

Summary
The three Min proteins spatially regulate Z ring positioning in E. coli and are dynamically associated with the membrane. MinD binds to vesicles in the presence of ATP and can recruit MinC or MinE. Biochemical and genetic evidence indicate the binding sites for these two proteins on MinD overlap. Here we solved the structure of a hydrolytic-deficient mutant of MinD truncated for the C-terminal amphipathic helix involved in binding to the membrane. The structure solved in the presence of ATP is a dimer and reveals the face of MinD abutting the membrane. Using a combination of random and extensive site-directed mutagenesis additional residues important for MinE and MinC binding were identified. The location of these residues on the MinD structure confirms that the binding sites overlap and reveals that the binding sites are at the dimer interface and exposed to the cytosol. The location of the binding sites at the dimer interface offers a simple explanation for the ATP-dependency of MinC and MinE binding to MinD.
Introduction
Cell division in bacteria such as E. coli occurs precisely at midcell due to spatial regulation of the positioning of the Z ring, which dictates the location of the divisome and thus, where division will occur (Rothfield et al., 2005, Lutkenhaus, 2007). Two negative effector systems, Min and Noc, cooperate to ensure placement of the Z ring at midcell. The Min system, consisting of MinC, MinD and MinE, prevents Z rings from forming near the poles and the Noc system prevents assembly of Z rings over the nucleoid (de Boer et al., 1989, Bernhardt & de Boer, 2005, Wu & Errington, 2004, Bi & Lutkenhaus, 1993). The Min proteins undergo a coupled oscillation that produces a time-averaged gradient of MinC, an antagonist of FtsZ assembly, on the membrane that is highest at the poles and lowest at midcell (Hale et al., 2001, Fu et al., 2001). The oscillation is driven by the interplay between MinD and MinE (Raskin & de Boer, 1999b). MinC is not required for the oscillation but is a passenger through interaction with MinD (Raskin & de Boer, 1999a, Hu & Lutkenhaus, 1999).
Underlying the oscillation is the dynamic association of the Min proteins with the membrane. MinD binds to the membrane through the C-terminal 10 amino acids, which form an amphipathic helix that inserts into the membrane bilayer (Szeto et al., 2002, Hu & Lutkenhaus, 2003, Zhou & Lutkenhaus, 2003). The binding is thought to require dimerization of MinD since one copy of the amphipathic helix is insufficient to tether GFP to the membrane whereas a tandem repeat is sufficient (Szeto et al., 2003). Although MinD undergoes ATP dependent dimerization in solution (Hu et al., 2003), it is not clear if MinD dimerizes in the cytoplasm before binding to the membrane since FRET data suggests that MinD oligomerization is promoted by the presence of lipid vesicles (Mileykovskaya et al., 2003).
MinE is a small protein of only 88 amino acids. It has been divided into two functional domains by deletion analysis (Pichoff et al., 1998, Zhao et al., 1995). Residues approximately 1–31 (MinE1–31) constitute an anti-MinCD domain. Examination of minE mutations that affect MinD binding suggests a model in which residues from ~9–30 form an α helix with residues lying on one side of the helix contacting MinD (Ma et al., 2003, Hu & Lutkenhaus, 2001). The other domain consists of residues 32–88 (MinE32–88) and constitutes a topological specificity domain (Pichoff et al., 1998, Zhao et al., 1995). Although MinE1–31 can counteract the inhibitory activity of MinC/MinD to prevent filamentation, it cannot spatially regulate division unless fused to MinE32–88. The recent structures of full length MinE reveal that critical residues in the MinE1–31 domain (around position 21) involved in MinD binding are buried in the structure, suggesting that MinE must undergo a conformational change to bind MinD (Ghasriani et al., 2010; Kang et al., 2010).
MinC also consists of two functional domains (Cordell et al., 2001, Hu & Lutkenhaus, 2000). The C-terminal domain (MinCC) is responsible for dimerization, binding to MinD and, in the presence of MinD, binding to the extreme C-terminal tail of FtsZ (Hu & Lutkenhaus, 2000, Shen & Lutkenhaus, 2009). The MinCC domain consists of a triangular β helix with a hydrophobic face involved in dimerization (Cordell et al., 2001). The MinD binding site is located at the vertex of the triangular β helix opposite the dimer interface (Zhou & Lutkenhaus, 2005). In a MinC dimer the two MinD binding sites are far apart and cannot be in contact with the same MinD dimer. The N-terminal domain of MinC is connected to the C-terminal domain through a flexible linker. This domain contacts an FtsZ subunit in the filament and may fragment the filament (Shen & Lutkenhaus, 2010).
MinD is at the center of the Min system and binds to both MinE and MinC. Biochemical and genetic studies indicate that the binding sites for MinE and MinC overlap. In vitro MinD binds to vesicles in the presence of a nonhydrolyzable analog of ATP and recruits MinC (Hu & Lutkenhaus, 2000). If MinE is added, MinC is displaced consistent with the possibility that the binding sites overlap (Lackner et al., 2003, Hu et al., 2003). Stronger evidence for overlap of the binding sites comes from genetic studies (Ma et al., 2004). Mutations altering residues in helix 7 of MinD affect either MinC or MinE binding or both (Ma et al. 2003). Residues S148, D154 and I159 are involved in binding to both MinC and MinE, whereas D152 is specifically required for MinE binding and L157, G158 and A161 are specifically required for MinC binding. D152 and D154 are highly conserved within the MinD subfamily, but are not conserved within the larger ParA family, consistent with a role specific to MinD.
The structures of three MinD proteins from various achaeal species have been determined (Cordell & Lowe, 2001, Hayashi et al., 2001, Sakai et al., 2001). Although two of them lack a C-terminal amphipathic helix, they contain several signature sequences indicating they are MinD homologues. The structures are monomers, even in the presence of a nonhydrolyzable analogue of ATP (Hayashi et al., 2001). In the monomer, residue D152 is one of three residues that interacts with residue K11, the signature lysine present in the deviant Walker A motif. Based on the observation that D152 is critical for MinE binding it was suggested that the anti-MinCD domain of MinE competes with K11 for interaction with D152, freeing K11 to interact with ATP (Ma et al., 2003). However, in the dimer structure of other proteins in the ParA family obtained in the presence of ATP or an analogue, including Soj and nitrogenase, the lysine corresponding to K11 interacts with ATP bound to the other monomer (Leonard et al., 2005, Schindelin et al., 1997). If MinD is similar, the K11-D152 interaction is disrupted when MinD binds ATP and dimerizes, a step preceding MinE binding. Consistent with this, MinD-D152A dimerizes with ADP suggesting that disrupting the K11-D152 linkage by mutation frees up K11, which promotes dimerization by interacting with ADP bound to the other monomer. Also, the MinD-D152A mutant is able to recruit MinE to lipid vesicles indicating residue D152 is not essential for MinE binding (Zhou et al., 2005). It should be noted, however, that the interaction of the signature lysine with an aspartic residue in the monomer is unique to MinD and is not observed in the ADP form of Soj or the iron protein of nitrogenase.
Although some residues in helix 7 of MinD have been implicated in binding to MinC and MinE, the results were largely interpreted on a MinD monomer structure (Ma et al., 2003, Zhou et al., 2005). Since the binding site for MinE on MinD is not well characterized, and it is now known that MinD dimerizes (Hu et al., 2003), we set out to further explore the region of MinD involved in binding to MinE and MinC. We started out with a random mutagenesis of minD and followed this up with extensive site-directed mutagenesis while pursuing the structure of MinD. Determining the structure of a hydrolysis-deficient derivative of MinD, along with the mutagenesis, confirms that the binding sites for MinC and MinE overlap and reveals that they are at the MinD dimer interface, offering a simple explanation for the ATP-dependency of binding. The structure also allowed determination of the orientation of MinD on the membrane.
Results
The bacterial 2-hybrid system for assessment of the MinD – MinE interaction
The region of MinE that interacts with MinD is defined by deletion and point mutations to residues ~9–30 of MinE (Zhao et al., 1995, Pichoff et al., 1995, Hu & Lutkenhaus, 2001, Ma et al., 2003), however, the region of MinD involved with MinE is not well defined. One study indicated residue D152 of MinD was a critical residue specifically required for interaction with MinE (Ma et al., 2003). This conclusion was based upon the MinD-D152A mutation eliminating the strong interaction observed between MinD and MinE1–31 in the yeast 2-hybrid system. MinE1–31, rather than full length MinE, was used since it displayed a strong interaction with MinD, whereas the interaction between MinD and full length MinE was barely detectable. Why that is the case is not completely clear since full length MinE is better than MinE1–31 in competing with MinC for MinD in this system. This latter result suggests that full length MinE has higher affinity for MinD than MinE1–31 or is more effective at removing MinC.
