

Abstract
Severe combined immunodeficiency (SCID) is one of the most severe forms of primary immunodeficiency characterized by absence of functional T lymphocytes. It is a paediatric emergency, which is life-threatening when recognized too late. The clinical presentation varies from the classical form of SCID through atypical SCID to Omenn syndrome. In addition, there is a considerable immunological variation, which can hamper the diagnosis. In this educational review, we describe the immunopathological background, clinical presentations and diagnostic process of SCID, as well as the therapeutic possibilities.
Keywords: Severe combined immunodeficiencies, Diagnosis, Lymphocytes, Therapy, Primary immunodeficiencies
Introduction
Severe combined immunodeficiency (SCID) is an inherited primary immunodeficiency, which is characterized by the absence or dysfunction of T lymphocytes affecting both cellular and humoral adaptive immunity [46]. It is one of the most severe forms of primary immunodeficiency (PID), which is life-threatening when recognized too late. Seven percent of PID patients suffer from a T cell deficiency, including SCID [18]. Depending on the genetic defect, B and natural killer (NK) cells may be present or absent. Conventionally, SCID can be classified as T−B+ and T−B− SCID with further subdivision based on the presence or absence of NK cells. However, the presentation is not always classic, and the presence or absence of NK cells may be misleading. Therefore, a phenotype describing NK cells no longer forms a part of the classification system of the International Union of Immunological Societies [47]. It has become clear that clinical presentation has wide phenotype variability with considerable immunological variation [43]. These aspects can impede the diagnosis of SCID. In this review, we address the immunological and clinical spectrum of SCID and provide some clues and tools for diagnosing SCID.
Clinical presentations of SCID
Classical SCID
A family history of unusual or fatal infective complications or unexplained infant death is important, particularly in consanguineous families; a history of affected male relatives suggests common gamma chain-deficient SCID. Affected infants generally appear well at birth, but within the first few months of life, demonstrate failure to clear infections and present with persistent respiratory tract or gastrointestinal infections, failure to thrive and, sometimes, apparent food intolerance (Table 1) [20]. Persistent respiratory tract infection is common, with failure to clear viruses accompanying persistent bronchiolitic-like signs. Insidiously progressive respiratory disease with radiological evidence of interstitial pneumonitis and hyperinflation suggests Pneumocystis jiroveci infection, which may be a co-pathogen with respiratory viruses (Fig. 1) [4]. Persistent viral diarrhoea with failure to thrive is an important sign. Although patients with SCID are often initially well and growing normally, they fall away from the growth centile after a few months when infection occurs because of intestinal villous atrophy, leading to malabsorption, which in severe cases results in malnutrition.
Table 1.
Presenting features of classical and atypical severe combined immunodeficiency and Omenn syndrome
| Classical SCID | Omenn syndrome | Atypical SCID |
|---|---|---|
| Present in infancy | Present in infancy | Present >12 months of age |
| Persistent viral respiratory +/− gastrointestinal infection | Erythroderma | Recurrent, severe, prolonged viral infection |
| Pneumocystis jiroveci pneumonitis | Alopecia | bronchiectasis |
| Disseminated BCG infection | Hepatosplenomegaly | Autoimmune cytopenias |
| Failure to thrive | Massive lymphadenopathy | Failure to thrive |
| Superficial candidiasis | Inflammatory pneumonitis/enteritis | Granulomatous cutaneous lesions |
| Maternofoetal graft versus host disease | Raised IgE | EBV-associated lymphoproliferation |
| Absent lymphoid tissue | Eosinophilia | Partial or restricted antigen-specific antibody responses |
| Absent immunoglobulins | Lymphocytosis | Lymphopenia |
| Absent T lymphocytes |
Fig. 1.

Chest radiograph from a 5-month-old infant with severe combined immunodeficiency showing bilateral patchy shadowing secondary to interstitial pnuemonitis due to infection with respiratory syncytial virus and Pneumocystis jiroveci. There is hyperinflation of the lungs, and the midline pleural borders of the upper lobes are visible because the thymic shadow is absent (courtesy of The Paediatric Immunology Unit, Great North Children’s Hospital, Newcastle upon Tyne)
Bacterial infections are less common in part because of the presence of maternal IgG in early infancy. However, prolonged otitis media and invasive bacterial infections, such as staphylococcal or pseudomonas septicaemia and pneumonia, may occur, which may respond poorly to appropriate treatment. However, patients with associated agranulocytosis, such as those with reticular dysgenesis due to adenylate kinase 2 (AK2) deficiency, generally present in the first few days of life with omphalitis or invasive bacterial sepsis [5].
Severe invasive fungal infection is rare, but often fatal. Extensive persistent superficial candidiasis is more common. Disseminated BCGosis occasionally may be the presenting feature in immunised infants. Skin lesions demonstrate acid fast bacilli on histological analysis. A mild reticular skin rash, which may be thickened and lichenoid, with or without slightly deranged liver function tests may be seen in maternofoetal graft versus host disease. As SCID infants lack functional T cells, they cannot reject foreign lymphocytes acquired from the mother in utero, and so the skin is infiltrated by abnormal maternal T lymphocyte clones [41]. A similar clinical picture may occur in patients who have received an unirradiated blood transfusion, due to viable donor lymphocytes in the red cell donation, although in these cases, the rash is more severe and lymphadenopathy and hepatosplenomegaly may be present.
Examination usually reveals a wasted child who has dropped through the weight centiles—head circumference is usually preserved. There may be abdominal distension and muscle wasting due to malabsorption and malnutrition. Respiratory signs may include tachypnoea, nasal flaring, subcostal and intercostal recession, with widespread crepitations and rales, and cyanosis. There may be evidence of oral or perineal candidiasis and other superficial infections. There is no clinically detectable lymphoid tissue, although detecting this in young infants is not easy because lymph nodes and tonsils in normal infants are often very small. There may be hepatomegaly, with or without splenomegaly, particularly when disseminated Bacille Calmette–Guerin (BCG) infection is present. Rare presentations include Hodgkin-like polymorphous lymphoproliferative disorder, with rapidly growing extranodal tumours [57]. Very rarely, erythrophagocytosis has been described, in association with maternal T lymphocyte engraftment.
