

Abstract
GRP78/BiP is a multifunctional protein which plays a major role in endoplasmic reticulum (ER) protein processing, protein quality control, maintaining ER homeostasis and controlling cell signaling and viability. Previously, using a transgene-induced mammary tumor model, we demonstrated that Grp78 heterozygosity not only impeded cancer growth through suppression of tumor cell proliferation and promotion of apoptosis, the Grp78+/− mice exhibited dramatic reduction (70%) in the microvessel density (MVD) of the endogenous mammary tumors while having no effect on the MVD of normal organs. This observation suggests that GRP78 may critically regulate the function of the host vasculature within the tumor microenvironment. In this report, we interrogated the role of GRP78 in the tumor microenvironment. In mouse tumor models where wild-type, syngeneic mammary tumor cells were injected into the host, we showed that Grp78+/− mice suppressed tumor growth and angiogenesis during the early but not late phase of tumor growth. Growth of metastatic lesions of wild-type, syngeneic melanoma cells in the Grp78+/− mice was potently suppressed. We created conditional heterozygous knockout of GRP78 in the host endothelial cells and demonstrated severe reduction of tumor angiogenesis and metastatic growth with minimal effect on normal tissue MVD. Furthermore, knockdown of GRP78 expression in immortalized human endothelial cells demonstrated that GRP78 is a critical mediator of angiogenesis by regulating cell proliferation, survival, and migration. Our findings suggest that concomitant use of current chemotherapeutic agents and novel therapies against GRP78 may offer a powerful dual approach to arrest cancer initiation, progression and metastasis.
Keywords: GRP78, tumor angiogenesis, conditional knockout, mouse model, metastatic growth, endothelial cells
Introduction
Glucose-regulated protein 78 (GRP78), also referred to as BiP or HSPA5, is a member of the HSP70 protein family with established function as an endoplasmic reticulum (ER) chaperone protein and regulator of the ER stress signaling pathway (1). As a multifunctional protein, GRP78 plays critical roles in physiologic and pathological stress (2). GRP78 is induced in a wide variety of tumors through intrinsic factors such as altered glucose metabolism in cancer cells, compounded by extrinsic factors such as glucose starvation and hypoxia in the microenvironment of poorly-perfused solid tumors (3). The induction of GRP78 leads to an increase of GRP78 in the ER compartment, as well as promotion of GRP78 to the cell surface, where it assumes a new function of co-receptor for cell surface signaling (4–7). While a large number of studies have established that GRP78 is required for tumor cell proliferation, survival and therapeutic resistance (3, 8, 9), little is known about the role of GRP78 in the tumor microenvironment, which is critical for support of tumor growth and metastasis.
Previously we established a heterozygous knockdown mouse model of GRP78 as homozygous deletion of Grp78 leads to early embryonic lethality (10). The heterozygous Grp78 (Grp78+/−) mice, despite reduced expression of GRP78 to about half the wild-type level in all the tissues, are phenotypically normal with respect to organ development and antibody production (11). However, through breeding of the wild-type and Grp78+/− mice with the transgenic mice expressing the middle T oncogene driven by the murine mammary tumor viral promoter, we discovered that Grp78 heterozygosity prolonged the latency period and significantly impeded cancer growth by suppressing tumor cell proliferation and promoting tumor cell apoptosis (11). Strikingly, the microvessel density (MVD) of the endogenous tumors in the Grp78+/− mice was reduced by 70%, while the vasculature of normal organs remained unaffected. This observation provided the initial evidence that GRP78 may selectively regulate the function and survival of endothelial cells associated with the tumor.
The tumor microenvironment contains a plethora of cells that support tumor growth and progression. Essential for tumor growth and metastasis is the tumor vasculature, which supplies the nutrients and oxygen crucial for the growth and maintenance of the tumor (12, 13). Endothelial cells are therefore the requisite members of the tumor microenvironment. Previous studies on human glioma-derived endothelial cells have demonstrated that these tumor-associated endothelial cells possess unique functional characteristics, including an activated phenotype (14). Tumors require an activated endothelial population to provide a constant new blood vessel formation at the growing border of the tumor, which allows for continued expansion and progression. Our recent investigations have also indicated that GRP78 expression is constitutively elevated within the tumor vasculature, emphasizing the constant state of activation of these tumor-associated endothelial cells (15).
In this report, we tested the role of GRP78 in host cells supporting tumor growth using syngeneic cancer models. Our studies revealed that neovascularization during early tumor growth and tumor metastasis was potently suppressed by host Grp78 heterozygosity. We further created an endothelial cell specific Grp78 heterozygous knockout mouse model (Grp78F/+;Tie2-Cre) and demonstrated severe reduction of tumor angiogenesis and growth of metastatic lesions with minimal effect on normal tissue MVD. Furthermore, knockdown of GRP78 in immortalized human endothelial cells results in suppression of endothelial cell proliferation, promotion of apoptosis and decreased migration. These findings reveal GRP78 is a regulator of endothelial cell-specific angiogenic functions and provides a mechanistic explanation for the requirement of GRP78 in the microenvironment during tumor growth and metastasis.