In contrast to the elimination of the interaction between MinD and the MinE1–31 by the MinD-D152A mutation we observed that the MinD-D152A mutant recruited full length MinE to lipid vesicles in vitro (Zhou et al., 2005). We observed that the MinD-D152A mutant bound to lipid vesicles and recruited MinE, even though the ATPase activity of this mutant was not stimulated by MinE. However, we observed that MinC displaced MinE bound to a MinD-D152A vesicle complex, which is the opposite of what is observed with a wild type MinD-vesicle complex suggesting that MinD-D152A is altered for MinE or MinC binding. Support for an interaction between MinD-D152A and MinE was also obtained from in vivo studies, which revealed that the MinD-D152A protein recruited MinE to the membrane, although not as effectively as wild type MinD (Zhou et al., 2005). These results were somewhat at odds to those obtained with the yeast 2-hybrid system so we investigated this further using the bacterial 2-hybrid system, which has also been used by Hsieh et al. (2010) to examine the MinD-MinE interaction.
A strong interaction between MinD and MinE1–31 was observed in this system (Table 1). A similar strong interaction was also observed between MinD and full length MinE. Why these two interactions are nearly equivalent in the bacterial 2-hybrid system and yet vastly different in the yeast 2-hybrid system is not known. The MinD-D152A mutation reduced the interaction between MinD and MinE by ~50% and the interaction between MinD and MinE1–31 by 90%. Thus, the MinD-D152A mutation reduces the binding of MinE but has a stronger affect on the interaction between MinD and MinE1–31 than on the interaction with full length MinE. In addition, these results indicate that the bacterial 2-hybrid system is more useful than the yeast 2-hybrid system in examining the interaction between MinD and MinE.
Table 1.
Bacterial 2-hybrid system for analysis of MinD-MinE interaction.
| MinE-MinD | 100% | (330)1 |
| MinE1-31-MinD | 120% | (398) |
| MinE-MinD-D152A | 44% | (145) |
| MinE1-31-MinD-D152A | 8% | (28) |
The numbers in parentheses are units of β-galactosidase activity.
Isolation of MinD mutants unresponsive to MinE
The above studies revealed that the MinD-D152A mutation reduced but did not eliminate the interaction between MinD and MinE. To obtain more information about the region of MinD involved in the interaction with MinE we screened for MinD mutants that fail to respond to MinE while retaining the ability to activate MinC. This approach should eliminate MinD mutants that do not fold properly since interaction with MinC is required. To do this screen pSEB104CDE (Para::minC minD minE) was mutagenized by passage through the mutagenic strain XL-1 Red and transformed into the min deletion strain JS964 (Δmin::kan). This strain carrying the WT plasmid displays a minicell phenotype in the absence of arabinose whereas in the presence of arabinose it displays a wild type morphology due to expression of the min operon. Colonies were patched onto plates containing 0.1% arabinose and those that did not grow were candidates for having a mutation.
Out of approximately 500 colonies screened 30 were obtained that did not grow when the min operon was induced. The inability to grow under inducing conditions correlated with filamentation and could be due to: 1) mutations that inactivate MinE, or 2) mutations that alter MinD such that it does not respond to MinE even though it must still interact with MinC. To differentiate between these two possibilities a compatible plasmid (pJPB216 [Plac::minE]) carrying MinE was introduced into each of the candidates to determine if it could rescue growth. Of the 30 candidates 13 could be rescued by expressing MinE in trans indicating they contained a mutation in MinE. Sequencing these candidates confirmed mutations in MinE including A15T, R21W, R31A, P36S and stop codons at codons 20, 23, 35, 55 and 65. The stop codon mutations are likely to produce a nonfunctional MinE due to truncation and a MinE-A15R mutant was previously shown to result in a reduced interaction with MinD. R21 is conserved in all bacterial MinEs and is likely to be important for MinD-MinE interaction.
The mutants that could not be rescued by MinE in trans were expected to have mutations in MinD. Sequence analysis revealed that several of the mutants contained multiple mutations and others that contained single mutations. The latter included MinD-E53K, -D192Y, -M193L, and -G224C. To confirm these mutations they were introduced by site-directed mutagenesis into a plasmid containing MinC/MinD under arabinose promoter control (pSEB104CD [Para::minC minD]). The resultant plasmids were transformed into JS964 (Δmin) along with a compatible plasmid (pJPB216[Plac::minE]) carrying MinE. Although MinE rescued the lethality due to expression of WT MinD (along with MinC) it could not rescue the lethality caused by the four MinD mutants (Fig. 1, results with MinD-M193L, but results were the same for all four; gel analysis revealed all four were stable [data not shown]).
Fig. 1.
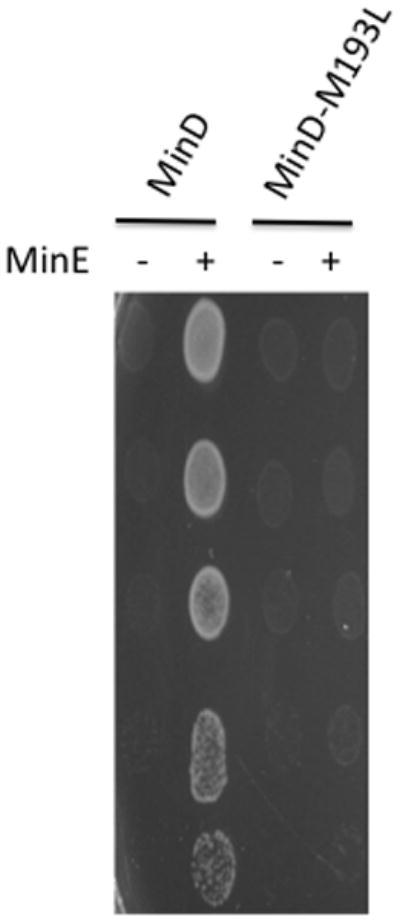
MinD-M193L does not respond to MinE. JS964 (Δmin) containing pSEB104CD (Para::minC minD) carrying WT minD or minD-M193L was transformed with a plasmid carrying minE (pJPB216 [Plac::minE]) or the corresponding vector. A colony from each transformation was picked into 500 μl of LB, serially diluted tenfold and 5 μl of each dilution was spotted on plates containing appropriate antibiotics, 0.1% arabinose (to induce MinCD) and 100μM IPTG (to induce MinE).
To check these results in vitro we attempted to purify the four mutants. Since MinD-M193L and MinD-G224C could be readily purified they were tested in a vesicle-binding assay. Similar to wild type MinD, MinD-M193L and MinD-G224C bind to vesicles in an ATP-dependent manner and recruit MinC (Fig. 2; only MinD-M193L is shown). The addition of MinE removes wild-type MinD and MinC from the vesicles, however, MinE is unable to displace MinD-M193L and the associated MinC from the vesicles. These results confirm that MinD-193L binds MinC but is unable to respond to MinE. The same results were obtained with MinD-G224C (data not shown).
Fig. 2.
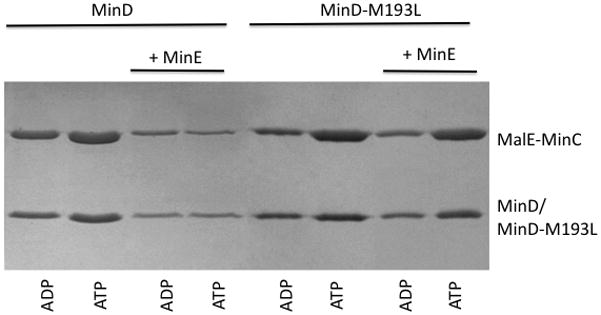
MinD-M193L recruits MinC to vesicles and is unresponsive to MinE. MinD or MinD-M193L was mixed with MalE-MinC in the presence of ADP or ATP and vesicles. MinE was added as indicated. Vesicles were collected by centrifugation and the bound proteins were examined by analyzing the pellets on SDS-PAGE.
Since the other two MinD mutants were more difficult to purify we checked if the failure of MinE to rescue these MinD mutants was due to loss of interaction in the bacterial 2-hybrid system. MinD-E53K, -D192Y, -M193L and -G224C all showed a dramatic decrease in interaction with full length MinE (Fig. 3). The controls included MinD-D152A, which was positive (also Table 1), MinD-D154A, which was only very weakly positive as shown previously (Ma et al., 2004), and MinD-K16Q, which cannot bind ATP (Hu et al., 2002), and served as a negative control. Thus, all four of the MinD mutants we isolated failed to bind to MinE even though they activate MinC.
Fig. 3.
