group approach is predominantly a method to generate hypotheses about specific genes that require future validation. However, we note that any physiological process that can be described via a transcriptomic response would be amenable to this exact same approach. For instance, drought networks could be queried for group response QTLs that may be predictive of the actual drought QTLs measured physiologically.
Conclusions
We assessed circadian CT phase groups as a measure of the circadian clock output, allowing us to use single time point data to analyze connections between the circadian clock and metabolism within Arabidopsis. This approach allowed us to identify circadian clock output QTL from a previously conducted RIL microarray experiment. This single-point expression analysis was already costly and labor intensive; it would not have been possible to carry out a conventional CT course experiment on these RILs. We identified numerous links between circadian clock outputs and plant metabolism; however, the identification of the molecular mechanisms underlying these connections will require intensive future efforts. Importantly, our study shows that natural variation in plant defense metabolism can influence the circadian clock, thus raising new questions about how to properly interpret studies involving plant defense metabolism. The CT phase group approach described here allows the circadian clock to be analyzed in a new manner that will potentially lead to the identification of clock function in many aspects of plant physiology and plant/environment interactions.
METHODS
Circadian CT Phase Group Generation
Previously published analysis of microarray gene expression data from wild-type Arabidopsis thaliana using the COSOPT (Cosine Optimization) algorithm identified 3975 genes as having circadian oscillations of their expression (Covington et al., 2008). COSOPT fits cosine waves of varying periods to gene expression data and determines the best match with the data using 101 different phases for each period considered. The best-fitting curve is then determined using a least-squares analysis and statistical significance determined by empirical resampling (Straume, 2004). We used the predicted phases from the best fit cosine waves to classify the clock-regulated genes into 24 phase groups based on the CT of peak expression. Phase group CT0 was defined by all transcripts whose peak expression fell between CT0 and CT1. This was repeated to obtain CT phase groups from 0 to 23 (see Supplemental Data Set 1 online).
Parental CT Phase Group Analysis
Previously published microarray data for the Bay-0 and Sha parental accessions was obtained to analyze the potential for using CT phase group analysis to detect natural variation in circadian expression (West et al., 2007). Within this experiment, the tissue was harvested between CT ~6 and 7 in a randomized design to minimize any potential bias. Transcripts were classified by their membership in the 24 CT phase groups, and the average log2 expression was obtained across the transcripts within each phase group as previously described (Kliebenstein et al., 2006; Kliebenstein, 2009c). Additionally, a standard normal (z) distribution, N(0,1), was used to individually standardize each gene’s expression across the individual samples. The expression value (x) for each transcript for each sample was transformed to the corresponding z score by subtracting the transcripts average across the arrays (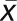 ) and then dividing by the transcripts standard deviation across the arrays (σ) using the equation
) and then dividing by the transcripts standard deviation across the arrays (σ) using the equation 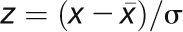 (Kliebenstein et al., 2006; Kliebenstein, 2009b). This functionally gives all genes an average of 0 and standard deviation of 1 across all of the arrays being used. This has previously been shown to eliminate the influence of highly expressed genes on a network average approach (Kliebenstein et al., 2006; Kliebenstein, 2009b). For presentation in Figures 2B and 2D, the Z scaling was done separately within each treatment. The average CT phase group Z value was also obtained. Nested analysis of variance (ANOVA) was then conducted to assess if there were statistically significant changes between the accessions for the CT phase groups. Differences between Bay-0 and Sha for individual CT phase groups were conducted within the ANOVA via partial F tests.
(Kliebenstein et al., 2006; Kliebenstein, 2009b). This functionally gives all genes an average of 0 and standard deviation of 1 across all of the arrays being used. This has previously been shown to eliminate the influence of highly expressed genes on a network average approach (Kliebenstein et al., 2006; Kliebenstein, 2009b). For presentation in Figures 2B and 2D, the Z scaling was done separately within each treatment. The average CT phase group Z value was also obtained. Nested analysis of variance (ANOVA) was then conducted to assess if there were statistically significant changes between the accessions for the CT phase groups. Differences between Bay-0 and Sha for individual CT phase groups were conducted within the ANOVA via partial F tests.