Materials and Methods
Cell cultures
The E0771 mouse breast cancer cell line was generously provided by Dr. Enrico Mihich (Roswell Park Cancer Institute) and was maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 10 mmol/L HEPES, and 1% penicillin antibiotics as described (16). The immortalized human microvascular endothelial cells (HMEC) were kindly provided by Dr. Betty Wu-Hsieh (Taiwan National University). HMEC were grown in Medium 200 (Invitrogen) supplemented with Low Serum Growth Supplement (Invitrogen). All cells were maintained at 37°C and 5% CO2.
Generation of the B16-Fluc-A1 melanoma cell line
B16 F0 cells (ATCC) were transduced with a lentiviral construct containing the firefly luciferase gene driven by an internal CMV promoter. Clonal cell lines were selected by limiting dilutions in 96 well plates and expanded and screened for high levels of luciferase expression using a Promega luciferase detection kit per the manufacturer’s instructions. The cells were maintained in high-glucose DMEM containing 4.5 mg/ml glucose supplemented with 10% fetal bovine serum, 2 mmol/L glutamine, and 1% penicillin antibiotics.
Generation of genetically altered Grp78 mouse model
The Grp78+/− mice were generated as described (10) and were backcrossed into the C57BL/6 genetic background for eight generations. To create endothelial cell specific Grp78 heterozygous knockout mice, mice carrying the Grp78 floxed allele (in C57BL6 and 129/Sv background) (17) were crossed with Tie2-Cre transgenic mice (Tek-Cre in C57BL6 background, the Jackson Laboratory) (18). Genotyping for the WT, floxed and KO alleles were performed by PCR using genomic DNA extracted from mouse tails biopsies as described (17). Genotyping was also performed using genomic DNA extracted from enriched primary brain endothelial cells as previously described (19) with modifications (20). The Tie2-Cre transgene was identified with forward primer: 5′-AAGAACCTGATGGACATGTTCAGGGA-3′ and reverse primer: 5′-ACGAACCTGGTCGAAATCAGTGCGTTC-3′. Three month old mice were used for the tumor model studies. All animal protocols were conducted with the approval of the USC University Animal Care and Use Committee.
Generation of tumor models
The generation and monitoring of endogenous mammary tumors driven by the MMTV-PyVT transgene in Grp78+/+ or +/− mice have been described (11). In the syngeneic tumor models, E0771 cells (4×106/mice) were resuspended in 200 μL PBS and injected into the mammary fat pad of 4-week-old female mice; B16-Fluc-A1 melanoma cells (amount as indicated) were resuspended in 200 μL PBS and injected through the lateral tail vein. Lung metastasis was monitored by luminescence imaging (Xenogen) weekly. Following sacrifice, the lungs were removed and the number of surface metastases was counted.
Immunofluorescent and immunohistochemical staining
Immunostaining was performed on paraffin-embedded tumor sections as previously described (11). The primary antibodies used were the following: mouse anti-PCNA (1:100), goat anti-VEGF (1:100) from Santa Cruz Biotechnology; rabbit anti-CD31 (1:50) from Thermo Fisher Scientific. The processed sections were visualized using a fluorescence microscope.
Microvessel density measurement
Mouse tumor tissues were processed for microvessel density (MVD) analysis as previously described (11). The tissues were stained with rat anti-mouse CD31 antibody (BD PharMingen), and quantified using the imaging processing program ImageJ (NIH).
Small interfering RNA transfection
HMECs were seeded at a density of 6×104 per well in 6-well plates and transfected with siRNA using Lipofectamine 2000 Transfection Reagent (Invitrogen) per the manufacturer’s instructions. The siRNA against Grp78 (siGrp78) is 5′-ggagcgcauugauacuagatt-3′ as described (21). The control siRNA (siCtrl) is 5′-aaggagacguauagcaacggu-3′, comprised of a 21 base pair scrambled sequence without significant homology to any known gene sequences from mouse, rat, or human. The experiments were repeated 3 times.
Detection of cell surface GRP78 protein
The biotinylation and detection of cell surface GRP78 by Western blot in HMECs 48 hours post transfection were performed as previously described (4). The experiments were repeated 3 times.
MTT cell viability assays
HMECs were seeded in quadruplicate at a density of 3×103 per well in 96-well plates 48 hours post-transfection and grown for an additional 48 hours. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed according to the manufacturer’s instructions (Sigma-Aldrich) and as previously described (15).
BrdU cell proliferation assays
HMEC were seeded in quadruplicate at a density of 3×103 per well in 96-well plates 48 hours post-transfection and grown for an additional 24, 48, or 72 hours. The bromodeoxyuridine (BrdU) cell proliferation assay (Roche Diagnostics Corp., Indianapolis, IN) was conducted according to the manufacturer’s instructions.