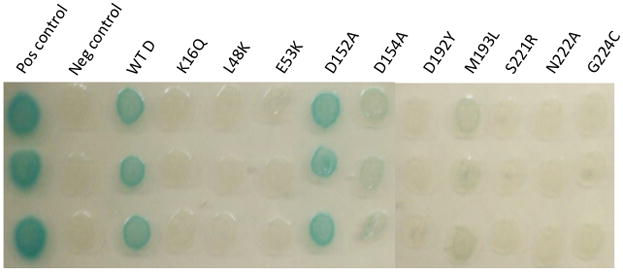
Bacterial 2 hybrid analysis of MinD mutants that fail to respond to MinE but activate MinC. MinD and MinE were fused to the C-termini of the T18 and T25 fragments of adenyl cyclase. Plasmids containing these fusions were cotransformed into BTH101 (Δmin). Three transformants for each were picked into LB and spotted on LB plates supplemented with X-gal and antibiotics. The appearance of blue color indicates a positive reaction. The positive control (containing bZIP in both vectors) and the negative control (plasmids expressing T25-MinD and the T18 empty vector) were included for reference. The plates were incubated overnight at 30°C.
Structure of MinD
MinD dimerizes in the presence of ATP but is not soluble at high concentrations (Hu & Lutkenhaus, 2000). In contrast, a truncated version of MinD (MinDΔ10) missing the carboxy 10 amino acids is very soluble (Hu & Lutkenhaus, 2003). This protein is only missing the amphipathic helix so it is possible that the structure might still give information about how the protein is positioned on the membrane. Since, several ATP analogues (ATPγS and AMPPCP) are unable to support MinD dimerization (Hu et al., 2002 and data not shown; implying that for MinD, these analogues are not good mimics of ATP), two approaches were used to try and capture MinD in the dimeric state. The first involved crystallization of MinDΔ10-D152A in the presence of ADP, since ADP promotes dimerization of this mutant as well as the binding of both MinC and MinE (Zhou et al., 2005). One condition was found in which crystals were obtained, however, they did not diffract well. Nonetheless, this condition proved useful for obtaining crystals of a second hydrolysis-deficient mutant of MinD. This mutant was obtained by introducing the D40A mutation. Mutating the equivalent residue in the closely related Soj protein produced a hydrolysis-deficient version of the protein that was crystallized as a dimer (Leonard et al., 2005).
A MinD mutant deficient in ATP hydrolysis would be expected to bind the membrane, MinC and MinE but would not be able to undergo oscillation and spatially regulate division. To determine if MinD-D40A behaved as expected we checked if it was able to activate MinC and respond to MinE. The growth of JS964 (Δmin) containing pSEB104CD-D40A (Para::minC minD-D40A) was inhibited by arabinose indicating MinD-40A activated MinC (Fig. 4A). Addition of MinE (pJPB216[Plac::minE]) allowed growth and prevented filamentation indicating that MinD-D40A also responded to MinE, however, the strain had a minicell phenotype indicating a defect in spatial regulation of division. GFP-MinD-D40A displayed a peripheral localization and did not undergo oscillation in the presence of MinE (data not shown; the lack of filamentation indicates MinD-D40A has a preference for MinE over MinC similar to WT MinD).
Fig. 4.
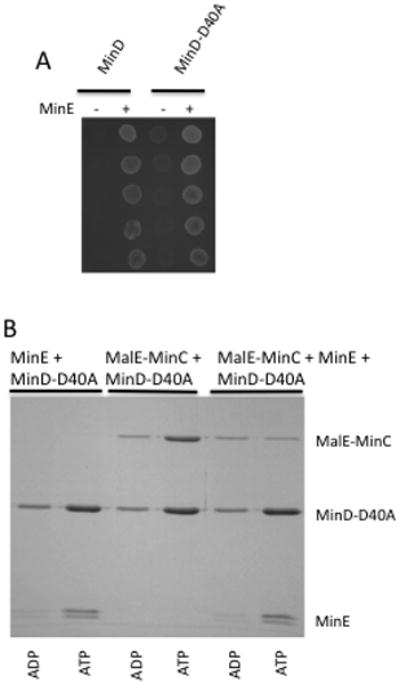
Characterization of MinD-D40A. (A) The ability of MinD-D40A to activate MinC and respond to MinE was determined. JS964 (Δmin) containing pSEB104CD expressing WT MinD or MinD-D40A was transformed with a plasmid carrying minE, or the corresponding vector. A colony from each transformation was picked into 500 μl of LB, serially diluted tenfold and 5 μl of each dilution was spotted on plates containing 0.1% arabinose (to induce MinCD) and 5 μM IPTG (to induce MinE). (B) MinD-D40A binds MinC and MinE in vitro. MinD-D40A was incubated with vesicles in the presence of ATP or ADP. MinE, MalE-MinC or both were added and vesicles recovered by centrifugation. Bound proteins were determined by analyzing the pellets on SDS-PAGE. Although MinD-D40A recruits MinC to vesicles, it is less effective than WT MinD or MinD-M193L. The two bands of MinE are due to proteolysis during purification and storage leading to loss of some N-terminal residues.
Purification of the full length version of MinD-D40A revealed that it bound to vesicles in an ATP-dependent fashion (Fig. 4B). It was able to recruit MinC to the vesicles and the MinC was displaced by MinE. Importantly, MinE was unable to cause release of MinD-D40A from the vesicles but bound to the MinD-D40A-vesicle complex. Although MinE was recruited to vesicles by MinD-40A it was unable to stimulate its ATPase activity (Table 2). Thus, MinD-D40A interacts with its partners and behaved as expected for a hydrolysis-deficient mutant. A C- terminal truncated version (MinDΔ10-D40A) was purified and size exclusion chromatography confirmed it underwent ATP-dependent dimerization (data not shown).
Table 2.
ATPase activity of MinD-D40A*.
| − MinE | +MinE | |
|---|---|---|
| MinD | 0.9 | 18.6 |
| MinD-D40A | 0.5 | 2.1 |
The ATPase activity is nmoles of Pi released/mg/min
MinDΔ10-D40A crystallized in the presence of ATP using the same condition that promoted crystallization of MinDΔ10-D152A. Determination of the structure at a resolution of 2.4 Angstroms confirmed that MinD formed a nucleotide sandwich dimer similar to Soj and nitrogenase (Fig. 5A). Examination of the structure confirms that the signature lysine in the deviant Walker A motif does not interact with D152 but reaches across the interface to interact with the phosphates of ATP bound to the other monomer (Fig. 5A and 5B). Thus dimerization, and not MinE, is responsible for breaking the D152-K11 interaction ruling out the previous model (Ma et al., 2004) for the role of MinE in the activation of the MinD ATPase.
Fig. 5.
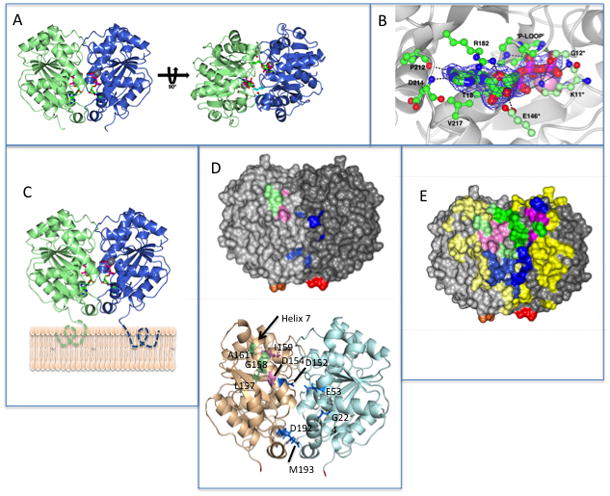
The dimeric structure of MinD and the location of residues involved in MinD and MinE binding. (A) Each monomer is colored differently and the ATPs (stick diagram) and Mg ions (gray spheres) are indicated. In the structure on the right the side chains for lysine 11, which reach across the dimer interface, are depicted.
(B) Residues interacting with ATP. Residues on one subunit important for ATP binding are indicated by a darker shade of green. The three residues making contact with ATP across the dimer interface (G12 and E146 in addition to K11) are indicated with an * and are a lighter shade of green. (C) Model of MinD bound to the membrane. The residues missing from the structure (KKGFLKRLFGG) that the form an an amphipathic helix involved in membrane binding are indicated by dotted lines extending from C-terminus of MinD. The orientation of MinD is the same as the structure on the left in panel A. (D) Location of the 4 residues identified in the random screen that are important for MinE interaction, but not for MinC, (E53, D192, M193 and G224; indicated in blue) relative to previously identified residues important for MinE or MinC binding. Based upon the model in panel (C) these residues are closer to the membrane. The location of D152 (blue), D154 and I159 (pink; involved in both MinC and MinE interaction) and L157, G158 and A161 (green; specifically required for MinC interaction) are also indicated. The C-terminal glutamate residues present in the structure are colored red and orange. The orientation of MinD is the same as in panel C.
