

Abstract
Expression of the c-Myc oncoprotein is affected by conserved Threonine 58 (T58) and Serine 62 (S62) phosphorylation sites that help to regulate c-Myc protein stability, and altered ratios of T58 and S62 phosphorylation have been observed in human cancer. Here, we report the development of three unique c-myc knock-in mice that conditionally express either c-MycWT, or the c-MycT58A or c-MycS62A phosphorylation mutant, from the constitutively active ROSA26 locus in response to Cre recombinase to study the role of these phosphorylation sites in vivo. Using a mammary-specific Cre model, we found that expression of c-MycWT resulted in increased mammary gland density but normal morphology and no tumors at the level expressed from the ROSA promoter. In contrast, c-MycT58A expression yielded enhanced mammary gland density, hyperplastic foci, cellular dysplasia, and mammary carcinoma, associated with increased genomic instability and suppressed apoptosis relative to c-MycWT. Alternatively, c-MycS62A expression reduced mammary gland density relative to control glands, and this was associated with increased genomic instability and normal apoptotic function. Our results indicate that specific activities of c-Myc are differentially affected by T58 and S62 phosphorylation. This model provides a robust platform to interrogate the role that these phosphorylation sites play in c-Myc function during development and tumorigenesis.
Introduction
The c-Myc oncoprotein is a pleiotropic transcription factor involved in controlling many cellular functions, including cell proliferation, cell growth, and cell differentiation, as well as pathways that regulate genome stability and cell death (1). c-Myc is constitutively highly expressed in most human tumors and high expression of c-Myc in animal models can drive tumorigenesis (2–4). c-Myc-induced tumorigenesis is tempered by its activation of intrinsic tumor suppressor pathways involving engagement of cell cycle checkpoint and apoptotic cell death pathways (5). Activation of these intrinsic tumor suppressor pathways induces cell death in normal cells (6), but in tumor cells these fail-safe mechanisms are bypassed by secondary lesions that likely evolve from genomic instability associated with high c-Myc expression. Thus, in order to maintain normal cell function, c-Myc expression is tightly controlled at the level of transcription, mRNA stability, translation, and protein stability. In breast cancer, c-MYC gene amplification has been reported in approximately 16% of cases and increased mRNA expression in approximately 22% (7, 8). However, elevated expression of c-Myc protein is reported in approximately 70% of human breast tumors, arguing for a potentially critical role for post-translational regulation of c-Myc expression (2, 9, 10).
Phosphorylation of c-Myc at conserved residues Serine 62 (S62) and Threonine 58 (T58) can regulate c-Myc protein stability in response to mitogen signalling (11). Phosphorylation of S62 by ERK or CDK kinases transiently increases c-Myc stability while phosphorylation of T58 by GSK3β triggers dephosphorylation of S62 by protein phosphatase 2A (PP2A), ubiquitination by the SCF-Fbw7 E3 ligase and proteasomal degradation (12). The scaffold protein Axin1 coordinates c-Myc degradation through this pathway (13). Mutations in PP2A subunits, FBW7 and AXIN1 have been reported in many human cancers (14–16), suggesting that this c-Myc degradation pathway can be deregulated in cancer cells leading to altered levels of T58 and S62 phosphorylation and increased c-Myc stability.
In cell culture experiments, mutation of S62 to Alanine (S62A) reduces c-Myc’s transforming activity while mutation of T58 to Alanine (T58A) enhances c-Myc’s transforming activity, suggesting that phosphorylation changes at these sites can affect c-Myc function (17–20). Furthermore, decreased T58 and increased S62 phosphorylation have been observed in human cancer cell lines associated with increased c-Myc protein stability (13, 21). This phosphorylation ratio is partially mimicked in the c-MycT58A mutant, which has no T58 and constitutive high S62 phosphorylation (18, 22, 23).
To carefully address the role of T58 and S62 phosphorylation in c-Myc’s activity in vivo, we have generated three conditional c-myc knock-in mice that express c-MycWT, c-MycT58A, or c-MycS62A (which lacks phosphorylation at both sites due to their hierarchical nature) from the endogenous ROSA26 locus in response to Cre-mediated recombination. In this study, we used Wap-Cre to drive expression in the mammary gland as it provides an elegant system to interrogate multiple activities of c-Myc. In normal mammary development, c-Myc expression is important for both pregnancy-associated proliferation and apoptosis during involution (24, 25). High-level expression of c-Myc can also drive mammary gland tumorigenesis in transgenic mouse models (3, 26, 27). The extended mammary tumor latencies in these studies suggest that additional lesions are required for mammary tumorigenesis. Results from this study indicate that lesions affecting phosphorylation at T58 and S62 could contribute to tumorigenesis, as the phosphorylation status of these sites substantially affected c-Myc function and tumorigenic potential in the mammary gland.
Methods
Generating RFS-mycWT, RFS-mycT58A and RFS-mycS62A mice
ROSA-Floxed-Stop (RFS)-mycWT, RFS-mycT58A and RFS-mycS62A mice were generated using an established gene knockin strategy (28) (see Suppl. Material). Briefly, murine c-mycWT-HA or phosphorylation mutant c-mycT58A-HA or c-mycS62A-HA cDNAs (29) were cloned into targeting vectors and electroporated into 129 ES cells. Correctly targeted and sequence-confirmed ES clones were injected into C57BL/6 blastocysts to obtain chimeric mice. Germline transmission was obtained by crossing with C57BL/6 mice to establish homozygous knock-in RFS-mycWT, RFS-mycT58A and RFS-mycS62A strains. RFS-mycWT, RFS-mycT58A and RFS-myc S62A were crossed with Wap-Cre transgenic mice (C57BL/6), obtained from the NCI Mouse Models of Human Cancer Consortium (MMHCC).
Genotyping and detection of recombination
RFS-mycWT, RFS-mycT58A and RFS-myc S62A knock-in mice and the presence of Wap-Cre were identified by PCR analysis using tail DNA and specific primer sets (see Suppl. Material). Cre-mediated recombination in RFS-myc(WT,T58A,S62A)/Wap-Cre mice was detected by PCR analysis and specified primers (see Suppl. Material).
RNA analysis
RNA was isolated from mammary glands using TRIzol reagent (Invitrogen) according to manufacturer’s protocol. cDNA was prepared from isolated RNA and analyzed by PCR or quantitative PCR (see Suppl. Material).
Antibodies and western blotting
Antibodies are listed in supplemental material. Mouse mammary gland samples were lysed by homogenizing in EBC buffer with protease and phosphatase inhibitors and subject to western analysis (see Suppl. Material).
