

Abstract
Integrins are essential adhesion receptors found on the surfaces of all metazoan cells. As regulators of cell migration and extracellular matrix assembly, these membrane-spanning heterodimers are critical for embryonic development, tissue repair and immune responses. Signals transmitted by integrins from outside to inside the cell promote cell survival and proliferation, but integrin affinity for extracellular ligands can also be controlled by intracellular cues. This bidirectional signaling is mediated by the short cytoplasmic tails of the two integrin subunits. Recent structural and functional studies of various integrin fragments and complexes between the cytoplasmic tails and intracellular proteins, such as talin, have provided new insight into the signaling processes centered around the tails, particularly inside-out integrin activation.
Integrins in Biology
The diverse characteristics of tissues in multicellular organisms arise from specific interactions between cells and their environment. Cells create those environments by laying down extracellular matrix (ECM) components to support the development of various tissue types. Integrins (Box 1) are the major cell surface receptors used to assemble and recognize a functional ECM and to facilitate cell migration to the correct tissue location. Integrins are unusual in their ability to respond both to extracellular and intracellular stimuli, thus signaling bidirectionally across the membrane. The essential roles of integrins in cell development and tissue organization, as well as their potential as therapeutic targets, are now well established [1, 2]. The specificity of integrins for various extracellular ligands, including ECM components, such as fibronectin, laminin and collagen, and receptors on other cells, such as ICAMs (inter-cellular adhesion molecules) has also been reviewed elsewhere [3, 4]. Here we focus on recent experimental evidence, from structural and functional studies, that has clarified integrin signaling mechanisms, especially the unusual process of inside-out signaling which depends on numerous interactions with the cytoplasmic tail of the integrin β-subunit.
Box 1. Integrin structure and conformational flexibility.
Integrins are membrane-spanning heterodimers composed of, in humans, 18 α and 8 β subunits that can combine in at least 24 different ways to make receptors with varying substrate specificity and expression patterns (Box 3) [1, 2]. Their size varies, but typically the α- and β-subunits contain around 1000 and 750 amino acids, respectively. Since the breakthrough crystal structure of the ectodomain αVβ3, published nearly 10 years ago [68], our understanding of the details of integrin structure and function has increased substantially; see reviews [4, 69, 70]. The α-subunit has a β-propeller head, a thigh, two calf domains, a single trans-membrane (TM) domain and a short cytoplasmic tail (Figure ID). Nine of the 18 integrin α chains have an α-I domain, homologous to the β-I domain, inserted between blades 2 and 3 of the β-propeller; apart from the recently solved αxβ2 structure [71], most available ectodomain structures have been of integrins without an α-I domain. The β-subunit starts with a plexin-semaphorin-integrin (PSI) domain followed by a hybrid domain, in which a β-I domain is inserted, followed by four cysteine-rich epidermal growth factor (EGF) modules, a β-tail domain, a TM domain and a cytoplasmic tail. β-I and α-I domains are similar to von Willebrand A domains. The β-leg is apparently more flexible than the α-leg [71, 72]. Figure I shows possible conformational states of integrins, generated using available PDB coordinates. The bent structure is usually taken to represent the inactive integrin state, although it maintains some ability to bind ligand [73]. The upright structures (Figures IB and IC) can be obtained by rotating a few bonds in linker regions, especially those between the calf and thigh domains and between EGF domains 1 and 2. The fully open state (Figure IC) is believed to correspond to the more adhesive, ‘activated’ state. Evidence for conformational flexibility comes from structurally sensitive antibodies [3] and several electron microscopy studies, the latest of which used single copies of intact integrins in membrane nanodiscs to detect structures resembling both the on and off states [9]. There is also evidence for conformational changes in the β-I/hybrid region, where movement of the α7-helix in β-I causes the hybrid domain to swing out [74] (Figure IC). This conformational flexibility of integrins is central to integrin function.
Integrin signaling and activation
Bidirectional integrin signaling involves conformational changes in the heterodimer (Box 1), integrin clustering, the assembly of a large intracellular adhesion complex and, eventually, a return to the resting, inactive, state [5]. These processes depend on various factors including the type of integrin involved and the nature and mechanical properties of the environment, including the ECM [6]. Added complexity arises from extensive cross-talk with other signaling pathways initiated by growth factors and G protein-coupled receptors (GPCRs) [7, 8].