Omenn syndrome
Omenn syndrome is characterised by a generalised thickened erythematous rash, often with scaling and erythematous exfoliating, protein-losing erythroderma, developing a “leathery” consistency (Fig. 2). Hair, including eyebrows and eyelashes, is usually lost as the rash evolves. The rash may be present at birth or evolve over the first few weeks of life. There is an associated lymphadenopathy, particularly of the axillary and inguinal nodes. Hepatosplenomegaly is a frequent finding. There are raised serum IgE levels with a marked eosinophilia and combined immunodeficiency [71]. Children usually suffer from diarrhoea, failure to thrive and persistent infection as seen in other forms of SCID—staphylococcal or pseudomonas skin infection are particularly common. Affected infants are often miserable because of the high levels of circulating inflammatory cytokines. Pneumonitis and enteritis may be predominantly inflammatory rather than infective. The clinical picture may resemble SCID with maternofoetal engraftment; molecular genetic studies to identify the origin of the dermal infiltrative T lymphocytes can differentiate the two disorders [2]. Originally described in patients with mutations in recombinase activation genes (RAG) 1 and 2, mutations in a number of different genes have subsequently been described, including artemis, IL7Ra, RMRP, 22q11 deletion, CHD7, DNA ligase IV (LIG4), adenosine deaminase (ADA) and interleukin 2 receptor, gamma (IL2RG) [71].
Fig. 2.
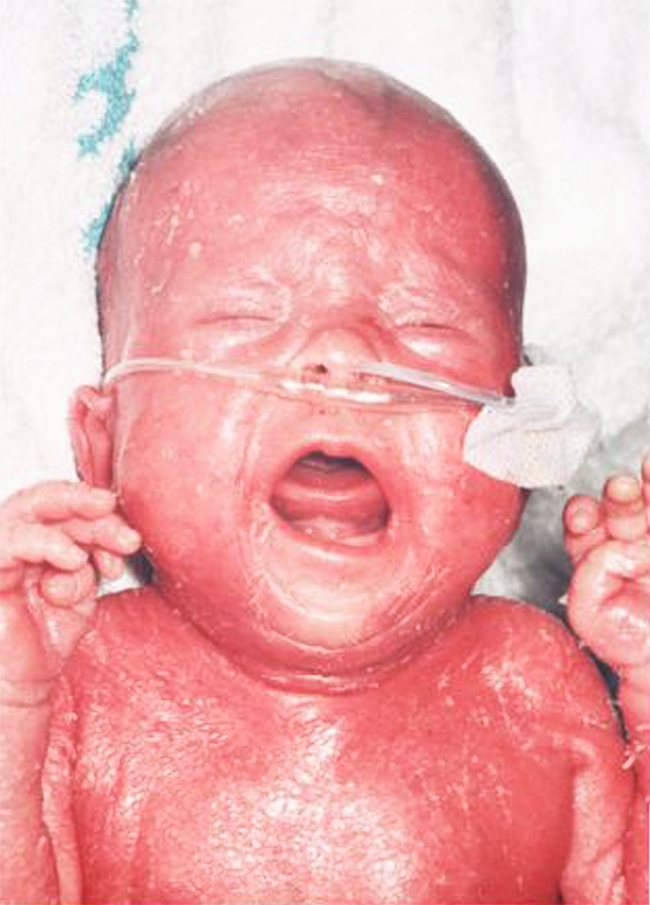
A newborn infant with Omenn’s syndrome due to a mutation in the RAG 1 gene. Note the confluent erythematous exfoliating, thickened rash with a “leathery” consistency and loss of hair and eyebrows (courtesy of The Paediatric Immunology Unit, Great North Children’s Hospital, Newcastle upon Tyne)
Atypical SCID
Patients may present with atypical forms of SCID or Omenn syndrome. Previously described as profound combined immunodeficiency, these patients usually survive beyond 12 months of age. Increasingly, hypomorphic mutations in genes normally associated with classical SCID are identified, thus retaining some protein function. Alternative mechanisms of demonstrating partial immunity include spontaneous gene reversion in early lymphoid progenitors [52, 59, 60, 74]. Such patients present with severe, prolonged infection, which may slowly resolve. Partial antibody responses can be demonstrated to restricted antigens. Other presentations include autoimmune manifestations, particularly with autoimmune cytopenias, and EBV-driven lymphoproliferative tumours. Rarely, cutaneous granulomatous lesions have been described [16, 19, 30, 32, 55]. It is important to consider atypical SCID presentations in children presenting beyond the first year of life so that appropriate antimicrobial treatment can be commenced and the patient considered for curative therapy (vide infra).
Other forms of SCID
In addition to SCID caused by developmental defects, SCID can also be caused by mutations affecting lymphocyte survival, as seen in patients with reticular dysgenesis due to mutations in AK2 [48] and in the enzyme deficiencies ADA and purine nucleoside phosphorylase (PNP), involved in nucleotide metabolism and salvage [3, 12]. As a result of the deficiency, toxic metabolites are formed, to which lymphocytes are exquisitely sensitive. Consequently, ADA and PNP deficiencies usually lead to profound lymphopenia [54]. Finally, several deficiencies have been described that can give rise to a clinical phenotype of SCID, but only affect a subset of T cells, e.g. MHC class II deficiency, ZAP 70 kinase deficiency [47] ([62] #1755). Additionally, defects in CD154 (CD40 ligand) and CD40 may present in infancy with P. jiroveci pneumonia. These types of SCID will not be further discussed in this review.