Quantification and Statistics
Western blots were visualized and quantified using LI-COR Odyssey Infrared software version 1.2, which is linear over 4 orders of magnitude. Statistical significance was determined by student’s t-test.
Cycloheximide half-life
Primary Mouse Embryo Fibroblasts (MEFs) or mammary epithelial cells (MECs) (see Suppl. Material) (1–2 passages) were infected with Ad-Cre for 48 hours and then starved in 0.1% FBS for another 48 hours. Cells were treated with cycloheximide and harvested at each time point (see Suppl. Material). HA tagged WT, T58A or S62A Myc was immunoprecipitated with HA antibody and western blotted with Y69 Myc antibody.
Pathologic assessment
Mice were anesthetized with avertin, sacrificed, and tissues were fixed in 10% formalin, embedded in paraffin, and 5µM-thick sections were stained with Mayer’s H&E. Pathologic findings were classified according to the Mouse Models of Human Cancers Consortium NIH/National Cancer Institute by MLT (30).
Apoptosis assay
Apoptosis was detected by terminal deoxynucleotidyltransferase-mediated dUTP nick end labelling (TUNEL) staining using the Apo Tag kit (Chemicon International) and the percentage of apoptotic cells was determined in 10 random fields per slide.
Whole mount analysis of mammary gland
Mammary gland tissues were dissected, spread on glass slides, and fixed immediately with Carnoy’s Fixative. Tissues were hydrated with serially diluted ethanol and stained with 0.6% carmine solution overnight at 4°C. Tissues were cleared of lipid with Xylene and mounted with permount.
Chromosome spreads and centrosome staining
Primary mammary epithelial cells, 80% confluent, were used for centrosome detection, and chromosome spreads after colcemid treatment (see Suppl. Material). Chromosome spreads were stained with Giemsa. Centrosomes were deteced by immunofluorescence for pericentrin. G-banding was performed by the OHSU Cytogenetics Laboratory.
Results
Generation of conditional ROSA-Floxed-Stop (RFS)-mycWT, RFS-mycT58A, and RFS-mycS62A mice
To study the effects of altering c-Myc phosphorylation at T58 and S62 on c-Myc’s activity in vivo, we utilized two phosphorylation mutants: c-MycT58A, which lacks T58 phosphorylation and has constitutively high S62 phosphorylation, as it is resistant to PP2A-mediated dephosphorylation (20), and c-MycS62A, which lacks phosphorylation at both sites due to their hierarchical nature (Suppl. Fig. 1A). We used an established gene knock-in strategy to insert HA-tagged cDNAs, either murine wild-type c-mycWT, or phosphorylation site mutants c-mycT58A or c-mycS62A, preceded by a transcription stop sequence flanked by loxP sites, into the constitutively active ROSA26 gene locus (Fig. 1A and Suppl. Fig. 1B). The ROSA26 locus ubiquitously drives gene expression in all embryonic and adult tissues; however, due to the transcription stop sequence, the knock-in c-myc genes will not be expressed unless Cre recombinase is present. We have established homozygous breeding pairs for each of these strains, termed ROSA-Floxed-Stop (RFS)-mycWT, RFS-mycT58A, and RFS-mycS62A (Suppl. Fig. 1C). Primary mouse embryo fibroblasts (MEFs) from each strain were infected with Ad-Cre and nuclear expression of HA-tagged c-Myc was verified by immunofluorescence (Suppl. Fig. 2A). The mean half-life of c-MycWT, c-MycT58A, and c-MycS62A in the primary MEFs was 19.5, 63.0, and 44.5 minutes, respectively, under serum starved conditions (Suppl. Fig. 2B). We did note variability in the half-life of the S62A mutant. Since we do not know what E3 ligase targets this mutant, the reason for this variability is currently unknown.
Figure 1. Generation and characterization of RFS-mycWT/WAP-Cre, RFS-mycT58A/WAP-Cre and RFS-mycS62A/WAP-Cre mice.
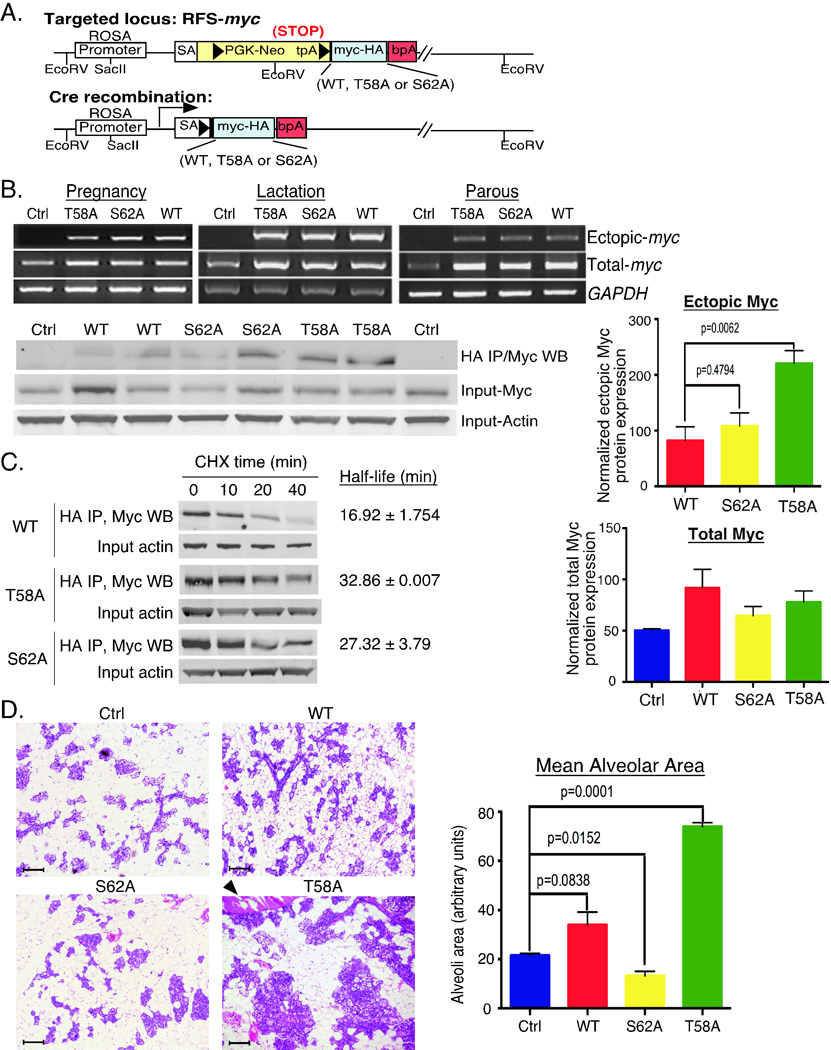
A. Knock-in strategy for conditional expression of c-mycWT-HA, c-mycT58A-HA or c-mycS62A-HA. Arrowheads represent loxP sites. tpA is a transcription stop sequence. Inserted c-myc cDNAs have a C-terminal HA tag. Cre recombination activates expression of the inserted c-myc cDNA driven from the ROSA promoter. RFS=ROSA Floxed Stop (see Suppl. Fig. 1 for more detail). B. Expression of ROSA-driven c-Myc. Upper panels: RNA was isolated from mammary glands from the indicated control (ctrl: no Wap-Cre) or RFS-myc/WAP-Cre strains (WT, T58A, S62A) at the indicated stages: pregnancy 18 days, lactation 18 days, parous 2 months. HA-tagged ectopic and total c-myc mRNA is shown by RT-PCR. Lower panels: lysates from mammary glands from 2 mice per indicated strain harvested at pregnancy day 17 were immunoprecipitated with anti-HA followed by western blotting with anti-c-Myc. Input total c-Myc and Actin are shown. Data is representative of 4 mice per genotype. Graphs: quantification of the Actin-normalized expression of HA-tagged ectopic and total c-Myc from 3 mice per genotype ± SD. P-values are given on graphs with significant differences. C. Analysis of ectopic c-Myc half-life in primary MECs from the indicated strains. Mean half-life ± SD was calculated from three independent experiments. D. Expanded lobule-alveolar areas with c-MycT58A expression. The 4th glands from the indicated mice on day 17 of the 3rd consecutive pregnancy were analyzed by H&E section staining. Arrowhead indicates enlarged duct. Scale bars are 50 µM. Data is representative of 4 mice per genotype. Mean alveolar area was analyzed by counting 10 areas each section and 3 mice per genotype by ImageJ and graphed ± SD.