Migration assays
HMEC were seeded in duplicate at a density of 4×104 per modified transwell Boyden chamber (BD BioCoat, Bedford, MA) 48 hours post-transfection. Each chamber housed a 6.5 mm-diameter polyethylene terephthalate filter with 8 μm pores. Migration was stimulated by adding complete growth media to the lower compartment of the experimental apparatus. After 6 hours, the filters were fixed and stained with Harleco Hemacolor staining solution (EMD Chemicals, Gibbstown, NJ). Cells that migrated to the underside of the filter were quantified under high-power magnification (400x). A 6 hour migration period was used to eliminate the effect of disparate rates of cell proliferation.
TUNEL assay
HMEC were seeded at a density of 1.5×104 per well on glass coverslips 48 hours post-transfection. The HMEC were mounted with ProLong gold anifade mounting medium with DAPI. The apoptotic cells detected by the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science, Indianapolis, IN) were visualized using a fluorescence microscope. A total of 1,000 cells were counted per treatment condition.
Statistical analysis
For the syngeneic E0771 mammary tumor model, a linear model was used to compare tumor volume over time, with slope, and quadratic and cubic terms for each mouse treated as random. The likelihood ratio test for the group × time interaction was used to indicate whether the tumor growth patterns were significantly different between the two genotypes. This analysis was based on the logarithm of tumor volume + 1. For the B16 melanoma tumor model, the log-rank test was used to compare time to lung metastasis between the two genotypes stratifying by experiment. The Pike estimates of relative hazard ratio were calculated using the observed and expected numbers of events based on the log-rank test statistic. Kaplan-Meier plots were graphed for time to lung metastasis. Two-way analysis of variance (ANOVA) was performed for comparison of pulmonary metastases and BrdU incorporation in HMEC with genotype and weeks as the two factors. Prior to ANOVA comparing the endothelial cells with and without GRP78 knockdown, logarithm was taken of the responses to render the data compatible with the assumptions of normality and homoscadesity. Pair-wise comparisons among the groups were performed using the least significance difference method if the overall p-value was <0.05. t-tests were used when only two groups were compared for a measurement.
Results
Grp78 heterozygosity in host environment suppresses early tumor growth
In the MMTV-PyT transgene driven mammary tumor model, tumors were formed in both the Grp78+/+ and Grp78+/− mice. Previously we reported the latency period for tumor formation was delayed and the size of the primary tumor at 15 weeks was reduced by about 60% in the Grp78+/− mice (11). Upon following the mouse cohorts for a longer period (after 18 weeks), secondary tumors became apparent. In this endogenous tumor model where both the tumor and host cells were heterozygous for Grp78, there were fewer total number of tumors (p<0.05) and the tumors were of smaller size (p<0.01) in the Grp78+/− PyT mice as compared to Grp78+/+ PyT mice (Fig. 1A and B).
Figure 1.
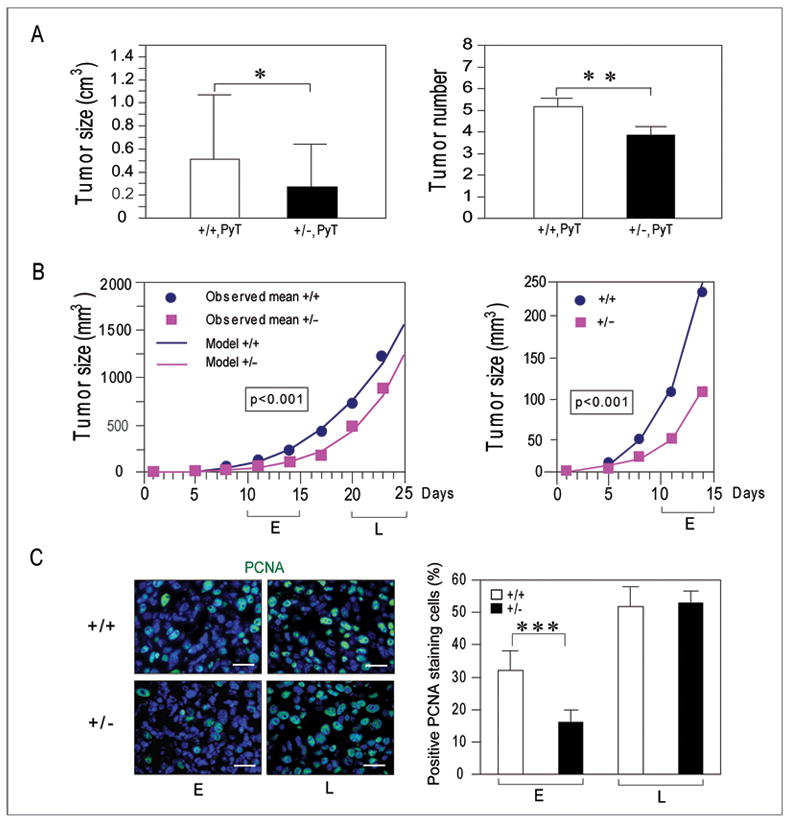
Grp78 host heterozygosity suppresses early phase tumor growth. A, comparison of the size and total number of endogenous tumors (primary and secondary) in female Grp78+/+, PyT mice and Grp78+/−, PyT mice (n=7 for each genotype) at 18 weeks old (size, *P<0.05; number, **P<0.01). B, left panel: cohorts of female Grp78+/+ mice (n=12) and Grp78+/− mice (n=15) were monitored for the time of appearance and size of the implanted wild-type EO771 tumor in each mouse. Dots, observed mean tumor volumes; lines, model-predicted tumor volumes. The likelihood ratio test based on random coefficient model comparing the two curves showed the significant reduction of EO771 tumor growth in heterozygous mice compared with the WT mice (P<0.001). E and L denote early (10 to 15 days) and late (20 to 25 days) phase following tumor implantation. Right panel: enlarged view of the initial tumor growth pattern for the two host genotypes. C, left panel: paraffin-embedded EO771 tumor sections were subjected to immunofluorescent staining for PCNA (green), and the nuclei were counterstained with DAPI (blue). The merged images are representative of six animals examined. Bar, 25μm. Right panel: quantitation of the percent of PCNA positive staining cells. The difference in early phase tumor between the two genotypes is significant (**P<0.001).