Stepwise diagnostics for SCID
Flow cytometric immunophenotyping of peripheral blood
The first step in the diagnostic process in a patient presenting with features consistent with SCID is ruling out HIV infection [17]. When HIV has been excluded, a blood smear differential may demonstrate lymphocytopenia, which is suggestive of SCID [28]. It should be noted, however, that a normal lymphocyte count on the differential white cell count does not exclude SCID because the absolute number of lymphocytes may be normal, but lymphocyte subsets may be severely reduced or absent. Therefore, flow cytometric immunophenotyping of lymphocyte subsets in peripheral blood is an important screening assay. In classical SCID, the various types can easily be discriminated (Fig. 3a) with a straightforward analysis of B, T and NK cells. For correct interpretation, reference values of age-matched controls should be used [13].
Fig. 3.

Flow cytometric analysis of peripheral blood and bone marrow of SCID patients. a Flow cytometric analysis of lymphocyte subsets in peripheral blood of SCID patients can be used for definition of the type of SCID and guides molecular diagnostics. b For B− SCID patients, flow cytometric analysis of the bone marrow precursor B cell compartment delineates the precursor B cell differentiation block, which can be helpful in candidate gene selection
Interpretation of results is more complicated in Omenn syndrome or atypical SCID. These patients present with high numbers of oligoclonal T cells [14], the presence of which may be misleading, and so detailed analysis of T cells in patients clinically suspected for typical or atypical SCID is of utmost importance [71].
Gene defects and disease mechanisms in T−B+ SCID
T−B+ SCID is caused by mutations in cytokine-mediated signalling. The majority of patients have X-linked SCID caused by mutations in the IL2RG gene encoding the common γ chain (γc). The γc chain is shared by the IL2, IL4, IL7, IL9, IL15 and IL21 cytokine receptors [31]. Cytokines mediate oligomerization of the γc chain with the appropriate cytokine receptor chain, which leads to Janus kinase 1 (JAK1) and Janus kinase 3 (JAK3) activation and phosphorylation of critical tyrosine residues in the receptor chains (Fig. 4) [23, 36]. JAK1 and JAK3 phosphorylate each other and phosphorylate STAT5. Upon phosphorylation, STAT5 dimerizes and translocates to the nucleus where it activates multiple genes [37]. Autosomal recessive forms of T−B+ SCID are less frequent and have been shown to be caused by mutations in the JAK3 or IL7RA genes [38, 50]. Mutations in the IL7RA gene abrogate T cell development, but do not interfere with NK cell development.
Fig. 4.

a γc/JAK3 signalling pathway (adapted from Gaspar et al. [23]). b T cell receptor with CD3 signalling complex
A separate category of T−B+ SCID patients have mutations in one of the four CD3 genes (CD3G, CD3D, CD3E and CD3Z) [21, 52]. The CD3 complex is composed of one CD3γ, CD3δ and CD3ε chain and two CD3ζ chains (Fig. 4b). The absence of one of the CD3 chains inhibits formation of the CD3 complex and consequently expression and signalling via (pre)T cell receptors.
Gene defects and disease mechanisms in T−B− SCID
Patients with T−B− SCID generally have a defect in V(D)J recombination [15]. This process takes place in developing B and T cells and is responsible for the rearrangement of the immunoglobulin and T cell receptor genes (Fig. 5). Different steps of V(D)J recombination can be discriminated. In the first step, proteins encoded by the recombination activating genes (RAG1 and RAG2) form a heterodimer and make a single-stranded nick between a coding element (Variable (V), Diversity (D), or Joining (J) gene segment and the recombination signal sequence (RSS) [69], resulting in the formation of a hairpin-sealed coding end at the side of the coding element and a blunt signal end at the RSS side (Fig. 4). In the second phase, which is referred to as the processing phase, the DNA–protein kinas (PK) complex, composed of Ku70, Ku80 and DNA–PKcs, binds to the hairpin-sealed coding end and phosphorylates Artemis, which subsequently opens the hairpin. Further processing of the DNA ends takes place, i.e. nucleotide deletions, random non-templated insertions of nucleotides by TdT before the ends, are ligated by LIG4/XRCC4 in conjunction with Cernunnos/XLF. The first phase of V(D)J recombination is lymphoid specific, while the processing and ligation phase are carried out by the ubiquitously expressed components of the non-homologous end-joining pathway (NHEJ) of DNA double-strand breaks [70]. If a mutation occurs in one of the NHEJ factors, the patients not only have defective V(D)J recombination but also a general DNA DSB repair defect, which results in increased sensitivity to ionizing radiation. Mutations have been identified in RAG1, RAG2, Artemis and DNA–PKcs giving rise to typical and atypical SCID [40, 56, 64]. Mutations in LIG4 can give either rise to T−B− SCID or the LIG4 syndrome, which is characterized by microcephaly and growth retardation [26, 65]. Mutations in XLF (Cernunnos) also give rise to these manifestations [9].
Fig. 5.

A schematic representation of the three steps of the V(D)J recombination process and the involved molecules
Detailed analysis of T cells potentially present in patients suspected for having typical or atypical SCID
There are several reasons for the presence of T cells in SCID patients. First, T cells can be engrafted transplacentally from the mother. In 50% of B− SCID and in 80% of B+ SCID, maternal T cells can be detected [41]. These T cells can be present at low frequencies, but can also exceed the upper limit of reference. The immunophenotype of these T cells can be diverse. Most have a mature (CD45RO+) phenotype, but this cannot be regarded as a golden rule. They may have a disturbed CD4/CD8 ratio or aberrant CD3 expression (van der Burg, unpublished observation). To prove that T cells are maternal, they can be analysed by human leukocyte antigen (HLA) typing or the origin determined by XY FISH in case of boys, or short tandem repeat analysis can be performed [41, 68].