Initial characterization of RFS-mycWT/Wap-Cre, RFS-mycT58A/Wap-Cre, and RFS-mycS62A/Wap-Cre mice
In order to address the consequences of altering c-Myc T58 and S62 phosphorylation on c-Myc activity in the mammary gland, we crossed our three RFS-c-myc strains with WAP (Whey Acidic Protein)-Cre transgenic mice, which express Cre in the secretory luminal and ductal epithelial cells of the mammary gland during late pregnancy and lactation (31), and experimental mice were subjected to three consecutive rounds of pregnancy/lactation. This resulted in approximately 50% Cre-mediated recombination in the mammary epithelial cells as detected by breeding with the R26R reporter mouse (Suppl. Fig. 1D). PCR analysis of genomic DNA from various tissues demonstrated that the recombined allele was primarily detected in mammary gland from RFS-myc mice with Wap-Cre, but not in control mice without Wap-Cre (Suppl. Fig. 1E). The advantage of this mouse model is that unlike other c-myc transgenic mouse models where c-Myc is expressed at very high levels, the ROSA26 promoter is relatively weak and ubiquitously drives expression of only 2 copies of the knocked-in c-myc genes at near physiological levels (Fig. 1B, Total-myc, Pregnancy) (32). We observe partial suppression of the endogenous c-myc gene with expression of ectopic c-MycWT, c-MycT58A and c-MycS62A in this system (data not shown), consistent with other reports (33, 34). As expected, expression of ectopic c-mycWT, c-mycT58A and c-mycS62A mRNA was equivalent and Cre-dependent (Fig. 1B, Ectopic-myc). Expression of ectopic c-MycT58A protein was on average higher than c-MycS62A, which was generally higher than c-MycWT (Fig. 1B, HA IP/Myc WB, and Ectopic Myc graph). However, total c-Myc levels measured in the IP input were not distinguishable across all of the strains during pregnancy, when the epithelial cells are stimulated to proliferate and endogenous c-Myc protein expression is high (Fig. 1B, Input-Myc and Total Myc graph). This result highlights the physiological expression level of this model. We also measured the half-life of the ROSA-driven c-Myc proteins in primary mammary epithelial cells (MECs) under serum starvation conditions (Fig. 1C). c-MycT58A had the longest half-life, followed by c-MycS62A and c-MycWT, consistent with their expression level (Fig. 1B), and consistent with the MEF data (Suppl. Fig. 2B), although differences in stability were not as pronounced as in the MEFs.
Although differences in expression of the ectopic Myc proteins was not dramatic, histological examination of mammary glands from the RFS-myc/Wap-Cre strains during the 3rd pregnancy showed significant alterations in the numbers of lobule-alveolar units and mean alveolar area. Specifically, MycS62A expressing glands showed significantly reduced numbers of alveoli compared to control, while MycT58A had the opposite effect, with substantially expanded alveolar areas and the presence of enlarged ducts containing secretion (Fig. 1D and graph). Analysis of proliferation by BrdU incorporation did not reveal any substantial differences between the strains during pregnancy or early parous stage (data not shown).
Delayed and incomplete mammary gland involution with c-MycT58A expression
c-Myc is a potent inducer of apoptosis under growth limiting conditions. Involution of the mammary gland following weaning represents a period of intense apoptotic activity associated with tissue remodelling, and c-Myc expression has been shown to play an important role in this process (25). We examined the effects of expressing c-MycT58A and c-MycS62A compared to c-MycWT on involution. Histological analysis of mammary glands three days post-weaning showed clear signs of involution with shrunken alveoli and the reappearance of adipose cells in control, MycWT and MycS62A expressing mice (Fig. 2A). In contrast, enlarged alveoli with prominent secretions predominated in MycT58A mammary glands. Analysis of apoptotic cells by TUNEL staining showed a dramatic reduction in apoptosis in MycT58A-expressing mammary glands (Fig. 2B–C). This reduction in apoptosis and impaired involution likely contributed to the expanded lobule-alveolar areas, as well as the persistence of enlarged secretion-filled ducts in MycT58A mammary glands (Fig. 1D). In contrast, mammary glands expressing MycWT showed an increase in apoptosis compared to control, consistent with wild-type c-Myc’s known apoptotic activity (Fig. 2C). Thus, expression of c-MycT58A, but not c-MycS62A, inhibits the wave of apoptosis in the mammary gland following weaning.