To examine the role of GRP78 in tumor growth in the host environment, the growth of tumors derived from syngeneic wild-type (WT) murine E0771 breast cancer cells injected subcutaneously into either Grp78+/+ (n=12) or +/− (n=15) mice was monitored. Thus, in these experiments, only the host environment was varied. The likelihood ratio test based on random coefficient model comparing the two curves showed a significant reduction of EO771 tumor growth in Grp78+/− mice as compared to the WT mice (P<0.001) (Fig. 1B, left panel). Tumor size was examined at different times during tumor development. The early (E) phase referred to the period between 10 to 15 days following tumor implantation, and the late (L) phase referred to the period at 20 to 25 days post-tumor injection. Our results showed that Grp78 heterozygosity in the host primarily affected the early phase of tumor growth (Fig. 1B, right panel); as after 20 days, the tumor growth rate between the two host genotypes was parallel to each other (Fig. 1A, left panel). The slower growth of the early tumors in the Grp78+/− mice associated with a 50% reduction (p<0.001) in tumor cell proliferation compared to the Grp78+/+ mice, as assayed by PCNA positive staining cells, whereas during the late phase, tumor cells in both host genotypes showed similar number of proliferating cells (Fig. 1C). Collectively, these data suggest that Grp78 heterozygosity in the host environment is sufficient to impede tumor growth and proliferation at the early phase of tumor growth, however at the late stage, the WT tumor cells are able overcome the host deficiency due to Grp78 heterozygosity.
Grp78 host heterozygosity suppresses tumor angiogenesis during early tumor growth
Initiation of tumor growth requires neovascularization. To examine the effects of Grp78 host heterozygosity on tumor MVD, E0771 breast tumors harvested from Grp78+/+ or +/− mice were snap-frozen, and stained with anti-CD31 antibody, which is specific for endothelial cells. Tumors from both early and late phase periods were analyzed. The early tumors of the Grp78+/− mice showed 70% reduction in MVD compared to Grp78+/+ mice (p<0.01), whereas the differences in the MVD between the two host genotypes were not significant in the late phase tumors (P=0.68) (Fig. 2A). These results were confirmed with fluorescence staining of CD31 in paraffin-embedded tumor sections (Fig. 2B). In contrast, staining for the vascular endothelial cell growth factor (VEGF) was similar between the two host genotypes, in both early and late phase tumors (Fig. 2C). This suggests that VEGF, primarily produced and released by the WT tumor cells (22), was not obviously affected by Grp78 host heterozygosity.
Figure 2.

Grp78 host heterozygosity inhibits tumor angiogenesis. A, left panel, representative CD31 staining pattern of snap frozen EO771 tumor sections during early (E) and late (L) tumor growth phase. The sections were lightly counterstained with hematoxylin. Reddish purple indicates positive staining. Right panel, quantitation of the microvessel density (MVD) in the tumor sections (n=6 from each group). One unit is defined as 1 μm2 for one x200 magnification field. The difference in MDV in early phase tumors is significant (**P<0.01). The difference in MDV from late phase tumors is not significant (P=0.68). Columns, mean MVD; bars, 95% CI. B and C, paraffin-embedded EO771 tumor sections from early (E) and late (L) phase tumors subjected to immunofluorescent staining for CD31 (B) or VEGF (C), denoted in green. Blue, nuclei were counterstained with DAPI. Representative merged staining patterns are shown from each host genotype. Bar, 50μm.