Patients with Omenn syndrome have hypomorphic mutations resulting in the presence of T cells that expanded in the periphery [72]. These T cells are autologous and generally oligoclonal. The clonality of the T cells can be determined by flow cytometry, e.g. by using a Vβ analysis kit [63]. Molecular clonality assays by heteroduplex analysis or spectratyping are alternative methods which reliably determine whether the T cells present are oligoclonal, polyclonal or oligoclonal in a polyclonal background [34, 35]. The latter would be predominantly due to infections.
A much rarer explanation for the presence of autologous T cells in SCID patients is the occurrence of somatic reversion mutations [73]. These reversion mutations have been described in a few X-linked SCID cases, in a single RAG deficiency and in patients with a CD3Z deficiency [52, 59, 60, 74]. In these patients, somatic reversion mutation occurred, probably in early T cells, and corrected the genetic defect. If somatic reversion occurs, the T cells have a selective growth advantage and the potential to develop normal function. The mechanism by which this somatic reversion arises is as yet unknown.
T cells in patients with suspected typical or atypical SCID should always be typed in detail. Analysis of TCR expression can be particularly helpful. In some patients with a partial V(D)J recombination defect, a high frequency of TCRγδ T cells is detected [19].
Analysis of protein expression of candidate genes
In T−B+ SCID, analysis of CD132 expression on lymphocytes and measurement of STAT5 phosphorylation upon IL2 stimulations and informative screening tests are advanced [25, 75]. If CD132 expression is absent, this is indicative of X-linked SCID, and in virtually all cases without CD132 expression, a mutation is found in the IL2RG gene. The same holds true for the analysis of IL7RA expression. This is somewhat more complicated because IL7Rα is mainly expressed on T cells, which are typically absent in these patients. Aberrant results in STAT5 phosphorylation [75] point toward defects downstream of the γc chain, and if aberrant, sequence analysis of JAK3 is a logical choice for molecular analysis.
Flow cytometric analysis of precursor B cell compartment in bone marrow
In the case of T−B− SCID, analysis of the precursor B cell compartment in bone marrow can give information whether or not there is an underlying defect in the V(D)J recombination process. A typical SCID patient with a V(D)J recombination defect due to mutations in RAG1, RAG2, Artemis or DNA–PKcs has a full block in precursor B cell differentiation before the cytoplasmic Igμ-positive pre-B-II cell stage (Fig. 3b) [44, 45, 64]. In a hypomorphic mutation, the differentiation block can be incomplete, implying that low frequencies of pre-B-II can be present. Alternatively, an incomplete precursor B cell differentiation block can be due to the type of gene defect. LIG4 and XLF deficiencies give rise to the presence of pre-B-II and immature B cells and even mature B cells in XLF deficiency [30, 65].
Sequence analysis of candidate genes
Based on the clinical presentation and the immunophenotype, a candidate gene is selected, and the gene is sequenced to identify a mutation [66]. The frequency of mutations in T−B+ and T−B− SCID are depicted in Fig. 6. For some genes, e.g. RAG1, RAG2 and Artemis, in vitro function tests can be used to determine the level of (reduced) enzymatic activity of the mutation [49, 67]. In T−B− SCID patients without a defect in the RAG1 or RAG2 gene, it is important to determine whether the patient is sensitive for ionizing radiation and consequently has a defect in the NHEJ pathway using a clonogenic survival assay on fibroblasts cultured from a skin biopsy [40, 56, 64]. Analysis of the coding joints of immunoglobulin gene rearrangements in bone marrow precursor B cells, in vivo V(D)J recombination studies, is a valuable tool in the diagnostic process of radiosensitive T−B− SCID patients because it can give a clue which step in the V(D)J recombination assay is affected [64, 65, 67]. The latter tests are not routinely done in a diagnostic setting, but can be of importance in more complicated cases.
Fig. 6.

Distribution of B+ SCID (n = 159 patients) and B− SCID (n = 136 patients) in Europe according to the ESID patient registry 2010 (http://www.esid.org/statistics.php?sub=2). ESID, European Society for Immunodeficiencies
Supportive management
Infants suspected of having a severe immunodeficiency disorder should be placed in protected isolation, limiting the numbers of persons involved with care; specifically, individuals with respiratory or gastrointestinal symptoms of infection should avoid contact. If the mother is cytomegalovirus (CMV)-negative, breastfeeding should be encouraged—otherwise, it should be discontinued to prevent neonatal CMV infection from being transmitted through breast milk. Strict handwashing procedures are critical to prevent infection. Blood products should be CMV-negative and irradiated to avoid the risk of transfusion GVHD [61]. Appropriate imaging of chest, abdominal organs and brain should be considered, guided by the clinical features. For those diagnosed later, particular attention needs to be paid to nutritional status and the management of dietary intolerances secondary to infectious or inflammatory gastrointestinal problems. Advice from paediatric gastroenterologists should be sought early, to minimise the impact of the disease on the gut and to institute modular formula milk feeds or parenteral nutrition as appropriate. Respiratory paediatricians should be consulted early to maximise supportive therapy and prevent further lung damage. Imaging to detect focal infiltration is important and may guide subsequent biopsy. Infection should be sought aggressively, and biopsy material may be required to demonstrate infection. Culture of appropriate tissue specimens, including bronchoalveolar lavage fluid, and PCR may be needed to identify infecting pathogens—serology is generally unhelpful. Infections should be vigorously treated—broad spectrum multi-agent antimicrobial therapy may be required. Co-trimoxazole as prophylaxis against P. jiroveci should be given. Antifungal prophylaxis should also be used, and antiviral prophylaxis with aciclovir is used in patients with a previous herpes simplex infection. Supporting the emotional needs of the family is also very important.
