

Abstract
The tumor microenvironment plays an important role in modulating tumor progression. We earlier showed that S100A8/A9 proteins secreted by myeloid-derived suppressor cells (MDSC) present within tumors and metastatic sites promote an autocrine pathway for accumulation of MDSC. In a mouse model of colitis-associated colon cancer, we also showed that S100A8/A9 positive cells accumulate in all regions of dysplasia and adenoma. Here we present evidence that S100A8/A9 interact with RAGE and carboxylated glycans on colon tumor cells and promote activation of MAPK and NF-κB signaling pathways. Comparison of gene expression profiles of S100A8/A9-activated colon tumor cells versus unactivated cells led us to identify a small cohort of genes upregulated in activated cells, including Cxcl1, Ccl5 and Ccl7, Slc39a10, Lcn2, Zc3h12a, Enpp2 and other genes, whose products promote leukocyte recruitment, angiogenesis, tumor migration, wound healing, and formation of premetastatic niches in distal metastatic organs. Consistent with this observation, in murine colon tumor models we found that chemokines were up-regulated in tumors, and elevated in sera of tumor-bearing wild-type mice. Mice lacking S100A9 showed significantly reduced tumor incidence, growth and metastasis, reduced chemokine levels, and reduced infiltration of CD11b+Gr1+ cells within tumors and premetastatic organs. Studies using bone marrow chimeric mice revealed that S100A8/A9 expression on myeloid cells is essential for development of colon tumors. Our results thus reveal a novel role for myeloid-derived S100A8/A9 in activating specific downstream genes associated with tumorigenesis and in promoting tumor growth and metastasis.
Keywords: S100A8/A9, gene expression, colon tumors, RAGE, glycans
Introduction
S100A8 and S100A9 belong to a family of more than 20 low molecular weight intracellular EF-hand motif calcium-binding proteins found exclusively in vertebrates [1–3]. They are expressed predominantly by myeloid cells, including granulocytes, monocytes, MDSC and other immature cells of myeloid lineage [4–7]. Although the proteins are products of distinct genes, they are often co-expressed and function mainly as heterodimer of S100A8/A9 (calprotectin). Expression is down-regulated during macrophage and dendritic cell differentiation [6, 8, 9], but can be induced in epithelial cells, osteoclasts and keratinocytes [10]. When these intracellular proteins are released into the extracellular medium in response to cell damage or activation they become danger signals (Damage Associated Molecular Pattern molecules or DAMP), which alert the host of danger by triggering immune responses and activating repair mechanisms through interaction with pattern recognition receptors [11–15]. Elevated S100A8/A9 is the hallmark of inflammatory conditions such as rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, cystic fibrosis and psoriasis [4, 11, 16]. Critical roles for these proteins in endotoxin-induced lethality and systemic autoimmunity have recently been recognized [17, 18]. In addition to expression within inflammatory milieu, strong up-regulation of these proteins has also observed in many tumors, including gastric, colon, pancreatic, bladder, ovarian, thyroid, breast and skin cancers [10, 19], and it is becoming increasingly clear that S100A8/A9 not only serve as markers of immune cells within the tumor microenvironment, but that they may also have independent pathogenic roles in cancer progression.
S100A8/A9 exhibit concentration-dependent dichotomy of function in tumors. At high concentrations (80–100 μg/mL), S100A8/A9 exert apoptotic effects on tumor cells [20], while at low concentrations (<25 μg/mL) promote tumor cell growth [21, 22]. S100A8/A9 also stimulate tumor cell migration at low concentrations [23–27]. More recently, other patho-physiological roles for these proteins in tumors have also been identified. Studies from our laboratories and others show that S100A8/A9 regulate the accumulation of MDSC [6, 7]. MDSC are immature myeloid cells that expand during inflammation and in tumors, and are potent suppressors of T-cell mediated immune responses [28–30]. Cheng et al showed that tumor derived factors promote sustained STAT3 dependent upregulation of S100A9 in myeloid precursors which results in inhibition of differentiation to DC and accumulation of MDSC [6]. We showed that S100A8/A9 are not only synthesized and secreted by MDSC, but they also have binding sites for S100A8/A9, and activate intracellular signaling that promote their migration [7]. These findings strongly suggest that the S100A8/A9 proteins support an autocrine feedback loop that sustains accumulation of MDSC in tumors [31]. S100A8/A9 are also involved in early metastatic processes. Expression of S100A8/A9 in myeloid and endothelial cells in premetastatic organs in response to soluble factors such as VEGF, TGFβ and TNFα expressed by distal primary tumors promotes homing of tumor cells to premetastatic niches [24].
Recent studies argue for prominent roles for two pattern recognition receptors, TLR4 and RAGE, in S100A8/A9 mediated pathological effects. The interaction with TLR4 promotes endotoxin-induced lethality and the development of systemic autoimmunity [17, 18]. S100A8/A9 mediated signaling through TLR4 also promotes premetastatic niches in lungs [32]. Interaction of S100A8/A9 with RAGE has been shown to promote tumor growth, and MDSC migration [7, 21, 33]. However, which receptor and signaling pathways are preferentially activated is not understood, and may depend on the pathological settings, cell types involved, ligand concentrations and other factors. Distinct epitopes on RAGE and TLRs recognized by the ligands may also impart specificity. We found that S100A8/A9 bind to a subpopulation of RAGE expressing carboxylated glycans [22]. These glycans show restricted expression on myeloid, endothelial and tumor cells [34]. Inhibiting carboxylated glycan-dependent interactions using mAbGB3.1, an anti-glycan antibody, blocked T cell mediated colitis [35], colitis-associated colon cancer [22] and accumulation of MDSC in a 4T1 model of metastatic mammary tumor [7]. RAGE-deficient mice show reduced tumors in the CAC model [22] and inflammation-induced skin cancer model [33], suggesting that RAGE and carboxylated glycans form important components of tumor and stromal cells promoting molecular communications leading to myeloid accumulation and tumor growth.
Our previous observation of the presence of S100A8 and S100A9-positive myeloid cells in the microenvironment of colitis-induced colon tumors [22] prompted us to examine possible interactions of these proteins with tumor cells. We investigated S100A8/A9 binding to colon tumor cells, and subsequent activation of signaling pathways and gene expression in vitro. The results led us to further investigate the contribution of these proteins in vivo to colon tumor growth, establishment of pre-metastatic niches in distal organs, and promotion of metastasis in tumor-bearing mice. Our findings uncovered several pro-tumorigenic genes activated in tumor cells by S100A8/A9, strongly supporting a novel role of S100A8/A9 and myeloid cells in tumor progression.
Materials and Methods
Mouse and human S100A8 and S100A9 heterodimers and homodimers were purified as described [37] and rendered endotoxin-free. MC38 cells and MC38 cells stably expressing GFP were kind gifts from Drs. Ajit and Nissi Varki, University of California, San Diego. Caco-2 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium containing 100 U/mL penicillin, and 100 μg/mL streptomycin, glutamine, 10% FBS, 0.1mM non-essential amino acids and 1mM sodium pyruvate (and 1mg/mL G418 for MC38 cells expressing GFP).
Flow cytometric analysis
To detect surface expression of RAGE or carboxylated glycans, tumor cells were incubated with rabbit polyclonal anti-RAGE (raised against a peptide corresponding to amino acids 39-58 of human RAGE and which recognizes human, bovine and mouse RAGE) or anti-carboxylated glycan antibody mAbGB3.1 [34] in HBSS containing 1% BSA, followed by PE-conjugated secondary antibodies and analyzed by flow cytometry with a FACScan (Becton Dickinson, Mountain View, CA) equipped with CellQuest software, and gated by the side scatter and forward scatter filters.
Immunoprecipitation
MC38 or Caco-2 cells were isolated using PBS containing 5mM EDTA, washed with HBSS and incubated in HBSS medium containing purified mouse S100A8, S100A9 or S100A8/A9 for MC38 cells (at 1μg/million cells in 100μl final volume) or corresponding purified human proteins for Caco-2 cells for 1h at 4°C. Cells were washed with cold PBS twice and lysed in 20mM Tris-HCl, pH7.4 with 150mM sodium chloride, 0.5% NP-40 and protease inhibitors, and centrifuged at 10,000×g for 15 minutes to remove cell debris. Lysates were precleared with Protein G Sepharose beads for 1hr at 4°C, and S100 proteins was immunoprecipitated by using rabbit polyclonal antibodies against the respective proteins or an irrelevant control antibody overnight at 4°C and Protein G Sepharose beads. The beads were washed of unbound proteins, and immunoprecipitated proteins were analyzed for RAGE or TLR4 by electrophoresis and Western blots as described below.
siRNA treatment
A target specific 20–25 nt siRNA duplex designed to knock down expression of mouse S100A9 mRNA (calgranulin B siRNA from Santa Cruz biotechnology Inc., Santa Cruz, CA) was used to transfect MC38 cells using transfection reagents and protocol provided by the manufacturer. Gene silencing was confirmed by Western blots of whole cell lysates using anti-S100A9. Cells were used for immunoprecipitation 48h after transfection.