Grp78 host heterozygosity suppresses growth of metastatic lesions
Formation of new tumor vasculature is critical for the establishment and growth of metastatic colonies. To test whether the inhibition of early phase tumor angiogenesis by Grp78 host heterozygosity negatively impacts tumor metastasis, we utilized the syngeneic B16 melanoma tumor model. A newly derived melanoma clonal cell line (B16-Fluc-A1) was generated through transduction with a lentivirus expressing luciferase, followed by serial dilution and screening. The B16-Fluc-A1 cell line was approximately five times stronger in luciferase expression than the original B16-Fluc cell line that also expressed the green fluorescent protein, with comparable in vitro and in vivo growth characteristics (23). Following tail vein injection of the WT tumor cells, (5×104/mice; n=10 for each host genotype), the progress of metastasis was first determined by whole body luminescence imaging and representative images are shown in Fig. 3A. Visible large tumor colonies were detected in the lungs of syngeneic Grp78+/+ mice beginning at 3 weeks and more were observed at 4 weeks; whereas Grp78+/− mice showed no visible tumor at 3 weeks and only one mice developed a small visible tumor at 4 weeks (Fig. 3A). Following sacrifice at 2, 3 and 4 weeks, the lungs of the Grp78+/+ mice showed considerably larger tumor and greater number of tumors as compared to Grp78+/− mice (Fig. 3B). The number of tumors per lung for the two genotypes is summarized in Fig. 3C. The result of stratified log-rank test showed that the risk of developing lung metastasis for Grp78+/− mice was 47% (95% CI 25%–89%) of that for Grp78+/+ mice (p-value<0.001) (Fig. 3D). Thus, consistent with the notion that GRP78 is required for tumor neovascularization, Grp78 heterozygosity in the host environment suppressed the number of visible metastatic lesions and slowed time to progression of metastatic tumors.
Figure 3.

Grp78 host heterozygosity inhibits pulmonary melanoma metastasis. A, representative luminescence images of Grp78+/+ and +/− mice injected with B16-Fluc-A1 melanoma cells (5×104/mice) at the 1 to 4 week post tail vein injection (n=10 for each host genotype). B, representative macroscopic pictures of lungs removed at 2, 3 and 4 weeks after tumor cell injection. Examples of metastatic lesions are indicated by blue arrows. C, lung surface metastases for each host genotype were quantitated and standard deviation was indicated. D, stratified log-rank test was used to compare time to lung metastasis between the two groups. The risk of developing lung metastasis for Grp78+/− mice was 47% (95% CI 25%–89%) of that for Grp78+/+ mice (p-value<0.001).
Endothelial cell specific Grp78 heterozygosity inhibits angiogenesis and proliferation in metastases
Endothelial cells, the principal cell component of the angiogenesis process, are a critical cellular component of the tumor microenvironment. Therefore to understand the role of GRP78 in endothelial cells, we created a new mouse model of heterozygous knockout of Grp78 specifically in endothelial cells by breeding the Grp78 floxed/floxed mice (17) with the Tie2Cre transgenic mice (18). The genotypes of the mouse strains were confirmed by PCR analysis (Supplemental Fig. 1A). The Cre-recombinase was driven by the receptor tyrosine kinase Tek promoter/enhancer, which is active predominantly in endothelial cells starting at embryonic E9.5 (24). The knockout of the Grp78 floxed allele in primary endothelial cells isolated from the Grp78F/+;Tie2Cre mice was confirmed by PCR analysis (Supplemental Fig. 1B). Mice with the Grp78F/+;Tie2Cre genotype, which is phenotypically normal served as the experimental group, with sibling Grp78F/+ mice as negative controls. A high dose of B16-Luc-A1 melanoma cells were injected via tail vein into Grp78F/+;Tie2Cre and Grp78F/+ mice (2×105/mice; n=21 for each genotype) to generate more metastatic lesions for comparison. Lungs were removed at 2, 3 and 4 week intervals (n=7 at each time point). The macroscopic inspection demonstrated clear reduction in metastasis in the Grp78F/+;Tie2Cre mice and representative images are shown in Fig. 4A. The visible pulmonary surface metastases were fewer in the Grp78F/+;Tie2Cre mice and the difference became greater as time increased (P<0.001) for the interaction between group and time. The mean difference (and associated 95% confidence interval) in number of colonies per lung between the two groups of mice was 60 (32–88), 167 (139–195) and 355 (327–383) at 2 weeks, 3 weeks and 4 weeks (all with P<0.001), respectively (Fig. 4B). Thus, Grp78 heterozygosity in endothelial cells causes significant delay and decrease in metastatic growth. In these same groups of mice, CD31 staining of cryostat sections of normal organs such as brain, liver and heart showed minimal difference in the blood vessel density (Fig. 4C and D). In contrast, immunofluorescence staining of paraffin-embedded lung sections of these mice with the CD31 antibody revealed fewer vessels at the border between the tumor and the adjacent normal tissue in the Grp78F/+;Tie2Cre tumor sections, compared to Grp78 F/+ controls (Fig. 5A), correlating with reduced PCNA staining in the Grp78F/+;Tie2Cre tumor sections (Fig. 5B). Thus endothelial cell specific heterozygous knockdown of GRP78 is sufficient to impede growth of metastatic lesions and vascularization at the growing edge of the tumor with minimal effect on normal organs.