Curative therapy
Hematopoeitic stem cell transplantation (HSCT) is the treatment of choice for patients with SCID. If HSCT with conditioning chemotherapy is embarked upon, isolation in facilities with positive-pressure-filtered air supply is necessary, mainly to reduce the risk of aspergillosis and droplet-borne viral infections. European data regarding outcome of HSCT for SCID Patient data are collected in the Stem Cell Transplantation for Immunodeficiencies in Europe registry, giving data on almost 700 patients, and have recently been published [24]. A broad repertoire of stem cell sources are used, including stem cells from marrow, mobilised peripheral blood stem cells or those harvested from umbilical cord blood. Best results are obtained using HLA-matched sibling donors, with survival of around 90% in the best circumstances. The molecular defect has a bearing on outcome, with B− SCID patients having an overall worse outcome than B+ forms of SCID [42]. The outcome is better in the absence of infection, arguing for the early identification of patients through neonatal screening programmes [8]. New chemotherapy conditioning regimens are increasingly utilised, with improved outcome. A successful procedure is generally curative, with patients leading normal lives off medication, but few long-term studies have demonstrated long-term sequelae for some patients [39, 58]. Particular problems relate to ongoing thymopoiesis, with failure leading to T lymphocyte senescence in the long term [7, 10]. Long-term immunoglobulin therapy is necessary for some B lymphocyte dysfunction or failure of donor engraftment. Chemotherapy may lead to infertility. Hypothyroidism, secondary to chemotherapy, affects about 10% of patients. Some sequelae relate to the specific genetic defect, for instance, human papillomavirus-associated warts in IL2RG/JAK3 SCID [33] and neurodevelopmental disorders in ADA deficiency [53].
For ADA deficiency, enzyme replacement therapy with polyethelyne-glycosylated ADA is an alternative treatment [29]. Treatment is required lifelong, is expensive and results in only partial immune reconstitution. Sequalae include the development of autoimmunity, but in the short term, it may allow some immune reconstitution and clearance of infection before proceeding to definitive therapy.
Gene therapy has been used for ADA- and IL2RG-deficient SCID [1, 6, 11]. Advantages include removal of the necessity for chemotherapy conditioning and available treatment despite lack of a matched donor. Earlier ADA trials were only partially successful, and the majority of patients required ongoing PEG-ADA therapy. More recently, the procedure has been more successful, although low doses of chemotherapy give the best results [22]. Some patients have an ongoing requirement for immunoglobulin replacement. XL-SCID gene therapy does not require chemotherapy and has led to complete immune reconstitution, but insertion of the retroviral vector close to oncogenes has led to the development of lymphoproliferation in some patients [27]. Development of new, probably safer, vectors and directed insertion of the mutated gene away from oncogenes promise improved outcome, and clinical trials are ongoing [51]. Treatment of other forms of SCID is at a pre-clinical phase.
Concluding remarks
SCID is one of the most severe forms of primary immunodeficiency and is a paediatric emergency, which is life-threatening if recognized too late. Therefore, early diagnosis and good clinical management are crucial. The clinical and immunological spectrum of SCID is broader than initially described, so (atypical) SCID should be considered as a potential diagnosis more often. Improved supportive care, detection of infection by molecular means and less toxic chemotherapy conditioning regimens have significantly improved survival, and patients should be referred urgently to centres specializing in the diagnosis and treatment of such patients to optimize outcome.
Learning points
▪ SCID is one of the most severe forms of PID and is a life-threatening paediatric emergency.
▪ The molecular basis of most forms of SCID is now recognized.
▪ Early careful liaison with the immunology laboratory will enable the most appropriate investigations to be performed.
▪ Atypical, later presentation of patients with partial gene function is increasingly described.
▪ Atypical SCID should be considered in patients presenting with unusual, severe or recurrent infections.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- GVHD
Graft versus host disease
- HSCT
Hematopoietic stem cell transplantation
- NHEJ
Non-homologous end joining
- PID
Primary immunodeficiency
- SCID
Severe combined immunodeficiency
References
- 1.Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo MG. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 2.Appleton AL, Curtis A, Wilkes J, Cant AJ. Differentiation of materno-fetal GVHD from Omenn’s syndrome in pre-BMT patients with severe combined immunodeficiency. Bone Marrow Transplant. 1994;14:157–159. [PubMed] [Google Scholar]
- 3.Benke PJ, Dittmar D. Purine dysfunction in cells from patients with adenosine deaminase deficiency. Pediatr Res. 1976;10:642–646. doi: 10.1203/00006450-197607000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Berrington JE, Flood TJ, Abinun M, Galloway A, Cant AJ. Unsuspected Pneumocystis carinii pneumonia at presentation of severe primary immunodeficiency. Arch Dis Child. 2000;82:144–147. doi: 10.1136/adc.82.2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertrand Y, Muller SM, Casanova JL, Morgan G, Fischer A, Friedrich W. Reticular dysgenesis: HLA non-identical bone marrow transplants in a series of 10 patients. Bone Marrow Transplant. 2002;29:759–762. doi: 10.1038/sj.bmt.1703531. [DOI] [PubMed] [Google Scholar]