CT Phase Group QTL Mapping
Previously published GC-RMA normalized microarrays for 211 individuals from the Bay-0 × Sha RIL population were from untreated samples. In addition, we obtained a salicylate-treated data set to act as a replicate to help confirm the presence of CT phase group QTLs (see Supplemental Figure 3 online). Each transcript was independently Z scaled across all of the arrays as described above, and the average CT phase group Z value was obtained across all the transcripts per CT phase group per individual, providing an estimation of the circadian pattern within each individual (Loudet et al., 2002; West et al., 2007). The calculated average CT phase group Z values are presented (see Supplemental Data Set 2 online). These CT phase group Z values were then used as phenotypic traits to map phase group eQTL as previously described (Kliebenstein et al., 2006). We used composite interval mapping for each CT phase group using Windows QTL Cartographer version 2.0 (Basten et al., 1999; Zeng et al., 1999; Wang et al., 2006). Significance thresholds for composite interval mapping analyses were generated using 500 permutations of the data with α = 0.10 (Doerge and Churchill, 1996). We settled on a lower threshold for significance on the assumption that the different CT phase groups would provide supportive evidence and any isolated marker × CT phase group significances would be ignored. QTL distribution across the Arabidopsis genome, including direction of allelic effects, was obtained from QTL Cartographer and visualized using R (R Development Core Team, 2008). We used the significance output to mask all additive values at marker × trait locations where there was no evidence of statistical significance (see Supplemental Figure 1 online). The resulting file was then visualized as a heat plot using a red/blue color scheme using the filled contour function.
One concern regarding eQTL analysis is the potential role that single feature polymorphisms may play in altering the estimated expression of an individual gene (Borevitz et al., 2007). Given the network approach that we used where the 24 phase groups each have at least 100 individual transcripts, we did not explicitly remove these polymorphisms prior to the analysis as their impact upon the network average would be minimal. Similarly, the network approach allows us to not a priori eliminate any large effect cis-eQTL that might also be presumed to influence the expression estimate (Kliebenstein et al., 2006; West et al., 2007). For instance, for the log2 approach, one single feature polymorphism or cis-eQTL would have to cause an allelic change in log2 of nearly 20 orders of magnitude to cause a 20% change in >100 genes. Given this, our network approaches are not inherently sensitive to single feature polymorphism or cis-eQTL complications at this scale (Kliebenstein et al., 2006; West et al., 2007).
Circadian Rhythm Analysis
Bay-0 and Sha plants were transformed with the CCR2:luc construct (Strayer et al., 2000). Two independent experiments were performed using three T2 families of each accession. Plants plated in Murashige and Skoog medium with the appropriate antibiotic were stratified for 4 d and entrained in 12-h white light/dark cycles (50 μmol m−2 s−1) for 7 d. Resistant plants were then transplanted to new Murashige and Skoog plates (Fisher Scientific), moved to constant red light (50 μmol m−2 s−1), and recorded every 2 h for 7 d using a cooled CCD camera. The data collected were analyzed for rhythmicity using the luciferase activity method described by Plautz et al. (1997). In total, we obtained luciferase data from 151 plants, for which 132 plants (36 Bay-0/12 Sha plants in experiment 1 and 33 Bay-0/51 Sha plants in experiment 2) presented robust oscillations (measured by a relative amplitude error [RAE] lower than 0.5). RAE is a measure of robustness of rhythmicity; a RAE of 0 indicates a very robust rhythm with an infinitely well-determined rhythmic component, whereas a RAE of 1 indicates that the error in the amplitude equals the amplitude value itself and thus no significant rhythm is detected.
