

Abstract
Cutaneous T cell lymphoma (CTCL) is characterized by constitutive activation of NFκB, which plays a crucial role in the survival of CTCL cells and their resistance to apoptosis. NFκB activity in CTCL is inhibited by the proteasome inhibitor bortezomib; however, the mechanisms remained unknown. In this study, we investigated mechanisms by which bortezomib suppresses NFκB activity in CTCL Hut-78 cells. We demonstrate that bortezomib and MG132 suppress NFκB activity in Hut-78 cells by a novel mechanism that consists of inducing nuclear translocation and accumulation of IκBα, which then associates with NFκB p65 and p50 in the nucleus and inhibits NFκB DNA binding activity. Surprisingly, however, while expression of NFκB-dependent anti-apoptotic genes cIAP1 and cIAP2 is inhibited by bortezomib, expression of Bcl-2 is not suppressed. Chromatin immunoprecipitation indicated that cIAP1 and cIAP2 promoters are occupied by NFκB p65/50 heterodimers, while Bcl-2 promoter is occupied predominantly by p50/50 homodimers. Collectively, our data reveal a novel mechanism of bortezomib function in CTCL and suggest that the inhibition of NFκB-dependent gene expression by bortezomib is gene specific and depends on the subunit composition of NFκB dimers recruited to NFκB-responsive promoters.
Keywords: Apoptosis; IκBα; leukemia, nuclear translocation; proteasome inhibition
Introduction
NFκB is a dimeric transcription factor that plays a key role in the expression of genes involved in cell survival, proliferation as well as immune responses (1-3). Since NFκB transcriptional activity is increased in many types of cancer and leukemia, inhibition of NFκB represents an important therapeutic target (4-8). In most unstimulated cells, NFκB proteins are localized in the cytoplasm bound to the inhibitory protein IκBα. Upon stimulation, IκBα is phosphorylated on Ser-32 and Ser-36 by the enzymes of IκB kinase complex (IKK), ubiquitinated and selectively degraded by the 26S proteasome (9,10). This results in the release of NFκB dimers from the inhibitory complex and in the translocation of NFκB to the nucleus, where it stimulates transcription of NFκB-dependent anti-apoptotic and pro-inflammatory genes.
The constitutive activation of NFκB observed in many types of cancer including different types of leukemia and lymphoma has been associated with increased cytoplasmic degradation of IκBα, resulting in the increased nuclear translocation of NFκB proteins and high levels of NFκB DNA binding activity (11-13). Proteasome inhibition results in the blockage of the cytoplasmic IκBα degradation, concomitant with the inhibition of NFκB nuclear translocation (14). Specifically, the 26S proteasome inhibitor bortezomib has been demonstrated to have anti-proliferative and pro-apoptotic properties in a wide range of hematological malignancies and has been widely used in the treatment of patients with multiple myeloma (15,16). In addition, it has shown promising results in cutaneous T cell lymphoma, non-Hodgkin’s lymphoma and other types of cancer and leukemia (17-20). It has been proposed that the pro-apoptotic and anti-proliferative effects of bortezomib on cancer cells result from the inhibition of the cytoplasmic IκBα degradation and inhibition of NFκB DNA binding activity (14). In cutaneous T cell lymphoma (CTCL) cells, where the constitutive activation of NFκB plays a crucial role in their survival and resistance to apoptosis, bortezomib inhibited the in vitro NFκB DNA binding activity and induced apoptosis (21-25). However, the molecular mechanisms of NFκB inhibition by bortezomib in CTCL have not been investigated.
We have recently demonstrated that in solid tumors such as the metastatic prostate cancer cells, the proteasome inhibitors MG132 and MG115 block NFκB activity by a novel mechanism that consists of inducing the nuclear translocation of IκBα (26). In this study, we tested hypothesis that the clinically used proteasome inhibitor bortezomib induces the nuclear translocation of IκBα in CTCL Hut-78 cells, thus inhibiting NFκB transcriptional activity and inducing apoptosis. Our results show that bortezomib induces the nuclear translocation and accumulation of IκBα, which then inhibits NFκB activity in CTLC cells. Surprisingly, however, our data indicate that the regulation of NFκB-dependent transcription by nuclear IκBα in CTCL is gene specific, and depends on the subunit composition of NFκB dimers recruited to the NFκB-responsive promoters.
Materials and Methods
Antibodies and reagents
Purified polyclonal antibodies against human IκBα (sc-371), NFκB-p65 (sc-372), NFκB-p50 (sc-7178), Bcl-2 (sc-492), and lamin B (sc-6216) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Purified polyclonal antibody against lactate dehydrogenase (LDH; 20-LG22) was from Fitzgerald Industries International (Concord, MA, USA), and actin antibody was from Sigma (St Louis, MO). cIAP1 (ab2399) and cIAP2 (ab32059) antibodies were from Abcam. Horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-mouse and anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
T4 polynucleotide kinase, poly (dI-dC), and Sephadex G25 spin columns were purchased from Pharmacia (Piscataway, NJ). CREB (sc-2504, sc-2517) and NFκB (sc-2505, sc-2511) gel shift oligonucleotides were from Santa Cruz Biotechnology (Santa Cruz, CA). [32P]-γ-ATP was purchased from Perkin Elmer (Boston, MA). Proteasome inhibitor MG132 was purchased from EMD Chemicals (San Diego, CA) and bortezomib was from ChemieTek (Indianapolis, IN). All other reagents were molecular biology grade and were purchased from Sigma (St Louis, MO).
Cell culture
Hut-78 human cutaneous T-cell lymphoma cells were obtained from American Type Culture Collection (ATCC; Rockville, MD). The cells were maintained at 37°C in RPMI 1640 medium, supplemented with 10% heat inactivated fetal bovine serum (FBS) and 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, in a humidified atmosphere with 5% CO2.
Transfection with si RNA and proteasome inhibition
Human IκBα (sc-29360) and non-silencing (sc-37007) small interfering RNAs (siRNAs) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Prior to transfection, Hut-78 cells were seeded into a 12-well plate and incubated in a humidified 5% CO2 atmosphere at 37°C in antibiotic-free RPMI medium supplement with 10% FBS for 24 h to 80% confluence. For each transfection, 60 μmol of either non-silencing siRNA-A control or IκBα siRNA (Santa Cruz Biotechnology, CA) were used. The cells were transfected for 6 h in transfection medium with siRNA transfection reagent according to manufacturer’s instructions (Santa Cruz Biotechnology; Santa Cruz, CA). After transfection, fresh RPMI medium supplemented with FBS and antibiotics was added and the cells were treated with proteasome inhibitors for 24 h.