Figure 2. Inhibition of mammary gland involution and apoptosis with c-MycT58A expression.

A. The 4th gland from the indicated mice was harvested after 12 days of lactation, 3 days post weaning after the 3rd pregnancy, and analyzed by H&E section staining. Scale bars are 50µM. Data is representative of 4–6 mice per genotype. B. c-MycT58A expression inhibits apoptosis during involution. Mammary gland sections as in A were analyzed by TUNEL assay. Scale bars are 50µM. C. TUNEL assays as in B were quantified to determine the percent apoptotic epithelial cells. Graph represents 4 mice per strain ± SD. D. Reduced expression of pro-apoptotic Bim in mammary glands expressing c-MycT58A. Protein lysates from the indicated mice (2 per genotype) 3 days post weaning were western blotted with the indicated antibodies and quantified. Data is representative of 4 mice per genotype. Average Actin-normalized BimEL and BimML expression relative to control is graphed ± SD.
Analysis of mammary glands at 3 days of involution for the expression of several proteins involved in apoptosis showed that pro-apoptotic Bim (both BimEL and BimML) is reduced in MycT58A expressing mammary glands relative to the other strains (Fig. 2D and graphs). No significant change in expression of anti-apoptotic Bcl2, or pro-apoptotic Bax or p53 was found across all four strains (data not shown). Together, these data demonstrate that c-MycT58A expression suppresses apoptosis in the mammary gland at least in part due to downregulation of Bim proteins, which have been shown to play an important role in mammary gland apoptosis (35).
Increased chromosomal instability and centrosome amplification in mammary epithelial cells with altered c-Myc phosphorylation
Aberrant c-Myc expression has been associated with increased genomic instability both in cell culture and in vivo, which can contribute to the cancer phenotype (36). To examine whether deregulated c-Myc T58 and S62 phosphorylation can affect genomic stability, we analyzed and quantified chromosome numbers in mitotic spreads from first passage primary MECs harvested at two months parous. We found the highest rates of polyploidy in the MycT58A MECs with a substantial number of cells showing greater than tetraploid karyotypes (Fig 3A). In addition, G-banding analyses revealed a high degree of aneuploidy in cells expressing both MycT58A and MycS62A, with nine out of twenty cells analyzed for each showing abnormalities including non-clonal trisomies, translocations, chromosomal breakages, and various extra and missing chromosomes (Fig. 3B). In contrast, MycWT expressing cells showed only four of twenty cells with abnormal karyotypes compared to control cells with two of twenty abnormal cells (data not shown).
Figure 3. Increased genomic instability with expression of c-Myc phosphorylation mutants.
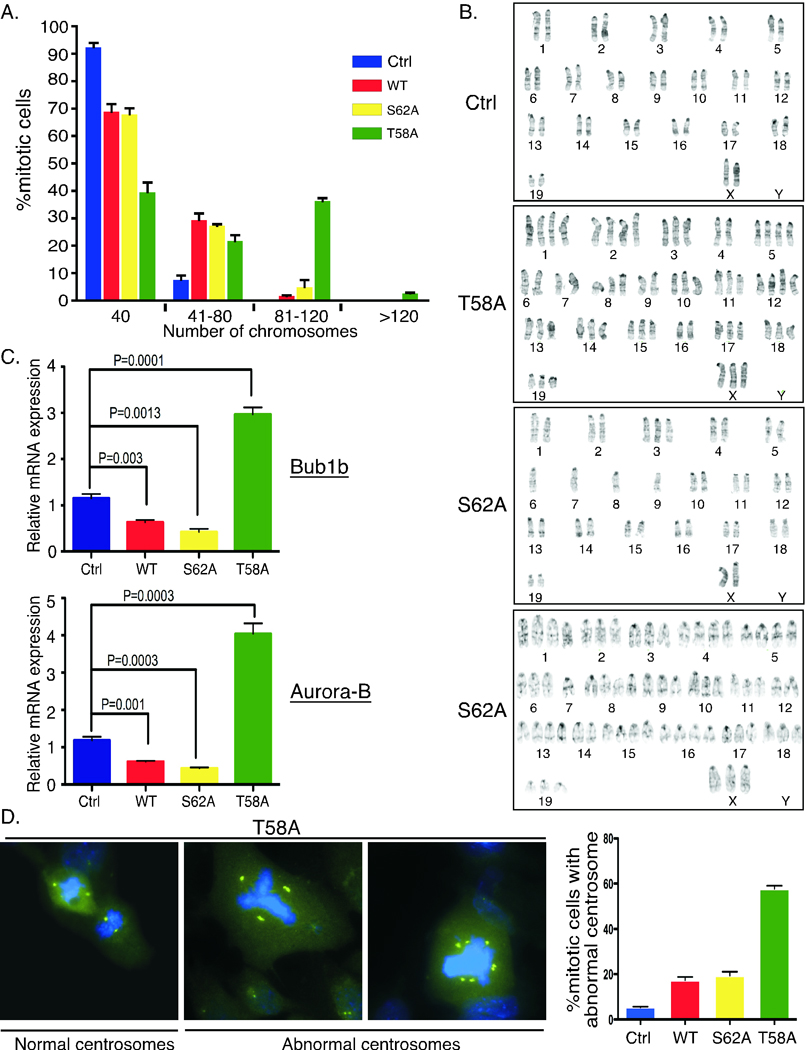
A. Mitotic spreads from primary MECs from the indicated strains were analyzed for chromosome numbers. Average percent mitotic cells with diploid (40 chromosomes) and hyperdiploid karyotypes from 3 mice per strain ± SD is graphed. B. Expression of c-MycT58A and c-MycS62A induces aneuploidy. Representative G-banding of mitotic cells from control mice or mice expressing c-MycT58A or c-MycS62A are shown. Cells were prepared as in A. Twenty mitotic cells were analyzed per strain. C. Substantial and opposing changes in expression of spindle checkpoint genes with c-MycT58A and c-MycS62A expression. RNA was isolated from 2-month parous mammary glands from the indicated strains. Quantitative RT-PCR analysis for the expression of Bub1b and Aurora-B genes relative to control mice are graphed ± SD. Data represents 4–5 mice per strain. D. Substantial centrosome amplification with expression of c-MycT58A. Centrosomes were visualized by anti-pericentrin immunofluorescence staining (green) and DNA by DAPI (blue). Cells were prepared as in A. Representative MycT58A cells are shown. Centrosome amplification (more than 2 per mitotic cell) was quantified from three mice per indicated strains and graphed ± SD.