(E) Location of MinD residues involved in interaction with MinE and MinC. The structure is of the MinD (same orientation as panel C) dimer with residues colored as in D. For all residues the lighter shade is the monomer to the left and the darker shade is the monomer to the right. Residues not involved in binding MinC or MinE are colored yellow. Three types of residues important for binding are indicated. Those specifically involved in MinE binding are blue, those specifically involved in MinC binding are green and those involved in both MinC and MinE binding are pink. The C-terminal glutamates are colored red and orange.
Three residues, D152, S148 and E146 interact with K11 in the monomer (Hayashi et al., 2001). In the dimer S148 and E146, like K11, make contacts across the dimer interface whereas D152 does not. S148 interacts with N45 in the other subunit and E146 makes contact with the ribose ring of the ATP bound to the other subunit. These two contacts are not essential for dimer formation since MinD-S148A and MinD-E146A mutants still dimerize (Zhou et al., 2005), although the ATPase activity of MinD-E146A is not stimulated by MinE. In contrast, the K11-ATP interaction is important since MinD-K11A does not dimerize (Zhou et al., 2005). G12 also makes contact with the γ-phosphate of ATP across the dimer interface (Fig. 5B).
Although the C-terminal 10 amino acids constituting the amphipathic helix were removed to obtain a more soluble version of MinD, the structure still gives information about the orientation of MinD at the membrane surface. MinD is ~20 residues longer than Soj or the achaeal MinD-like proteins that lack the amphipathic helix. The structures of Soj and these latter proteins terminate in a helix that angles away from the bottom of the structure (as drawn in Fig. 5C) towards the side. In the MinDΔ10 structure the final 10 amino acids, including the three glutamates that precede the amphipathic helix, are visible and extend from the end of the terminal helix in the structure to the bottom of the structure as drawn in Fig. 5C. Since only residues comprising the amphipathic helix, which would be in intimate contact with the membrane bilayer (Zhou & Lutkenhaus, 2003), are missing from the structure the bottom face of the dimer must be near the membrane (Fig. 5C).
The mutations isolated earlier (Ma et al., 2004) that affect MinC and/or MinE binding are located in helix 7 and map to the monomer on the left (colored green and pink in Fig. 5D with D152 colored blue in the middle of the structure). The residues altered by the four mutations from the random mutagenesis in this study expand the area involved in MinE binding. Two residues are beneath the residues located in helix 7 (Fig. 5D, colored blue). In contrast, the other two residues map nearby but to the subunit to the right. This result suggests that the possibility that the MinE binding site is at the MinD-dimer interface and is only formed upon dimerization. To further test this possibility we performed site- directed mutagenesis.
Site-directed mutagenesis to further characterize the binding sites
With the availability of the MinD structure and the location of the residues from the random mutagenesis we carried out additional site-directed mutagenesis to further characterize the MinE binding site. For this analysis, we used the two-plasmid system (pSEB104CD [Para::minC minD] and pJB216 Plac::minE]) used above to confirm the mutations from the random mutagenesis. Mutations were introduced into minD on pSEB104CD and their effect was determined by examining the ability of the plasmid to prevent the growth of JS964 (Δmin) when induced. If the mutant MinD was able to activate MinC, pJB216 was introduced to determine if the mutant was able to respond to MinE. Using this approach we found the following nine MinD mutants that were able to activate MinC but were unable to respond to MinE: L48K, D67R, V147E, L194D, D198R, I202D, L218E, S221R and N222A (Table S1). This response was tested at two levels of MinE, the basal level of expression and that produced by 100 μM IPTG. None of the mutants, including the four from the random mutagenesis, were rescued by the basal expression of MinE, which is sufficient to overcome lethality caused by wild type MinD. In addition, most of the mutants were not rescued by the higher level of MinE induced by 100 μM IPTG (Fig. 1, MinD- M193L). Only the MinD-D67R mutant was rescued by the induced level of MinE suggesting it binds MinE weakly. Examination of these mutants with the bacterial 2-hybrid system confirmed a reduction in interaction with MinE; D67R produced pale blue colonies whereas the others produced white colonies (Fig. 3, L48K, S221R, N222A and data not shown). The weak interaction observed between D67R and MinE is consistent with it responding to a high level of MinE.
A number of other MinD mutants were generated that can still activate MinC and respond to MinE indicating that the corresponding residues are not required for MinC or MinE binding: MinD-G51D, -C52A, -C52R, -R54E, -R55, -Q65R, -Q72R, -I75D, -K76D, K78A, R79D, -D93R, -D95R, -T98D, -R99D, -E100K, K104E, -S162K, -K163D, -A167K, E168A, -N169R, -G170R, -E172K, -K175D, -E176K, -R187A, -R190A, -G191E, -S195R, -E197K, -E201A, -R204A, -K206E, -D214A, -Q215R, -R219A, -E225K, -L229K, -N232A, -I246D, -F253D, (Table S1). Since these residues surround those that are required for MinE binding, a more complete picture of the surface of MinD involved in the interaction with MinE emerges (Fig. 5E; residues specifically required for binding to MinE are in blue and those not required are yellow; note residues on the left monomer are in a lighter shade). From this result it is clear that residues required for MinE binding come together upon dimerization.
During the above mutagenesis we also isolated several mutants in which the activation of MinC was lost but MinE binding, determined by the bacterial 2-hybrid system, was not affected indicating these residues appear specifically required for MinC binding (Table S1). These included MinD-R44A, -V57E, -Q90A, -T91K, -R92A, -R151A, -G158R, -I159D, -A161R and -P173R. Locating these residues on the MinD structure (Fig. 5E; residues in green), as well as those not required for MinC activation (Fig. 5E; residues in yellow), gives a more complete picture of the region of MinD involved in MinC binding. It is clear that the MinC binding site is also composed of residues coming together upon MinD dimerization.
During the site directed mutagenesis we isolated several mutations that affected the interaction of MinD with both MinE and MinC. Such MinD mutants were unable to kill JS964 (Δmin) when expressed from pSEB104CD and to interact with MinE in the bacterial 2-hybrid system. Such mutations included MinD-L43D, -D47R, -V56E, -Y58D, -D59R, -V61E, -K94D, -E146K, V150E, -L157D and -V188E. D154A and I159R, isolated earlier, also fall into this class (Zhou & Lutkenhaus, 2005, Ma et al., 2004). Since the new mutations affect the interaction of MinD with both MinC and MinE it was possible that they affect MinD dimerization.
The ability of these mutants to dimerize was assessed with the bacterial 2-hybrid system. MinD-K16Q, a mutant defective in interaction with ATP was used as a control. Surprisingly, we detected a strong interaction between MinD and MinD-K16Q, however, we did not detect an interaction between MinD-K16Q and MinD-K16Q (data not shown). Therefore, in testing the other mutants we used constructs carrying the relevant mutation in both components of the system. Using this approach we found five mutants that displayed no self-interaction (MinD-D47R, -D59R, -E146K, -V150E, and -V188E) and therefore are probably defective in dimerization or folding. However, six of the mutants (MinD-V56E, -Y58D, -V61E, -N62K, -K94D, -R155E and -L157D; Fig. 6, K94D is shown; Table S1), displayed strong self-interaction. D154A, previously shown to be deficient in interaction with both MinC and MinE, but able to self interact, served as a control (Ma et al., 2004, Zhou et al., 2005). These mutations are unlikely to affect the folding of MinD suggesting that the corresponding residues are required for interaction with both MinC and MinE (Fig. 5E, pink residues).
Fig. 6.
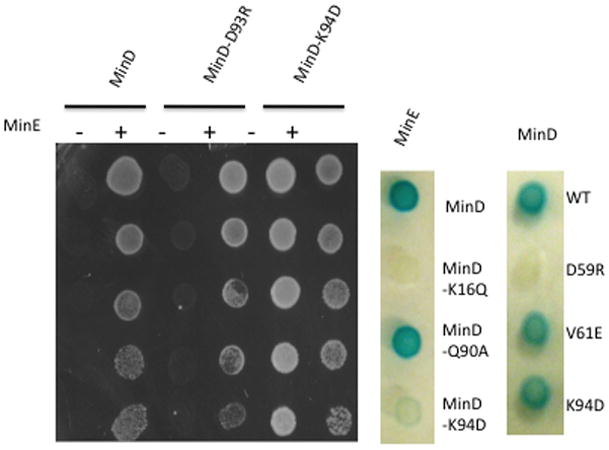
Example of tests of mutants obtained by site-directed mutagenesis of minD. Spot tests were used to determine if MinD mutants activated MinC (no growth in the absence of MinE). Mutants, such as MinD-D93R activated MinC and responded to MinE, whereas MinD-K94D did not activate MinC. To see if MinD-K94D interacts with MinE it was tested in the bacterial 2-hybrid system (minD alleles are shown to the right of the test strip). If, like minD-K94D, they did not interact with minE, they were tested for self-interaction (minD-K94D and minD-V161E did self-interact whereas minD-D159R did not). Results for all mutants are summarized in Table 1 of the supporting information.