Proteasome inhibitors MG132 and bortezomib were dissolved in DMSO and stored at −80°C. An equivalent volume of DMSO was used in all experiments as a solvent control.
Preparation of cytoplasmic and nuclear extracts
Nuclear (NE) and cytoplasmic extracts (CE) were prepared as described previously (27,28). Contamination of nuclear and cytoplasmic fractions by cytoplasmic and nuclear proteins, respectively, was determined by western analysis using LDH and lamin B as specific markers as described (27,28).
Electrophoretic mobility shift assay (EMSA)
EMSA assays of NFκB and CREB DNA binding protein complexes were performed in nuclear extracts as described (27-29). For competition or supershift experiments, binding reactions were performed in the presence of 30 M excess of unlabeled wild type (wt) or mutant (mut) oligonucleotide or 1 μg of specific polyclonal antibody. The resulting complexes were resolved on 7.5% nondenaturing polyacrylamide gels that had been pre-run at 150 V for 1 h in 0.5 X TBE buffer. Electrophoresis was conducted at 150 V for 3 h. After electrophoresis, gels were transferred to Whatman DE-81 paper, dried, and analyzed by Perkin-Elmer phosphoimager (Waltham, MA).
Immunoprecipitation
Nuclear extracts were prepared by using the Active Motif’s Nuclear Complex Co-IP Kit (Active Motif, 54001; Carlsbad, CA). The nuclear extracts were incubated (4°C, overnight) with IκBα antibody (sc-371) or control rabbit pre-immune IgG (sc-2027) as described (26). The immune complexes were immunoprecipitated on A/G Plus Agarose (sc-2003), washed four times with PBS buffer, resolved on 10% SDS gel and detected with IκBα, and NFκB p65 and p50 antibodies.
Apoptosis assay
Apoptosis was quantified with a cell death detection ELISA kit that quantifies release of nucleosomes into the cytoplasm (Cell Death Detection ELISAPLUS, Roche, Indianapolis, IN) as described (26). The assay was performed at the indicated time points as per the manufacturer’s instructions.
Real time PCR
Total RNA was isolated by using RNeasy mini-kit (Qiagen, Valencia, CA). The iScript one-step RT-PCR kit with SYBR Green (BioRad, Hercules, CA) was used as a supermix and 20 ng of RNA was used as template on a Bio-Rad MyIQ Single Color Real-Time PCR Detection System (BioRad). The primers used for quantification of cIAP1, cIAP2 and Bcl-2 mRNA were purchased from SA Biosciences (Frederick, MD).
Chromatin immunoprecipitation (ChIP)
ChIP analyses were performed by using the protocol from Upstate Biotechnology Inc., (Billerica, MA). Proteins and DNA were cross-linked by adding formaldehyde to the growth medium to a final concentration of 1% for 10 min at 37 °C and glycine was added at a final concentration of 0.125 M to neutralize formaldehyde. Cells were washed with PBS containing protease inhibitors and collected by centrifugation. Cells were then resuspended in SDS lysis buffer, incubated at 4 °C for 10 min, and sonicated. The lysates were centrifuged at 15,000 g for 10 min at 4 °C, and the supernatant extracts were incubated (4 °C, overnight) with ChIP dilution buffer and pre-cleared with Protein A/G agarose (Santa Cruz, CA) for 30 min at 4 °C. Immunoprecipitation was performed overnight at 4 °C, with p65 or p50 antibodies. Following immunoprecipitation, the samples were incubated with Protein A/G Agarose for 1 h, and the immune complexes were collected by centrifugation (150 g at 4 °C), washed, and extracted with 1% SDS-0.1 M NaHCO3. The cross-linking was reversed by heating with 5 M NaCl at 65 °C for 4 h. Proteins were digested with proteinase K, and the samples were extracted with phenol/chloroform, followed by precipitation with ethanol. The pellets were resuspended in nuclease-free water and subjected to real time PCR. Immunoprecipitated DNA was analyzed by real-time PCR (25 μl reaction mixture) using the iQ SYBR Green Supermix and the Bio-Rad MyIQ Single Color Real-Time PCR Detection System (Bio-Rad). Each immunoprecipitation was performed five times using different chromatin samples, and the occupancy was calculated by using the ChIP-qPCR Human IGX1A Negative Control Assay (SA Biosciences, Frederick, MD) as a negative control and corrected for the efficiency of the primers, which detect specific genomic DNA sequences within ORF-free intergenic regions or “promoter deserts” lacking any known or predicted structural genes. The primers used for real time PCR were the following: cIAP1: forward, 5′-TGACTGGCAGGCAGAAATGA-3′ and reverse, 5′-TTTGCCCGTTGAATCCGAT-3′; cIAP2: forward, 5′-TTCAGTAAATGCCGCGAAGAT-3′ and reverse, 5′-TGGTTTGCATGTGCACTGGT-3′; Bcl-2: forward, 5′-TGCATCTCATGCCAAGGG-3′ and reverse, 5′-CCCCAGAGAAAGAAGAGGAGTT-3′.
Statistical analysis
The results represent at least three independent experiments. Numerical results are presented as means ± SE. Data were analyzed by using an InStat software package (GraphPAD, San Diego, CA). Statistical significance was evaluated by using Mann-Whitney U test with Bonferroni correction for multiple comparisons, and p<0.05 was considered significant.