The term ‘integrin activation’ is used to describe processes arising from either outside-in or inside-out signaling. In inside-out activation, a signal generated within the cell leads to increased affinity of the integrin ectodomain for its ECM ligand (Figure 1); in the final stage of this process, talin (Box 2) binds the cytoplasmic tail of the integrin, causing tail separation and distinct conformational changes in the integrin ectodomain (Box 1) [9]. Outside-in activation, by contrast, is brought about when binding to an ECM ligand leads to integrin conformational changes and integrin clustering, or some combination of these, that results in intracellular changes, such as phosphorylation; the net result is increased cell viability, proliferation and growth [10]. Talin also plays a central role in outside-in signaling, by forming a direct linkage between the integrin and the actin cytoskeleton [11]. Mechanical force is known to be a factor in activation and can contribute to both outside-in and inside-out signaling [12, 13]. Despite differences, signaling pathways in both directions converge upon the cytoplasmic tail of the integrin β subunit.
Figure 1.

A simplified version of integrin inside-out activation pathways. A)Engagement of an agonist with a GPCR receptor increases the Ca2+ and DAG concentration, activating a GEF that activates Rap1. This induces Rap1 binding to RIAM, which is believed to recruit talin to the membrane. Talin can exist in both an autoinhibited and activated state (Box 2). Other possible activation pathways of talin include interactions with PIPKIγ/PIP2 and cleavage of the head by calpain--both of which serve to release inhibition of the talin head by the talin rod. However, the full details and relative importance of these pathways are not yet well worked out. (i) Tthe integrin ‘off’ state is stabilized by binding of filamin or other PTB-containing proteins, rather than talin, to the β-tail and by tyrosine phosphorylation of the β-tail by Src family kinases; phosphorylation promotes the binding of PTB-containing proteins such as Dok1, thus preventing the binding of talin F3. The binding of other PTB-containing talin competitors, such as ICAP, is not dependent upon integrin tyrosine phosphorylation. Members of the kindlin family can enhance the effect of talin in producing the integrin ‘on’ state (ii). B)The underlying domain structure in some of the cartoons shown in the main picture is illustrated for the talin head (Box 2), kindlin-1, RIAM and Src family kinases.
Box 2. Talin.
Talin is a large, 2541 amino acid, intracellular protein that is a key player in the activation of integrins. Vertebrates express two isoforms of talin; talin1 is widely expressed, whereas talin2 is found primarily in striated muscles and in the brain [75]. Talin has an N-terminal head region of ~50kDa and an elongated helical rod of ~220kDa [11]. The head contains a FERM domain [76] whose name derives from its presence in the four proteins, band four-point one, ezrin, radixin and moesin. FERM domains have around 300 amino-acids with three sub-domains, usually called F1, F2 and F3. The F1 sub-domain in talin has a 30 residue insertion and is preceded by an ‘F0’ sub-domain, which has an ubiquitin-like fold, like the F1 domain [46] . The talin head domain has a linear arrangement of sub-domains rather than the clover leaf structure usually found in most FERM domains [47]. The F3 sub-domain is homologous to PTB domains and binds directly to integrin β-tails, forming an interface with both an integrin NPxY motif (the canonical PTB domain binding site) [77] and an integrin membrane-proximal helix [41] (Figures 2 and 3). F3 also binds to a region of PIPKIγ [27] and layilin, a cell surface hyaluronan receptor [78]. The talin rod domain consists of bundles of α-helices that can interact with vinculin at multiple sites, especially if talin is subjected to mechanical stress [11]. Talin has an F-actin binding site near the C-terminal end and it can thus provide a direct link between the β-integrin tail and the actin cytoskeleton. There is evidence that talin exists in several conformational states: monomer and dimer as well as ‘open’ and auto-inhibited ‘closed’ states [11]. An autoinhibited state that can be relieved by PIP2 has been identified [79], and the structure of this inhibitory complex formed between the F3 sub-domain and a helical bundle in the rod has been presented [80]. Autoinhibited talin can be activated by the Rap1–RIAM and calpain pathways as well as PIP2 (Figure 1); the relative importance of these three pathways is not yet clear.
Outside-in and inside-out signaling involve the regulated assembly and disassembly of numerous components. These time-dependent adhesion complexes formed during signaling depend on various factors, including tension [14]. The constituents of these adhesion complexes, sometimes known as the integrin ‘adhesome’, form a network of at least 156 proteins, linked by many hundreds of protein–protein interactions [15, 16]. Many of the constituents are adapter proteins that bring together the various components of the dynamic assembly in the right place at the right time. These adapters are usually constructed from identifiable folded domains (e.g., LIM[R1], pleckstrin homology (PH), Src homology 2 (SH2), SH3, phosphotyrosine binding (PTB); see e.g. SMART database, http://smart.embl-heidelberg.de/) linked or terminated by stretches of disordered polypeptide regions designed to facilitate dynamic and flexible protein–protein interactions [17]. Other members of the complex include enzymes that can, for example, phosphorylate or dephosphorylate residues to modulate the binding properties (see below). Talin, paxillin, filamin, integrin linked kinase (ILK) and focal adhesion kinase (FAK) are prominent members of the adhesome [5, 10, 15].