Epistatic Network Analysis
We used a previously described ANOVA analysis to test for epistatic interactions between metabolomic QTLs for impacts upon CT phase group using the control sample data only (Rowe et al., 2008). Briefly, this used an ANOVA model containing all previously identified CT phase group/metabolomic loci as individual main effect terms using the markers most closely associated with each significant QTL cluster. Additionally, we tested all 55 possible pairwise interactions between these markers. For each metabolite, the average CT phase group expression in lines of genotype g at marker m was shown as ygm. The model for each CT phase group was
where g = Bay or Sha; m = 1, …,11; and n was the identity of the second marker for an interaction. The main effect of the 11 markers was denoted as M. The error, εgmn, was assumed to be normally distributed with mean 0 and variance σε2. An automated SAS script was developed to test all CT phase groups within this ANOVA and return all P values, Type III sums-of-squares for the complete model, and each individual term and QTL pairwise-effect estimates in terms of allelic substitution values. Significance values were corrected for multiple testing within a model using false discovery rate (<0.05) in the automated script. Additionally, the CT phase group expression data were permuted across the RILs 500 times to test for any potential impact of segregation distortion upon epistatic interaction detection power.
Glucosinolate Mutant CT Phase Group Analysis
Previously published microarray data investigating transcript accumulation in 35S:AOP2 versus wild-type Col-0 were obtained (Wentzell et al., 2007). The 35S:AOP2 introduction into Col-0 mimics the AOP2 allele within the Bay-0 × Sha RIL population (Kliebenstein et al., 2001; Wentzell et al., 2007; Chan et al., 2010). For further testing of a link between glucosinolates and circadian network expression, we obtained previously published microarray data from experiments querying the effect of knockouts in two of the major transcription factors for glucosinolates, MYB28 and MYB29 (Sønderby et al., 2007, 2010). Similar microarray data for the myb28 (SALK_136312), myb29 (SM.31316), myb28/myb29 double knockout, and the corresponding wild-type Col-0 controls were also obtained (Sønderby et al., 2007, 2010). All plants from these experiments were grown in the same conditions and growth chambers as the Bay-0 × Sha RIL population, enhancing the ability to conduct the comparison (West et al., 2007). GC-RMA normalized transcript values for all transcripts in the 24 CT phase groups and the average CT phase group log2 values were obtained as described above and tested via ANOVA for significant differences within each CT phase group. Each mutant-to-wild type comparison had at least three independent biological replicates as described (Sønderby et al., 2007, 2010; Wentzell et al., 2007). The average CT phase group log2 values were then used within ANOVA to test for differences between the mutant and the wild type. For the MYBs, an interaction between the two genes was included within the ANOVA. Differences between the wild type and mutants for individual CT phase groups were obtained by conducting partial F tests within the ANOVA. The CT phase group Z scale values were also used and showed identical statistical responses.
Glucosinolate Mutant Circadian Period Analysis
The circadian periods of the four mutants and wild-type Col-0 were estimated from DF data (Gould et al., 2009). After sterilization, clusters of ~15 to 20 seeds were plated on half-strength Murashige and Skoog medium with 1.5% agar and subsequently stratified for 2 to 4 d at 4°C. Plants were entrained to 12/12-h light/dark cycles at 22°C and grown for 16 d prior to assaying their circadian periods. At subjective dawn on the 16th day, plants were transferred to an imaging chamber and put under constant RB (red and blue) light from an LED array set to ~20 μmol m−2 s−1. Illumination was interrupted to acquire DF images once every hour for 63 s, 3 s of preexposure darkness, followed by 60 s of image exposure. Fluorescence was captured using an iKon-M DU-934N-BV low-light CCD camera (Andor Technology) cooled to −80°C. The LED array and CCD camera were controlled with a custom LabVIEW (National Instruments) program. DF values were extracted from CCD camera images with ImageJ (W.S. Rasband, U.S. National Institutes of Health) using the Multi Measure plug-in and the circle tool macro set to measure integrated pixel density. These data were then analyzed using BRASS (available at http://www.amillar.org) to estimate circadian period and RAE by fast Fourier transformed nonlinear least squares (Plautz et al., 1997). The data were normalized and detrended for amplitude and baseline differences prior to analysis. Period estimates with an RAE >0.6 were excluded from downstream analyses. A linear model was fit to the circadian period data from three separate trials to determine if the mutants were significantly different than Col-0; the nlme package (Pinheiro and Chao, 2006) in R (R Development Core Team, 2008) was used for this analysis.