Results
Bortezomib and MG132 induce nuclear translocation of IκBα in leukemia Hut-78 cells, resulting in the inhibition of the constitutive NFκB DNA binding activity
The cutaneous T cell lymphoma Hut-78 cells are characterized by high levels of nuclear expression of NFκB p65 and p50 proteins, resulting in the constitutive NFκB DNA binding activity (21-24). To test the hypothesis that the proteasome inhibition induces nuclear IκBα translocation in these cells, Hut-78 cells were treated for 24 h with increasing concentrations of bortezomib or MG132, and the cytoplasmic and nuclear fractions were prepared and analyzed by western blotting. As a control of the purity of cytoplasmic and nuclear extract fractions, we used lactate dehydrogenase (LDH) and lamin B as specific cytoplasmic and nuclear markers, respectively. In untreated Hut-78 cells, IκBα was localized predominantly in the cytoplasm, while NFκB p65 and p50 proteins were both in the cytoplasm and in the nucleus (Figure 1). Cell treatment with bortezomib (Figure 1A) or MG132 (Figure 1B) decreased IκBα levels in the cytoplasm and induced its dose-dependent translocation to the nucleus; the nuclear translocation of IκBα was induced by 10 nM bortezomib (Figure 1A) and 5 μM MG132 (Figure 1B). Figures 1C and D illustrate the densitometric evaluation of western analysis of the nuclear IκBα levels induced by bortezomib and MG132, respectively. The nucleo-cytoplasmic distribution of NFκB p50 and p65 proteins was not changed by 1-100 nM bortezomib (Figure 1A) or 1-10 μM MG132 (Figure 1B), and there was no pronounced effect on the nuclear levels of p50 and p65 NFκB proteins within these concentrations.
Figure 1. Proteasome inhibitors bortezomib and MG132 induce nuclear translocation of IκBα in Hut-78 cells.
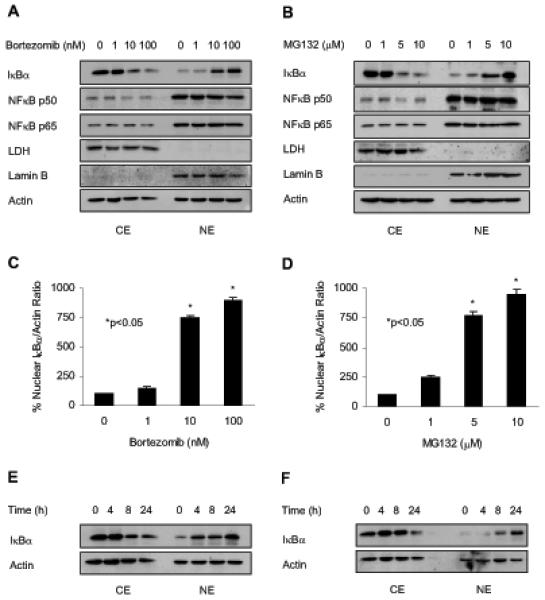
Hut-78 cells were treated with increasing concentrations of bortezomib (A) or MG132 (B) for 24 h; cytoplasmic (CE) and nuclear extracts (NE) were prepared and analyzed by western blotting using IκBα, p50 and p65 NFκB antibodies. To confirm equal protein loading, the membranes were stripped and re-probed with actin antibody. The purity of cytoplasmic and nuclear fractions was monitored using lactate dehydrogenase (LDH) and lamin B antibodies. The nuclear IκBα bands in cells treated with bortezomib (C) or MG132 (D) were scanned and the densities were normalized to densities of nuclear actin. The value corresponding to 0 proteasome inhibitor concentration was arbitrarily set to 100%, and the other values are presented relative to this value; the asterisks denote a statistically significant (p<0.05) change compared to 0 nM inhibitor. Panels (E) and (F) illustrate western blotting of CE and NE prepared from Hut-78 cells treated with 10 nM bortezomib or 10 μM MG132 for 0-24 h, respectively. Each lane corresponds to approximately 5×104 cells.
To determine whether the nuclear translocation of IκBα in response to bortezomib and MG132 is time dependent, we analyzed IκBα levels in cytoplasmic and nuclear extracts of Hut-78 cells treated with 10 nM bortezomib or 5 μM MG132 for 0 to 24 h. In bortezomib treated cells, the nuclear IκBα translocation appeared 4 h after incubation (Figure 1E), while in MG132 treated cells, IκBα translocated to the nucleus 8 to 24 h after treatment with MG132 (Figure 1F).
To determine whether the nuclear translocation of IκBα induced by proteasome inhibition is associated with the inhibition of NFκB activity, we measured NFκB DNA binding activity in nuclear extracts prepared from Hut-78 cells treated 24 h with increasing concentrations of bortezomib or MG132. As shown in Figure 2, the constitutive NFκB DNA-binding activity in Hut-78 cells was significantly reduced by 10 nM bortezomib (Figures 2A and C) or 5 μM MG132 (Figures 2B and D), which also induced the nuclear translocation and accumulation of IκBα (Figures 1A-D). Supershift analysis using NFκB p65 and p50 antibodies indicated that the NFκB DNA binding complex in Hut-78 cells contained both p65 and p50 NFκB proteins (Figure 2E). In contrast to NFκB, DNA binding activity of another, IκBα-independent transcription factor, CREB, was not significantly affected by increasing concentrations of bortezomib (Figures 2A and C) or MG132 (Figures 2B and D), indicating specificity for NFκB. Figure 2F confirms the CREB DNA binding specificity.
Figure 2. The bortezomib- and MG132-induced nuclear translocation of IκBα is associated with the inhibition of NFκB activity.
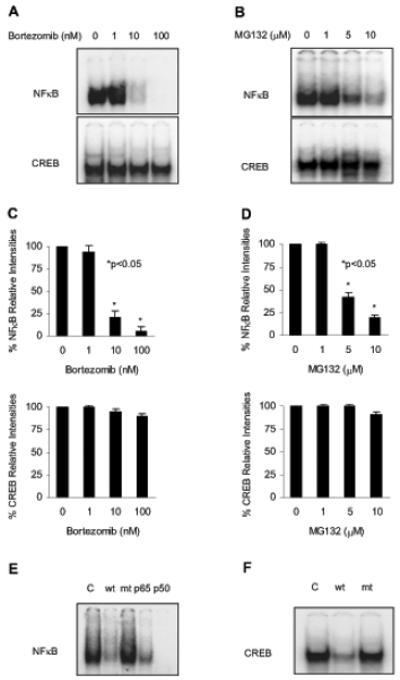
EMSA of NFκB and CREB DNA binding activities analyzed in nuclear extracts from Hut-78 cells treated for 24 h with increasing concentrations of bortezomib (A) or MG132 (B). Panels (C) and (D) show the densitometric evaluation of EMSA of NFκB (top panel) and CREB (bottom panel) DNA binding activities analyzed in nuclear extracts from Hut-78 cells treated 24 h with increasing concentrations of bortezomib or MG132, respectively. The value corresponding to 0 proteasome inhibitor concentration was arbitrarily set to 100%, and the other values are presented relative to this value; the asterisks denote a statistically significant (p<0.05) change compared to no inhibitor. Panels (E) and (F) illustrate subunit and specificity characterization of the constitutive NFκB and CREB DNA binding activities in Hut-78 cells, as described in Materials and Methods.