Signaling assays
MC38 or Caco-2 cells were treated with mouse or human S100A8, S100A9 or S100A8/A9 (10 μg/mL) for 0, 15, 30, 60 min, after overnight (16 hr) starvation in medium containing 0.1% serum. After indicated periods of incubation, cells were washed with cold PBS, harvested and lysed at 4°C, and lysates were analyzed by Western blot using respective MAPK or IκB antibodies as described below. For studies using mAbGB3.1 or anti-RAGE, following starvation, cells were preincubated for 2h with 20 μg/mL of mAbGB3.1 or rabbit polyclonal αRAGE prior to activation.
Electrophoresis and Western blots
Tumor cell lysates, immunoprecipitated proteins from tumor cells, or cell lysates from signaling assays were electrophoresed on denaturing and reducing 10% polyacrylamide gels, and transferred to nitrocellulose membranes. The blots were blocked with 10% dry skimmed milk. To detect RAGE or TLR4 in lysates or immunoprecipitates, blots were incubated with goat polyclonal anti-mouse S100A9 or anti-mouse RAGE (R&D Systems, Minneapolis, MN), rat monoclonal anti-human RAGE (kind gift from Novartis foundation) or rabbit polyclonal anti-TLR4 antibody (Imgenex Corporation, San Diego, CA) followed by respective peroxidase-conjugated secondary antibody. Phosphorylation of ERK1/ERK2, p38, SAPK/JNK and IκB in activated cell lysate proteins was detected using respective rabbit polyclonal or mouse monoclonal phospho-specific antibodies (Cell Signaling Technology, Danvers, MA) followed by peroxidase conjugated secondary antibodies. As loading controls, separate lanes with lysate proteins were incubated with rabbit polyclonal antibodies for total ERK1/ERK2, p38, SAPK/JNK, IκB or β-actin (Cell Signaling Technology, Danvers, MA) followed by peroxidase-conjugated secondary antibody. Bands were visualized using ECL detection system (GE Healthcare, Piscataway, NJ).
Measurement of NF-κB binding
Nuclear extracts isolated from MC38 cells or Caco-2 cells treated with respective S100 proteins were assayed for NF-κB-p65 binding activity using TransAM NF-κB assay kit (ActiveMotif, Carlsbad, CA) according to the manufacturer’s instructions.
Isolation of total RNA and gene expression profiling
Subconfluent cultures of MC38 cells were serum-starved for 16 hrs and activated with 10μg/mL S100A8/A9 for 6 hrs. Total RNA was extracted from unactivated or activated cells using an RNeasy kit (Qiagen, Valencia, CA) and biotinylated cRNA was prepared using the Illumina RNA Amplification Kit (Applied Biosystems/Ambion, Austin, TX). Hybridization to the Sentrix Mouse-6 Expression BeadChip containing >45,000 transcript-specific probe sequences/array (Illumina Incorporated, San Diego, CA) followed by washing and scanning were performed according to manufacturer’s instructions. The resulting images were analyzed using GenomeStudio (Illumina Incorporated, San Diego, CA) and GeneSpringGX11 (Agilent Technologies, Santa Clara, CA) image processing software. Experiments were performed in duplicates.
RQ-PCR Analysis of chemokine genes
SYBR Green oligonucleotide primers for the RQ-PCR analyses were designed using Primer 3 software. Mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. Following RT using Roche Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, Indianapolis, IN) PCR (45 cycles) was performed using Roche Lightcycler 480 as follows: pre-incubation at 95°C for 5 min, denaturation at 95°C for 10s, annealing at 60°C for 10s, and elongation at 72°C for 10s.
Quantitation of CXCL1
CXCL1 in culture supernatants and mouse sera were measured using a commercial ELISA kit (R&D Systems, Minneapolis, MN).
Mouse tumor models
S100A9 null mice were generated as described [38]. They were backcrossed to C57BL/6 mice for more than 10 generations. 6–8 week old S100A9 null mice, their wild type littermates or age-matched wild type mice were used for experiments. All animal protocols were approved by the Sanford-Burnham Medical Research Institute Animal Care and Use Committee and were in compliance with NIH policies.
CAC model
CAC was induced in separate groups of wild type or S100A9 null mice using AOM and DSS essentially as described [39] except that mice were subject to two cycles of DSS two weeks apart. Animals were constantly monitored for clinical signs of illness, and were sacrificed at the end of 2 wks, 6 wks, 12 wks or 20 wks after DSS. Blood samples were collected by retro-orbital bleeding prior to induction of disease and at time points as above. At each time point, colons were removed and fixed as “Swiss-rolls” in 4% buffered formalin. Stepwise sections were cut and stained with H&E. Colonic inflammation, dysplasia and neoplasms were graded based on described criteria [39].
CAC in bone marrow chimeric mice
Bone marrow cells were aseptically isolated from the femur and tibia of wild type or S100A9 null mice and each injected intravenously into either wild type or S100A9 null mice at 6 million cells per mouse. Recipient mice were lethally irradiated using a Gammacell 40 Exactor (9 Gy from a 137Cs source) before injection. Reconstitution of leukocyte populations was comparable in these groups. We confirmed successful engraftment by measurement of S100A8/A9 in serum. Mice were subjected to the AOM/DSS protocol 4 weeks later, sacrificed 12 weeks after DSS and blood and tissues were collected.
MC38 ectopic tumor model
To generate primary tumors, single cell suspensions of 1×106 MC38 cells in logarithmic phase of growth were injected subcutaneously into the flank of wild type or S100A9 null mice and allowed to grow for 10–20 days. To evaluate the role of carboxylated glycans, separate groups of wild type mice were treated with 10μg/gm of mAbGB3.1 weekly starting from 2 days prior to injection of tumor cells. Tumor growth was measured using calipers over the experimental period, and tumor volume estimated. Lungs, liver (primary metastatic organs) and tumors were frozen for further analysis. BM responses to tumor growth were evaluated as follows: Bone marrow cells were isolated from femur and tibia of mice and RBCs lysed according to standard protocols. Myeloid cells were stained with either differentiation marker CD11b and co-stained with mAbGB3.1, anti-RAGE or anti-Gr-1 (Ly6C and Ly6G, BD-Pharmingen, San Diego, CA). 7-AAD (Invitrogen, Carlsbad, CA) or propidium iodide (BD-Pharmingen, San Diego, CA) was included to identify dead cells and analyzed by flow cytometry. Peripheral blood hematology profile was obtained on EDTA samples using a VetScan HMII hematology system (Abaxis, Union City, CA).
Liver metastasis model
S100A9 null mice and age-matched wild type C57BL/6 mice were anesthetized using ip injection of avertin. Under aseptic conditions, a small longitudinal incision was made in the left upper flank to visualize the spleen, and 1× 106 MC38 cells in 50 μl of serum-free medium were injected under the spleen capsule with a 27-gauge needle. The spleen was then inserted back into the abdominal cavity and the peritoneum and abdominal walls were sutured with silk. Animals were sacrificed 2 wks later, and livers were isolated, fixed in buffered formalin and paraffin-embedded. Tumors were enumerated by visual inspection and by examining liver sections stained by H&E. Slides were scanned using an automatic high throughput ScanScope, viewed and tumor areas measured using Aperio software (Aperio, Vista, CA).
Immunosuppression assay
Spleen CD4+ T cells isolated from OTII transgenic mice (kindly provided by the Rickert and Bradley labs, Sanford-Burnham Medical Research Institute) were co-cultured in 48-well plates at 37°C in RPMI-1640 medium containing 10% FBS, penicillin, streptomycin and 2-ME) with CD11b+Gr1+ cells isolated from the spleens of tumor-bearing mice by MACS, at increasing ratios of MDSC: T cells in the presence of 10 μg/mL OVA peptide (OVA323–339). Cells were pulsed with 1 μCi [3H] thymidine/well on day 3, and 18 h later the cells were harvested and counted. Proliferation in the absence of MDSC was considered 100%.