Glucosinolate Mutant Flowering Time Analysis
The same mutants and wild-type Col-0 were placed in three chambers that differed in their light cycles (constant light, 16/8-h light/dark, and 10/14-h light/dark). Flowering time was measured in six to 12 plants per genotype per chamber using the time to bolting as determined by the inflorescence being > 1 cm (Clarke et al., 1995; Jansen et al., 1995).
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL database or the Arabidopsis Genome Initiative database under the following accession numbers: CCR2 (At2g21660), CCA1 (At2g46830), GI (At1g22770), LHY (At1g01060), PRR3 (At5g60100), PRR7 (At5g02810), PRR9 (At2g46790), TOC1 (At5g61380), ZTL (At5g57360), MYB28 (At5g61420), MYB29 (At5g07690), AOP2 (At4g03060), AOP3 (At4g03050), MAM1 (At5g23010), MAM3(At5g23020), and ELF3 (At2g32150). Accession numbers for MYB28 and MYB29 knockout mutants are SALK_136312 (myb28) and Sail SM.31316 (myb29).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Analysis of CT Phase Group Expression across a Circadian Microarray Experiment.
Supplemental Figure 2. Analysis of CT Phase Group Expression across a Circadian Microarray Experiment.
Supplemental Figure 3. Analysis of Z Scaled Expression for All Genes within the CT 0 Phase Group across a Circadian Microarray Experiment.
Supplemental Figure 4. Circadian Rhythms in Bay-0 and Sha.
Supplemental Figure 5. Additive QTL Estimates across the Genome for Salicylate-Treated RILs.
Supplemental Figure 6. Additive QTL Estimates across Chromosome II.
Supplemental Figure 7. Modeled QTL Effects Using a Single Time Point.
Supplemental Figure 8. Circadian Oscillation in Chlorophyll Delayed Fluoresence.
Supplemental Figure 9. Circadian Oscillation in Chlorophyll Delayed Fluoresence.
Supplemental Data Set 1. CT Phase Group Definition.
Supplemental Data Set 2. Average CT Phase Group Expression in 211 Bay × Sha RILs.
Acknowledgments
This work was funded by National Science Foundation awards DBI 0820580 and DBI 064281 to D.J.K., National Institutes of Health award R01 GM06418 to S.L.H., and National Science Foundation awards IOS 0923752 and IOS 0227103 to J.N.M.
References
- Atwell S., et al. (2010). Genome-wide association study of 107 phenotypes in a common set of Arabidopsis thaliana inbred lines. Nature 465: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basten C.J., Weir B.S., Zeng Z.-B. (1999). QTL Cartographer, Version 1.13. (Raleigh, NC: North Carolina State University; ). [Google Scholar]
- Beekwilder J., et al. (2008). The impact of the absence of aliphatic glucosinolates on insect herbivory in Arabidopsis. PLoS ONE 3: e2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benderoth M., Textor S., Windsor A.J., Mitchell-Olds T., Gershenzon J., Kroymann J. (2006). Positive selection driving diversification in plant secondary metabolism. Proc. Natl. Acad. Sci. USA 103: 9118–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidart-Bouzat M.G., Kliebenstein D.J. (2008). Differential levels of insect herbivory in the field associated with genotypic variation in glucosinolates in Arabidopsis thaliana. J. Chem. Ecol. 34: 1026–1037 [DOI] [PubMed] [Google Scholar]
- Borevitz J.O., et al. (2007). Genome-wide patterns of single-feature polymorphism in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 12057–12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R.B., Yvert G., Clinton R., Kruglyak L. (2002). Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755 [DOI] [PubMed] [Google Scholar]
- Bussemaker H.J., Ward L.D., Boorsma A. (2007). Dissecting complex transcriptional responses using pathway-level scores based on prior information. BMC Bioinformatics 8 (suppl. 6): S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.K.F., Rowe H.C., Kliebenstein D.J. (2010). Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics 185: 991–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christley S., Nie Q., Xie X.H. (2009). Incorporating existing network information into gene network inference. PLoS ONE 4: e6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.H., Mithen R., Brown J.K., Dean C. (1995). QTL analysis of flowering time in Arabidopsis thaliana. Mol. Gen. Genet. 248: 278–286 [DOI] [PubMed] [Google Scholar]