The bortezomib-induced nuclear IκBα accumulation is irreversible, and is caused by the nuclear association of IκBα with p65 and p50 NFκB
To determine whether the bortezomib-induced nuclear translocation of IκBα is reversible, or whether IκBα remains bound in the nucleus even after bortezomib is removed, Hut-78 cells were incubated for 24 h with control DMSO (Figure 3A) or 10 nM bortezomib (Figure 3B), washed extensively, and incubated another 0-48 h in the medium. The cytoplasmic and nuclear levels of IκBα were then analyzed by western blotting. Interestingly, once IκBα translocated to the nucleus in response to bortezomib treatment, it stayed there regardless of bortezomib removal (Figure 3B). As expected, DMSO itself did not induce nuclear IκBα accumulation, and IκBα remained in the cytoplasm (Figure 3A). These data indicate that the bortezomib-induced nuclear accumulation of IκBα is irreversible, and once IκBα translocates to the nucleus in Hut-78 cells, it binds to intranuclear proteins or structures.
Figure 3. Irreversibility of the nuclear translocation of IκBα and its association with the nuclear p65 and p50 NFκB in Hut-78 cells.
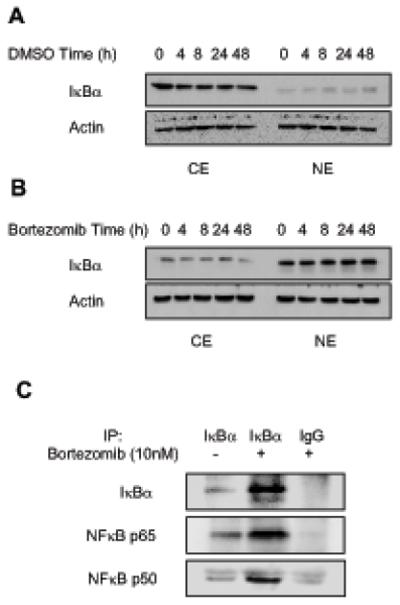
(A) Western blotting of CE and NE prepared from Hut-78 cells treated with DMSO (A) or 10 nM bortezomib (B) for 24 h, washed, incubated for another 0-48 h in medium with FBS, and analyzed by using IκBα and actin antibodies. Each lane corresponds approximately to 5×104 cells. (C) Co-immunoprecipitation experiment from nuclear extracts of untreated or bortezomib-treated (10 nM, 24h) Hut-78 cells by using pre-immune IgG or IκBα-specific antibody. The western blots were analyzed with IκBα and NFκB p65 and p50 antibodies.
To determine whether the nuclear IκBα binds to NFκB p65 and p50 proteins in the nucleus of Hut-78 cells, we performed a co-immunoprecipitation experiment using IκBα antibody and nuclear extracts prepared from untreated as well as bortezomib-treated (10 nM, 24 h) Hut-78 cells. As shown in Figure 3C (top panel), IκBα was immunoprecipitated from the nuclear extracts of bortezomib-treated cells, but not from the nuclear extracts of untreated cells, or from bortezomib-treated cells immunoprecipitated by using control pre-immune IgG. Immunoblotting using p65 NFκB antibody revealed the presence of p65 NFκB in the nuclear extracts of bortezomib-treated cells immunoprecipitated with IκBα antibody, but not with pre-immune IgG (Figure 3C, middle panel). Similarly, immunoblotting using p50 NFκB antibody demonstrated p50 NFκB presence in bortezomib-treated nuclear extracts immunoprecipitated with IκBα, but not control pre-immune IgG antibody (Figure 3C, bottom panel). Low levels of p65 and p50 NFκB signals were detected in the IκBα-immunoprecipitates prepared from the nuclear extracts of untreated cells, even though both NFκB proteins are highly expressed in the nucleus of untreated Hut-78 cells (Figure 1), demonstrating specificity for the IκBα-binding proteins. Together, these data demonstrate that the bortezomib-induced nuclear translocation of IκBα is irreversible, and that the nuclear IκBα binds to p65 and p50 NFκB proteins present in the nucleus.
Bortezomib-induced nuclear accumulation of IκBα results in the induction of apoptosis in leukemia Hut-78 cells
To determine whether the inhibition of NFκB DNA binding by bortezomib is directly caused by the bortezomib-induced nuclear IκBα, we hypothesized that suppression of IκBα nuclear levels should increase the NFκB DNA binding in Hut-78 cells. To test this, we suppressed IκBα expression by IκBα-specific siRNA, and then treated cells with increasing concentrations of bortezomib for 24 h. As expected, IκBα siRNA greatly reduced the cellular protein levels of IκBα compared to cells transfected with non-silencing siRNA, resulting in barely detectable IκBα in the nucleus of bortezomib-treated cells (Figure 4A). The decreased nuclear levels of IκBα in cells transfected with IκBα siRNA, compared to cells transfected with non-silencing siRNA, resulted in a substantially increased NFκB DNA binding activity, both in untreated Hut-78 cells, and in cells treated with increasing concentrations of bortezomib (Figures 4B, C).
Figure 4. The bortezomib-induced nuclear translocation of IκBα results in the suppression of NFκB DNA binding activity and induction of apoptosis in Hut-78 cells.
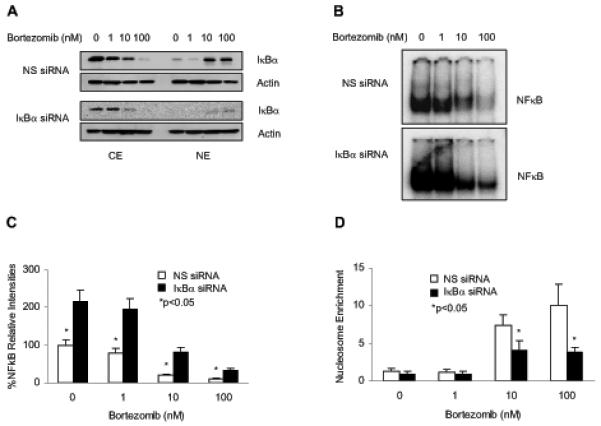
(A) Western blotting of CE and NE prepared from Hut-78 cells transfected with non-silencing (NS) or IκBα siRNA, followed by treatment with increasing concentrations of bortezomib for 24 h. The membranes were analyzed by using IκBα and actin antibodies. (B) Representative EMSA assay of NFκB DNA binding activity measured in Hut-78 cells transfected with NS (top panel) or IκBα (bottom panel) siRNA, followed by incubations with increasing concentrations of bortezomib for 24 h. (C) Densitometric evaluation of the EMSA assays of NFκB DNA binding activity measured in Hut-78 cells transfected with NS (empty columns) or IκBα (full columns) siRNA as described above in Fig. 4B. (D) Apoptosis measured by the nucleosome enrichment assay in Hut-78 cells transfected with NS (empty columns) or IκBα (full columns) siRNA, followed by 24 h treatment with increasing concentrations of bortezomib. The values shown in panels (C) and (D) represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) change compared to control si RNA transfected cells.