The cytoplasmic tails have no enzymatic or actin-binding activity themselves but provide a hub for protein complex assembly. More than 19 of the 90 core components in the adhesome bind directly to α-integrin tails [15] (a recent review lists 42 proteins that have been reported to bind to β-tails [18]). The α–subunit cytoplasmic tails are also involved in interactions e.g. α4 binds paxillin [19], but β-tail interactions are better characterized and the β-tails are more highly conserved than α-tails [18, 20]. The α-subunit seems to be a primary determinant of extracellular ligand binding specificity, whereas the β-tail is the main moderator of intracellular interactions [1] (Box 3). The β-tail also functions as a hub for adhesome interactions that lead to outside-in signaling [18, 21], but here we will mainly focus on the role of integrin β-tail interactions that are associated with inside-out signaling.
Box 3. Integrin heterogeneity.
There are at least 24 unique integrin αβ heterodimers found in mammals; these occupy a spectrum of biological niches, with differing tissue specificities and extracellular ligand preferences [1, 2]. Although they share a conserved architecture and general mechanism of action, some clear functional differences have emerged, particularly between integrins that play specialized roles in the circulatory system (e.g. the platelet integrin αIIbβ3) and those in adherent cells (e.g. most β1 integrins). Whereas αIIbβ3, for example, exists in a default ‘off’ state, only to be activated at specific biological moments, the integrins of adherent cells often exist in a more active state. These differences are manifested in the differing phenotypes observed in knock-out and knock-in studies in mice, involving β1 [81, 82] and β3 [83, 84]. Functionally, interesting differences have emerged in how these integrins are affected by key partners, including talin [45], kindlin [51] and filamin [85]. Interesting differences have also been noted recently in the fine details of integrin–talin interactions and how these appear to be correlated with biological niche; for example, striated muscle–where there is a need for strong attachment–utilizes the isoforms β1D and talin2, which have an unusually high affinity for each other; see main text and [43]. The current state of our understanding of integrin activation has largely been informed by studies of β3 integrins; until the recent structure of αXβ2 [71], for example, all atomic resolution structures of full integrin ectodomains involved β3. It will be important to keep in mind these functional and structural differences as the fine details of integrin activation are explored.
The initial signaling steps in inside-out integrin activation
Integrins are normally expressed on the cell surface in an inactive state, unable to bind to their receptors. This inactivity can be an essential attribute; in blood platelets, for example, inappropriate integrin activation will lead to thrombosis; excessive activation in adherent cells can also disrupt function [22]. Integrin activation in platelets and leukocytes has been the focus of most studies, but the phenomenon is important in many tissues where ECM remodeling, angiogenesis and cell migration are involved. There are several known triggers for integrin activation; these include chemokine–chemokine receptor interactions in leukocytes [23], thrombin– ‘protease activated receptor’ interactions in platelets [24] and T-cell receptor engagement in T-cells [25]. An early step in a simplified activation pathway is an increase in Ca2+ and diacyl glycerol (DAG) concentration, leading to activation of a guanine nucleotide exchange factor (GEF); this activates Rap1, a member of the small GTPase family, by promoting GDP/GTP exchange (Figure 1). Active Rap1 then interacts with RIAM (Rap1–GTP-interacting adapter molecule; a member of the MIG10, RIAM and lamellipodin (MRI) family), an adapter protein that contains an RA (Ras association)-like domain, a PH domain, and proline-rich sequences (Figure 1). There is evidence that RIAM acts as a scaffold that connects membrane targeting sequences in Rap1 to talin, resulting in the recruitment of talin to the plasma membrane (Box 2) [24, 26].
Related but parallel activation pathways involve phosphatidylinositol (4,5)-bisphosphate (PIP2) and calpain. The local concentration of PIP2 is increased by stimulated activity of the phosphatidylinositol phosphate kinase type Iγ (PIPKIγ) enzyme, leading to the association of talin with the integrin tail [11]. PIPK1γ also targets talin to the membrane by a direct interaction between a C-terminal peptide region of PIPK1γ and the talin F3 domain [27]. Another aspect of the multifaceted role of PIPK1γis the suggestion that it plays an inhibitory role in αLβ2 integrin activation [28, 29]. Cleavage by the calcium-stimulated protease, calpain, is also believed to release an active talin head domain [30], although such cleavage also primes talin head for ubiquitylation and degradation by increasing its affinity for SMAD specific E3 ubiquitin protein ligase 1 (SMURF1). Phosphorylation of talin head by cyclin-dependent kinase 5 (CDK5) inhibits this degradation [31].