Immunochemical analysis
Swiss rolls of colons from the AOM/DSS model were deparafinized and endogenous peroxidase blocked by incubating with 0.36% beta-glucose, 0.01% glucose oxidase and 0.013% sodium azide in PBS for 60 minutes at 37°C. The sections were stained with 1:50 dilution of anti-mouse CXCL1, or CCL7 (Santa Cruz Biotechnology, Santa Cruz, CA), followed by anti-goat peroxidase (1:100) and developed using DAB substrate. To characterize macrophage populations, frozen sections of tumors and pre-metastatic livers and lungs from the MC38 tumor model were stained with 1:50 dilution of anti-mouse CD11b and anti-mouse Gr-1 (BD-Pharmingen, San Diego, CA) followed by Alexa-488 and Alexa-594 conjugated secondary antibodies (Invitrogen, Carlsbad, CA), and cover-slipped with VectaShield DAPI mounting medium (Vector Laboratories, Burlingame, CA). MC38 tumor sections were also separately blocked with glucose oxidase as above and stained with anti-mouse S100A9 (R&D Systems, Minneapolis, MN), anti-mouse CD31 (BD-Pharmingen, San Diego, CA), or F4/80 (Invitrogen, Carlsbad, CA), followed by 1:100 dilution of respective peroxidase-conjugated secondary antibodies and developed with DAB substrate. Slides were scanned using an automatic high throughput ScanScope and viewed using Aperio software. They were also examined using an Inverted TE300 Nikon Wide Field and Fluorescence Microscope and images were acquired with a CCD SPOT RT Camera (Diagnostic Instruments Inc. Sterling Heights, MI) using SPOT advanced software.
Statistics
Statistical comparisons were performed using paired t test, and p values calculated using GraphPad Prism (San Diego, CA). Differences were considered statistically significant when p < 0.05.
Results
RAGE is the receptor for S100A8/A9 on colon tumor cells
We earlier found S100A8/A9+CD11b+Gr1+ myeloid progenitor cells in the tumor microenvironment of colitis-induced colon tumors, while they were absent from the normal adjacent colon tissue [22]. Our subsequent study in a 4T1 model of mammary carcinoma showed that these were MDSC [7]. Given that activated myeloid cells and MDSC can secrete S100A8/A9, we postulated that there might be crosstalk between myeloid cells and colon tumor cells, and that S100A8/A9 might interact with the tumor epithelium. In support of this, we earlier found that CT-26 mouse colon tumor cells expressed binding sites for S100A8/A9, and that a subpopulation of RAGE expressed on these cells is modified by carboxylated glycans [22]. The proteins have been earlier shown to bind to RAGE on human prostate and breast cancer cells [21, 23]. To further study cell-signaling pathways mediated by S100A8/A9 interactions, in this study we used MC38 colon tumor cells that are syngeneic to C57BL/6 strain, since S100A9 null mice and RAGE null mice have been backcrossed to this strain. We first confirmed cell surface expression of RAGE and carboxylated glycans on MC38 colon tumor cells by flow cytometry (Fig 1A). Examination of whole cell lysates also showed that MC38 cells constitutively express both RAGE and TLR4 (Fig 1B and 1C). To investigate whether RAGE or TLR4 provided binding sites for S100A8/A9 on MC38 colon tumor cells, we performed co-immunoprecipitation assays. Lysates from MC38 cells incubated with S100A8 or S100A9 homodimers or S100A8/A9 heterodimer were immunoprecipitated with anti-S100A8 or anti-S100A9 antibodies and the immunoprecipitated proteins were separated by electrophoresis and immunoblotted with anti-RAGE or anti-TLR4. We could only detect RAGE in lysates of cells incubated with S100A8 or S100A9 homodimers or the heterodimer, but did not detect TLR4 (Fig 1B and C). RAGE was also present in immunoprecipitates from cells not exposed to S100A8 or S100A9 suggesting that some RAGE on the surface of colon tumor cells could exist as a complex with S100A8/A9 possibly from endogenous sources. To confirm this, we undertook immunoprecipitation of MC38 cells in which endogenous S100A9 was silenced using specific siRNA (knockdown was confirmed by Western blots using anti-S100A9, supplement Fig S1), and found that RAGE in the immunoprecipitate was significantly reduced in S100A9-silenced cells compared to control cells (Fig 1B). No RAGE was detected in control immunoprecipitates obtained using an irrelevant control rabbit antibody. Subsequently, we also found RAGE in immunoprecipitates of Caco-2 human colon cancer cells incubated with human S100A8, S100A9 or the heterodimer, but not TLR4 (not shown). These results suggested that RAGE could be the predominant receptor for S100A8/A9 on colon tumor cells.
Figure 1.
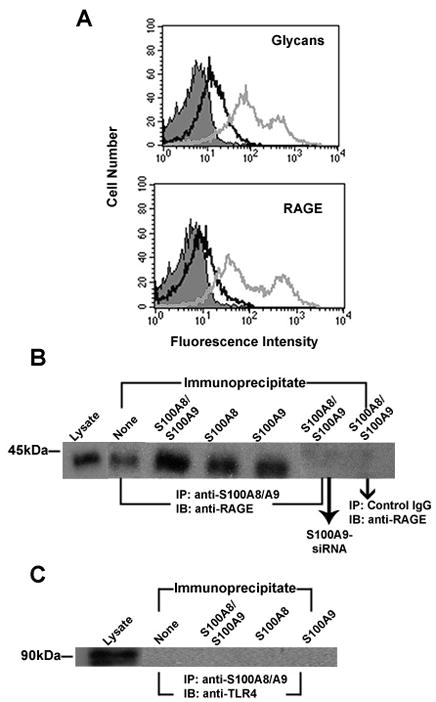
A. RAGE and mAbGB3.1 glycans are expressed on colon tumor cells. MC38 colon tumor cells in culture were analyzed for surface expression of mAbGB3.1 glycans and RAGE by flow cytometry. Cells were stained with mAbGB3.1 or anti-RAGE followed by FITC-conjugated anti-mouse or anti-rabbit Ig. Unstained cells (filled) and cells stained with secondary antibody alone (dark line) served as negative control. B and C. Receptor on colon tumor cells for S100A8/A9 identified by co-immunoprecipitation. MC38 cells were incubated with purified mouse S100A8/A9, S100A8 or S100A9 and bound proteins were immunoprecipitated with anti-S100A8 and/or anti-S100A9, or an irrelevant control rabbit IgG. To confirm potential interaction of RAGE with endogenous S100 proteins, immunoprecipitation was also performed using MC38 cells in which endogenous S100A9 was silenced using target-specific siRNA. Whole cell lysates and immunoprecipitated proteins were separated on SDS-PAGE gels, transferred and immunoblotted with anti-RAGE (B) or anti-TLR4 (C).
RAGE and carboxylated glycan-dependent binding of S100A8/A9 promotes MAPK and NF-κB signaling
Since RAGE ligation activates all members of the MAPK cascades, including the p38, ERK, and the JNK families, and promotes NF-κB activation [13], we next examined S100A8/A9 activated signaling pathways in colon tumor cells. We analyzed lysates of MC38 cells stimulated with low concentrations (10μg/mL) of S100A8/A9 for varying periods of time by immunoblotting using specific phospho-MAPK antibodies. This concentration was chosen for stimulation since our earlier studies and that of Ghavami et al show that S100A8/A9 at 1–10μg/mL induced tumor cell growth [21, 22]. Ghavami et al also showed that S100A8/A9 at 10μg/mL stimulated intracellular signaling in human breast cancer cell lines. In tumor-bearing mice, we found serum levels of S100A8/A9 in the order of ~500ng/mL [7]. However local concentrations in inflamed and tumor tissues could be several micrograms per mL, for example, as shown in exudates of carrageenan-induced inflammation, where S100A9 levels are reported to be ~1–4mg/mL [40].
S100A8/A9 stimulated rapid phosphorylation of ERK1/ERK2 and SAPK/JNK in MC38 cells within 15 min, with reduced but sustained phosphorylation up to 60 min, while there was no detectable phosphorylation of p38 (Fig 2A). This effect was different from RAGE-dependent S100A8/A9 induced activation triggered in human prostate and breast cancer cells [21, 23], where ERK1/ERK2 and p38 are activated, but not SAPK/JNK, suggesting that S100A8/A9 preferentially activate different MAPK pathways depending upon tumor cell type. Phosphorylation of ERK1/ERK2 was also seen in Caco-2 human colon tumor cells (Fig 2B). ERK1/ERK2 phosphorylation was inhibited in both MC38 and Caco-2 cells when they were pre-incubated with mAbGB3.1 or anti-RAGE, suggesting that the effects were mediated through RAGE and carboxylated glycans (Fig 2B). Since S100A8 and S100A9 homodimers also bind to RAGE, we investigated whether homodimers would individually stimulate activation. We found that S100A8 and S100A9 promoted a more delayed activation of ERK1/ERK2 in MC38 cells when compared to the heterodimers (Fig 2C).