- Covington M.F., Maloof J.N., Straume M., Kay S.A., Harmer S.L. (2008). Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome Biol. 9: R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah C., Taylor B.L., Edwards K.D., Brown P.E., Hall A., McWatters H.G. (2006). Analysis of phase of LUCIFERASE expression reveals novel circadian quantitative trait loci in Arabidopsis. Plant Physiol. 140: 1464–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd A.N., Gardner M.J., Hotta C.T., Hubbard K.E., Dalchau N., Love J., Assie J.M., Robertson F.C., Jakobsen M.K., Gonçalves J., Sanders D., Webb A.A.R. (2007). The Arabidopsis circadian clock incorporates a cADPR-based feedback loop. Science 318: 1789–1792 [DOI] [PubMed] [Google Scholar]
- Doerge R.W., Churchill G.A. (1996). Permutation tests for multiple loci affecting a quantitative character. Genetics 142: 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duez H., Staels B. (2008). Rev-erb alpha gives a time cue to metabolism. FEBS Lett. 582: 19–25 [DOI] [PubMed] [Google Scholar]
- Edwards K.D., Anderson P.E., Hall A., Salathia N.S., Locke J.C.W., Lynn J.R., Straume M., Smith J.Q., Millar A.J. (2006). FLOWERING LOCUS C mediates natural variation in the high-temperature response of the Arabidopsis circadian clock. Plant Cell 18: 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards K.D., Lynn J.R., Gyula P., Nagy F., Millar A.J. (2005). Natural allelic variation in the temperature-compensation mechanisms of the Arabidopsis thaliana circadian clock. Genetics 170: 387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer D.S., Mackay T.F.C. (1996). Introduction to Quantitative Genetics. (Essex, UK: Longman, Harlow; ). [Google Scholar]
- Fukushima A., Kusano M., Nakamichi N., Kobayashi M., Hayashi N., Sakakibara H., Mizuno T., Saito K. (2009). Impact of clock-associated Arabidopsis pseudo-response regulators in metabolic coordination. Proc. Natl. Acad. Sci. USA 106: 7251–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamoustaris A., Mithen R. (1995). The effect of modifying the glucosinolate content of leaves of oilseed rape (Brassica napus Ssp oleifera) on its interaction with specialist and generalist pests. Ann. Appl. Biol. 126: 347–363 [Google Scholar]
- Gigolashvili T., Engqvist M., Yatusevich R., Müller C., Flügge U.I. (2008). HAG2/MYB76 and HAG3/MYB29 exert a specific and coordinated control on the regulation of aliphatic glucosinolate biosynthesis in Arabidopsis thaliana. New Phytol. 177: 627–642 [DOI] [PubMed] [Google Scholar]
- Gigolashvili T., Yatusevich R., Berger B., Müller C., Flügge U.I. (2007). The R2R3-MYB transcription factor HAG1/MYB28 is a regulator of methionine-derived glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 51: 247–261 [DOI] [PubMed] [Google Scholar]
- Gould P.D., Diaz P., Hogben C., Kusakina J., Salem R., Hartwell J., Hall A. (2009). Delayed fluorescence as a universal tool for the measurement of circadian rhythms in higher plants. Plant J. 58: 893–901 [DOI] [PubMed] [Google Scholar]
- Grennan A.K. (2006). Genevestigator. Facilitating web-based gene-expression analysis. Plant Physiol. 141: 1164–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel T., Zeier J. (2008). Light regulation and daytime dependency of inducible plant defenses in Arabidopsis: Phytochrome signaling controls systemic acquired resistance rather than local defense. Plant Physiol. 147: 790–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R.A., Stokes T.L., Thum K., Xu X., Obertello M., Katari M.S., Tanurdzic M., Dean A., Nero D.C., McClung C.R., Coruzzi G.M. (2008). Systems approach identifies an organic nitrogen-responsive gene network that is regulated by the master clock control gene CCA1. Proc. Natl. Acad. Sci. USA 105: 4939–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer S.L. (2009). The circadian system in higher plants. Annu. Rev. Plant Biol. 60: 357–377 [DOI] [PubMed] [Google Scholar]