Discussion
The interaction of MinD with MinE is essential to achieve Min oscillation whereas the interaction of MinD with MinC is essential to effectively antagonize FtsZ assembly (Raskin & de Boer, 1999b, de Boer et al., 1992, Shen & Lutkenhaus, 2009, Hu et al., 2002). In this study we determined the structure of MinD lacking the C-terminal amphipathic helix and found that it formed a nucleotide sandwich dimer similar to Soj. This structure, along with the extensive site-directed mutagenesis, confirmed that the binding sites for MinE and MinC on MinD overlap and revealed that the sites are at the MinD dimer interface. The binding site for MinE appears more extensive and involves residues that extend from the bottom to the top of the dimer interface whereas the site for MinC involves residues limited to the upper half of the dimer interface. Since the binding sites overlap the dimer interface it offers a simple explanation for the ATP-dependent binding of MinC and MinE to MinD; the complete binding sites are only present upon dimerization of MinD, a process known to be ATP-dependent. In the model of the MinD the dimer bound to the membrane the binding sites for MinE and MinC are exposed to the cytosol.
MinD structure
MinDΔ10-D40A formed a nucleotide sandwich dimer in the presence of ATP as expected based upon its known ability to dimerize in solution and homology to Soj. Nonetheless, several important features of MinD were revealed by solving the structure and are unique to MinD. The role of MinE in stimulating the ATPase activity of MinD was previously postulated to arise from MinE disrupting the K11-D152 interaction freeing K11 to bind to ATP and stimulate ATP hydrolysis (Ma et al., 2004). An argument against this was that the residue equivalent to K11 interacts with ATP in the dimer structures of Soj and the iron protein of nitrogenase indicating that ATP binding and/or dimerization disrupts the K11-D152 interaction (Leonard et al., 2005, Schindelin et al., 1997). Since Soj and nitrogenase do not have residues equivalent to D152 this remained a possibility for MinD. However, the MinD dimer structure obtained in this study confirmed that K11 interacts with ATP leading to the conclusion that dimerization, and not MinE, disrupts the D152-K11 interaction. The interaction between K11 and ATP probably stabilizes the dimer since a K11A mutant does not dimerize (Zhou et al., 2005). Comparison of the structure of the monomer of MinD from Pyrococcus furiosus (Hayashi et al., 2001) with the E. coli MinD dimer did not reveal significant structural changes indicating that dimerization may be primarily due to the K11-ATP interaction. This possibility is consistent with ADP promoting dimerization of the MinD-D152A mutant (Zhou et al., 2005).
The MinD structure also revealed the likely orientation of MinD on the membrane. In the dimer of MinDΔ10 the C-terminal 10 residues of each monomer are fixed in position, extending from the C-terminal helix towards the bottom of the MinD dimer (Fig. 5A and C). Since both C-termini end up on the same face of the dimer it suggests that this face of the MinD dimer comes near the membrane. Furthermore, the absence of only the amphipathic helix from the structure suggests that this face of MinD is in close contact with the membrane surface. Interestingly, this face of MinD is analogous to the face of Soj that is involved in nonspecific DNA binding (Hester & Lutkenhaus, 2007). Thus, these two related proteins bind to completely different substrate surfaces with the same face of the protein using very different mechanisms. Soj uses conserved arginines present on this face, whereas MinD uses strategically located amphipathic helices. In this model of MinD on the membrane, the binding sites for MinE and MinC are fully exposed and available for interaction. This is especially important for MinC, which is a relatively large molecule. One complication is that there is some evidence that MinD forms larger oligomers and it is possible that the binding sites could be occluded in the oligomer (Hu et al., 2002).
MinC-MinD
Each MinC dimer consists of a C-terminal dimerization domain fused through a flexible linker to an N-terminal domain responsible for disrupting FtsZ filaments (Cordell et al., 2001). The extensive site-directed mutagenesis in this study limits the MinC binding site to the top half of the MinD dimer. Thus, it is likely that the C-terminal domain of MinC binds near the top of MinD in such an orientation that the N-terminal domain is extending away from the membrane (Fig. 7). In this orientation MinC would be in position to make the two known contacts with FtsZ filaments. The MinD/MinCC complex on the membrane would be in position to compete with membrane bound FtsA and ZipA for interaction with the carboxy tail of FtsZ subunits present in polymers (Dajkovic et al., 2008, Shen & Lutkenhaus, 2009). MinCN would extend further out away from the membrane where it would be in position to contact the body of FtsZ subunits in the filament (Shen & Lutkenhaus, 2010) (Fig. 7). Previous mutagenesis of the MinC C-terminal domain indicated the MinD binding sites on the MinC dimer (containing the conserved sequence RSGQ) are located far apart on opposite ends of the MinC dimer (Zhou & Lutkenhaus, 2005, Ramirez-Arcos et al., 2004). Each C-terminal domain consists of a triangular beta helix with one of the faces involved in dimerization (Cordell et al., 2001). The MinD interaction site is at a vertex of the triangular beta helix farthest away from the dimer interface. Thus, it is possible that a MinD dimer could be sandwiched between two MinC dimers and/or a MinC dimer could be sandwiched between two MinD dimers.
Fig. 7.
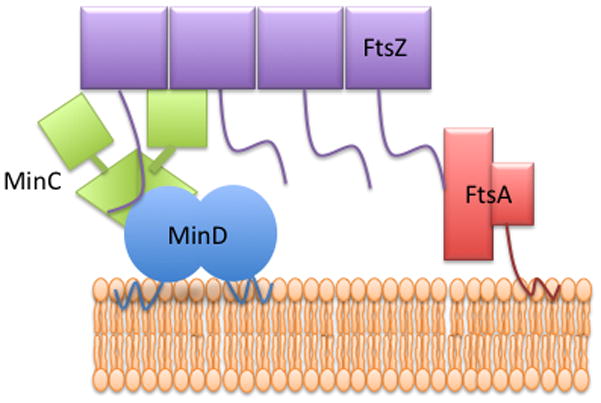
Model of the interaction of Min proteins and FtsZ. MinD and FtsA are known to bind to the membrane through carboxy-terminal amphipathic helices. The C-terminal domain of MinC (MinCC) binds to MinD whereas the N-terminal domain of MinC extends away from the membrane and is free to contact FtsZ. The conserved tail of FtsZ binds to FtsA or to the MinD-MinCC complex.
MinD-MinE
Locating the residues altered by the four mutations isolated from the random mutagenesis, which are deficient in MinE binding but proficient in MinC activation, was the first indication that the binding site for MinE forms upon dimerization. More extensive site-directed mutagenesis indicates the MinE binding site on MinD extends from the bottom of the MinD dimer interface (i.e. membrane proximal) to the top of the structure (membrane distal) (Fig. 5C & 5E). The residues that are involved in MinE binding line a cleft that extends along the side of the MinD dimer interface (Fig. 5E). The anti-MinCD domain of MinE, composed of residues ~9–30, is proposed to assume an alpha helix that would fit into this cleft (Ma et al., 2004). However, the recent structure of full length MinE reveals that a portion of the anti-MinCD domain is occluded and unavailable for interaction suggesting that, for this model to be true, MinE would have to undergo major rearrangement (Ghasarini et al., 2010; Kang et al., 2010). The structure of MinD is similar to the iron protein of the nitrogenase complex (Hu & Lutkenhaus, 2003, Lutkenhaus & Sundaramoorthy, 2003). Notably, the cleft in the iron protein is occupied by a helix that arises from the carboxy domain of the iron protein itself. The structure of Soj is similar to MinD with a cleft running along each side of the dimer interface (Leonard et al., 2005). Similar to MinD, the ATPase activity of Soj is stimulated by a partner protein containing an unstructured N-terminal domain that is critical for stimulation (SpoOJ for Soj). It is possible that this unstructured region of SpoOJ also assumes an alpha helix upon contact with Soj that fits in the cleft and that this is common theme among ParA/MinD family members.