Figure 2.
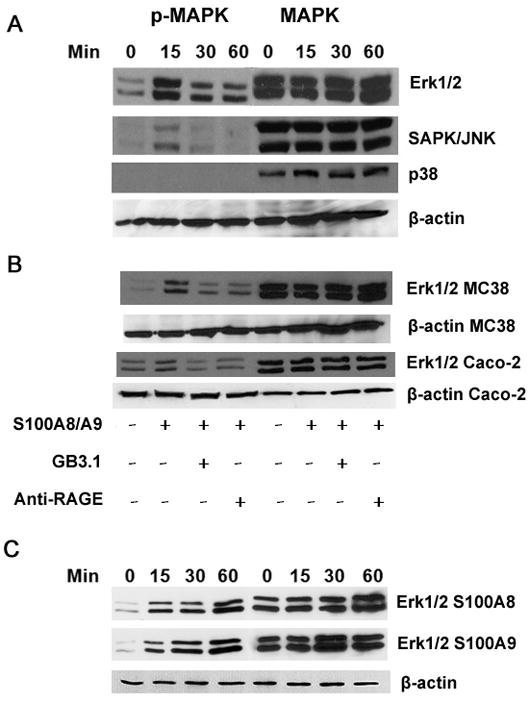
S100A8/A9 activates MAPK signaling pathways in colon tumor cells. (A) MC38 cells were incubated with purified endotoxin-free S100A8/A9 for different periods of time, and cell lysates were analyzed by Western blotting using respective antibodies against phosphorylated MAPK. As loading controls, separate lanes with lysate proteins were incubated with rabbit polyclonal antibodies for total ERK1/ERK2, p38, SAPK/JNK or β-actin B. MC38 or Caco-2 cells were incubated with purified endotoxin-free mouse or human S100A8/A9 for 15 minutes in the presence or absence of mAbGB3.1 or RAGE, and cell lysates were analyzed by Western blot using respective antibodies against phosphorylated or total ERK1/ERK2 or β-actin. C. MC38 cells were incubated with purifed mouse S100A8 or S100A9 homodimers for different periods of time, and cell lysates were analyzed by Western blot using phosphorylated or total ERK1/ERK2 or β-actin.
S100A8/A9 also induced phosphorylation of IκBα in MC38 and Caco-2 cells within 30min (Fig 3A), suggesting activation of NF-κB pathway. This was accompanied by IκBα degradation in Caco-2 cells evident by 30–60 min, but not in MC38 cells. Native colonic epithelial cells, and many intestinal epithelial cells, have delayed or incomplete IκBα degradation following stimulation, despite evidence for concomitant IκBα phosphorylation and NF-κB activation [41]. This has been attributed to impaired stimulation of an upstream IKK activator, and an altered steady state level of IκBα, which is dependent on rate of re-synthesis, and strength of the inducer [42].
Figure 3.
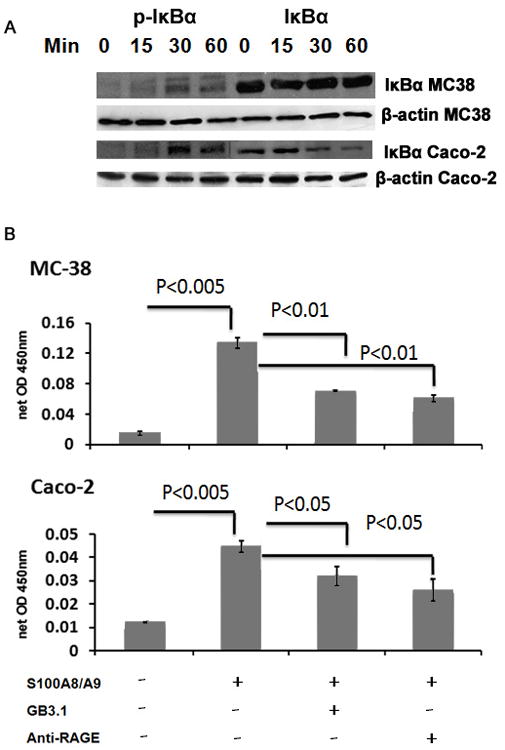
S100A8/A9 activates NF-κB in colon tumor cells. A. MC38 or Caco-2 cells were incubated with purified endotoxin-free mouse or human S100A8/A9 for different periods of time, and cell lysates were analyzed by Western blot using respective antibodies against phosphorylated or total IκBα or β–actin. B. MC38 or Caco-2 cells were treated with respective purified S100A8/A9 for 6 h in the presence or absence of mAbGB3.1 or anti-RAGE and nuclear extracts were analyzed for NF-κBp65 using DNA-binding ELISA.
Therefore, in order to further confirm S100A8/A9-induced activation of NF-κB pathway, we examined nuclear extracts isolated from MC38 and Caco-2 colon tumor cells and found considerable NF-κBp65 in the extracts from S100A8/A9 stimulated cells compared to unstimulated cells (Fig 3B). This effect was significantly inhibited when cells were pretreated with mAbGB3.1 or anti-RAGE IgG (Fig 3B). These results indicated that RAGE and carboxylated glycan-dependent binding of S100A8/A9 to RAGE on tumor cells resulted in activation of MAPK signaling pathways and nuclear translocation of NF-κB within the cells.
Stimulation of colon tumor cells by S100A8/A9 promotes pro-tumorigenic gene expression
MAPK pathways link extracellular signals with intracellular responses promoting cell growth, proliferation, differentiation and migration [43]. Activation of NF-κB dependent genetic programs in tumor cells and macrophages is critical for development of inflammation-based tumors [44–46]. We therefore reasoned that gene expression studies of activated colon tumor cells might provide valuable insight into the consequences of S100A8/A9 activation. To identify whether S100A8/A9 activated signaling pathways promoted gene transcription, we isolated total RNA from S100A8/A9 stimulated and unstimulated MC38 colon tumor cells and performed global gene expression analysis. Surprisingly, we found only a small cohort of 28 differentially expressed genes (p<0.01), of which 24 were up regulated and 4 were down regulated in stimulated cells compared to unstimulated cells (Fig 4A). Of these, the expression of 14 genes was induced two-fold or more when compared with non-stimulated cells. Most of the upregulated genes encoded proteins with either well-known or more recently recognized functions in leukocyte migration and recruitment, inflammation, proliferation of tumor cells, tumor invasion, angiogenesis and wound healing (Table 1). These genes included four chemokines (Cxcl1, Ccl5, Ccl2, Ccl7), NF-κB family member (Nfkbiz), zinc transporter (Slc39a10 or Zip10), lipocalin-2 (Lcn2), Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 (Enpp2), zinc finger protein (Zc3h12a), guanylate binding protein 4 (Gbp4), anti-leukoproteinase (Slpi), proliferin-2 (plf2) and apoptotic gene Fas. Many are up regulated in human tumors, their expression correlating with poor prognosis [47–52]. RQ-PCR analysis of chemokine genes Cxcl1, Ccl5 and Ccl7 further confirmed elevated transcript levels in S100A8/A9-stimulated cells (~6-fold increase in Cxcl1, 2-fold increase in Ccl7 and 2.5 fold increase in Ccl5) compared to unstimulated cells (Fig 4B). We next examined whether up-regulation of the Cxcl1 gene in cell cultures stimulated with S100A8/A9 led to corresponding secretion of CXCL1 (GROα or KC) protein. We analyzed culture supernatants of MC38 cells stimulated with S100A8/A9 for varying periods of time for CXCL1 by ELISA. We found that unstimulated MC38 cells constitutively secreted CXCL1 (Fig 4C). In addition, in line with the gene expression data, S100A8/A9 activation led to a 3-fold increase in secretion of CXCL1 compared to unstimulated cells within 6h of stimulation (Fig 4C). This S100A8/A9-induced secretion was significantly diminished when cells were pretreated with anti-RAGE or mAbGB3.1.
Figure 4.