- Harmer S.L., Hogenesch J.B., Straume M., Chang H.S., Han B., Zhu T., Wang X., Kreps J.A., Kay S.A. (2000). Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290: 2110–2113 [DOI] [PubMed] [Google Scholar]
- Hirai M.Y., et al. (2007). Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. USA 104: 6478–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper K.R. (1999). Risk-spreading and bet-hedging in insect population biology. Annu. Rev. Entomol. 44: 535–560 [DOI] [PubMed] [Google Scholar]
- Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. (2001). A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98: 4569–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R.C., Vanooijen J.W., Stam P., Lister C., Dean C. (1995). Genotype-by-environment interaction in genetic-mapping of multiple quantitative trait loci. Theor. Appl. Genet. 91: 33–37 [DOI] [PubMed] [Google Scholar]
- Jen C.H., Manfield I.W., Michalopoulos I., Pinney J.W., Willats W.G.T., Gilmartin P.M., Westhead D.R. (2006). The Arabidopsis co-expression tool (ACT): A WWW-based tool and database for microarray-based gene expression analysis. Plant J. 46: 336–348 [DOI] [PubMed] [Google Scholar]
- Jiménez-Gómez J.M., Wallace A.D., Maloof J.N. (2010). Network analysis identifies ELF3 as a QTL for the shade avoidance response in Arabidopsis. PLoS Genet. 6: e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes J.J.B., Fu J.Y., Terpstra I.R., Garcia J.M., van den Ackerveken G., Snoek L.B., Peeters A.J.M., Vreugdenhil D., Koornneef M., Jansen R.C. (2007). Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. USA 104: 1708–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein D. (2009a). Quantitative genomics: Analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol. 60: 93–114 [DOI] [PubMed] [Google Scholar]
- Kliebenstein D.J. (2009b). Quantification of variation in expression networks. In Plant Systems Biology, Belostotsky D., (Totowa, NJ: Humana Press; ), pp. 227–245 [DOI] [PubMed] [Google Scholar]
- Kliebenstein D.J. (2009c). A quantitative genetics and ecological model system: Understanding the aliphatic glucosinolate biosynthetic network via QTLs. Phytochem. Rev. 8: 243–254 [Google Scholar]
- Kliebenstein D.J., Lambrix V.M., Reichelt M., Gershenzon J., Mitchell-Olds T. (2001). Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 13: 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein D.J., West M.A., van Leeuwen H., Loudet O., Doerge R.W., St Clair D.A. (2006). Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinformatics 7: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac J., Husse J., Oster H. (2009). A time to fast, a time to feast: The crosstalk between metabolism and the circadian clock. Mol. Cells 28: 75–80 [DOI] [PubMed] [Google Scholar]
- Kroymann J., Donnerhacke S., Schnabelrauch D., Mitchell-Olds T. (2003). Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc. Natl. Acad. Sci. USA 100(suppl. 2): 14587–14592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann J., Mitchell-Olds T. (2005). Epistasis and balanced polymorphism influencing complex trait variation. Nature 435: 95–98 [DOI] [PubMed] [Google Scholar]
- Kroymann J., Textor S., Tokuhisa J.G., Falk K.L., Bartram S., Gershenzon J., Mitchell-Olds T. (2001). A gene controlling variation in Arabidopsis glucosinolate composition is part of the methionine chain elongation pathway. Plant Physiol. 127: 1077–1088 [PMC free article] [PubMed] [Google Scholar]
- Lankau R.A. (2007). Specialist and generalist herbivores exert opposing selection on a chemical defense. New Phytol. 175: 176–184 [DOI] [PubMed] [Google Scholar]
- Lankau R.A., Kliebenstein D.J. (2009). Competition, herbivory and genetics interact to determine the accumulation and fitness consequences of a defence metabolite. J. Ecol. 97: 78–88 [Google Scholar]
- Lankau R.A., Strauss S.Y. (2007). Mutual feedbacks maintain both genetic and species diversity in a plant community. Science 317: 1561–1563 [DOI] [PubMed] [Google Scholar]