To examine potential molecular alterations that could contribute to chromosomal instability we examined the expression of several genes important for the spindle assembly checkpoint (SAC) in 2-month parous mammary glands. Interestingly, we found that expression of Bub1b, a key player in this pathway, was substantially downregulated in MycS62A expressing mammary glands while it was substantially upregulated in MycT58A expressing glands (Fig. 3C). We also found that expression of Aurora kinase B, a chromosomal passenger serine/threonine protein kinase involved in the SAC, was dramatically upregulated with expression of MycT58A while it was substantially downregulated with expression of MycS62A (Fig. 3C), and Aurora A showed a similar trend, but less dramatic (data not shown). Since precise levels of these proteins are critical for proper SAC function (37–40), these results may partially explain the enhanced genomic instability with MycT58A and MycS62A expression.
Abnormalities in the SAC leading to failed cytokinesis can result in centrosome amplification, as well as overexpression of Aurora kinases (38). Immunofluorescent staining for centrosomes revealed clear examples of abnormal numbers of centrosomes in MycT58A MECs (Fig. 3D). Quantification revealed a high degree of centrosome amplification in MycT58A MECs, compared to MycS62A and MycWT, which were still significantly higher than control cells (Fig. 3D, graph). Together, these results reveal a potential for c-Myc-associated genomic instability in our mouse model, and expression of c-MycT58A and c-MycS62A appear to exacerbate this in different ways.
Altered mammary gland morphology with expression of the Myc phosphorylation mutants
We assessed the phenotypic effects of expressing ROSA-driven c-MycWT, c-MycT58A and c-MycS62A in the mammary gland two and five months parous after the third pregnancy and found substantial differences in morphology at both time points (Fig. 4A and Supp. Fig. 3). Expression of MycWT showed a consistent increase in mammary ductal branching relative to control mammary glands, while MycS62A showed a reduction, particularly when compared to MycWT. In contrast, expression of MycT58A resulted in dramatically increased mammary ductal branching and alveolar budding (Fig. 4A) and the appearance of hyperplastic foci resembling hyperplastic alveolar nodules by 5 months (Suppl. Fig. 3A and Fig. 5A). H&E sections revealed normal architecture in all strains except MycT58A, where expanded alveolar regions with increased stromal and epithelial cells were detected (Fig. 4B and Suppl. Fig. 3B). Mean alveolar area reflected the observed changes in density with expression of the three c-Myc isoforms (Fig. 4C). Further analyses of mammary gland sections from the MycT58A mice at five to eight months parous revealed hyperplastic foci (Fig. 5A, middle panel) with high proliferation (Fig. 5A, right panel), and areas of dysplasia including atypical nuclei with multiple nucleoli (Fig. 5B, left panel), disorganized alveolar structures with hyperchromatic crowded nuclei, loss of polarity and mitotic figures (Fig. 5B, middle panel), and abnormal foci with increased stromal cells, atypical nuclei, and immune cell infiltration including mast cells (Fig. 5B, right panel). Loss of polarity and alveolar disorganization was also apparent in co-staining for the luminal epithelial marker Keratin8/18 (green) and myoepithelial marker Keratin14 (red), which normally form a single layer of supporting cells around the polarized luminal epithelial cells (Fig. 5C, left panel, arrow). Moreover, alveoli in MycT58A glands showed down-regulation of E-cadherin expression (red) compared to alveoli in MycWT glands (Fig. 5C, middle vs right panel), suggesting loosened adhesion of intercellular junctions in the luminal cells. H&E sections and K8/18, K14 staining also revealed ductal hyperplasia (Fig. 5D).
Figure 4. Characterization of mammary glands expressing c-MycWT, c-MycT58A and c-MycS62A at two months parous.
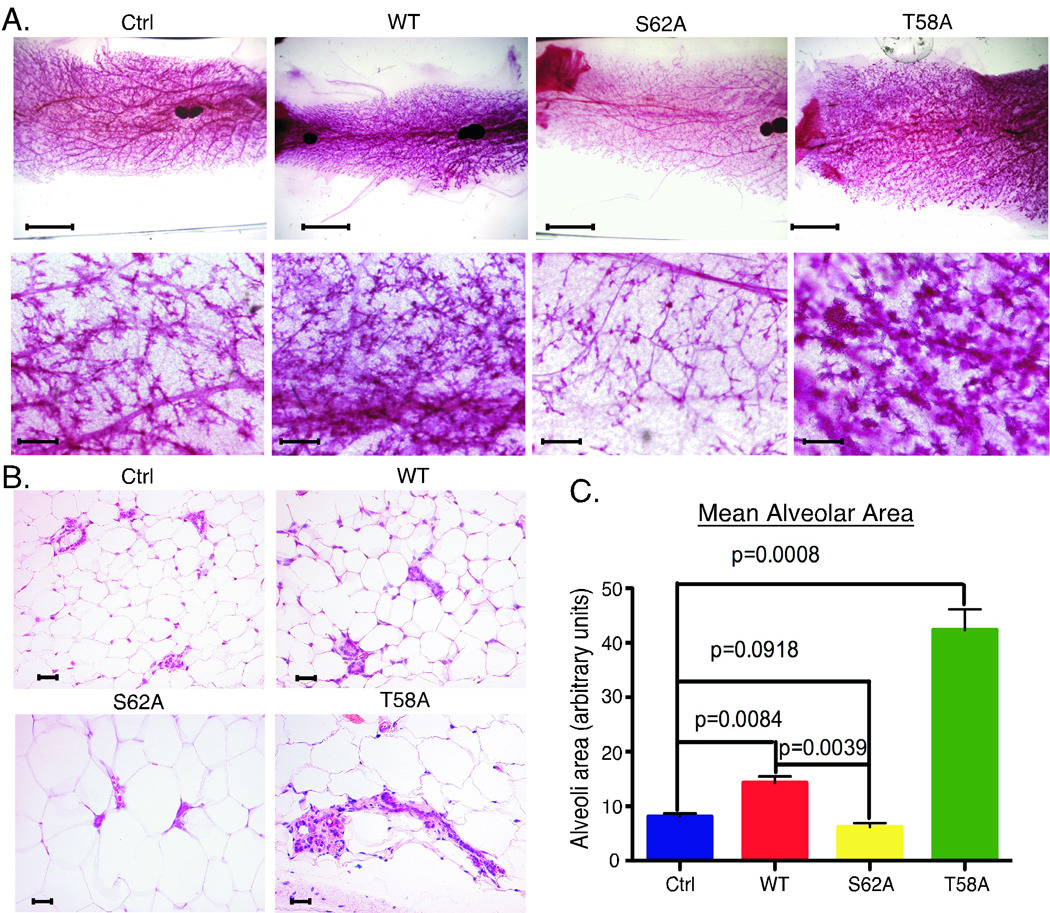
A. The 4th glands from the indicated strains of mice two months parous after the 3rd pregnancy were analyzed by whole mount. Solid, darkly staining areas are lymph nodes. Scale bars are 5 mM (top row) and 200 µM (bottom row). Images shown are representative of 3–4 mice per genotype. B. H&E sections from mice as in A. Scale bars are 50 µM. Images are representative of 4 mice per genotype. C. Expanded lobule-alveolar areas with c-MycT58A expression. Mean alveolar area from mice as in A was analyzed by counting 10 areas each H&E section. 3 mice per genotype ± SD is graphed.