Next, we investigated whether the increased NFκB DNA binding activity in Hut-78 cells transfected with IκBα siRNA, would translate into an increased resistance to apoptosis in response to bortezomib treatment. To this end, Hut-78 cells were transfected with non-silencing or IκBα siRNA, treated with increasing concentrations of bortezomib as described above, and apoptosis was measured by a quantitative ELISA assay based on the detection of nucleosome release into the cytoplasm. As shown in Figure 4D, the decreased nuclear expression of IκBα in cells treated with 10 and 100 nM bortezomib and transfected with IκBα siRNA, compared to cells transfected with non-silencing siRNA, resulted in a significantly reduced apoptosis (p<0.05). Thus, these results show that the increased apoptosis observed in bortezomib-treated Hut-78 cells is directly caused by the increased nuclear levels of IκBα.
Bortezomib-induced nuclear IκBα differentially regulates NFκB-dependent anti-apoptotic gene expression in Hut-78 cells
Since the NFκB-regulated anti-apoptotic genes involve Bcl-2 as well as cIAP1 and cIAP2, we speculated that all these NFκB-responsive genes would be inhibited by the nuclear IκBα in bortezomib-treated Hut-78 cells. To test this, Hut-78 cells were treated 24 h with 10 nM bortezomib or control DMSO, and mRNA levels were analyzed by quantitative real time RT-PCR. Surprisingly, however, while expression of cIAP1 and cIAP2 was significantly reduced by 10 nM bortezomib, expression of Bcl-2 was not suppressed (Figure 5A). To confirm these results also on a protein level, we analyzed the total cellular protein levels of Bcl-2, cIAP1, cIAP2, as well as IκBα and control actin, in whole cell extracts prepared from Hut-78 cells treated 24 h with increasing concentrations of bortezomib (Figure 5B). Similarly as mRNA expression, protein levels of cIAP1 and cIAP2 were decreased by 10 and 100 nM bortezomib, while Bcl-2 levels were not changed. These data suggested that the bortezomib-induced nuclear IκBα might regulate NFκB-dependent transcription in a gene-specific manner.
Figure 5. Bortezomib-induced nuclear IκBα differentially regulates NFκB-dependent anti-apoptotic gene expression in Hut-78 cells.
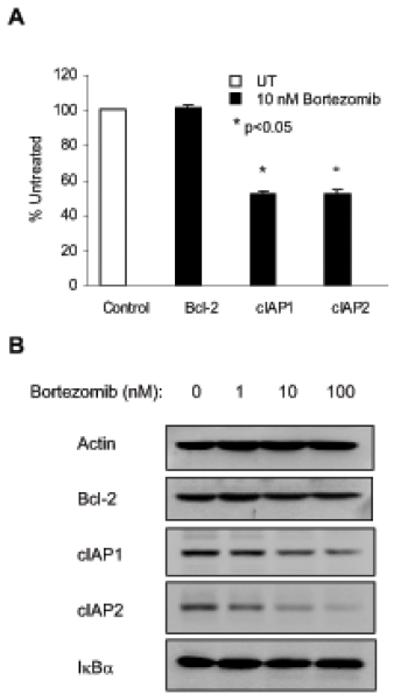
(A) Real time RT-PCR analysis of Bcl-2, cIAP1 and cIAP2 mRNA levels in Hut-78 cells treated 24 h with control DMSO (empty column) or 10 nM bortezomib (full columns). The values represent the mean +/−SE of five experiments; asterisks denote a statistically significant (p<0.05) inhibition compared to control untreated (UT) cells. (B) Western analysis of actin, Bcl-2, cIAP1, cIAP2 and IκBα protein expression in whole cell extracts of Hut-78 cells treated 24 h with increasing concentrations of bortezomib.
Of note, increased concentrations of bortezomib did not change the total cellular levels of IκBα (Figure 5B), which was in a good agreement with the data illustrated in Figure 1, showing that the net gain of IκBα in the nucleus equals its net loss in the cytoplasm in cells treated with proteasome inhibitors. These results indicate that the rate of IκBα degradation in Hut-78 cells equals the rate of IκBα resynthesis, which is regulated by NFκB. However, since NFκB activity is inhibited by the bortezomib-induced nuclear IκBα, IκBα resynthesis is suppressed, and thus the total IκBα cellular levels remain constant, despite the inhibited IκBα degradation.
To confirm the cIAP1 and cIAP2 regulation by nuclear IκBα, we analyzed Bcl-2, cIAP1 and cIAP2 mRNA and protein levels in cells that were transfected with non-silencing or IκBα siRNA and treated with increasing concentrations of bortezomib (Figure 6) or MG132 (data not shown). As expected, Bcl-2 mRNA (Figure 6A) and protein (Figure 6B) levels did not change between cells transfected with non-silencing and IκBα siRNA, and there was no substantial change in response to bortezomib treatment. In contrast, both cIAP1 and cIAP2 mRNA and protein levels decreased with increasing bortezomib concentrations; however, transfection with IκBα siRNA reduced this bortezomib-induced decrease. Similar data were obtained when cells were treated with MG132 instead of bortezomib (not shown). These data correlate well with the increased levels of NFκB DNA binding activity in cells transfected with IκBα siRNA (Figure 4), and indicate that the nuclear IκBα regulates transcription of cIAP1 and cIAP2, but not Bcl-2, in Hut-78 cells.
Figure 6. Analysis of Bcl-2, cIAP1 and cIAP2 expression in IκBα siRNA transfected Hut-78 cells treated with increasing concentrations of bortezomib.