- Lankau R.A., Strauss S.Y. (2008). Community complexity drives patterns of natural selection on a chemical defense of Brassica nigra. Am. Nat. 171: 150–161 [DOI] [PubMed] [Google Scholar]
- Li Y., Huang Y., Bergelson J., Nordborg M., Borevitz J.O. (2010). Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107: 21199–21204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudet O., Chaillou S., Camilleri C., Bouchez D., Daniel-Vedele F. (2002). Bay-0 x Shahdara recombinant inbred line population: A powerful tool for the genetic dissection of complex traits in Arabidopsis. Theor. Appl. Genet. 104: 1173–1184 [DOI] [PubMed] [Google Scholar]
- Loudet O., Michael T.P., Burger B.T., Le Metté C., Mockler T.C., Weigel D., Chory J. (2008). A zinc knuckle protein that negatively controls morning-specific growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 105: 17193–17198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Walsh B. (1998). Genetics and Analysis of Quantitative Traits. (Sunderland, MA: Sinauer Associates; ). [Google Scholar]
- Martínez C., Pons E., Prats G., León J. (2004). Salicylic acid regulates flowering time and links defence responses and reproductive development. Plant J. 37: 209–217 [DOI] [PubMed] [Google Scholar]
- Mauricio R., Rausher M.D. (1997). Experimental manipulation of putative selective agents provides evidence for the role of natural enemies in the evolution of plant defense. Evolution 51: 1435–1444 [DOI] [PubMed] [Google Scholar]
- Michael T.P., Salomé P.A., Yu H.J., Spencer T.R., Sharp E.L., McPeek M.A., Alonso J.M., Ecker J.R., McClung C.R. (2003). Enhanced fitness conferred by naturally occurring variation in the circadian clock. Science 302: 1049–1053 [DOI] [PubMed] [Google Scholar]
- Nozue K., Covington M.F., Duek P.D., Lorrain S., Fankhauser C., Harmer S.L., Maloof J.N. (2007). Rhythmic growth explained by coincidence between internal and external cues. Nature 448: 358–361 [DOI] [PubMed] [Google Scholar]
- Obayashi T., Kinoshita K., Nakai K., Shibaoka M., Hayashi S., Saeki M., Shibata D., Saito K., Ohta H. (2007). ATTED-II: a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 35(Database issue): D863–D869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul-Victor C., Züst T., Rees M., Kliebenstein D.J., Turnbull L.A. (2010). A new method for measuring relative growth rate can uncover the costs of defensive compounds in Arabidopsis thaliana. New Phytol. 187: 1102–1111 [DOI] [PubMed] [Google Scholar]
- Pinheiro J.C., Chao E.C. (2006). Efficient Laplacian and adaptive Gaussian quadrature algorithms for multilevel generalized linear mixed models. J. Comput. Graph. Statist. 15: 58–81 [Google Scholar]
- Plautz J.D., Straume M., Stanewsky R., Jamison C.F., Brandes C., Dowse H.B., Hall J.C., Kay S.A. (1997). Quantitative analysis of Drosophila period gene transcription in living animals. J. Biol. Rhythms 12: 204–217 [DOI] [PubMed] [Google Scholar]
- Potokina E., Druka A., Luo Z., Wise R., Waugh R., Kearsey M. (2008). Gene expression quantitative trait locus analysis of 16 000 barley genes reveals a complex pattern of genome-wide transcriptional regulation. Plant J. 53: 90–101 [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2008). R: A language and environment for statistical computing. (Vienna: R Foundation for Statistical Computing; ). [Google Scholar]
- Roden L.C., Ingle R.A. (2009). Lights, rhythms, infection: The role of light and the circadian clock in determining the outcome of plant-pathogen interactions. Plant Cell 21: 2546–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe H.C., Hansen B.G., Halkier B.A., Kliebenstein D.J. (2008). Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20: 1199–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J., Reick M., McKnight S.L. (2002). Metabolism and the control of circadian rhythms. Annu. Rev. Biochem. 71: 307–331 [DOI] [PubMed] [Google Scholar]