Figure 5. Preneoplastic changes with expression of c-MycT58A at five to eight months parous.

A. Hyperplastic foci in RFS-mycT58A/WAP-Cre mammary glands 5–8 months parous after the 3rd pregnancy. Whole mount, H&E section, and IHC for BrdU labelling are shown. Arrow in whole mount indicates hyperplastic foci. In H&E, dark blue/purple represents calcifications in acinar lumens. B. Areas of cellular dysplasia in MycT58A mammary glands. H&E sections from MycT58A mice as in A. Arrows (from left to right) indicate atypical nuclei with multiple nucleoli, mitotic figure, and mast cell. C. Alveolar disorganization and reduced adhesion in MycT58A mammary glands. Immunofluorescence co-staining for luminal marker K8/18 (green) and myoepithelial marker K14 (red) in mice as in A. Arrow indicates disorganized alveolar region. E-cadherin staining (red), Dapi stained nuclei (blue) in MycT58A (middle panel) and MycWT (right panel) mammary glands. D. Ductal hyperplasia with c-MycT58A expression. H&E staining and immunofluorescence K8/18 (green)/K14 (red) in mice as in A. Dilated ducts with proliferative epithelium (tufting) are shown. All whole mount and H&E images are from different MycT58A mice and are representative of 8 RFS-mycT58A/WAP-Cre mice 5–8 months parous. Immunofluorescence images are representative of 3 mice per genotype. All scale bars are 50µM.
RFS-mycT58A/Wap-Cre mice develop mammary gland tumors
To study potential affects of altering c-Myc phosphorylation on tumorigenesis, we followed cohorts of RFS-mycWT, T58A and S62A/Wap-Cre and control RFS-myc (no Cre) female mice that had undergone three cycles of pregnancy (25–28 mice per genotype). Neither the control nor RFS-mycWT/Wap-Cre mice developed mammary gland tumors out to 1.5 years, although the latter expressed deregulated c-Myc. In contrast, RFS-mycT58A/Wap-Cre mice developed mammary adenocarcinomas between 7 and 13 months of age. These were poorly differentiated carcinomas of solid, cribriform, papillary and/or adenosquamous architecture, many with necrosis (Fig. 6A and data not shown), and all with a high proliferation index (Fig. 6B and data not shown). Invasion was demonstrated by absence or disruption of myopepithelial smooth muscle actin (SMA) and K14 staining in most of the tumors (Fig. 6C and data not shown) (41). Several were accompanied by in situ carcinoma at the periphery (data not shown). Moreover, one of these tumors metastasized to lung, liver and spleen, and the metastatic lesions also showed high rates of proliferation (Fig. 6D and data not shown). Of the 28 RFS-mycT58A/Wap-Cre mice, five mammary gland tumors developed in four mice (Table 1). Unfortunately, but of interest, the remainder of the RFS-mycT58A/Wap-Cre cohort succumbed to brain tumors with a median survival of approximately 10.5 months (46 weeks) (Table 1). This corresponded with spurious expression of Wap-Cre in the newborn mouse brain (Suppl. Fig. 4) as previously reported (31). Choroid plexus and pituitary tumors were observed. Interestingly, RFS-mycS62A/Wap-Cre mice also developed the same spectrum of brain tumors with a median survival of 12.5 months (54 weeks) (Table 1). Of the RFS-mycT58A/Wap-Cre and RFS-mycS62A/Wap-Cre mice with pituitary tumors, one each also developed a mammary tumor. Due to potential confounding effects from prolactin in mice with pituitary hyperplasia or tumors, any mice exhibiting these features were excluded from data presented in this paper. Analysis of the seven MycT58A mice that succumbed to choroid plexus tumors without evidence of abnormal pituitary, showed that 80% had atypical mammary gland changes similar to those shown in Figure 5, suggesting that given more time these mice may also have developed mammary gland tumors (data not shown). In contrast, MycS62A mammary glands from mice succumbing to choroid plexus tumors only, displayed reduced density similar to that shown in Figure 4 and Supplemental Figure 3 (data not shown). Together, these results indicate that phosphorylation of c-Myc at T58 and S62 has profound and differential affects on c-Myc’s oncogenic potential in the mammary gland.
Figure 6. Expression of c-MycT58A induces mammary adenocarcinoma with an average latency of 9 months.

A. Examples of mammary tumors in MycT58A expressing glands. Gross photograph of 5th gland tumor in 10-month old RFS-mycT58A/Wap-Cre mouse (upper left), H&E section showing the cribriform architecture of same (upper right), adenosquamous tumor from 7-month RFS-mycT58A/Wap-Cre mouse (lower left), adenocarcinoma with solid architecture in different 10-month RFS-mycT58A/Wap-Cre mouse (lower right). B. High proliferation in MycT58A-driven tumor. Ki-67 staining of tumor shown in A, lower right. C. Invasive features of tumor from MycT58A gland. Immunohistochemistry for smooth muscle actin (SMA) in normal duct (upper panel) close to the periphery of tumor shown in A, upper right, and loss of SMA expression in the tumor (lower panel). D. Metastasis to liver and lung of MycT58A-induced mammary tumor shown in A, lower right. All scale bars are 50 µM.
Table 1.