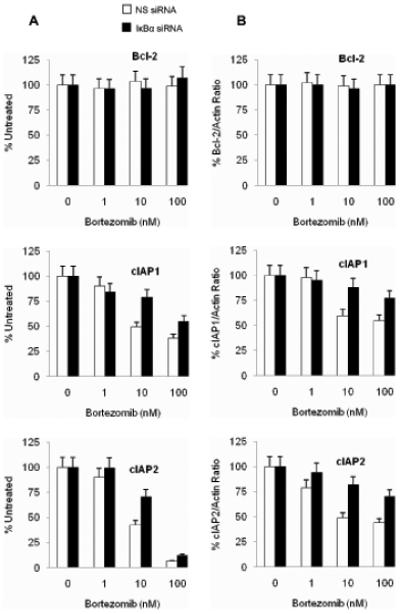
Bcl-2, cIAP1 and cIAP2 mRNA expression analyzed by real time RT-PCR (A) and total cellular protein levels analyzed by western blotting and densitometry normalized to actin (B) in Hut-78 cells transfected with NS (empty column) or IκBα siRNA (full columns) and treated 24 h with increasing concentrations of bortezomib.
The gene specific inhibition of NFκB-dependent transcription by bortezomib in Hut-78 cells depends on the subunit composition of recruited NFκB proteins
To analyze the mechanisms regulating transcription of NFκB-responsive genes in Hut-78 cells, we used chromatin immunoprecipitation to measure the in vivo recruitment of NFκB p65 and p50 subunits to promoters of Bcl-2, cIAP1 and cIAP2 genes. Hut-78 cells were treated 24 h with 10 nM bortezomib or control DMSO, cells were cross-linked with formaldehyde, lysed, chromatin was sheared by sonication, and p65 and p50 NFκB proteins were immunoprecipitated. The binding of p65 and p50 NFκB proteins to promoter regions of Bcl-2, cIAP1 and cIAP2 genes was measured by quantitative real time PCR.
As shown in Figure 7A, while p65 NFκB was heavily recruited to promoter regions of cIAP1 and cIAP2 genes, its recruitment to Bcl-2 promoter was only marginal. The p65 recruitment to cIAP1 and cIAP2 promoters was significantly reduced by the bortezomib-induced nuclear IκBα, whereas its recruitment to Bcl-2 promoter was not affected. As shown in Figure 7B, in contrast to p65 NFκB, the p50 NFκB subunit was recruited to all tested promoters, including Bcl-2, and this recruitment was inhibited by the bortezomib-induced nuclear IκBα. Thus, these data indicate that in Hut-78 cells, the cIAP1 and cIAP2 promoters are occupied predominantly by p65/50 NFκB heterodimers, whereas the promoter of Bcl-2 is occupied mainly by p50/50 NFκB homodimers (Table 1). However, while the bortezomib-induced nuclear IκBα removes both p65 and p50 NFκB from the gene promoters (Figures 7A, B), p65/50-regulated transcription of cIAP1 and cIAP2 is inhibited, whereas the Bcl-2 promoter occupied by p50/50-homodimers is not regulated by IκBα. Together, these data indicate that in Hut-78 cells, the inhibition of NFκB-dependent transcription by bortezomib is gene specific, and depends on the subunit composition of NFκB proteins recruited to the gene promoters (Table 1).
Figure 7. Recruitment of NFκB p65 and p50 proteins to NFκB-dependent promoters in Hut-78 cells.
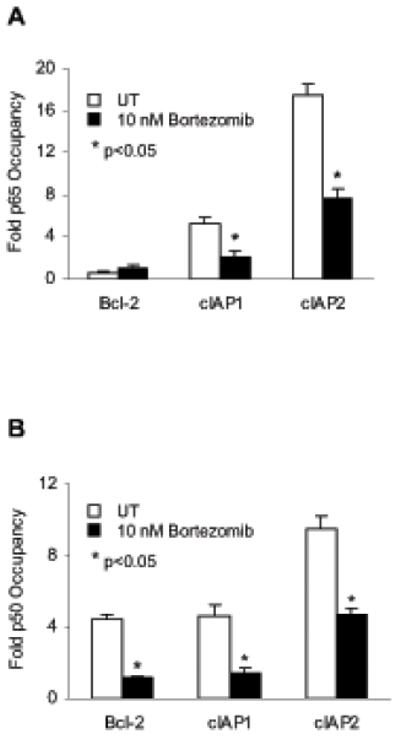
The recruitment of NFκB p65 (A) and p50 (B) proteins to NFκB-dependent promoters of Bcl-2, cIAP1 and cIAP2 genes in Hut-78 cells treated 24 h with DMSO (empty columns) or 10 nM bortezomib (full columns) was analyzed by chromatin immunoprecipitation and quantified by real time PCR. The data are presented as the change in occupancy over the human IGX1A (SA Biosciences) sequence control and represent the mean +/−SE of five experiments. Asterisks denote a statistically significant (p<0.05) inhibition compared to control untreated (UT) cells.
Table I.
NFκB sequences in EMSA and anti-apoptotic gene promoters, composition of the bound NFκB dimers, and regulation by the nuclear IκBα
| Gene | NFκB site | NFκB proteins | Inhibition by IκBα |
|---|---|---|---|
| EMSA consensus sequence | GGGACTTTCC | p50/65 | + |
| cIAP1 | GGAATTCCCC | p50/65 | + |
| cIAP2 | GGAAATCCCC | p50/65 | + |
| Bcl-2 | GAAATCCTCC | p50/50 | − |
Discussion
The proteasome inhibitor bortezomib, which is approved by the FDA for treatment of multiple myeloma and mantel cell lymphoma, acts by targeting the catalytic 20S core of the proteasome and induces apoptosis in cancer cells (15-20). One of the mechanisms consists of inhibiting the cytoplasmic degradation of IκBα, resulting in the suppression of NFκB DNA binding activity and decreased expression of NFκB-dependent anti-apoptotic genes (14,30). NFκB is constitutively activated in CTCL and many other forms of cancer and leukemia, where it plays a crucial role in cell survival and resistance to apoptosis (21-23). Recently, bortezomib has been evaluated in CTCL and exhibited promising antitumor effects in vitro and in vivo (18,19).