- 6.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 7.Borghans JA, Bredius RG, Hazenberg MD, Roelofs H, Jol-van der Zijde EC, Heidt J, Otto SA, Kuijpers TW, Fibbe WE, Vossen JM, Miedema F, van Tol MJ. Early determinants of long-term T-cell reconstitution after hematopoietic stem cell transplantation for severe combined immunodeficiency. Blood. 2006;108:763–769. doi: 10.1182/blood-2006-01-009241. [DOI] [PubMed] [Google Scholar]
- 8.Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, Veys P, Gennery AR, Gaspar HB. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117(11):3243–3246. doi: 10.1182/blood-2010-08-300384. [DOI] [PubMed] [Google Scholar]
- 9.Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 10.Cavazzana-Calvo M, Carlier F, Le Deist F, Morillon E, Taupin P, Gautier D, Radford-Weiss I, Caillat-Zucman S, Neven B, Blanche S, Cheynier R, Fischer A, Hacein-Bey-Abina S. Long-term T-cell reconstitution after hematopoietic stem-cell transplantation in primary T-cell-immunodeficient patients is associated with myeloid chimerism and possibly the primary disease phenotype. Blood. 2007;109:4575–4581. doi: 10.1182/blood-2006-07-029090. [DOI] [PubMed] [Google Scholar]
- 11.Cavazzana-Calvo M, Hacein-Bey S, de Saint BG, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, Bousso P, Deist FL, Fischer A. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 12.Cohen A, Doyle D, Martin DW, Jr, Ammann AJ. Abnormal purine metabolism and purine overproduction in a patient deficient in purine nucleoside phosphorylase. N Engl J Med. 1976;295:1449–1454. doi: 10.1056/NEJM197612232952603. [DOI] [PubMed] [Google Scholar]
- 13.Comans-Bitter WM, De Groot R, Van den Beemd R, Neijens HJ, Hop WCJ, Groeneveld K, Hooijkaas H, Van Dongen JJM. Immunophenotyping of blood lymphocytes in childhood. J Pediatr. 1997;130:388–393. doi: 10.1016/S0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 14.de Saint-Basile G, Le Deist F, de Villartay JP, Cerf-Bensussan N, Journet O, Brousse N, Griscelli C, Fischer A. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome) J Clin Invest. 1991;87:1352–1359. doi: 10.1172/JCI115139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Villartay JP. V(D)J recombination deficiencies. Adv Exp Med Biol. 2009;650:46–58. doi: 10.1007/978-1-4419-0296-2_4. [DOI] [PubMed] [Google Scholar]
- 16.de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, Gougeon ML, Lemainque A, Eidenschenk C, Jouanguy E, Abel L, Casanova JL, Fischer A, Le Deist F. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115:3291–3299. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Vries E. Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. 2006;145:204–214. doi: 10.1111/j.1365-2249.2006.03138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Vries E, Driessen G. Educational paper: primary immunodeficiencies in children: a diagnostic challenge. Eur J Pediatr. 2011;170:169–177. doi: 10.1007/s00431-010-1358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kuhr J, Mascart F, Schmitt-Graeff A, Niemeyer C, Fisch P. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. 2005;115:3140–3148. doi: 10.1172/JCI25221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer A. Severe combined immunodeficiencies (SCID) Clin Exp Immunol. 2000;122:143–149. doi: 10.1046/j.1365-2249.2000.01359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer A, de Saint BG, Le Deist F. CD3 deficiencies. Curr Opin Allergy Clin Immunol. 2005;5:491–495. doi: 10.1097/01.all.0000191886.12645.79. [DOI] [PubMed] [Google Scholar]
- 22.Gaspar HB, Bjorkegren E, Parsley K, Gilmour KC, King D, Sinclair J, Zhang F, Giannakopoulos A, Adams S, Fairbanks LD, Gaspar J, Henderson L, Xu-Bayford JH, Davies EG, Veys PA, Kinnon C, Thrasher AJ. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505–513. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Gaspar HB, Gilmour KC, Jones AM. Severe combined immunodeficiency—molecular pathogenesis and diagnosis. Arch Dis Child. 2001;84:169–173. doi: 10.1136/adc.84.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, Amrolia PJ, Gaspar HB, Davies EG, Friedrich W, Hoenig M, Notarangelo LD, Mazzolari E, Porta F, Bredius RG, Lankester AC, Wulffraat NM, Seger R, Gungor T, Fasth A, Sedlacek P, Neven B, Blanche S, Fischer A, Cavazzana-Calvo M, Landais P. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126(602–610):e601–e611. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 25.Gilmour KC, Cranston T, Loughlin S, Gwyther J, Lester T, Espanol T, Hernandez M, Savoldi G, Davies EG, Abinun M, Kinnon C, Jones A, Gaspar HB. Rapid protein-based assays for the diagnosis of T-B+ severe combined immunodeficiency. Br J Haematol. 2001;112:671–676. doi: 10.1046/j.1365-2141.2001.02578.x. [DOI] [PubMed] [Google Scholar]
- 26.Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 27.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint BG, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 28.Hague RA, Rassam S, Morgan G, Cant AJ. Early diagnosis of severe combined immunodeficiency syndrome. Arch Dis Child. 1994;70:260–263. doi: 10.1136/adc.70.4.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hershfield MS. Enzyme replacement therapy of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase (PEG-ADA) Immunodeficiency. 1993;4:93–97. [PubMed] [Google Scholar]