A. Profile of differentially expressed genes from S100A8/A9-activated MC38 cells compared to unstimulated cells obtained by global gene expression analysis. Fold variation represent the mean of replicate (n=2) analysis. B. Cellular mRNA levels of chemokines. mRNA levels of Cxcl1, Ccl7 and Ccl5 were measured by RQ-PCR using RNA samples isolated from control MC38 cells, MC38 cells starved overnight and either untreated or stimulated with S100A8/A9. The expression values were normalized relative to GAPDH. The levels of mRNA in unstimulated or stimulated cells are shown relative to control non-starved MC38 cells considered as 100%. Each value is the mean level ± SD in two different samples for each condition, each sample assayed in duplicate. C. CXCL1 is secreted into medium from activated MC38 cultures. MC38 cultures were activated with S100A8/A9 in presence or absence of mAbGB3.1 or anti-RAGE, and CXCL1 in culture supernatants harvested at different time points was measured by ELISA.
Table 1.
Pro-tumorigenic genes activated in colon tumor cells by S100A8/A9
| Gene | Accession Number | Known Functions |
|---|---|---|
| Cxcl1 | NM_008176.1 | chemokine, promote angiogenesis, mobilization of leukocytes including MDSC |
| C3 | NM_009778.1 | complement component 3, mobilization of HSC into tumor stroma |
| Slc39a10 | NM_172653.2 | zinc transporter, belongs to Zip family (Zip10), upregulated in endometrial carcinoma, promote migration of breast tumor cells |
| Nfkbiz | NM_030612 | IκB family, IL-6 activator, modulates NF-κb transcription |
| Lcn2 | NM_008491.1 | lipocalin 2, upregulated in inflammation, has pro and anti-tumor effects |
| Fas | NM_007987.1 | apoptosis, TNF family member with the death domain |
| Zc3h12a | NM_153159.1 | zinc finger family, RNase, controls stability of inflammatory genes, mediates CCL2 induced angiogenesis, macrophage activation |
| Enpp2 | NM_015744.1 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 2, autotaxin, promotes tumor cell migration (invadopodia), angiogenesis, and its expression is upregulated in several kinds of carcinomas |
| Ccl5 | NM_013653.1 | chemokine, stimulate angiogenesis |
| Ccl2 | NM_013653.2 | chemokine, potent stimulator of angiogenesis |
| Ccl7 | NM_013653.3 | chemokine, promote macrophage infiltration into tumors |
| Gbp4 | NM_018734.2 | guanylate binding protein 4; gene family induced during macrophage activation, IFN gamma inducible, GTPase |
| Slpi | NM_011414.1 | secretory leucocyte peptidase inhibitor, upregulated in tumors, secreted inhibitor which protects epithelial tissues from serine proteases, implicated in wound healing |
| Plf2 | NM_011118.1 | proliferin-2, HSC growth factor, associated with angiogenesis and wound healing |
Mouse models of colon tumors
Our earlier studies and those of Cheng et al have shown that S100A8 and S100A9 play a critical role in tumor growth and metastasis through increased accumulation MDSC [6, 7]. A role in growth and migration of tumor cells has also been described by many studies [21–27]. However, molecular signature of S100A8/A9 activated cells revealed by gene expression analysis as shown above strongly implied that RAGE and carboxylated-glycan dependent activation of tumor cells by S100A8/A9 differentially altered expression of genes whose products could mediate many pro-tumorigenic effects, predicting that S100A8/A9 could have other novel roles in tumor progression. To further elucidate pro-tumorigenic and pro-metastatic roles of S100A8/A9 in vivo, we subjected S100A9 null mice to different colon tumor models and compared responses to those observed in wild type mice. Deletion of S100A9 in mice leads to a complete lack of S100A8 and a functional S100A8/A9 complex in cells of peripheral blood and bone marrow, despite normal mRNA levels of S100A8, suggesting that S100A9 expression is important for the stability of S100A8 protein [38, 53]. Induction of tumors in S100A9 null mice thus provided us an excellent opportunity to test the importance of both proteins in tumorigenesis and malignancy.
Reduced tumor incidence and chemokine expression in S100A9 null mice in the CAC model
We induced in CAC in S100A9 null mice and wild type mice by azoxymethane (AOM) injection followed by two cycles of dextran sodium sulfate (DSS) treatment as described earlier [22]. DSS causes epithelial damage and triggers an innate immune response that recruits activated macrophages and induces an acute colitis evident within 2 weeks after DSS. This initial response progresses to chronic inflammation by about 6 weeks by activation of adaptive immune responses. Wild type mice develop dysplasia, adenoma and adenocarcinoma within 12–20 weeks of combined administration of AOM and DSS [44, 54–56] with 100% penetrance. Both S100A9 null mice and age-matched C57BL/6 wild type mice lost up to 10% of body weight after DSS treatment before recovery, and colons showed inflammation in both S100A9 null mice and wild type mice (not shown), suggesting that S100A8/A9 do not contribute to DSS-induced colon inflammation. However, there was a significant reduction in tumor incidence in S100A9 null mice at 12 and 20 wks after AOM/DSS (Fig 5A and 5B). In contrast, all the wild type mice developed adenomas (5–8 tumors per mouse) by 12–20 wks, with a few adenocarcinomas by 20 wks, suggesting that S100A8/A9 could exert independent roles in the tumorigenic phase of CAC.
Figure 5.

A. Representative H&E stained colon “Swiss rolls” obtained from wild type and S100A9 null mice subjected to the AOM-DSS protocol 12 weeks after initiation (10× magnification). Arrows indicate regions of dysplasia and adenoma. B. Colonic tumor incidence in S100A9 null and wild type mice 12 and 20 weeks after AOM-DSS (n=5 mice per group per time point). C. Representative sections of tumor regions and normal adjacent tissue in colons of wild type and S100A9 null mice subjected to the AOM-DSS protocol 20 weeks after initiation stained for CXCL1 or CCL7 (400×) D. CXCL1 in sera of wild type and S100A9 null mice before and 12 weeks after AOM-DSS (n=5 mice). E. Colonic tumor incidence in bone marrow chimeric mice 12 weeks after AOM-DSS (n=4 recipient mice per group).
We had earlier shown that S100A8/A9+ and CD11b/Gr1+ myeloid cells infiltrate all regions of dysplasia and tumors in this model [22]. Since chemokines are upregulated in colon tumor cells in vitro in response to S100A8/A9, we examined whether chemokines CXCL1 and CCL7 were also induced in the colon tumors in this model. We found moderate to intense staining for CXCL1 and CCL7 in most epithelial and some stromal cells in tumor regions but not in adjacent normal tissues. Staining was reduced in tumor regions from S100A9 null mice (Fig 5C). We also measured serum CXCL1 as a marker of S100A8/A9-induced activation of tumor cells. Serum CXCL1 was elevated 2–3 fold compared to pre-tumor levels in all wild type mice, at 12 weeks of disease initiation, when the tumors are not invasive, while CXCL1 levels were minimally altered in tumor-bearing S100A9 null mice (Fig 5D). This further substantiated our in vitro findings that S100A9/A9 promoted expression of pro-tumorigenic downstream effectors in early tumors.
Bone marrow derived cells in the tumor microenvironment contribute S100A8/A9
Epithelial cells can express S100A8/A9. To investigate whether S100A8/A9 expressed by tumor cells or infiltrating bone-marrow derived myeloid cells within the tumor microenvironment is required for disease progression, we evaluated tumorigenesis in chimeric mice after bone marrow transplantation. Bone marrow cells from wild type or S100A9 null mice were injected into lethally irradiated groups of recipient wild type or S100A9 null mice. Chimerism was confirmed by measurement of S100A8/A9 in serum (not shown). Mice were subjected to the AOM/DSS protocol 4 weeks later and sacrificed 12 weeks after initiation of disease. Tumor incidence in wild type mice reconstituted with wild type bone marrow cells (WT→WT) and S100A9 null mice reconstituted with S100A9 null bone marrow cells (S100A9 null→S100A9 null) was similar to responses seen earlier in wild type mice and S100A9 null mice (Fig 5E). However, S100A9 null mice reconstituted with bone marrow cells from wild type mice (WT→S100A9 null) showed higher incidence of tumors, compared to wild type mice reconstituted with bone marrow cells from S100A9 null (S100A9→WT) mice. This strongly indicated that S100A8/A9 expressed by bone marrow derived cells in the tumor microenvironment is essential for the promotion of tumorigenesis.