Tumorigenesis table of RFS-MycWT/Wap-Cre, RFS-MycT58A/Wap-Cre and RFS-MycS62A/Wap-Cre mice
| WT/Cre (n=27) |
T58A/Cre (n=28) |
S62A/Cre (n=28) |
Control (n=25) |
|
|---|---|---|---|---|
| Mice with Mammary Tumor | 0 | 4 | 0 | 0 |
| Mean Mammary Tumor Latency | 38 weeks | |||
| Mice with Brain Tumor | 0 | 24 | 28 | 0 |
| Mean Brain Tumor Latency | 46 weeks | 54 weeks |
Cohorts of the indicated numbers of RFS-MycWT/Wap-Cre, RFS-MycT58A/Wap-Cre, and RFS-MycS62A/Wap-Cre or Control (RFS-Myc(WT,T58AorS62A) no Cre) were maintained for 24 months or until moribund with tumor. Numbers of mice with the indicated tumor types along with mean latency are indicated.
Discussion
Although c-MYC is a well-known oncogene, many questions remain as to how deregulation of c-Myc leads to tumorigenesis. c-Myc is constitutively overexpressed in the majority of human tumors, and it is clear that the expression level of c-Myc helps to determine its oncogenic activity (32, 34). Specifically, deregulated low levels of c-Myc increase cell proliferation without engaging tumor suppressor pathways to induce apoptosis (32). Higher levels of c-Myc activate both proliferation and apoptosis (3, 32), and high levels of c-Myc are associated with genomic instability, which can contribute to cancer development (36). c-Myc expression level is in part controlled by phosphorylation at T58 and S62 that affect c-Myc stability. In this study, we investigated the consequences of altering T58 and S62 phosphorylation in vivo for mammary gland development and tumorigenesis. We found that the level of T58 and S62 phosphorylation can have dramatic effects on specific activities of c-Myc in the mammary gland. These results have important implications for the oncogenic function of c-Myc with altered phosphorylation that we find expressed in human breast cancer ((13) and Sears lab, unpublished).
T58 and S62 phosphorylation affects c-Myc activity in the mammary gland
Previous reports have indicated that mutation of T58 or S62 to Alanine affects c-Myc-mediated cell transformation in tissue culture where T58A Myc is more transforming and S62A less transforming compared to c-MycWT (18–20). In addition, a study using retrovirally transduced hematopoietic stem cells expressing c-MycWT or c-MycT58A demonstrated more robust lymphomagenesis with c-MycT58A expression (42). While these studies all point to a role for these phosphorylation sites in regulating c-Myc function, we have found that high expression of c-Myc, whether wild-type or phosphorylation mutant, can swamp out or dilute differences in their activity, making interpretation difficult (data not shown). In this regard, expressing c-MycWT, c-MycT58A or c-MycS62A from the constitutive, but weak, ROSA promoter is an ideal system to study how these phosphorylation sites affect specific activities of c-Myc in vivo at physiologic levels both in development and for tumorigenesis. We focused on the mammary gland as it is a highly dynamic organ, which undergoes repeated cycles of proliferation and apoptosis, and c-Myc is involved in both of these processes (24, 25). We found that expression of c-MycT58A inhibited the apoptotic program of involution associated with reduced pro-apoptotic Bim expression, in agreement with a previous report (42), while c-MycWT increased apoptosis and did not impair involution even though c-MycWT is expressed at lower protein levels in our system and thus should be less likely to induce apoptosis (32). Moreover, c-MycS62A, which is expressed at intermediate levels between c-MycWT and c-MycT58A was also competent for apoptosis and involution. These results point to differential specific activities of c-Myc with altered phosphorylation, although, since expression of the different c-Myc proteins in this system is not equivalent due to differences in their stability (see Fig. 1), we cannot readily distinguish quantitative versus qualitative effects.
We also found that changes in the phosphorylation of T58 and S62 affected genomic stability in primary MECs isolated from the mice. While expression of c-MycWT showed some increase in genomic instability over control, expression of c-MycT58A substantially increased chromosomal instability associated with dramatically increased expression of spindle assembly checkpoint proteins, Bub1b and Aurora B, which are overexpressed in many human cancers associated with chromosomal instability (37, 38). Interestingly, the expression of c-MycS62A was also associated with significant chromosomal instability, particularly evident in the G-banding studies, but it had the opposite effect on the expression of Bub1b and Aurora B, which showed substantially reduced expression. Decreased expression of these proteins is also associated with aneuploidy, but generally results in an aging phenotype, growth arrest, and apoptosis (39, 40), which could contribute to the reduced mammary gland density observed in the MycS62A mice.
Analysis of mammary glands over time revealed that MycWT expressing mammary glands had increased ductal branching, but maintained phenotypically normal alveoli, while MycT58A expressing glands had increased ductal branching and developed expanded and disorganized alveoli. We speculate that increased genomic instability in the face of reduced apoptotic potential in the MycT58A mammary glands contributes to their dysplastic phenotype. In contrast, MycS62A mammary glands generally showed reduced ductal branching with fewer alveoli. In this case, genomic instability in the face of competent activation of apoptotic programs could lead to elimination of aneuploid cells, potentially explaining the reduction in mammary gland density in the MycS62A glands. In addition, recent publications report that phosphorylation at S62 is important to inhibit Ras-induced cellular senescence and that reducing pS62 through elimination of Cdk2 increases c-Myc-driven senescence (43, 44). Increased cellular senescence in the MycS62A expressing glands could also contribute to their reduced density, and future experiments will address this. In addition, future ChIP-Seq experiments to identify global differences in DNA binding among the three isoforms of c-Myc in MECs from our model may provide insight into how the T58 and S62 phosphorylation sites affect c-Myc function.
Phosphorylation at T58 and S62 impacts c-Myc tumorigenic potential
Mice with mammary gland expression of c-MycT58A, but not c-MycWT or c-MycS62A, developed mammary carcinoma. In addition to increased genomic instability and suppressed apoptosis contributing to this, MycT58A glands showed disorganization of alveolar glandular architecture. Studies have shown that expression of c-Myc does not induce proliferation in polarized epithelial cells in 3-D culture, but disruption of polarity allows c-Myc driven proliferation (45); consistent with our observation of mitotic figures in dysplastic foci in MycT58A glands. We also observed that c-MycT58A expressing mammary glands had increased STAT3 activation (data not shown), which is common in human breast cancer and contributes to the disruption of epithelial adhesion and polarity (46). Thus, disruption of alveolar architecture could allow for inappropriate proliferation with accompanying genomic instability, promoting tumorigenesis in MycT58A glands.