The integrin ‘off’ state, and the role of the TM domains
The current view in the field is that association of integrin α and β transmembrane (TM) and membrane proximal (MP) cytoplasmic segments results in an inactive, resting receptor (Box 1, Figure IA and Figure 2A). Evidence for this comes from disulfide cross-linking [32], activating mutations [33] and fluorescent resonance energy transfer (FRET) of labeled cytoplasmic tails [34]. Recent studies have provided new insight into the structure of the resting state. The structures of β3 and αIIb TM segments in phospholipid bicelle model membranes have been solved by NMR separately and in complex [35]. A similar structure was obtained for the TM region in intact αIIbβ3 using disulfide-based distance restraints combined with protein modeling [36].
Figure I.

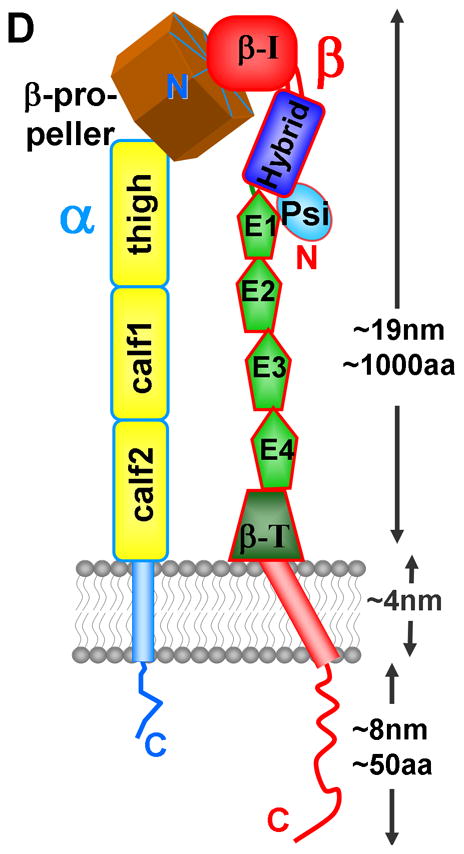
Schematic views of integrin conformations. A) A compact extracellular structure of the type seen in X-ray structures, together with the TM domains and the cytoplasmic tails (this image was generated from PDB coordinates of an ectodomain [72], 3IJE, and a complex of an α/β TM complex [37], 2K9J ; B) a possible intermediate conformation, generated from the view in IA by rotating bonds in the integrin ‘legs’ and separating the tails; C) a possible conformation of the ‘on’ state where the hybrid domain(blue) has swung out after engagement with a ligand (green cylinder). (This image was generated using PDB coordinates 1TYE [74] as well as 3IJE and 2K9J.) D) illustrates the location of the various domains in many integrins, using the same color code as A-C. E1–E4 are EGF-like domains and β-T is the β–tail domain. The cytoplasmic tails are truncated in B and C. The images in IA–C were generated using PyMOL (Delano Scientific).
Figure 2.