In this study, we have shown that the proteasome inhibitors bortezomib and MG132 suppress the constitutive NFκB DNA binding activity in CTCL Hut-78 cells by a new mechanism that consists of inducing the nuclear translocation and accumulation of IκBα. Once in the nucleus, the nuclear IκBα then binds to NFκB p65 and 50 proteins and removes them from the promoters of NFκB-dependent genes. Importantly, however, our data show that the ability of nuclear IκBα to inhibit NFκB-dependent transcription in Hut-78 cells is gene specific. While expression of NFκB-dependent anti-apoptotic genes cIAP1 and cIAP2 is inhibited by bortezomib, expression of Bcl-2 is not suppressed. Analysis of the in vivo binding of NFκB proteins to cIAP and Bcl-2 promoters by chromatin immunoprecipitation showed that both p65 and p50 NFκB are recruited to cIAP1 and cIAP2 promoters, while the Bcl-2 promoter is occupied only by p50 NFκB. Thus, these results indicate that the cIAP1 and cIAP2 promoters associate with NFκB p65/50 heterodimers, and this binding and transcription are inhibited by the bortezomib-induced nuclear IκBα. In contrast, Bcl-2 promoter is occupied predominantly by p50/50 NFκB homodimers, and its transcription is not inhibited by the bortezomib-induced nuclear IκBα.
Compared to p65 NFκB, p50 lacks the transactivation domain and, therefore, p50/50 homodimers, which retain their ability to bind DNA, were thought to function only as transcriptional repressors (1-3). However, recent studies have shown that p50/50 homodimers may be transcriptionally active as well, especially if bound to trans-activating elements (31-35). Indeed, increased constitutive DNA binding activity of p50/50 homodimers has been observed in several types of lymphoma and leukemia, and has been associated with the increased expression of Bcl-2 (31-35). Interestingly, while Bcl-2 expression was shown to be suppressed by bortezomib in some solid tumors, such as lung or prostate cancer (36,37), its suppression in other cells has not been consistently observed (38-40). These differences in Bcl-2 regulation by bortezomib – and by the nuclear IκBα – could be caused by a differential expression and regulation of NFκB subunits. In this model, in cells expressing p50 NFκB, the Bcl-2 promoter would be occupied predominantly by p50/50 homodimers, which would not regulate Bcl-2 transcription, and thus Bcl-2 expression would not be suppressed by the bortezomib-induced nuclear IκBα. Conversely, in cells that do not express p50 NFκB, the Bcl-2 promoter might be occupied by other NFκB dimers, which regulate Bcl-2 transcription, and thus in these cells, Bcl-2 transcription would be inhibited by bortezomib.
Alternatively, the regulation of Bcl-2 transcription by the bortezomib-induced nuclear IκBα might depend on a promoter-specific recruitment of other transcription factors, which might affect NFκB affinity for IκBα. Interestingly, the Bcl-2 NFκB binding site differs from cIAP promoters as well as from the oligonucleotide sequence used in EMSA assay by having A and C instead of G and T in the 2nd and 6th position, respectively (Table 1). It might be possible that the Bcl-2 promoter associates with transcriptional factors and/or regulators that decrease the in vivo NFκB affinity for IκBα. Since Bcl-2 plays a crucial role in cell survival and drug resistance (41-44), future studies should determine the mechanisms that regulate its transcription by bortezomib and by the bortezomib-induced nuclear IκBα.
In addition to IκBα, bortezomib controls the ubiquitin-proteasome mediated cytoplasmic degradation of other short-lived proteins that include p53, c-myc and N-myc, cyclins, and the cyclin-dependent kinase inhibitors p21 and p27 (45). Intriguingly, all these proteins function as transcriptional regulators and/or tumor suppressors, and their pro-apoptotic and cell cycle regulatory function is controlled by their nuclear-cytoplasmic translocation. Since bortezomib is being evaluated for the treatment of a wide range of human malignancies (45), it will be interesting to determine whether it induces the nuclear translocation and accumulation of these proteins as well.
NFκB activity and expression of NFκB-dependent anti-apoptotic genes are increased in many types of cancer and leukemia. Thus, the bortezomib-induced nuclear translocation of IκBα could provide a new therapeutic strategy aimed at the suppression of NFκB activity by nuclear IκBα and induction of apoptosis. However, the regulation of NFκB-dependent transcription by the bortezomib-induced nuclear IκBα is gene specific, and in CTCL Hut-78 cells, depends on the subunit composition of recruited NFκB complexes. These differences in the transcriptional regulation by the bortezomib-induced nuclear IκBα might hold the key for development of more specific therapies for cancers characterized by increased NFκB activity.
Acknowledgements
This work was supported by the National Institutes of Health research grants GM079581 and AI085497 to I. Vancurova.
Grant Support: This work was supported by the National Institutes of Health research grants GM079581 and AI085497 to I. Vancurova
Footnotes
Disclosure
No potential conflicts of interest were disclosed.