Reduced MC38 colon tumor growth and formation of premetastatic niches in S100A9 null mice
The CAC model described above allows us to understand the role of S100A8/A9 in early events in colon carcinogenesis under the setting of inflammation. However, the tumors rarely became invasive and malignant within the experimental period of 20 weeks. Therefore in order to define the role of S100A8/A9 and its downstream effectors in tumor invasion, myeloid cell migration, and formation of premetastatic niches in distal organs, we used a primary ectopic tumor model using MC38 colon tumor cells. We followed tumor growth in wild type and S100A9 null mice injected s.c. with 1 × 106 MC38 cells. Tumors were evident in all wild type mice (n=10) by 7–10 days after injection and continued to grow until 21 days when the mice were sacrificed. When S100A9 null mice were challenged with MC38 cells, tumors were significantly smaller in 6 out of 12 S100A9 null mice at 21 days after injection (Fig 6A). In addition, tumors were completely rejected in 2 out of the remaining six S100A9 null mice. Collectively, 8 out of 12 S100A9 null mice (67%) examined showed minimal tumor growth or tumor rejection. Tumor growth in wild type mice was accompanied by elevated serum CXCL1, but not in S100A9 null mice (Fig 6B).
Figure 6.

A. Tumor volumes of ectopic MC38 tumors in wild type (n=10) and S100A9 null mice (n=12) 3 weeks after sc injection of 1×106 cells. 6 out of 12 S100A9 null mice showed significantly reduced tumor growth shown here. In addition, 2 of the remaining six S100A9 null mice completely rejected the tumors. B. CXCL1 in sera of wild type and S100A9 null mice before and 3 weeks after MC38 tumor growth. C. Quantitation of CD11b+ cells co-staining with Gr1 or GB3.1 glycans or RAGE from bone marrow of MC38 tumor-bearing wild type mice D. Tumors were examined for infiltrating macrophages and tumor endothelial cells by immunochemical staining for S100A9+ cells, CD11b+Gr1+ cells (merged images of Alexa-488 stained CD11b+ cells and Alexa-594 stained Gr1+ cells showing double positive yellow cells), and CD31+ and F4/80+ cells (200×). E. Representative sections showing CD11b+Gr1+ cells in premetastatic livers of tumor-free and tumor-bearing mice. F. Quantitation of average number of CD11b+Gr1+ cells in premetastatic livers in 3 high power fields.
Since CXCL1 promotes MDSC and other myeloid cell recruitment within tumors and premetastatic organs, we measured bone marrow responses to the ectopic MC38 tumors and found significantly increased CD11b+ populations co-expressing Gr1, carboxylated glycans (as stained by mAbGB3.1) or RAGE, in all of the tumor-bearing wild type mice at 18–21 days after transplantation compared to tumor-free control mice (Fig 6C). CD11b+Gr1+ cells were also found within the tumors, and substantially reduced in tumors from S100A9 null mice, while the levels of F4/80+ macrophages and CD31+ endothelial cells were unchanged, suggesting that S100A8/A9 do not alter intra-tumoral infiltration of other tumor-associated, angiogenic macrophages, while affecting infiltration of CD11b+Gr1+ cells (Fig 6D). To confirm that these were in fact MDSC, we isolated CD11b+Gr1+ cells from the spleens of MC38 tumor-bearing mice and co-cultured them at varying ratios with CD4+ T cells from OTII transgenic mice and OVA peptide (OVA323–339) and measured T cell proliferation by uptake of 3H-thymidine. With increasing ratios of MDSC: T cells, splenic CD11b+Gr1+ cells from tumor-bearing mice progressively reduced T cell proliferation (Supplement Fig S2).
A more recent study by Connolly et al shows that the expansion of CD11b+Gr1+ MDSC in premetastatic sites in liver in response to intra-abdominal tumors is contingent upon the expression of CXCL1 [57]. In keeping with observation, and with elevated serum CXCL1 levels, we found markedly increased accumulation of CD11b+/Gr1+ cells in the premetastatic lungs and liver of tumor-bearing mice 21 days after tumor initiation, compared to tumor-free mice (Figure 6E shown for liver). To exclude the possibility that any micrometastasis of MC38 cells in liver and lungs induced the accumulation of CD11b+Gr1+ cells, we injected separate groups of wild type mice with MC38 cells stably expressing GFP. No GFP+ cells were detected in lungs or livers at 21 days after tumor initiation (not shown). MC38 tumor challenge in mice lacking S100A8/A9, or wild type mice treated with mAbGB3.1 significantly diminished accumulation of CD11b+Gr1+ positive niches in liver (Fig 6E and 6F). This finding is consistent with the studies of Hiratsuka et al who showed S100A8/A9 promote the formation of premetastatic niches in distal organs in response to primary tumors [24].
The liver is the primary site for colorectal carcinoma metastasis. Since the CAC model and ectopic MC38 tumor models did not show any evidence of distal metastasis, we chose a liver metastasis model to further understand the role of S100A8/A9 and S100A8/A9-induced proteins in promoting metastasis. We injected S100A9 null mice and age-matched C57BL/6 wild type mice with 1 × 106 MC38 cells by the intra-splenic route. MC38 cells generated tumors within the spleen (primary) and in the liver (metastasis). Multiple hepatic tumor nodules, detectable by gross inspection, were evident by 2 wks. Livers were isolated and the incidence of hepatic metastases was evaluated. Livers from S100A9 null mice showed significantly reduced numbers of metastatic tumors, smaller tumor foci, and decreased tumor-occupied area compared to livers from tumor-bearing wild type mice (Fig 7). These results further indicate that S100A8/A9 play a critical role in promoting metastasis.
Figure 7.

S100A9 null mice exhibit reduced metastatic tumors. A. Representative livers from wild type and S100A9 null mice 2 wks after intrasplenic injection of MC38 cells. Arrows indicate visible tumors B. Histology of representative livers stained by hematoxylin and eosin (25× magnification). C. Numbers of metastatic nodules in the livers, and total tumor area represented as % of liver tissue, 2 wks after intrasplenic injection of MC38 cells (wild type (n=6) and S100A9 null mice (n=5).
Taken together, our observations strongly support the notion that S100A8/A9 activate signaling pathways that promote tumor growth and metastasis by inducing expression of multiple downstream pro-tumorigenic effector proteins, and suggest that strategies that target S100A8/A9 in the tumor microenvironment could provide effective therapeutic approaches to treating patients with colorectal cancer.
Discussion
Cells of the tumor microenvironment contribute to tumor growth and metastasis through complex interactions with tumor cells [58–60]. The presence of S100A8/A9 in many human tumors, along with recent recognition of their roles in tumorigenesis and MDSC accumulation, warrants a more detailed understanding of the molecular mechanisms involved in their interactions within the tumor microenvironment. Our earlier studies provided evidence that S100A8/A9 promote accumulation of MDSC [7]. Here we show that S100A8/A9 expressed by myeloid cells interacts with RAGE and carboxylated glycans expressed on colon tumor cells promoting intracellular signaling pathways and pro-tumorigenic gene expression, and that S100A9 null mice show reduced tumor growth and metastasis, thus defining yet another novel role for S100A8/A9 in tumor progression.
Although many studies implicate both TLR4 and RAGE in S100A8/A9 mediated pathological effects, the relative contribution of each receptor to downstream effects is unknown. Based on our earlier studies and immunoprecipitation results shown here, we surmise that RAGE is the principal receptor of S100A8/A9 on tumor cells, and this is consistent with the finding that S100A8/A9-mediated responses in human tumor cells involves RAGE [21, 23]. However, studies implicating S100A8/A9 in endotoxin-induced lethality and systemic autoimmunity show that TLR4, rather than RAGE, could play a more prominent role as receptor for these ligands on macrophages [17, 18]. This suggests that cell types and pathological settings could dictate which receptor predominates. Besides cell types, the differential effects could also be mediated by carboxylated glycans, which are expressed on RAGE and not on TLR4. Also, TLR4 is only functional active in the presence of myeloid differentiation factor-2 (MD2) protein for both LPS and S100A9-mediated interactions on macrophages [17]. TLR4 expressed on MC38 cells is functional, since it has been shown to respond to LPS, as determined by LPS-induced expression of IL-6 by MC38 cells, which is reduced upon TLR4 gene silencing [61].