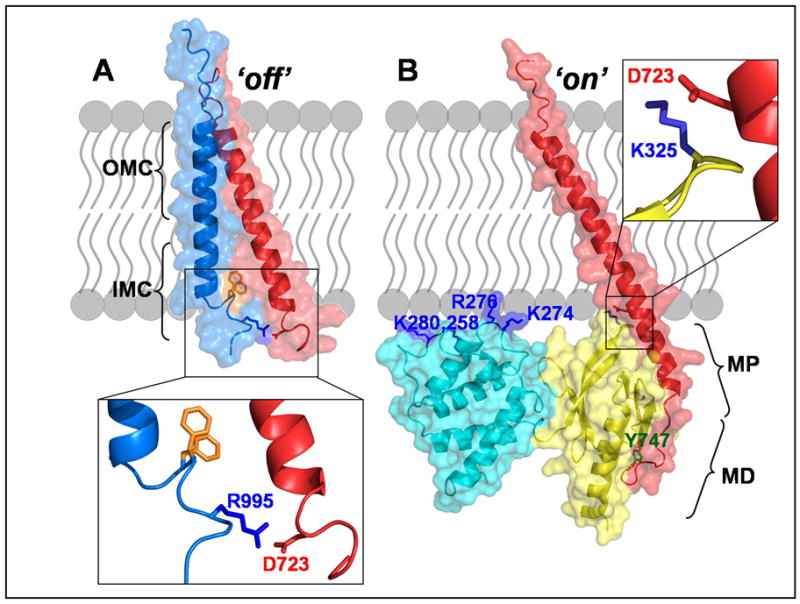
Integrin activation at the membrane-cytoplasm interface. A) The inactive state of the integrin is represented by the complex formed between the transmembrane segments of the α (light blue) and β (red) integrin subunits (PDB coordinates 2K9J). This interaction consists of both an outer membrane clasp (OMC) and an inner membrane clasp (IMC), which includes a pair of α tail phenylalanines highlighted in orange. B) The complex formed between the β-cytoplasmic tail (red) and the F2 (cyan) and F3 (yellow) sub-domains of the talin head illustrates the activated integrin. This model was constructed from PDB coordinates 2RMZ and 3G9W. The membrane proximal (MP) and membrane distal (MD) regions of the tail are shown, as is the key MD residue, Y747 (green) which is in the first NPxY sequence of the β-tail. A cluster of positively charged residues is also indicated on the talin F2-domain. In both panels, the expanded boxes show the salt bridge that forms between the β tail residue D723 and either the α tail residue R995 (A) or the talin residue K325 (B). (Notes: the residue numbering in this figure refers to αIIb, β3 and talin2; the disordered β-tail residues after position ~750 are not shown here.)
In the NMR structure of the αIIbβ3 TM complex, which is probably a good representation of the resting state, the αIIb helix is approximately perpendicular to the membrane whereas the β3 helix is tilted [37] (Figure 2A). Within the membrane, the α/β helix-helix interface forms the ‘outer membrane clasp’, which is mediated by glycines that allow close helix packing. An ‘inner membrane clasp’ (IMC) is also made from an unusual αIIb backbone reversal that packs a consecutive pair of Phe residues against the β3 TM helix; this arrangement promotes an electrostatic interaction between αIIb(D723) and β3(R995), which also contributes to the IMC. This D723–R995 salt bridge had previously been proposed to explain the stabilizing effect of these residues on the integrin inactive state [38].
The two TM segments have essentially the same structure when studied separately or in a complex, suggesting that the topological features of the TM segments will remain unchanged in the separated ‘active’ state [35]. How the separation of TM domains couples to the state of the ectodomains is still unclear, particularly given that recent structures suggest that the linkers between them are flexible [36, 37].
The role of talin in activation
In vertebrates, the binding of talin (Box 2) to the cytoplasmic tail of integrin-β subunits is a key step in integrin activation [39]. Characterization of the structural details of the integrin–talin interaction was comparatively difficult because of the low affinity and the poor behavior of integrin peptides in solution; thus extensive protein engineering was required. The first insight into the interface between the β3 NPxY motif and the talin F3 domain emerged from a crystal structure of a short membrane distal (MD) fragment of the β3 tail covalently tethered to the talin1 F2–F3 fragment [40]. Further features of the interface between the β3 MP region and the talin1 F3 domain came from an NMR structure that employed a chimeric peptide of the β3 MP helix attached to a sequence from PIPK1γ, which binds tightly to talin [41]. The later discovery that β1D made a relatively tight complex with talin2 led to the first structure determination of an authentic β-tail in complex with a talin fragment [42]. It is now clear that the talin F3 sub-domain binds to the MD portion of the integrin tail via a typical PTB domain–NPxY motif interaction and to the MP helix of the integrin tail in a manner that is apparently unique to the talin F3 domain (Figure 2). The structure of full-length β1D integrin tail bound to the F2–F3 domains of talin2 revealed that residues M755-N788 of the β1D tail form an elongated interface with the F3 domain of talin2. The MP interface, which is largely hydrophobic, and the MD interface, which contains numerous hydrogen bonds, appear to act relatively independently in terms of protein binding [42, 43] (Figure 2).
A working activation model is that formation of the talin F3–β MP interaction destabilizes interactions between the integrin transmembrane and cytoplasmic domains. One contribution to the destabilization is disruption of the αIIb(D723)/β3(R995) salt bridge by the formation of a new salt bridge between D723 and a lysine in talin F3 (Figure 2). Full integrin activation requires additional action by talin, possibly by inducing an altered tilt in the β-TM helix [42]. Although binding of the F3 domain to the β tail is sufficient for integrin activation [44], other domains in the talin head contribute to activation [45]. Interactions between negatively charged membrane phospholipids, such as PIP2, with positively charged patches on F3 [41], F2 [42] (Figure 2) and a loop in F1 [46] (see inset in Figure 1) also are important for integrin activation and cell migration. The various positively charged clusters of residues in the linear arrangement of talin head domains are well suited to make extensive interactions with a membrane bilayer [47] and thus exert a force that tilts the β-TM helix to promote tail separation.