Experimental procedures
Bacterial strains and growth conditions
E. coli strain JS964 (MC1061 malP::lacIq Δmin::kan) has been described previously (Pichoff et al., 1995). BTH101Δmin strain (F−cya-99, araD139, galE15, galK16, rpsL1 (Strr), hsdR2, mcrA1, mcrB1)(Karimova et al., 2000)has the entire minB operon consisting of three genes, minC, minD, and minE, deleted and replaced with a kan-resistance cassette. It was made by P1 transduction with P1 grown on JS964 (Δmin::kan). LB (Luria-Bertani) medium containing 0.5% NaCl and relevant antibiotics at 37 C was used for most experiments unless otherwise indicated.
Plasmid constructions
Plasmid pSEB104CD (PBAD::minCD)and pJB216 (Plac::minE)were described earlier (Zhou & Lutkenhaus, 2005). pSEB104CDE (PBAD:minCDE) was created by ligation of the small BstXI/HindIII fragment from pJBP210(Pichoff et al., 1995)into BstXI/HindIII-digested pSEB104CD. pZH115 (pJF118EH [Ptac::minD]) has been described before (Hu & Lutkenhaus, 2001). The same strategy that yieldedpZH115 was employed to construct pZH115-10(pJF118EH Ptac::minDΔ10) in which MinD deleted of the C-terminal 10 codons (Hu & Lutkenhaus, 2003). The minD-D40A or minD-D152A mutations were introduced into this plasmid by site-directed mutagenesis to give pZH115-40 and pZH115-152, respectively. The construction of pZH112(PBAD::malE-minC116–231)was detailed in an earlier study (Hu et al., 2003)
pKT25 and pUT18 were described before (G. Karimova et al., 1998). pCT25 (CmR) wasconstructed by ligating the PvuII/HindIII fragment of pKT25(KmR) with the HincII/HindIII fragment of pACYC184 (Chang & Cohen, 1978). pCT25-MinD (cyaT25-minD) was generated by ligating PCR-amplified minD from pSEB104CDinto BamHI/KpnI–digestedpCT25 (cyaT25 ). The PCR amplification of minE followed by digestion with BamHI/KpnI and ligation into BamHI/KpnI site of pUT18 vector (cyaT18) yielded pUT18-MinE (cyaT18-MinE). Various minE and minD mutations were introduced into these plasmids by site-directed mutagenesis. Mutations in minD were introduced by site directed mutagenesis.
Bacterial two-hybrid analysis and β-Galactosidase Assay
A cya-null strain BTH101Δmin was transformed with plasmids pCT25-MinD and pUT18-MinE, respectively carrying wild-type or mutant minD and minE alleles, and grown overnight at 37 C on LB plates containing 0.2% glucose, chloramphenicol (20μg/ml) and ampicilin (100 μg/ml). For plate-based assay, colonies from the LB plate were diluted in 300 μl volume of LB broth and spotted onto fresh LB plates supplemented with chloramphenicol (20 μg/ml), ampicilin (100 μg/ml), 5-bromo- 4-chloro-3-indoyl-β-D-galactopyranoside (X-Gal) at 40 μg/ml, and 0.5 mM IPTG. Observation was usually made after 14–18 hours of incubation at 30 C.
For the quantitative β-galactosidase assay, three colonies were picked from each plate and cultured overnight at 30 C in LB medium containing0.2% glucose, 20μg/ml chloramphenicol, and 100μg/ml ampicillin. The overnight cultures were then diluted 1/100 into fresh LB medium containing 0.5mM IPTG, 20μg/ml chloramphenicol, and 100μg/ml ampicillin and cultured for 3–4 h at 30 C followed by measurement of OD600. Cells were permeabilized with the addition of 0.0016% SDS [w/v] and 2.5% chloroform [v/v]) and vigorously vortexed. Then0.4ml of permeabilized cells weremixed with 0.6ml of Z buffer (60 mM Na2HPO4, 40mM NaH2PO4 [pH 7.5], 1 mM MgSO4, 50 mM β-mercaptoethanol) and0.25ml of ONPG (o-nitrophenyl β-D-galactopyranoside, 4 mg/ml)(Sigma) was added, the reactions wereincubated for additional 20 min at 30 C and stopped with 400mM Na2CO3. OD420 values were recorded and converted into Miller activity units as described.
Mutagenesis using XL-Red strain
Plasmid pSEB104CDE was transformed into E. coli XL1-Red strain (F- endA1 gyrA96 (nalR) thi-1 relA1 lac glnV44 hsdR17 (rK− mK+) mutS mutT mutD5 Tn10)(Stratagene) containing 25 mM β-mercaptoethanoland plated onto LBplates containing 0.2% glucose, spectinomycin (100 μg/ml), and kanamycin (50 μg/ml). The colonies from the LB plates were pooled and the plasmid DNAs were isolated using a Plasmid Miniprep Kit(Qiagen). This mutagenized plasmid library was then transformed into strain JS964 (Δmin) and grown in LB plates containing 0.2% glucose, spectinomycin (100 μg/ml), and kanamycin (50 μg/ml). To look for mutations affecting min function, approximately 500 colonies were picked and streaked onto LB plates containing 0.2%arabinose and spectinomycin (100 μg/ml) to induce expression of MinCDE. The colonies manifesting filamentous phenotypes were selected and cultured prior to plasmid DNA isolation and sequencing confirmation. Additional information is available in the main text.
Site-directed mutagenesis
The specific mutations within MinD and MinE ORFs were introduced into the various plasmids using the QuickChange site-directed mutagenesis kit according to the manufacturer’s instruction (Stratagene). The plasmids used as templates and primers are available upon request.
Protein purification
MinDΔ10-D40A and MinDΔ10-D152A were purified from JS964 (Δmin) containing pZH115-40 and pZH115-152, respectively. MinDΔ10-D40A labeled with Se-methionine was purified from B834 (DE3)(Δmin) grown in minimal medium. Cells were collected from 1 liter cultures grown in LB with ampicillin (100 μg/ml), resuspended in buffer A (25 mM Tris-HCl [pH 7.5]), 20 mM NaCl, 1 mM EDTA, 2 mM DTT and 10 % glycerol) and lysed with a French press. The clarified lysate was loaded on a DEAE column and MinDΔ10-D40A eluted with a 60–120 mM NaCl gradient in buffer B (10 mM HEPES-NaOH pH 7.2, 20 mM NaCl, and 10% glycerol). The peak fractions were pooled and ran over a HiLoad Superdex 200 column in bufferA. The peak fractions were collected and concentrated with a Vivaspin 20 (MW cutoff of 10 kDa) to ~20 mg/ml.
The purification of full length wild-type and mutant MinD proteins were previously described (Hu & Lutkenhaus, 2001). MalE-MinCCwas purified using the method reported previously (Dajkovic et al., 2008).
Vesicle binding assay
The preparation of multilamellar vesicles (MLV)has been described previously (Hu & Lutkenhaus, 2001). MinD, MinD-M193L or MinDΔ10-D40A (4 μM) were incubated with vesicles (400 μg/ml) in the presence of ADP or ATP (1 mM) and MinE (4 μM) or MalE-MinC (4 μM) or both were added. After a 10 minute incubation at room temperature, the vesicles were collected by centrifugation and the pellets analyzed by SDS-PAGE.
Crystallization
Crystals of the MinD 10-D40A-Mg-ATP complex were grown by the hanging drop vapor diffusion method at 4°C. 4 μL MinD-D40A (10 mg/ml) in 10 mM HEPES pH 7.0, 200mM NaCl,2 mM DTT,5 mM ATP, 10 mM MgSO4 was mixed with 2μl1.5–1.7 M (NH4)2SO4, 0.1M Hepes, pH 7.4. The crystals were subsequently cryoprotected by transfer to a solution of 60% sodium malonate (Holyoak et al., 2003)and cryocooled by immersion in liquid nitrogen.
Data Collection
Data on the cryocooled crystalsmaintained at 100K were collected at the Stanford Synchrotron Radiation Laboratory, Beamline 11-1, Menlo Park, CA. All data were integrated and scaled with HKL-2000 (Otwinowski & Minor, 1997).