Figure 4.

Endothelial cell specific Grp78 heterozygosity suppresses pulmonary melanoma metastasis. A, representative macroscopic pictures of lungs from Grp78 F/+ and Grp78 F/+;Tie2Cre mice at 2, 3 and 4 weeks following tail vein injection of B16- Fluc-A1 melanoma cells (2×105/mice; n=21 for each genotype). B, quantitation of average pulmonary surface metastases at 2, 3 and 4 weeks. Significant differences between the two host genotypes are detected for all time points (***P<0.001). The means and associated 95% confidence intervals are indicated. C. Representative CD31 staining pattern of sections of frozen brain tissue from Grp78 F/+ and Grp78 F/+;Tie2Cre mice. Reddish purple indicates positive staining. D. Summary of microvessel density of brain, liver, and heart from Grp78 F/+ and Grp78 F/+;Tie2Cre mice. The standard error is shown.
Figure 5.

Reduction of tumor angiogenesis and proliferation in mice with endothelial cell specific Grp78 heterozygous knockout mice. Paraffin-embedded pulmonary B16-Fluc-A1 melanoma sections from the Grp78 F/+ and Grp78 F/+;Tie2Cre mice were subjected to immunofluorescent staining for CD31 (A) and PCNA (B) denoted in green, with nuclei counterstained with DAPI (blue). Representative hematoxylin and fluorescence staining patterns are shown for each host genotype. Bar, 50μm.
Knockdown of GRP78 suppresses proliferation, migration and promotes apoptosis in immortalized endothelial cells
In order to identify the functional consequence resulting from decreased GRP78 levels in endothelial cells mimicking those in the tumor microenvironment, in vitro studies were performed with HMECs. The immortalized HMEC are SV40 transformed endothelial cells (25) which constitutively express high levels of GRP78, as observed in tumor-associated endothelial cells (14, 15). The HMECs were transiently transfected with siRNA specifically targeting Grp78 (siGrp78) or control siRNA (siCtrl). Western blot analysis of whole-cell lysates indicated that a greater than 85% knockdown of GRP78 expression was achieved (Fig. 6A). In the HMECs, biotinylation of cell surface protein followed by avidin agarose pull down and Western blot revealed that approximately 1% of total intracellular GRP78 was present on the cell surface, and following siGrp78 treatment, cell surface GRP78 was below detection limits (Fig. 6A). Investigation of cell overall viability using the MTT assay showed a 55% [95% confidence interval (CI) from 49% to 61%; P<0.001] decrease in the number of viable HMECs at 48 hours following GRP78 knockdown (Fig. 6B). Analysis of cell proliferation via incorporation of BrdU demonstrated that knockdown of GRP78 reduced cell proliferation and the reduction became greater as time increased (P<0.001 for the interaction between cell type and time). Knockdown of GRP78 reduced cell proliferation by 24% (95% CI from 4% to 40%; P=0.024), 61% (95% CI from 50% to 69%; P<0.001), and 79% (95% CI from 73% to 83%; P<0.001) when compared to siCtrl-transfected HMEC at 24, 48, and 72 hours, respectively (Fig. 6B). In addition to proliferation, within 6 hours of the knockdown of GRP78, HMEC showed reduced overall cell migration by 31% (95% CI from 16% to 43%; P=0.001) cells per high-powered field (Fig. 6B). Partial loss of GRP78 led to cell death, however, it required at least 48 hours. TUNEL staining revealed that apoptosis in siGrp78-transfected cells at 48 hours post transfection was significantly increased, from 2.5% in siCtrl transfected cells to 23% (P<0.001) (Fig. 6C). Thus, reduced GRP78 expression in immortalized endothelial cells suppresses cell proliferation, promotes apoptosis and decreases migration.
Figure 6.