References
- 1.Baeuerle PA, Baltimore D. NFκB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin AS., Jr. Series introduction: the transcription factor NFκB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh S, Karin M. Missing pieces in the NFκB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NFκB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–42. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilmore TD. The Re1/ NFκB /IκB signal transduction pathway and cancer. Cancer Treat Res. 2003;115:241–65. [PubMed] [Google Scholar]
- 6.Aggarwal BB. NFκB: the enemy within. Cancer Cell. 2004;6:203–08. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NFκB is the lynchpin. Trends Immunol. 2005;26:318–25. doi: 10.1016/j.it.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Naugler WE, Karin M. NFκB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verma IM, Stevenson J. IκB kinase: beginning, not the end. Proc Natl Acad Sci USA. 1997;94:11758–60. doi: 10.1073/pnas.94.22.11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NFκB activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 11.Jost PJ, Ruland J. Aberrant NFκB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109:2700–07. doi: 10.1182/blood-2006-07-025809. [DOI] [PubMed] [Google Scholar]
- 12.Packham G. The role of NFκB in lymphoid malignancies. Br J Haematol. 2008;143:3–15. doi: 10.1111/j.1365-2141.2008.07284.x. [DOI] [PubMed] [Google Scholar]
- 13.Saitoh Y, Yamamoto N, Dewan MZ, et al. Overexpressed NFκB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood. 2008;111:5118–29. doi: 10.1182/blood-2007-09-110635. [DOI] [PubMed] [Google Scholar]
- 14.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. 2008;11:164–79. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–11. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 16.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–79. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barr P, Fisher R, Friedberg J. The role of bortezomib in the treatment of lymphoma. Cancer Invest. 2007;25:766–75. doi: 10.1080/07357900701579570. [DOI] [PubMed] [Google Scholar]
- 18.Zinzani PL, Musuraca G, Tani M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:4293–97. doi: 10.1200/JCO.2007.11.4207. [DOI] [PubMed] [Google Scholar]
- 19.Horwitz SM. Novel therapies for cutaneous T-cell lymphomas. Clin Lymphoma Myeloma. 2008;(Suppl. 5):S187–92. doi: 10.3816/CLM.2008.s.015. [DOI] [PubMed] [Google Scholar]
- 20.Heider U, Rademacher J, Lamottke B, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol. 2009;82:440–49. doi: 10.1111/j.1600-0609.2009.01239.x. [DOI] [PubMed] [Google Scholar]
- 21.Izban KF, Ergin M, Qin JZ, et al. Constitutive expression of NFκB is a characteristic feature of mycosis fungoides: implications for apoptosis resistance and pathogenesis. Hum Pathol. 2000;31:1482–90. doi: 10.1053/hupa.2000.20370. [DOI] [PubMed] [Google Scholar]
- 22.Sors A, Jean-Louis F, Pellet C, et al. Down-regulating constitutive activation of the NFκB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood. 2006;107:2354–63. doi: 10.1182/blood-2005-06-2536. [DOI] [PubMed] [Google Scholar]
- 23.Sors A, Jean-Louis F, Begue E, et al. Inhibition of IκB kinase subunit 2 in cutaneous T-cell lymphoma down-regulates NFκB constitutive activation, induces cell death, and potentiates the apoptotic response to antineoplastic chemotherapeutic agents. Clin Cancer Res. 2008;14:901–11. doi: 10.1158/1078-0432.CCR-07-1419. [DOI] [PubMed] [Google Scholar]
- 24.Kiessling MK, Klemke CD, Kaminski MM, Galani IE, Krammer PH, Gulow K. Inhibition of constitutively activated NFκB induces reactive oxygen species and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res. 2009;69:2365–74. doi: 10.1158/0008-5472.CAN-08-3221. [DOI] [PubMed] [Google Scholar]
- 25.Ri M, Iida S, Ishida T, et al. Bortezomib-induced apoptosis in mature T-cell lymphoma cells partially depends on upregulation of Noxa and functional repression of Mcl-1. Cancer Sci. 2008;100:341–48. doi: 10.1111/j.1349-7006.2008.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu HY, Juvekar A, Ghosh C, Ramaswami S, Le DH, Vancurova I. Proteasome inhibitors induce apoptosis of prostate cancer cells by inducing nuclear translocation of IκBα. Arch Biochem Biophys. 2008;475:156–63. doi: 10.1016/j.abb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vancurova I, Miskolci V, Davidson D. NFκB activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase C-delta. Correlation to nuclear IκBα. J Biol Chem. 2001;276:19746–52. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 28.Castro-Alcaraz S, Miskolci V, Kalasapudi B, Davidson D, Vancurova I. NFκB regulation in human neutrophils by nuclear IκBα: correlation to apoptosis. J Immunol. 2002;169:3947–53. doi: 10.4049/jimmunol.169.7.3947. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh CC, Vu HY, Mujo T, Vancurova I. Analysis of nucleocytoplasmic shuttling of NFκB proteins in human leukocytes. Methods Mol Biol. 2008;457:279–92. doi: 10.1007/978-1-59745-261-8_21. [DOI] [PubMed] [Google Scholar]
- 30.Prasad S, Ravindran J, Aggarwal BB. NFκB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurland JF, Kodym R, Story MD, Spurgers KB, McDonnell TJ, Meyn RE. NFκB (p50) homodimers contribute to transcription of the bcl-2 oncogene. J Biol Chem. 2001;276:45380–86. doi: 10.1074/jbc.M108294200. [DOI] [PubMed] [Google Scholar]
- 32.Viatour P, Bentires-Alj M, Chariot A, et al. NFκB B2/p100 induces Bcl-2 expression. Leukemia. 2003;17:1349–56. doi: 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- 33.Mathas S, Johrens K, Joos S, et al. Elevated NFκB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106:4287–93. doi: 10.1182/blood-2004-09-3620. [DOI] [PubMed] [Google Scholar]
- 34.Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–50. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paz-Priel I, Ghosal AK, Kowalski J, Friedman AD. C/EBPalpha or C/EBPalpha oncoproteins regulate the intrinsic and extrinsic apoptotic pathways by direct interaction with NFκB p50 bound to the bcl-2 and FLIP gene promoters. Leukemia. 2009;23:365–74. doi: 10.1038/leu.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fahy BN, Schlieman MG, Mortenson MM, Virudachalam S, Bold RJ. Targeting BCL-2 overexpression in various human malignancies through NFκB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother Pharmacol. 2005;56:46–54. doi: 10.1007/s00280-004-0944-5. [DOI] [PubMed] [Google Scholar]
- 37.Mortenson MM, Schlieman MG, Virudachalam S, et al. Reduction in BCL-2 levels by 26S proteasome inhibition with bortezomib is associated with induction of apoptosis in small cell lung cancer. Lung Cancer. 2005;49:163–70. doi: 10.1016/j.lungcan.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 38.Goktas S, Baran Y, Ural AU, et al. Proteasome inhibitor bortezomib increases radiation sensitivity in androgen independent human prostate cancer cells. Urology. 2010;75:793–98. doi: 10.1016/j.urology.2009.07.1215. [DOI] [PubMed] [Google Scholar]
- 39.Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101:1530–34. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 40.Fennell DA, Chacko A, Mutti L. Bcl-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene. 2008;27:1189–97. doi: 10.1038/sj.onc.1210744. [DOI] [PubMed] [Google Scholar]
- 41.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The Bcl-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 43.Neuzil J, Weber T, Schröder A, et al. Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 2001;15:403–15. doi: 10.1096/fj.00-0251com. [DOI] [PubMed] [Google Scholar]
- 44.Neuzil J, Wang XF, Dong LF, Low P, Ralph SJ. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006;580:5125–9. doi: 10.1016/j.febslet.2006.05.072. [DOI] [PubMed] [Google Scholar]
- 45.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–9. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]