Other factors contributing to integrin activation
Inside-out integrin signaling appears to involve more interactions than simply those between talin and integrin. Recently, the kindlin family of proteins has emerged as key players in assisting talin activation of integrins [48]. Kindlins have FERM domains with talin-like features, such as an N-terminal F0 domain and a large flexible F1 loop [49]. The PTB-like sub-domain within the kindlin FERM domain is similar to that of talin but binds to the second NPxY motif in β-integrin tails [50, 51], in contrast to talin, which binds to the first motif. Despite the substantial evidence in favor of kindlin playing a key role in integrin activation (particularly of β3 integrins) the details remain uncertain, and kindlin can inhibit as well as activate β1 integrins in some circumstances [51].
The mechano-transduction properties of integrins are known to be important [6], and plausible models have been presented for how force might influence the generation of the ‘on’ state (Box 1, Figure IA) [13]. However, a recent cryo-electron microscopy study on intact integrins in membrane nanodiscs showed that the talin head can induce extension of unclustered integrins in the absence of force or other membrane proteins [9]. Thus, although mechanical force and kindlin binding can contribute to integrin activation, the central molecular events in triggering integrin activation are the interactions between the talin F3 domain and the β-integrin tail and between the talin head and negatively charged lipid head groups.
Regulation of the concentration of the tail–talin complex
The formation of a precise complex between the β-tail, talin and the membrane is thus the key final step in inside–out activation. The cellular environment carefully regulates the amount of that complex that forms; as mentioned above, the local concentration of active talin can be modulated by PIP2, Rap1 and calpain (Box 2 and Figure 1). A variety of other mechanisms are also in place to control integrin activation by changing the amount of talin–tail complex in the cell, as discussed below.
Affinity control
The affinity of the β-tail for talin is carefully adjusted for different environments: the higher the affinity, the more activating complex there will be for a given amount of activated talin. The sequences of some cytoplasmic β-tails (Figure 3) indicate that they are quite similar; however, their talin affinities can vary widely. For example, the affinity ofβ3 for talin2 is ~450μM, whereas the affinity of two splice variants of β1 are ~35μM (β1D) and ~650μM (β1A). Because β1D and talin2 are expressed primarily in striated muscle cells, the relatively high affinity of these two isoforms for each other might fulfill a need to have a high fraction of integrins switched on in a tissue with high stresses [42, 43] (Box 3).
Figure 3.

Aligned sequences of some selected β-integrin cytoplasmic tails (those that are highly studied and share a large degree of sequence homology). The numbering of the most studied tail, β3, is also shown. Note the conserved Asp (D) at position 723 in β3 and the two NPxY/F sequences in each tail (see also Figure 2). There is evidence that the Tyr residues (shown in green) can be phosphorylated, much like some of the Ser and Thr residues. Regions of β3 that bind to different proteins are also shown. The length of the shaded ‘linker’ region has a strong influence on affinity for talin.
Before forming a complex with talin, the β tails are essentially disordered, as shown by NMR dynamics studies, although the MP region has propensity to form an α-helix. Disordered protein regions often act as ‘hubs’ for promiscuous interactions with several different partners; the disorder gives rise to weak but specific interactions because of an entropic cost in forming the complex [17]. This characteristic appears to facilitate the dynamic control of integrin adhesion. The subtle control that can be exerted in the formation of a complex with a partially disordered partner was illustrated recently by a study of differences between β3 and β1 complexes [43]. β3 integrins are more readily switched on than β1 integrins [45], yet the overall affinities of β3 and β1 for talin (arising from contribution from both MP and MD regions – Figure 2) are similar. An explanation for this observation was provided by experiments showing that the affinity of the MP region for talin is much higher in β3 than inβ1 because the helix in that region is preformed, thus the entropic cost of binding to talin is reduced[43]. It was also demonstrated that the weak affinity observed in talin–tail complexes has been tuned by biology to be optimal, given that removal of two-residues in the tail increased its talin affinity roughly 1,000-fold [43]. The β2 tail, which is one residue shorter than other tails in the same MD-MP linker region (Figure 3), also has a higher affinity for talin [52]. (The length of a flexible loop in the ectodomain has also been shown recently to modulate the amount of integrin in the inactive state in a similar fashion [53].)