Structure determination and refinement
The structure of MinD 10-D40A was solved using Se-methionine substituted enzyme and a three-wavelength MAD approach. Incorporation of Se-methionine was carried out using the methionine auxotroph B834(DE3) and near 100% incorporation was verified using mass spectrometry (data not shown). The programs SOLVE and RESOLVE (Terwilliger, 2003b, Terwilliger & Berendzen, 1999, Terwilliger, 2000, Terwilliger, 2003a)as incorporated into the Phenix program suite (Adams et al., 2010)were utilized for the determination of phases from the experimental diffraction data and initial automated model building. All ten of the selenium sites present in the dimeric structure were located and utilized by Solve/Resolve in the phasing solution. Despite the presence of a lattice translocation defect (LTD) in the crystals (see below and supplementary Figures 1–3) the phases were of excellent quality and allowed the automated building of 468 of the 516 residues with 343 side chains. Only in those regions that exhibited overlap with the LTD fraction was automated model building not successful (supplementary Figure 3). Phases were subsequently extended to a native dataset that diffracted to 2.4 Å. As mentioned above, despite an exhaustive search of crystallization conditions, all crystals of MinD 10-D40A suffer from a lattice translocation defect that results from the random translocation of some layers in the crystal by a fixed constant with respect to the other layers comprising the crystal. Crystal defects of this type were first described by Bragg and Howells in 1954 (Bragg and Howells, 1954) and the identification of this type of defect and the correction of diffraction data suffering from the LTD defect has been previously described (Hare et al., 2009, Tsai et al., 2009; Wang et al., 2005a, Wang et al., 2005b, Zhu et al., 2008). Reflection intensities for the MinD 10-D40A crystals were corrected by these published procedures using the program LTDcorrect generously provided by Dr. Jimin Wang, Center for Structural Biology, Yale University and an iterative modulation of the defect fraction. Determination of the final defect fraction was performed via inspection of the native Patterson maps for the elimination of the cross peak due to the lattice translocation defect fraction (Hare et al., 2009). In the case of the MinD diffraction data, this cross peak is observed at ~(0,0.3,0) and has a peak height that is ~27% of the origin peak height (supplementary Fig. 1). The final defect fraction was also verified by minimization of the Rfree value for a model refined against data corrected with varying defect fractions. Based upon Patterson map inspection and the Rfree values, the final defect fraction for the 2.4 Ådataset was determined to be 35%. While the correction procedure does not perfectly remove the contribution from the lattice defect fraction, a significant improvement in map quality in the overlap regions is observed allowing for complete model building, and a significant improvement in R and Rfree is observed. Prior to correction the R/Rfree is 34.7/39.9, while the corrected data results in a final R/Rfree of 30.6/34.0, After data correction, a model building and refinement were carried out using COOT() and Phenix. ATP, magnesium and water addition and validation were also performed in COOT and Phenix. NCS restraints between the two molecules of MinD were superimposed thoughout refinement. Refinement of the data against the final model results in R/Rfree values of 26.6/30.7. Final model statistics are presented in Table 3.
Table 3.
Crystallographic data and model statistics for the 2.4Å structure of the MinD 10-D40A-Mg-ATP complex.
| Data Statistics | Model Statistics | ||
|---|---|---|---|
| Wavelength (Å) | 0.9 | Resolution Range | 68.2 – 2.34 |
| Space group | P212121 | Completeness % (after LTD correction) | 96(92) |
| Unit cell | a = 83.7 Å | No. of ASU molecules | 2 |
| b = 86.6 Å | Rfree | 30.7(43.3) | |
| c = 110.7 Å | Rwork | 26.6(35.3) | |
| α = β = γ = 90.0° | Estimated coordinate error based on maximum likelihood, Å | 0.4 | |
| Resolution Limits, Å | 100–2.35 | Bond length rmsd, Å | 0.01 |
| No. of unique reflections | 33366 | Bond angle rmsd, | 1.35 |
| Completeness, % (all data) | 96.8 (94.6) | Ramachandran Statistics (preferred, allowed, outliers) % | 95, 4, 1 |
| Redundancy | 3.8 (3.6) | Average B factors (Å2) Protein | 37.4 |
| I/σ(I) | 10.4(2.0) | Water | 37.9 |
| Rmerge | 0.04 (0.34) | ATP | 27.3 |
| Magnesium ions | 25.7 | ||
| Refined Residues/Waters/ATP/Magnesium ions | 516/240/2/2 |
Supplementary Material
Acknowledgments
This research was supported by NIH grant GM27964 to JL. We thank Jessica Kuecker for helping with the mutagenesis. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We would like to thank Dr. Jimin Wang for providing us with his source code for the LTDcorrect program.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S. The Ccp4 Suite -Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over Chromosomes in E. coli. Mol Cell. 2005;18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J Bacteriol. 1993;175:1118–1125. doi: 10.1128/jb.175.4.1118-1125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg WL, Howells ER. X-Ray Diffraction by Imidazole Methaemoglobin. Acta Crystallographica. 1954;7:409–411. [Google Scholar]
- Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell SC, Anderson RE, Lowe J. Crystal structure of the bacterial cell division inhibitor MinC. EMBO J. 2001;20:2454–2461. doi: 10.1093/emboj/20.10.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell SC, Lowe J. Crystal structure of the bacterial cell division regulator MinD. FEBS Lett. 2001;492:160–165. doi: 10.1016/s0014-5793(01)02216-5. [DOI] [PubMed] [Google Scholar]
- Dajkovic A, Lan G, Sun SX, Wirtz D, Lutkenhaus J. MinC spatially controls bacterial cytokinesis by antagonizing thescaffolding function of FtsZ. Curr Biol. 2008;18:235–244. doi: 10.1016/j.cub.2008.01.042. [DOI] [PubMed] [Google Scholar]
- de Boer PA, Crossley RE, Rothfield LI. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell. 1989;56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- de Boer PA, Crossley RE, Rothfield LI. Roles of MinC and MinD in the site-specific septation block mediated by the MinCDE system of Escherichia coli. J Bacteriol. 1992;174:63–70. doi: 10.1128/jb.174.1.63-70.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fu X, Shih YL, Zhang Y, Rothfield LI. The MinE ring required for proper placement of the division site is a mobile structure that changes its cellular location during the Escherichia coli division cycle. Proc Natl Acad Sci U S A. 2001;98:980–985. doi: 10.1073/pnas.031549298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasriani H, Ducat T, Hart CT, Hafizi F, Chang N, Al-Baldawi A, Ayed SH, Lundstrom P, Dillon JA, Goto NK. Appropriation of the MinD protein-interaction motif by the dimeric interface of the bacterial cell division regulator MinE. Proc Natl Acad Sci U S A. 2010;107:18416–18421. doi: 10.1073/pnas.1007141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Meinhardt H, de Boer PA. Dynamic localization cycle of the cell division regulator MinE in Escherichia coli. EMBOJ. 2001;20:1563–1572. doi: 10.1093/emboj/20.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Cherepanov P, Wang J. Application of general formulas for the correction of a lattice-translocation defect in crystals of a lentiviral integrase in complex with LEDGF. Acta Crystallogr D. 2009;65:966–973. doi: 10.1107/S0907444909023695. [DOI] [PubMed] [Google Scholar]
- Hayashi I, Oyama T, Morikawa K. Structural and functional studies of MinD ATPase: implications for the molecular recognition of the bacterial cell division apparatus. EMBO J. 2001;20:1819–1828. doi: 10.1093/emboj/20.8.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester CM, Lutkenhaus J. Soj (ParA) DNA binding is mediated by conserved arginines and is essential for plasmid segregation. Proc Natl Acad Sci U S A. 2007;104:20326–20331. doi: 10.1073/pnas.0705196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoak T, Fenn TD, Wilson MA, Moulin AG, Ringe D, Petsko GA. Malonate: a versatile cryoprotectant and stabilizing solution for salt-grown macromolecular crystals. Acta Crystallogr D Biol Crystallogr. 2003;59:2356–2358. doi: 10.1107/s0907444903021784. [DOI] [PubMed] [Google Scholar]
- Hsieh CW, Lin TY, Lai HM, Lin CC, Hsieh TS, Shih YL. Direct MinE-membrane interaction contributes to the proper localization of MinDE in E. coli. Mol Microbiol. 2010;75:499–512. doi: 10.1111/j.1365-2958.2009.07006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Gogol EP, Lutkenhaus J. Dynamic assembly of MinD on phospholipid vesicles regulated by ATP and MinE. Proc Natl Acad Sci U S A. 2002;99:6761–6766. doi: 10.1073/pnas.102059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Lutkenhaus J. Topological regulation of cell division in Escherichia coli involves rapid pole to pole oscillation of the division inhibitor MinC under the control of MinD and MinE. Mol Microbiol. 1999;34:82–90. doi: 10.1046/j.1365-2958.1999.01575.x. [DOI] [PubMed] [Google Scholar]