GRP78 regulates function and viability of endothelial cells. A, Western blot analysis of total and cell surface GRP78 expression level in HMECs transfected with control siRNA (siCtrl) or siRNA against Grp78 (siGrp78), with β-actin serving as loading control for the total lysate samples, and control free of cytoplasmic contamination for the cell surface protein samples. The amount of lysate applied onto the gel represented 20% of input. B, left panel, analysis of cell viability at 48 hours post-transfection by the MTT assay showed knockdown of GRP78 significantly reduced cell survival (***P<0.001). Middle panel, BrdU incorporation in HMECs showed knockdown of GRP78 significantly decreased cell proliferation at 24, 48, and 72 hours (**P=0.024 at 24 hour time point and ***P<0.001 for 48 and 72 hour time point). Data are expressed as the mean OD of the siGRP78 group relative to the mean OD of the siCtrl group at each time point. The 95% confidence intervals are indicated. Data are based on two independent experiments. Right panel, HMEC migration was assessed by the modified transwell Boyden chamber assay. After a 6 hour migration period, the number of migrated HMECs was significantly lower in the siGrp78-transfected group as compared to siCtrl-transfected group (***P<0.001). Data represent the mean number of cells per high-powered field that migrated to the underside of the porous filter and are representative of two independent experiments. The 95% confidence intervals are indicated. C, left panel: detection of apoptotic cells (in red indicated by arrows) by TUNEL assay in the transfected HMECs. Bar, 50 μm. Right panel: quantitation of apoptosis cells in the transfected groups with significant differences detected (***P<0.001).
Discussion
In an array of tissue culture and tumor models, GRP78 has been established to be important for tumor progression and chemotherapeutic drug resistance; correlating with clinical studies that patients with high GRP78 expression levels in their tumors generally have a poorer prognosis and experience earlier relapse (3, 9, 11, 21, 26, 27). These discoveries suggest that GRP78 can be a potent therapeutic target for anti-cancer therapy. Previous studies on GRP78 function in cancer progression have largely focused on tumor cells and how GRP78 exerts its protective anti-apoptotic effect to allow them to escape immune surveillance and stress associated with tumor progression (3, 9). Here we examined the role of GRP78 in the tumor microenvironment. In two syngeneic tumor models, WT tumors in the Grp78 heterozygous host environment were significantly impeded in tumor angiogenesis and proliferation. Through the creation of a new mouse model with endothelial cell specific heterozygous knockout of Grp78, and coupled with in vitro studies on GRP78 knockdown in immortalized endothelial cells, we further demonstrated that GRP78 is an important mediator of endothelial cell proliferation, survival, and migration and provide a new mechanistic explanation for the requirement of GRP78 in the microenvironment during tumor growth and metastasis.
How might GRP78 regulate function and promote survival of endothelial cells supporting tumor growth? Expression of GRP78, with well established chaperone function (28), is induced in hypoxic endothelial cells (29). Induction of GRP78 leads to increase in both intracellular form of GRP78 as well as cell surface of GRP78 (4). Upregulation of GRP78 in the ER can protect endothelial cells against ER stress, maintain ER calcium homeostasis and suppress onset of stress induced apoptosis through suppression of caspase 7 and CHOP induction which mediate cytotoxicity in cells undergoing ER (17, 30). On the other hand, cell surface GRP78 is reported to colocalize with pro-angiogenic growth factor receptors, promoting growth signaling, proliferation and cell migration (5, 6, 8). GRP78 cell surface expression is induced by VEGF in human umbilical vein endothelial cells (HUVEC) and is required for VEGF-induced proliferation and angiogenic signaling (31). Therefore, the observed decrease in HMEC proliferation and migration, two critical angiogenic functions, correlate well with the consequences of GRP78 knockdown in these cells. Interestingly, our studies indicate that the affects of Grp78 host heterozygosity is most evident in a minimal growth factor environment at the early stage of tumor growth. A possible explanation is that as the tumor grows bigger, sufficient levels of proangiogenic, growth factors are produced and/or secreted by the wild-type tumor cells to stimulate tumor endothelial cell growth, hence able to compensate for the effects of GRP78 loss in the host microenvironment.
Given the critical role of GRP78 in tumor cell survival as well as in endothelial cells supporting tumor growth, it represents a dual target for anti-cancer therapy. GRP78 is the receptor for Kringle 5, which induces apoptosis of both cancer cells and endothelial cells (29). Sensitization of Kringle 5-induced apoptosis of brain microvessel endothelial cells by radiation requires GRP78 (32). Proteomic studies suggest GRP78 reduces the efficacy of topoisomerase inhibitors to induce endothelial cell apoptosis (33). Furthermore, some tumor cells are capable of secreting GRP78, which confers bortezomib-resistance to endothelial cells (34). We demonstrate here reducing GRP78 affects several critical aspects of angiogenesis: endothelial cell number, and endothelial cell migration. Migration is especially important because blood vessel growth involves the migration of endothelial progenitor cells to the tumors, as well as the expansion of neighboring blood vessels (35). Furthermore, other anti-angiogenic drugs (i.e., bevacizumab, Avastin) alter the VEGF levels in the patient resulting in a change in the delicate balance of angiogenic growth factors. Such an imbalance can cause severe blood vessel regrowth and resistance to drugs (36). It is recently reported that angiogenesis inhibitors targeting the VEGF and other proangiogenic growth factor pathways, while demonstrating anti-tumor effects, concomitantly elicit tumor adaptation leading to accelerated metastasis (37, 38). Here we showed that Grp78 host heterozygosity does not appear to affect VEGF staining in the tumor sections, suggesting that VEGF production in the tumor tissue is not altered. Furthermore, the decrease in tumor metastatic growth in both Grp78+/− and Grp78F/+;Tie2Cre mice persisted throughout the entire course of the experiments which was terminated at 4 weeks when animals required euthanasia. One explanation is that altering the cytokine balance by decreasing VEGF activation is likely to induce a response that is different from decreasing GRP78 expression, as GRP78 regulates a much wider repertoire of cellular function (2, 7). Thus, concomitant use of current chemotherapeutic agents and novel therapies against GRP78 may offer a powerful new approach to arrest cancer initiation, progression and metastasis.