Competition and phosphorylation
The fraction of active talin–integrin tail complex can also be changed by other proteins that compete with talin for the β-integrin tail. Structural analyses have revealed an overlap between talin- and filamin-binding sites on β-integrin tails (Figure 3), thus explaining how filamin–talin competition can regulate integrin activation [54]. The phosphorylation state of the tyrosine residues within the NPxY motifs of β3 can also differentially regulate β3 interactions with proteins containing PTB domains. Phosphorylated tyrosine is required for high affinity binding to some PTB domains (e.g. in Src homology 2 domain containing transforming protein 1 (Shc) and docking protein 1, 62kDa (Dok1)), whereas ICAP1α (integrin beta 1 binding protein 1) [55], talin, and kindlin PTB domains have similar or higher affinity for non-phosphorylated peptides. Dok1, for example, binds phosphorylated Y747 on β3 with much higher affinity than talin, thus reducing the activation complex by competition [56, 57]. By contrast, phosphorylation reduces the affinity of integrins for kindlin [58], further contributing to the inactivating effect of phosphorylation. However, this effect involves the second NPxY position (β3 Y759) as opposed to the first (Y747), which is predominant in Dok1 and talin binding. Interestingly, the Shc PTB–β3 complex, which is involved in outside-in signaling is primarily defined by the phosphorylation of Y759 rather than Y747 [59]. Other types of phosphorylation have also been implicated; for example, in activated cells, Thr758 of β2 tails is phosphorylated, leading to 14-3-3 protein recruitment to β2 integrins. 14-3-3 proteins bind the phosphorylated integrin tail, whereas filamin only binds the unphosphorylated tail [60].
It is clear from these observations that cells go to considerable lengths to achieve the right level of integrin adhesion by subtle variations in the affinity of these complexes and by numerous competitive interactions that can be fine tuned by phosphorylation.
Concluding remarks and future perspectives
A number of powerful genetic, structural biology and imaging techniques have been used to good effect in the last 10 years to bring about a remarkable increase in our understanding of integrin activation. Our knowledge of integrin structure and integrin complexes has increased considerably, although some uncertainties remain; for example how are TM and ectodomain movements coupled to talin engagement with the β-tail? It will also be important to carry out more experiments to validate structural conclusions at the cellular level (see e.g. [61]). The role of talin is now relatively well understood although the precise role of kindlin as a helper is still uncertain. The regulated formation of dynamic integrin adhesions of various sizes and numerous proteins is an area where we are some way short of a full understanding, but developments such as a recent cryo-electron tomography view of an adhesion complex [62] will continue to improve our understanding. How integrin adhesions are broken down is another area of relative uncertainty, although it is believed that phosphorylation and calpain cleavage are contributors. The pathogenic effects of genetic defects in integrin adhesion proteins are quite well studied [10, 63], and the dysregulated expression of adhesome proteins such as FAK and ILK have been implicated in various pathologies including cancer [64]. The application of integrin based inhibitors has in some ways been disappointing, with some limited success targeting integrins of the vasculature and immune system, but the increased level of understanding in recent years is likely to lead to improved therapies [65–67].