- Saito K., Hirai M., Yonekura-Sakakibara K. (2008). Decoding genes with coexpression networks and metabolomics – ‘majority report by precogs.’ Trends Plant Sci. 13: 36–43 [DOI] [PubMed] [Google Scholar]
- Schadt E.E., et al. (2003). Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302 [DOI] [PubMed] [Google Scholar]
- Sønderby I.E., Burow M., Rowe H.C., Kliebenstein D.J., Halkier B.A. (2010). A complex interplay of three R2R3 MYB transcription factors determines the profile of aliphatic glucosinolates in Arabidopsis. Plant Physiol. 153: 348–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sønderby I.E., Hansen B.G., Bjarnholt N., Ticconi C., Halkier B.A., Kliebenstein D.J. (2007). A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2: e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straume M. (2004). DNA microarray time series analysis: Automated statistical assessment of circadian rhythms in gene expression patterning. Numerical Computer Methods, Part D. In Methods in Enzymology, Vol. 383, Brand L., Johnson M.L., (Burlington, MA: Academic Press/Elsevier; ), pp. 149–166 [DOI] [PubMed] [Google Scholar]
- Strayer C., Oyama T., Schultz T.F., Raman R., Somers D.E., Más P., Panda S., Kreps J.A., Kay S.A. (2000). Cloning of the Arabidopsis clock gene TOC1, an autoregulatory response regulator homolog. Science 289: 768–771 [DOI] [PubMed] [Google Scholar]
- Swarup K., Alonso-Blanco C., Lynn J.R., Michaels S.D., Amasino R.M., Koornneef M., Millar A.J. (1999). Natural allelic variation identifies new genes in the Arabidopsis circadian system. Plant J. 20: 67–77 [DOI] [PubMed] [Google Scholar]
- Thum K.E., Shin M.J., Gutierrez R.A., Mukherjee I., Katari M.S., Nero D., Shasha D., Coruzzi G.M. (2008). An integrated genetic, genomic and systems approach defines gene networks regulated by the interaction of light and carbon signaling pathways in Arabidopsis. BMC Syst. Biol. 2: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H.R., Chen W.B., Minami Y., Honma S., Honma K., Iino M., Hashimoto S. (2004). Molecular-timetable methods for detection of body time and rhythm disorders from single-time-point genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 101: 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening J.W., Smits W.K., Kuipers O.P. (2008). Bistability, epigenetics, and bet-hedging in bacteria. Annu. Rev. Microbiol. 62: 193–210 [DOI] [PubMed] [Google Scholar]
- Walley J.W., Coughlan S., Hudson M.E., Covington M.F., Kaspi R., Banu G., Harmer S.L., Dehesh K. (2007). Mechanical stress induces biotic and abiotic stress responses via a novel cis-element. PLoS Genet. 3: 1800–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Basten C.J., Zeng Z.-B. (2006). Windows QTL Cartographer 2.5. (Raleigh, NC: North Carolina State University; ). [Google Scholar]
- Wentzell A.M., Kliebenstein D.J. (2008). Genotype, age, tissue, and environment regulate the structural outcome of glucosinolate activation. Plant Physiol. 147: 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzell A.M., Rowe H.C., Hansen B.G., Ticconi C., Halkier B.A., Kliebenstein D.J. (2007). Linking metabolic QTL with network and cis-eQTL controlling biosynthetic pathways. PLoS Genet. 3: e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M.A.L., Kim K., Kliebenstein D.J., van Leeuwen H., Michelmore R.W., Doerge R.W., St Clair D.A. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175: 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood T.L., Bridwell-Rabb J., Kim Y.I., Gao T., Chang Y.G., LiWang A., Barondeau D.P., Golden S.S. (2010). The KaiA protein of the cyanobacterial circadian oscillator is modulated by a redox-active cofactor. Proc. Natl. Acad. Sci. USA 107: 5804–5809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.I., Lauga B., Charlesworth D. (2002). Rates and patterns of molecular evolution in inbred and outbred Arabidopsis. Mol. Biol. Evol. 19: 1407–1420 [DOI] [PubMed] [Google Scholar]
- Zeng Z.-B., Kao C.-H., Basten C.J. (1999). Estimating the genetic architecture of quantitative traits. Genet. Res. 74: 279–289 [DOI] [PubMed] [Google Scholar]