Supplementary Material
Acknowledgments
We thank Dr. Enrico Mihich of Roswell Park Cancer Institute for providing the E0771 cells, Sam Li, Grant Dagliyan and the USC Norris Cancer Center Small Animal Imaging Core Facility for assistance and advice in mouse imaging.
Footnotes
Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–51. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2010 doi: 10.1016/j.ceb.2010.09.007. org/10.1016/j.ceb.2010.09.007 in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–75. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Mol Cell Biol. 2008;28:666–77. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 7.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signaling and therapeutic targeting. Biochem J. 2011;434:181–8. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307–16. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 10.Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–97. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 12.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 13.Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 14.Charalambous C, Chen TC, Hofman FM. Characteristics of tumor-associated endothelial cells derived from glioblastoma multiforme. Neurosurg Focus. 2006;20:E22. doi: 10.3171/foc.2006.20.4.e22. [DOI] [PubMed] [Google Scholar]
- 15.Virrey JJ, Dong D, Stiles C, Patterson JB, Pen L, Ni M, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6:1268–75. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewens A, Luo L, Berleth E, Alderfer J, Wollman R, Hafeez BB, et al. Doxorubicin plus interleukin-2 chemoimmunotherapy against breast cancer in mice. Cancer Res. 2006;66:5419–26. doi: 10.1158/0008-5472.CAN-05-3963. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Ye R, Barron E, Baumeister P, Mao C, Luo S, et al. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2010;17:488–98. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–54. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Z, Hofman FM, Zlokovic BV. A simple method for isolation and characterization of mouse brain microvascular endothelial cells. J Neurosci Methods. 2003;130:53–63. doi: 10.1016/s0165-0270(03)00206-1. [DOI] [PubMed] [Google Scholar]
- 20.Perriere N, Demeuse P, Garcia E, Regina A, Debray M, Andreux JP, et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J Neurochem. 2005;93:279–89. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- 21.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA. 2008;105:19444–9. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 23.Craft N, Bruhn KW, Nguyen BD, Prins R, Liau LM, Collisson EA, et al. Bioluminescent imaging of melanoma in live mice. J Invest Dermatol. 2005;125:159–65. doi: 10.1111/j.0022-202X.2005.23759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;19:2054–65. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–90. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 26.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 27.Baumeister P, Dong D, Fu Y, Lee AS. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol Cancer Ther. 2009;8:1086–94. doi: 10.1158/1535-7163.MCT-08-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–97. [PubMed] [Google Scholar]
- 29.Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005;65:4663–72. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 30.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 31.Katanasaka Y, Ishii T, Asai T, Naitou H, Maeda N, Koizumi F, et al. Cancer antineovascular therapy with liposome drug delivery systems targeted to BiP/GRP78. Int J Cancer. 2010;127:2685–98. doi: 10.1002/ijc.25276. [DOI] [PubMed] [Google Scholar]
- 32.McFarland BC, Stewart J, Jr, Hamza A, Nordal R, Davidson DJ, Henkin J, et al. Plasminogen Kringle 5 induces apoptosis of brain microvessel endothelial cells: sensitization by radiation and requirement for GRP78 and LRP1. Cancer Res. 2009;69:5537–45. doi: 10.1158/0008-5472.CAN-08-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruneel A, Labas V, Mailloux A, Sharma S, Royer N, Vinh J, et al. Proteomics of human umbilical vein endothelial cells applied to etoposide-induced apoptosis. Proteomics. 2005;5:3876–84. doi: 10.1002/pmic.200401239. [DOI] [PubMed] [Google Scholar]
- 34.Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, et al. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. 2009;114:3960–7. doi: 10.1182/blood-2009-03-209668. [DOI] [PubMed] [Google Scholar]
- 35.Patenaude A, Parker J, Karsan A. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc Res. 2010;79:217–23. doi: 10.1016/j.mvr.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Duda DG, di Tomaso E, Ancukiewicz M, Chung DC, Lauwers GY, et al. Direct evidence that bevacizumab, an anti-VEGF antibody, up-regulates SDF1alpha, CXCR4, CXCL6, and neuropilin 1 in tumors from patients with rectal cancer. Cancer Res. 2009;69:7905–10. doi: 10.1158/0008-5472.CAN-09-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.