The activation of RAGE-mediated signaling pathways could also depend on the tumor cell involved. We found that S100A8/A9 induces RAGE and carboxylated-glycan dependent phosphorylation of ERK1/ERK2 and SAPK/JNK MAPK in colon tumor cells, but we did not observe significant phosphorylation for p38. In contrast, in human prostate and breast cancer cells, S100A8/A9 activate p38, but not SAPK/JNK [21, 23]. In this context, it is interesting that S100A8/A9 activate SAPK/JNK in macrophages through TLR-dependent pathway [62]. In support of our finding, it was recently shown that treatment of tumor cells with a JNK inhibitor blocked RAGE ligand-induced cellular invasion [63]. p38 and SAPK/JNK MAPK proteins are known to function in cell context and cell type-specific manner to co-ordinate signaling pathways mediating tumor cell proliferation, survival and migration, and may even exert antagonistic effects, depending on signal duration and cross-talk with other signaling pathways [64]. Their expression is altered in many human tumors, and it is therefore important to consider the tumor type before modulations of the pathways are attempted for therapeutics.
S100A8/A9 binding to colon tumor cells stimulates RAGE and carboxylated glycan-dependent activation of NF-κB pathway. NF-κB provides a critical link between inflammation and cancer [44, 45]. Since S100A8/A9 binding to cells stimulates NF-κB transcription, and proximal promoter regions of S100A8 and S100A9 have binding sites for NF-κB [10], ligation of cell surface receptors by S100A8/A9 in inflammation could lead to a positive feedback loop and sustained cellular activation promoting tumor development. In support of this, S100A8/A9 proteins have been identified as novel NF-κB target genes in hepatic carcinoma cells during inflammation-mediated liver carcinogenesis [65].
Our gene expression analysis revealed for the first time the molecular signature of S100A8/A9 activation in tumor cells. Some of the genes that are activated represent known NF-κB target genes, and are directly associated with tumorigenesis. Most notable are the chemokines CXCL1 (GROα or KC), CCL2 (MCP-1), CCL5 (RANTES) and CCL7 (MCP-3). While many previous studies have focused on the role of chemokines in immune responses, recent studies show that they also promote chemotaxis and leukocyte recruitment to tumors, angiogenesis, bidirectional cross-talk between tumor cells and tumor-associated fibroblasts, tumor invasion, and migration of tumor cells to distal organs [48, 49, 66]. CXCL1, CCL2 and CCL5 have been also identified in human colorectal tumors and expression correlates with poor prognosis [47, 67, 68]. CCL2 is a crucial mediator of CAC in mice [69] and CXCL1 mediates pro-angiogenic effects of PGE2 in colorectal cancer [70]. More recent studies shows that the expansion of CD11b+Gr1+ MDSC in premetastatic sites in liver in response to intra-abdominal tumors is contingent upon the expression of CXCL1, and self-seeding of circulating tumor cells promote tumor growth, angiogenesis and tumor recruitment through mediators such as CXCL1 [57, 71]. We found that CXCL1 is secreted by colon tumor cells in response to interaction with S100A8/A9, and is up-regulated in colon tumors. It is elevated in sera of not only the MC38 tumor-bearing mice, but also in mice with colitis-induced tumors within 12 weeks of AOM-DSS treatment, at which time point the tumors are early, well-contained and non-invasive, suggesting that CXCL1 could provide an early marker of metastatic tumor progression. We also found that tumor-bearing mice lacking S100A8/A9 show marginal or no elevation of CXCL1 and significantly diminished CD11b+Gr1+ cells in tumors and premetastatic niches in liver and lungs.
In addition, we found other new S100A8/A9-induced genes that are implicated in tumor progression. Zc3h12a, which encodes a zinc finger protein, is an RNase and a downstream effector of CCL2-mediated angiogenesis [72]. Enpp2 encodes autotaxin, a lysophospholipase D enzyme that hydrolyzes extracellular lysophospholipids to produce lysophosphatidic acid (LPA). LPA receptors are overexpressed in many tumors [51] and LPA2 receptor knock-out mice show markedly reduced tumor incidence and progression of colon adenocarcinomas associated with reduced tumor-infiltrating macrophages [73]. More recently, Liu et al showed a causal link between autotaxin-LPA receptor signaling and mammary tumor progression [74]. Autotaxin is also implicated in the formation of invadopodia by various human cancer cell types [75]. Slpi encodes a secretory leukocyte peptidase inhibitor that is upregulated in tumors. It protects epithelial tissues from serine proteases and has been implicated in wound healing [76]. Neutrophil gelatinase-associated lipocalin-2 encoded by NF-κB inducible Lcn2 gene, has paradoxically both pro and anti-tumor effects [52]. Plf2 encodes proliferin-2, a hematopoietic stem cell growth factor associated with angiogenesis and wound healing [77, 78].
The induction of these downstream effector genes in tumors would thus greatly amplify tumor growth, migration and invasion, induction of myeloid cells, and metastatic progression promoted by S100A8/A9. Consistent with this, we found that S100A9 null mice showed markedly reduced tumor incidence and progression of AOM-DSS induced colon adenomas, and reduced ectopic MC38 tumor growth and tumor metastasis. Since CD11b+Gr1+ cells in the tumor-bearing mice are MDSC, it is likely that reduced tumor growth and metastasis in S100A9 null mice are due to combined effects of lack of immune suppression and reduced induction of pro-tumorigenic genes. The effects of S100A8/A9 in tumorigenesis in the AOM-DSS model could be mediated through RAGE and carboxylated glycans, since we earlier showed that RAGE null mice and wild type mice receiving mAbGB3.1 treatment showed reduced AOM-DSS induced tumor incidence. However, the contribution of TLR4 in S100A8/A9 mediated effects in tumors cannot be overlooked, since TLR4 null mice are protected markedly from CAC [79] and TLR4 mediates the formation of premetastatic niches promoted by S100A8/A9 [32].
Colorectal cancer is one of the most common malignancies affecting both sexes and a common cause of mortality worldwide. Each year about 50,000 people die from the disease in the US alone. Our present findings, along with earlier studies, show that S100A8/A9 function at multiple stages in disease progression. S100A8/A9, their receptors and signaling pathways therefore provide important targets for development of pharmacological interventions and for the identification of early-stage disease biomarkers.
Supplementary Material
Acknowledgments
We thank Dr. Hudson Freeze for his long-standing support and collaboration, and for his critical reading of the manuscript. We also thank Dr. Dirk Foell and Dr. Johannes Roth at the University of Muenster, Germany for their collaboration. We gratefully acknowledge the invaluable help from Adriana Charbono, Buddy Charbono and Larkin Slater with the animal experiments and colony maintenance, Robbin Newlin and Gia Garcia for histology expertise, Kang Liu and Jian Xing for microarray and RQ-PCR analysis, and Harish Khandrika for illustrations. This work was generously supported by National Institutes of Health grant R21-CA127780 (to GS).