- Hu Z, Lutkenhaus J. Analysis of MinC reveals two independent domains involved in interaction with MinD and FtsZ. J Bacteriol. 2000;182:3965–3971. doi: 10.1128/jb.182.14.3965-3971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Lutkenhaus J. Topological regulation of cell division in E. coli. spatiotemporal oscillation of MinD requires stimulation of its ATPase by MinE and phospholipid. Mol Cell. 2001;7:1337–1343. doi: 10.1016/s1097-2765(01)00273-8. [DOI] [PubMed] [Google Scholar]
- Hu Z, Lutkenhaus J. A conserved sequence at the C-terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol Microbiol. 2003;47:345–355. doi: 10.1046/j.1365-2958.2003.03321.x. [DOI] [PubMed] [Google Scholar]
- Hu Z, Saez C, Lutkenhaus J. Recruitment of MinC, an inhibitor of Z-ring formation, to the membrane in Escherichia coli: role of MinD and MinE. J Bacteriol. 2003;185:196–203. doi: 10.1128/JB.185.1.196-203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang GB, Song HE, Kim MK, Youn HS, Lee JG, An JY, Chun JS, Jeon H, Eom SH. Crystal structure of Helicobacter pylori MinE, a cell division topological specificity factor. Mol Microbiol. 2010;76:1222–1231. doi: 10.1111/j.1365-2958.2010.07160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Ullmann A, Ladant D. A bacterial two-hybrid system that exploits a cAMP signaling cascade in Escherichia coli. Methods Enzymol. 2000;328:59–73. doi: 10.1016/s0076-6879(00)28390-0. [DOI] [PubMed] [Google Scholar]
- Lackner LL, Raskin DM, de Boer PA. ATP-dependent interactions between Escherichia coli Min proteins and the phospholipid membrane in vitro. J Bacteriol. 2003;185:735–749. doi: 10.1128/JB.185.3.735-749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard TA, Butler PJ, Lowe J. Bacterial chromosome segregation: structure and DNA binding of the Soj dimer--a conserved biological switch. EMBO J. 2005;24:270–282. doi: 10.1038/sj.emboj.7600530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhaus J. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu Rev Biochem. 2007;76:539–562. doi: 10.1146/annurev.biochem.75.103004.142652. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J, Sundaramoorthy M. MinD and role of the deviant Walker A motif, dimerization and membrane binding in oscillation. Mol Microbiol. 2003;48:295–303. doi: 10.1046/j.1365-2958.2003.03427.x. [DOI] [PubMed] [Google Scholar]
- Ma L, King GF, Rothfield L. Positioning of the MinE binding site on the MinD surface suggests a plausible mechanism for activation of the Escherichia coli MinD ATPase during division site selection. Mol Microbiol. 2004;54:99–108. doi: 10.1111/j.1365-2958.2004.04265.x. [DOI] [PubMed] [Google Scholar]
- Ma LY, King G, Rothfield L. Mapping the MinE site involved in interaction with the MinD division site selection protein of Escherichia coli. J Bacteriol. 2003;185:4948–4955. doi: 10.1128/JB.185.16.4948-4955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskaya E, Fishov I, Fu X, Corbin BD, Margolin W, Dowhan W. Effects of phospholipid composition on MinD-membrane interactions in vitro and in vivo. J Biol Chem. 2003;278:22193–22198. doi: 10.1074/jbc.M302603200. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Cryst. 2005;D61:465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D. 2006a;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006b;39:109–111. [Google Scholar]
- Pichoff S, Alibaud L, Guedant A, Castanie MP, Bouche JP. An Escherichia coli gene (yaeO) suppresses temperature-sensitive mutations in essential genes by modulating Rho-dependent transcription termination. Mol Microbiol. 1998;29:859–869. doi: 10.1046/j.1365-2958.1998.00981.x. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Vollrath B, Touriol C, Bouche JP. Deletion analysis of gene minE which encodes the topological specificity factor of cell division in Escherichia coli. Mol Microbiol. 1995;18:321–329. doi: 10.1111/j.1365-2958.1995.mmi_18020321.x. [DOI] [PubMed] [Google Scholar]
- Ramirez-Arcos S, Greco V, Douglas H, Tessier D, Fan D, Szeto J, Wang J, Dillon JR. Conserved glycines in the C terminus of MinC proteins are implicated in their functionality as cell division inhibitors. J Bacteriol. 2004;186:2841–2855. doi: 10.1128/JB.186.9.2841-2855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin DM, de Boer PA. MinDE-dependent pole-to-pole oscillation of division inhibitor MinC in Escherichia coli. J Bacteriol. 1999a;181:6419–6424. doi: 10.1128/jb.181.20.6419-6424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin DM, de Boer PA. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc Natl Acad Sci U S A. 1999b;96:4971–4976. doi: 10.1073/pnas.96.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfield L, Taghbalout A, Shih YL. Spatial control of bacterial division-site placement. Nat Rev Microbiol. 2005;3:959–968. doi: 10.1038/nrmicro1290. [DOI] [PubMed] [Google Scholar]
- Sakai N, Yao M, Itou H, Watanabe N, Yumoto F, Tanokura M, Tanaka I. The three-dimensional structure of septum site-determining protein MinD from Pyrococcus horikoshii OT3 in complex with Mg-ADP. Structure. 2001;9:817–826. doi: 10.1016/s0969-2126(01)00638-4. [DOI] [PubMed] [Google Scholar]
- Schindelin H, Kisker C, Schlessman JL, Howard JB, Rees DC. Structure of ADP x AIF4(-)-stabilized nitrogenase complex and its implications for signal transduction. Nature. 1997;387:370–376. doi: 10.1038/387370a0. [DOI] [PubMed] [Google Scholar]
- Shen B, Lutkenhaus J. The conserved C-terminal tail of FtsZ is required for the septal localization and division inhibitory activity of MinC(C)/MinD. Mol Microbiol. 2009;72:410–424. doi: 10.1111/j.1365-2958.2009.06651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Lutkenhaus J. Examination of the interaction between FtsZ and MinCN in E. coli suggests how MinC disrupts Z rings. Mol Microbiol. 2010;75:1285–1298. doi: 10.1111/j.1365-2958.2010.07055.x. [DOI] [PubMed] [Google Scholar]
- Szeto TH, Rowland SL, Habrukowich CL, King GF. The MinD membrane targeting sequence is a transplantable lipid-binding helix. J Biol Chem. 2003;278:40050–40056. doi: 10.1074/jbc.M306876200. [DOI] [PubMed] [Google Scholar]
- Szeto TH, Rowland SL, Rothfield LI, King GF. Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea, and chloroplasts. Proc Natl Acad Sci U S A. 2002;99:15693–15698. doi: 10.1073/pnas.232590599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003a;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003b;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YS, Sawaya MR, Yeates TO. Analysis of lattice-translocation disorder in the layered hexagonal structure of carboxysome shell protein CsoS1C. Acta Crystallographica Section D-Biological Crystallography. 2009;65:980–988. doi: 10.1107/S0907444909025153. [DOI] [PubMed] [Google Scholar]
- Wang J, Kamtekar S, Berman AJ, Steitz TA. Correction of X-ray intensities from single crystals containing lattice-translocation defects. Acta Crystallogr D Biol Crystallogr. 2005a;61:67–74. doi: 10.1107/S0907444904026721. [DOI] [PubMed] [Google Scholar]
- Wang J, Rho SH, Park HH, Eom SH. Correction of X-ray intensities from an HslV-HslU co-crystal containing lattice-translocation defects. Acta Crystallogr D Biol Crystallogr. 2005b;61:932–941. doi: 10.1107/S0907444905009546. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Errington J. Coordination of cell division and chromosome segregation by a nucleoid occlusion protein in Bacillus subtilis. Cell. 2004;117:915–925. doi: 10.1016/j.cell.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Zhao CR, de Boer PA, Rothfield LI. Proper placement of the Escherichia coli division site requires two functions that are associated with different domains of the MinE protein. Proc Natl Acad Sci U S A. 1995;92:4313–4317. doi: 10.1073/pnas.92.10.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lutkenhaus J. Membrane binding by MinD involves insertion of hydrophobic residues within the C-terminal amphipathic helix into the bilayer. J Bacteriol. 2003;185:4326–4335. doi: 10.1128/JB.185.15.4326-4335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lutkenhaus J. MinC mutants deficient in MinD-and DicB-mediated cell division inhibition due to loss of interaction with MinD, DicB, or a septal component. J Bacteriol. 2005;187:2846–2857. doi: 10.1128/JB.187.8.2846-2857.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Schulze R, Cox S, Saez C, Hu Z, Lutkenhaus J. Analysis of MinD mutations reveals residues required for MinE stimulation of the MinD ATPase and residues required for MinC interaction. J Bacteriol. 2005;187:629–638. doi: 10.1128/JB.187.2.629-638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Xu X, Wilson IA. Structure determination of the 1918 H1N1 neuraminidase from a crystal with lattice-translocation defects. Acta Crystallogr D Biol Crystallogr. 2008;D64:843–850. doi: 10.1107/S0907444908016648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.