- 30.IJspeert H, Lankester AC, Van den Berg JM, Wiegant W, Van Zelm MC, Weemaes CMR, Warris A, Pan-Hammarström Q, Pastink A, Van Tol MJD, Van Dongen JJM, Van Gent DC, Van der Burg M (2011) Artemis splice defects cause atypical SCID and can be restored in vitro by an antisense oligonucleotide. Genes Immun (in press) [DOI] [PubMed]
- 31.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 32.Kumaki S, Villa A, Asada H, Kawai S, Ohashi Y, Takahashi M, Hakozaki I, Nitanai E, Minegishi M, Tsuchiya S. Identification of anti-herpes simplex virus antibody-producing B cells in a patient with an atypical RAG1 immunodeficiency. Blood. 2001;98:1464–1468. doi: 10.1182/blood.V98.5.1464. [DOI] [PubMed] [Google Scholar]
- 33.Laffort C, Le Deist F, Favre M, Caillat-Zucman S, Radford-Weiss I, Debre M, Fraitag S, Blanche S, Cavazzana-Calvo M, de Saint BG, de Villartay JP, Giliani S, Orth G, Casanova JL, Bodemer C, Fischer A. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common gammac cytokine receptor subunit or JAK-3 deficiency. Lancet. 2004;363:2051–2054. doi: 10.1016/S0140-6736(04)16457-X. [DOI] [PubMed] [Google Scholar]
- 34.Langerak AW, Szczepanski T, van der Burg M, Wolvers-Tettero ILM, van Dongen JJM. Heteroduplex PCR analysis of rearranged T cell receptor genes for clonality assessment in suspect T cell proliferations. Leukemia. 1997;11:2192–2199. doi: 10.1038/sj.leu.2400887. [DOI] [PubMed] [Google Scholar]
- 35.Langerak AW, van Den Beemd R, Wolvers-Tettero IL, Boor PP, van Lochem EG, Hooijkaas H, van Dongen JJ. Molecular and flow cytometric analysis of the Vbeta repertoire for clonality assessment in mature TCRalphabeta T-cell proliferations. Blood. 2001;98:165–173. doi: 10.1182/blood.V98.1.165. [DOI] [PubMed] [Google Scholar]
- 36.Leonard WJ, Lin JX. Cytokine receptor signaling pathways. J Allergy Clin Immunol. 2000;105:877–888. doi: 10.1067/mai.2000.106899. [DOI] [PubMed] [Google Scholar]
- 37.Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000;19:2566–2576. doi: 10.1038/sj.onc.1203523. [DOI] [PubMed] [Google Scholar]
- 38.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, Vezzoni P, Notarangelo LD. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 39.Mazzolari E, de Martiis D, Forino C, Lanfranchi A, Giliani S, Marzollo R, Airo P, Imberti L, Porta F, Notarangelo LD. Single-center analysis of long-term outcome after hematopoietic cell transplantation in children with congenital severe T cell immunodeficiency. Immunol Res. 2009;44:4–17. doi: 10.1007/s12026-008-8022-4. [DOI] [PubMed] [Google Scholar]
- 40.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/S0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 41.Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood. 2001;98:1847–1851. doi: 10.1182/blood.V98.6.1847. [DOI] [PubMed] [Google Scholar]
- 42.Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, Mahlaoui N, Debre M, Casanova JL, Dal Cortivo L, Madec Y, Hacein-Bey-Abina S, de Saint BG, de Villartay JP, Blanche S, Cavazzana-Calvo M, Fischer A. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood. 2009;113:4114–4124. doi: 10.1182/blood-2008-09-177923. [DOI] [PubMed] [Google Scholar]
- 43.Niehues T, Perez-Becker R, Schuetz C. More than just SCID—the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. 2010;135:183–192. doi: 10.1016/j.clim.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 44.Noordzij JG, De Bruin-Versteeg S, Verkaik NS, Vossen JMJJ, De Groot R, Bernatowska E, Langerak AW, Van Gent DC, Van Dongen JJM. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood. 2002;100:2145–2152. [PubMed] [Google Scholar]
- 45.Noordzij JG, Verkaik NS, Van der Burg M, Van Veelen LR, De Bruin-Versteeg S, Wiegant W, Vossen JMJJ, Weemaes CMR, De Groot R, Zdzienicka MZ, Van Gent DC, Van Dongen JJM. Radiosensitive SCID patients with Artemis gene mutations show a complete B-cell differentiation arrest at the pre-B-cell receptor checkpoint in bone marrow. Blood. 2003;101:1446–1452. doi: 10.1182/blood-2002-01-0187. [DOI] [PubMed] [Google Scholar]
- 46.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–S194. doi: 10.1016/j.jaci.2009.07.053. [DOI] [PubMed] [Google Scholar]
- 47.Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Hammartrom L, Nonoyama S, Ochs HD, Puck J, Roifman C, Seger R, Wedgwood J. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–1178. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pannicke U, Honig M, Hess I, Friesen C, Holzmann K, Rump EM, Barth TF, Rojewski MT, Schulz A, Boehm T, Friedrich W, Schwarz K. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009;41:101–105. doi: 10.1038/ng.265. [DOI] [PubMed] [Google Scholar]
- 49.Poinsignon C, Moshous D, Callebaut I, de Chasseval R, Villey I, de Villartay JP. The metallo-beta-lactamase/beta-CASP domain of Artemis constitutes the catalytic core for V(D)J recombination. J Exp Med. 2004;199:315–321. doi: 10.1084/jem.20031142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 51.Qasim W, Gaspar HB, Thrasher AJ. Progress and prospects: gene therapy for inherited immunodeficiencies. Gene Ther. 2009;16:1285–1291. doi: 10.1038/gt.2009.127. [DOI] [PubMed] [Google Scholar]
- 52.Rieux-Laucat F, Hivroz C, Lim A, Mateo V, Pellier I, Selz F, Fischer A, Le Deist F. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. N Engl J Med. 2006;354:1913–1921. doi: 10.1056/NEJMoa053750. [DOI] [PubMed] [Google Scholar]
- 53.Rogers MH, Lwin R, Fairbanks L, Gerritsen B, Gaspar HB. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J Pediatr. 2001;139:44–50. doi: 10.1067/mpd.2001.115023. [DOI] [PubMed] [Google Scholar]
- 54.Sauer AV, Aiuti A. New insights into the pathogenesis of adenosine deaminase-severe combined immunodeficiency and progress in gene therapy. Curr Opin Allergy Clin Immunol. 2009;9:496–502. doi: 10.1097/ACI.0b013e3283327da5. [DOI] [PubMed] [Google Scholar]