Abbreviations
- AOM
azoxymethane
- CAC
colitis-associated cancer
- DSS
dextran sulfate sodium
- DAMP
Damage Associated Molecular Pattern
- ERK1/ERK2
Extracellular signalregulated kinases 1 and 2
- MAPK
mitogen-activated protein kinase
- MDSC
myeloid derived suppressor cells
- NF-κB
nuclear factor kappa B
- RAGE
Receptor for Advanced Glycation End Products
- SAPK/JNK
Stress-activated protein kinase/c-Jun N-terminal kinase
- TLR4
Toll-like Receptor 4
References
- 1.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. International Journal of Biochemistry & Cell Biology. 2001;33(7):637–68. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 2.Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002;7:d1356–68. doi: 10.2741/A846. [DOI] [PubMed] [Google Scholar]
- 3.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322(4):1111–22. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 4.Roth J, et al. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003;24(4):155–8. doi: 10.1016/s1471-4906(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 5.Goyette J, Geczy CL. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids. 2010 doi: 10.1007/s00726-010-0528-0. [DOI] [PubMed] [Google Scholar]
- 6.Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha P, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181(7):4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagasse E, Clerc RG. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol. 1988;8(6):2402–10. doi: 10.1128/mcb.8.6.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odink K, et al. Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature. 1987;330(6143):80–2. doi: 10.1038/330080a0. [DOI] [PubMed] [Google Scholar]
- 10.Gebhardt C, et al. S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol. 2006;72(11):1622–31. doi: 10.1016/j.bcp.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 11.Foell D, et al. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81(1):28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 12.Srikrishna G, Freeze HH. Endogenous Damage Associated Molecular Pattern (DAMP) molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11(7):615–28. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donato R. RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med. 2007;7(8):711–24. doi: 10.2174/156652407783220688. [DOI] [PubMed] [Google Scholar]
- 14.Leclerc E, et al. Binding of S100 proteins to RAGE: An update. Biochim Biophys Acta. 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 15.Ehrchen JM, et al. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86(3):557–66. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 16.Roth J, Goebeler M, Sorg C. S100A8 and S100A9 in inflammatory diseases. Lancet. 2001;357(9261):1041. doi: 10.1016/S0140-6736(05)71610-X. [DOI] [PubMed] [Google Scholar]
- 17.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–9. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 18.Loser K, et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med. 2010;16(6):713–7. doi: 10.1038/nm.2150. [DOI] [PubMed] [Google Scholar]
- 19.Salama I, et al. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008;34(4):357–64. doi: 10.1016/j.ejso.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Ghavami S, et al. Mechanism of apoptosis induced by S100A8/A9 in colon cancer cell lines: the role of ROS and the effect of metal ions. J Leukoc Biol. 2004;76(1):169–75. doi: 10.1189/jlb.0903435. [DOI] [PubMed] [Google Scholar]
- 21.Ghavami S, et al. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol. 2008;83(6):1484–92. doi: 10.1189/jlb.0607397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turovskaya O, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29(10):2035–43. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hermani A, et al. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006;312(2):184–97. doi: 10.1016/j.yexcr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Hiratsuka S, et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–75. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 25.Moon A, et al. Global gene expression profiling unveils S100A8/A9 as candidate markers in H-ras-mediated human breast epithelial cell invasion. Mol Cancer Res. 2008;6(10):1544–53. doi: 10.1158/1541-7786.MCR-08-0189. [DOI] [PubMed] [Google Scholar]
- 26.Saha A, et al. Lack of an endogenous anti-inflammatory protein in mice enhances colonization of B16F10 melanoma cells in the lungs. J Biol Chem. 2010;285(14):10822–31. doi: 10.1074/jbc.M109.083550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ang CW, et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis. 2010 doi: 10.1093/carcin/bgq137. [DOI] [PubMed] [Google Scholar]
- 28.Dolcetti L, et al. Myeloid-derived suppressor cell role in tumor-related inflammation. Cancer Lett. 2008;267(2):216–25. doi: 10.1016/j.canlet.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrand-Rosenberg S. Cancer and complement. Nat Biotechnol. 2008;26(12):1348–9. doi: 10.1038/nbt1208-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiratsuka S, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349–55. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 33.Gebhardt C, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205(2):275–85. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srikrishna G, et al. A novel anionic modification of N-glycans on mammalian endothelial cells is recognized by activated neutrophils and modulates acute inflammatory responses. Journal of Immunology. 2001;166:624–632. doi: 10.4049/jimmunol.166.1.624. [DOI] [PubMed] [Google Scholar]
- 35.Srikrishna G, et al. Carboxylated Glycans Mediate Colitis through Activation of NF-{kappa}B. J Immunol. 2005;175(8):5412–22. doi: 10.4049/jimmunol.175.8.5412. [DOI] [PubMed] [Google Scholar]
- 36.Egger B, et al. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology. 1997;113(3):825–32. doi: 10.1016/s0016-5085(97)70177-x. [DOI] [PubMed] [Google Scholar]
- 37.Vogl T, et al. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim Biophys Acta. 2006;1763(11):1298–306. doi: 10.1016/j.bbamcr.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 38.Manitz MP, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23(3):1034–43. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki R, et al. Sequential observations on the occurrence of preneoplastic and neoplastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci. 2004;95(9):721–7. doi: 10.1111/j.1349-7006.2004.tb03252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata F, et al. Fibroblast growth-stimulating activity of S100A9 (MRP-14) Eur J Biochem. 2004;271(11):2137–43. doi: 10.1111/j.1432-1033.2004.04129.x. [DOI] [PubMed] [Google Scholar]
- 41.Jobin C, et al. Evidence for altered regulation of I kappa B alpha degradation in human colonic epithelial cells. J Immunol. 1997;158(1):226–34. [PubMed] [Google Scholar]
- 42.Russo MP, et al. NF-kappaB-inducing kinase restores defective IkappaB kinase activity and NF-kappaB signaling in intestinal epithelial cells. Cell Signal. 2004;16(6):741–50. doi: 10.1016/j.cellsig.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 45.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 46.Pikarsky E, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 47.Erreni M, et al. Expression of chemokines and chemokine receptors in human colon cancer. Methods Enzymol. 2009;460:105–21. doi: 10.1016/S0076-6879(09)05205-7. [DOI] [PubMed] [Google Scholar]
- 48.Mantovani A. Chemokines in neoplastic progression. Semin Cancer Biol. 2004;14(3):147–8. doi: 10.1016/j.semcancer.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Hembruff SL, Cheng N. Chemokine signaling in cancer: Implications on the tumor microenvironment and therapeutic targeting. Cancer Ther. 2009;7(A):254–267. [PMC free article] [PubMed] [Google Scholar]
- 50.Mishra P, Banerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukoc Biol. 2010 doi: 10.1189/jlb.0310182. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, et al. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle. 2009;8(22):3695–701. doi: 10.4161/cc.8.22.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolignano D, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: a new protein enters the scene. Cancer Lett. 2010;288(1):10–6. doi: 10.1016/j.canlet.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 53.Hobbs JA, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23(7):2564–76. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka T, et al. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94(11):965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dieleman LA, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114(3):385–91. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulphate sodium progresses into chronicity in C57BL/6 but not in BALB/c mice - correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005 doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 57.Connolly MK, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87(4):713–25. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mantovani A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 60.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Earl TM, et al. Silencing of TLR4 decreases liver tumor burden in a murine model of colorectal metastasis and hepatic steatosis. Ann Surg Oncol. 2009;16(4):1043–50. doi: 10.1245/s10434-009-0325-8. [DOI] [PubMed] [Google Scholar]
- 62.Pouliot P, et al. Myeloid-related proteins rapidly modulate macrophage nitric oxide production during innate immune response. J Immunol. 2008;181(5):3595–601. doi: 10.4049/jimmunol.181.5.3595. [DOI] [PubMed] [Google Scholar]
- 63.Kalea AZ, et al. Alternatively spliced RAGEv1 inhibits tumorigenesis through suppression of JNK signaling. Cancer Res. 2010;70(13):5628–38. doi: 10.1158/0008-5472.CAN-10-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 65.Nemeth J, et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology. 2009;50(4):1251–62. doi: 10.1002/hep.23099. [DOI] [PubMed] [Google Scholar]
- 66.Mantovani A, et al. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14(3):155–60. doi: 10.1016/j.semcancer.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 67.Danese S, Mantovani A. Inflammatory bowel disease and intestinal cancer: a paradigm of the Yin-Yang interplay between inflammation and cancer. Oncogene. 2010;29(23):3313–23. doi: 10.1038/onc.2010.109. [DOI] [PubMed] [Google Scholar]
- 68.Horst D, et al. Invasion associated up-regulation of nuclear factor kappaB target genes in colorectal cancer. Cancer. 2009;115(21):4946–58. doi: 10.1002/cncr.24564. [DOI] [PubMed] [Google Scholar]
- 69.Popivanova BK, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69(19):7884–92. doi: 10.1158/0008-5472.CAN-09-1451. [DOI] [PubMed] [Google Scholar]
- 70.Wang D, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203(4):941–51. doi: 10.1084/jem.20052124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim MY, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–26. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsushita K, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458(7242):1185–90. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- 73.Lin S, et al. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology. 2009;136(5):1711–20. doi: 10.1053/j.gastro.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu S, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15(6):539–50. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harper K, et al. Autotaxin promotes cancer invasion via the lysophosphatidic acid receptor 4: participation of the cyclic AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer Res. 2010;70(11):4634–43. doi: 10.1158/0008-5472.CAN-09-3813. [DOI] [PubMed] [Google Scholar]
- 76.Devoogdt N, et al. Secretory leukocyte protease inhibitor in cancer development. Ann N Y Acad Sci. 2004;1028:380–9. doi: 10.1196/annals.1322.044. [DOI] [PubMed] [Google Scholar]
- 77.Choong ML, et al. A novel role for proliferin-2 in the ex vivo expansion of hematopoietic stem cells. FEBS Lett. 2003;550(1–3):155–62. doi: 10.1016/s0014-5793(03)00844-5. [DOI] [PubMed] [Google Scholar]
- 78.Fassett JT, Nilsen-Hamilton M. Mrp3, a mitogen-regulated protein/proliferin gene expressed in wound healing and in hair follicles. Endocrinology. 2001;142(5):2129–37. doi: 10.1210/endo.142.5.8132. [DOI] [PubMed] [Google Scholar]
- 79.Fukata M, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–81. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.