In contrast, while c-MycWT expressing mammary glands showed increased density, they appeared morphologically normal, suggesting that the deregulated, but near physiologic expression of c-MycWT from the ROSA locus was able to achieve some sort of homeostatic, non-tumorigenic balance. This provides a potentially important new model for examining collaborative interactions with c-Myc in vivo, which will be important to exploit in future studies. Interestingly, c-MycS62A expressing mammary glands, which generally showed higher c-Myc protein expression compared to c-MycWT glands, had reduced density; nevertheless, expression of c-MycS62A was tumorigenic in the choroid plexus and pituitary. This finding is difficult to reconcile with both our data showing decreased mammary gland density with c-MycS62A expression and recent reports suggesting that decreased S62 phosphorylation may preferentially drive senescence (43). One possibility is that neural epithelial tissues are much less sensitive to drivers of apoptosis and senescence, and in this setting the increased genomic instability with c-MycS62A is pro-tumorigenic. We also cannot rule out the possibility that c-MycS62A expression in the mammary gland could eventually be tumorigenic, similar to the Bub1b−/− mice, which show increased genomic instability and aging phenotypes, with cancer predisposition (39).
Supplementary Material
Acknowledgements
We thank our lab colleagues for comments and feedback. This study was supported by the Department of Defense Breast Cancer research program award BC061306 to RCS, the Susan. G. Komen Breast Cancer Foundation awards BCTR0201697 and BCTR0706821 to RCS, and the NIH award 1 R01 CA129040-01 to RCS.
Footnotes
Conflict of Interests
The authors declare that they have no conflict of interest.
References
- 1.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 2.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 3.D'Cruz CM, Gunther EJ, Boxer RB, et al. c-MYC induces mammary by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 4.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 5.Khan M, Evan G, Pelengaris S. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Nature Reviews Cancer. 2002;2:764–776. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 6.Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 7.Bieche I, Laurendeau I, Tozlu S, et al. Quantitation of MYC gene expression in sporadic breast tumors with a real-time reverse transcription-PCR assay. Cancer Res. 1999;59:2759–2765. [PubMed] [Google Scholar]
- 8.Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83:1688–1695. doi: 10.1054/bjoc.2000.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agnantis NJ, Mahera H, Maounis N, Spandidos DA. Immunohistochemical study of ras and myc oncoproteins in apocrine breast lesions with and without papillomatosis. Eur J Gynaecol Oncol. 1992;13:309–315. [PubMed] [Google Scholar]
- 10.Chrzan P, Skokowski J, Karmolinski A, Pawelczyk T. Amplification of c-myc gene and overexpression of c-Myc protein in breast cancer and adjacent non-neoplastic tissue. Clin Biochem. 2001;34:557–562. doi: 10.1016/s0009-9120(01)00260-0. [DOI] [PubMed] [Google Scholar]
- 11.Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004;3:1133–1137. [PubMed] [Google Scholar]
- 12.Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnold HK, Zhang X, Daniel CJ, et al. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. Embo J. 2009;28:500–512. doi: 10.1038/emboj.2008.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev. 2005;15:34–41. doi: 10.1016/j.gde.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol. 2005;58:225–236. doi: 10.1136/jcp.2003.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang DW, Claassen GF, Hann SR, Cole MD. The c-Myc transactivation domain is a direct modulator of apoptotic versus proliferative signals. Mol Cell Biol. 2000;20:4309–4319. doi: 10.1128/mcb.20.12.4309-4319.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene. 1994;9:59–70. [PubMed] [Google Scholar]
- 19.Thibodeaux CA, Liu X, Disbrow GL, et al. Immortalization and transformation of human mammary epithelial cells by a tumor-derived Myc mutant. Breast Cancer Res Treat. 2009;116:281–294. doi: 10.1007/s10549-008-0127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeh E, Cunningham M, Arnold H, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 21.Malempati S, Tibbitts D, Cunningham M, et al. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia. 2006;20:1572–1581. doi: 10.1038/sj.leu.2404317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol. 1994;14:5510–5522. doi: 10.1128/mcb.14.8.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sodir NM, Evan GI. Nursing some sense out of Myc. J Biol. 2009;8:77. doi: 10.1186/jbiol181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutherland KD, Lindeman GJ, Visvader JE. The molecular culprits underlying precocious mammary gland involution. J Mammary Gland Biol Neoplasia. 2007;12:15–23. doi: 10.1007/s10911-007-9034-8. [DOI] [PubMed] [Google Scholar]
- 26.Schoenenberger CA, Andres AC, Groner B, van der Valk M, LeMeur M, Gerlinger P. Targeted c-myc gene expression in mammary glands of transgenic mice induces mammary tumours with constitutive milk protein gene transcription. Embo J. 1988;7:169–175. doi: 10.1002/j.1460-2075.1988.tb02797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38:627–637. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- 28.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 29.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardiff RD, Anver MR, Gusterson BA, et al. The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene. 2000;19:968–988. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- 31.Wagner KU, Wall RJ, St-Onge L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy DJ, Junttila MR, Pouyet L, et al. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blakely CM, Sintasath L, D'Cruz CM, et al. Developmental stage determines the effects of MYC in the mammary epithelium. Development. 2005;132:1147–1160. doi: 10.1242/dev.01655. [DOI] [PubMed] [Google Scholar]
- 34.Smith DP, Bath ML, Metcalf D, Harris AW, Cory S. MYC levels govern hematopoietic tumor type and latency in transgenic mice. Blood. 2006;108:653–661. doi: 10.1182/blood-2006-01-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell. 2007;12:221–234. doi: 10.1016/j.devcel.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scintu M, Vitale R, Prencipe M, et al. Genomic instability and increased expression of BUB1B and MAD2L1 genes in ductal breast carcinoma. Cancer Lett. 2007;254:298–307. doi: 10.1016/j.canlet.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 39.Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 40.Hardwicke MA, Oleykowski CA, Plant R, et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther. 2009;8:1808–1817. doi: 10.1158/1535-7163.MCT-09-0041. [DOI] [PubMed] [Google Scholar]
- 41.Sternlicht MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48:37–46. doi: 10.1016/s0306-9877(97)90022-0. [DOI] [PubMed] [Google Scholar]
- 42.Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campaner S, Doni M, Hydbring P, et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol. 2010;12(54-9) sup:1–14. doi: 10.1038/ncb2004. [DOI] [PubMed] [Google Scholar]
- 44.Hydbring P, Bahram F, Su Y, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A. 2010;107:58–63. doi: 10.1073/pnas.0900121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Partanen JI, Nieminen AI, Makela TP, Klefstrom J. Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci U S A. 2007;104:14694–14699. doi: 10.1073/pnas.0704677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo W, Pylayeva Y, Pepe A, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126:489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.