- 55.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, Schneider DT, Manfras B, Pannicke U, Willemze R, Knuchel R, Gobel U, Schulz A, Borkhardt A, Friedrich W, Schwarz K, Niehues T. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358:2030–2038. doi: 10.1056/NEJMoa073966. [DOI] [PubMed] [Google Scholar]
- 56.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W, Seger RA, Hansen-Hagge TE, Desiderio S, Lieber MR, Bartram CR. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 57.Slatter MA, Angus B, Windebank K, Taylor A, Meaney C, Lester T, Norbury G, Hambleton S, Abinun M, Flood TJ, Cant AJ, Gennery AR. Polymorphous lymphoproliferative disorder with Hodgkin-like features in common gamma-chain-deficient severe combined immunodeficiency. J Allergy Clin Immunol. 2011;127:533–535. doi: 10.1016/j.jaci.2010.09.036. [DOI] [PubMed] [Google Scholar]
- 58.Slatter MA, Brigham K, Dickinson AM, Harvey HL, Barge D, Jackson A, Bown N, Flood TJ, Cant AJ, Abinun M, Gennery AR. Long-term immune reconstitution after anti-CD52-treated or anti-CD34-treated hematopoietic stem cell transplantation for severe T-lymphocyte immunodeficiency. J Allergy Clin Immunol. 2008;121:361–367. doi: 10.1016/j.jaci.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 59.Speckmann C, Pannicke U, Wiech E, Schwarz K, Fisch P, Friedrich W, Niehues T, Gilmour K, Buiting K, Schlesier M, Eibel H, Rohr J, Superti-Furga A, Gross-Wieltsch U, Ehl S. Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency. Blood. 2008;112:4090–4097. doi: 10.1182/blood-2008-04-153361. [DOI] [PubMed] [Google Scholar]
- 60.Stephan V, Wahn V, Le Deist F, Dirksen U, Broker B, Muller-Fleckenstein I, Horneff G, Schroten H, Fischer A, de Saint BG. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335:1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 61.Treleaven J, Gennery A, Marsh J, Norfolk D, Page L, Parker A, Saran F, Thurston J, Webb D. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol. 2011;152:35–51. doi: 10.1111/j.1365-2141.2010.08444.x. [DOI] [PubMed] [Google Scholar]
- 62.Turul T, Tezcan I, Artac H, de Bruin-Versteeg S, Barendregt BH, Reisli I, Sanal O, van Dongen JJ, van der Burg M. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur J Pediatr. 2009;168:87–93. doi: 10.1007/s00431-008-0718-x. [DOI] [PubMed] [Google Scholar]
- 63.van den Beemd R, Boor PP, van Lochem EG, Hop WC, Langerak AW, Wolvers-Tettero IL, Hooijkaas H, van Dongen JJ. Flow cytometric analysis of the Vbeta repertoire in healthy controls. Cytometry. 2000;40:336–345. doi: 10.1002/1097-0320(20000801)40:4<336::AID-CYTO9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 64.van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, van Dongen JJ, van Gent DC. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–98. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, Brugmans L, Raams A, Jaspers NG, Zdzienicka MZ, van Dongen JJ, van Gent DC. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116:137–145. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van der Burg M, van Zelm MC, van Dongen JJ. Molecular diagnostics of primary immunodeficiencies: benefits and future challenges. Adv Exp Med Biol. 2009;634:231–241. doi: 10.1007/978-0-387-79838-7_19. [DOI] [PubMed] [Google Scholar]
- 67.van der Burg M, Verkaik NS, den Dekker AT, Barendregt BH, Pico-Knijnenburg I, Tezcan I, vanDongen JJ, van Gent DC. Defective Artemis nuclease is characterized by coding joints with microhomology in long palindromic-nucleotide stretches. Eur J Immunol. 2007;37:3522–3528. doi: 10.1002/eji.200737624. [DOI] [PubMed] [Google Scholar]
- 68.van der Burg M, Weemaes CM, Preijers F, Brons P, Barendregt BH, van Tol MJ, Hoogerbrugge P, van Dongen JJ. B-cell recovery after stem cell transplantation of Artemis-deficient SCID requires elimination of autologous bone marrow precursor-B-cells. Haematologica. 2006;91:1705–1709. [PubMed] [Google Scholar]
- 69.Van Gent DC, McBlane JF, Ramsden DA, Sadofsky MJ, Hesse JE, Gellert M. Initiation of V(D)J recombinations in a cell-free system by RAG1 and RAG2 proteins. Curr Top Microbiol Immunol. 1996;217:1–10. doi: 10.1007/978-3-642-50140-1_1. [DOI] [PubMed] [Google Scholar]
- 70.van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene. 2007;26:7731–7740. doi: 10.1038/sj.onc.1210871. [DOI] [PubMed] [Google Scholar]
- 71.Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082–1086. doi: 10.1016/j.jaci.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 72.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, Gatta LB, Ochs HD, Schwarz K, Notarangelo LD, Vezzoni P, Spanopoulou E. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93:885–896. doi: 10.1016/S0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]
- 73.Wada T, Candotti F. Somatic mosaicism in primary immune deficiencies. Curr Opin Allergy Clin Immunol. 2008;8:510–514. doi: 10.1097/ACI.0b013e328314b651. [DOI] [PubMed] [Google Scholar]
- 74.Wada T, Toma T, Okamoto H, Kasahara Y, Koizumi S, Agematsu K, Kimura H, Shimada A, Hayashi Y, Kato M, Yachie A. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood. 2005;106:2099–2101. doi: 10.1182/blood-2005-03-0936. [DOI] [PubMed] [Google Scholar]
- 75.Walshe D, Gaspar HB, Thrasher AJ, Cale CM, Gilmour KC. Signal transducer and activator of transcription 5 tyrosine phosphorylation for the diagnosis and monitoring of patients with severe combined immunodeficiency. J Allergy Clin Immunol. 2009;123:505–508. doi: 10.1016/j.jaci.2008.11.041. [DOI] [PubMed] [Google Scholar]