Acknowledgments
IDC thanks BBSRC, MRC and the NIH cell migration consortium for financial support. NJA is currently supported by an NIH post-doctoral grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Barczyk M, et al. Integrins. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humphries JD, et al. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo BH, et al. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009;122:159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker EL, Zaman MH. The biomechanical integrin. J Biomech. 2010;43:38–44. doi: 10.1016/j.jbiomech.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 8.Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 9.Ye F, et al. Recreation of the terminal events in physiological integrin activation. J Cell Biol. 2010;188:157–173. doi: 10.1083/jcb.200908045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Legate KR, et al. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397–418. doi: 10.1101/gad.1758709. [DOI] [PubMed] [Google Scholar]
- 11.Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu Rev Biophys. 2009;38:235–254. doi: 10.1146/annurev.biophys.050708.133744. [DOI] [PubMed] [Google Scholar]
- 12.Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. doi: 10.1016/j.immuni.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Zhu J, et al. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfenson H, et al. The heel and toe of the cell's foot: a multifaceted approach for understanding the structure and dynamics of focal adhesions. Cell Motil Cytoskeleton. 2009;66:1017–1029. doi: 10.1002/cm.20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaidel-Bar R, et al. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphries JD, et al. Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci Signal. 2009;2:ra51. doi: 10.1126/scisignal.2000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tompa P, et al. Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays. 2009;31:328–335. doi: 10.1002/bies.200800151. [DOI] [PubMed] [Google Scholar]
- 18.Legate KR, Fassler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009;122:187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 19.Deakin NO, et al. An integrin-alpha4-14-3-3zeta-paxillin ternary complex mediates localised Cdc42 activity and accelerates cell migration. J Cell Sci. 2009;122:1654–1664. doi: 10.1242/jcs.049130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wegener KL, Campbell ID. Transmembrane and cytoplasmic domains in integrin activation and protein-protein interactions (review) Mol Membr Biol. 2008;25:376–387. doi: 10.1080/09687680802269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nieves B, et al. The NPIY motif in the integrin beta1 tail dictates the requirement for talin-1 in outside-in signaling. J Cell Sci. 2010;123:1216–1226. doi: 10.1242/jcs.056549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shattil SJ, Ginsberg MH. Integrin signaling in vascular biology. J Clin Invest. 1997;100:S91–95. [PubMed] [Google Scholar]
- 23.Evans R, et al. Integrins in immunity. J Cell Sci. 2009;122:215–225. doi: 10.1242/jcs.019117. [DOI] [PubMed] [Google Scholar]
- 24.Shattil SJ, et al. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker RG, Koretzky GA. Regulation of T cell integrin function by adapter proteins. Immunol Res. 2008;42:132–144. doi: 10.1007/s12026-008-8047-8. [DOI] [PubMed] [Google Scholar]
- 26.Lee HS, et al. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Pereda JM, et al. Structural basis for phosphatidylinositol phosphate kinase type Igamma binding to talin at focal adhesions. J Biol Chem. 2005;280:8381–8386. doi: 10.1074/jbc.M413180200. [DOI] [PubMed] [Google Scholar]
- 28.Wernimont SA, et al. PIPKI gamma 90 negatively regulates LFA-1-mediated adhesion and activation in antigen-induced CD4+ T cells. J Immunol. 2010;185:4714–4723. doi: 10.4049/jimmunol.1001445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wernimont SA, et al. Calpain 4 is not necessary for LFA-1-mediated function in CD4+ T cells. PLoS One. 2010;5:e10513. doi: 10.1371/journal.pone.0010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- 31.Huang C, et al. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat Cell Biol. 2009;11:624–630. doi: 10.1038/ncb1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo BH, et al. Locking the beta3 integrin I-like domain into high and low affinity conformations with disulfides. J Biol Chem. 2004;279:10215–10221. doi: 10.1074/jbc.M312732200. [DOI] [PubMed] [Google Scholar]
- 33.Partridge AW, et al. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J Biol Chem. 2005;280:7294–7300. doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 34.Kim M, et al. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 35.Ulmer TS. Structural basis of transmembrane domain interactions in integrin signaling. Cell Adh Migr. 2010;4:243–248. doi: 10.4161/cam.4.2.10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, et al. The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol Cell. 2009;34:234–249. doi: 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau TL, et al. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. Embo J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hughes PE, et al. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 39.Tadokoro S, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 40.Garcia–Alvarez B, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 41.Wegener KL, et al. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 42.Anthis NJ, et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. Embo J. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anthis NJ, et al. Structural diversity in integrin/talin interactions. Structure. 2010:18. doi: 10.1016/j.str.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calderwood DA, et al. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–21758. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- 45.Bouaouina M, et al. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J Biol Chem. 2008;283:6118–6125. doi: 10.1074/jbc.M709527200. [DOI] [PubMed] [Google Scholar]
- 46.Goult BT, et al. Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 2010;29:1069–1080. doi: 10.1038/emboj.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elliott PR, et al. The Structure of the talin head reveals a novel extended conformation of the FERM domain. Structure. 2010;18:1289–1299. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meves A, et al. The Kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol. 2009;19:504–513. doi: 10.1016/j.tcb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Goult BT, et al. The structure of the N-terminus of kindlin-1: a domain important for alphaiibbeta3 integrin activation. J Mol Biol. 2009;394:944–956. doi: 10.1016/j.jmb.2009.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moser M, et al. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 51.Harburger DS, et al. Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem. 2009;284:11485–11497. doi: 10.1074/jbc.M809233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhunia A, et al. NMR solution conformations and interactions of integrin alphaLbeta2 cytoplasmic tails. J Biol Chem. 2009;284:3873–3884. doi: 10.1074/jbc.M807236200. [DOI] [PubMed] [Google Scholar]
- 53.Smagghe BJ, et al. Modulation of integrin activation by an entropic spring in the {beta}-knee. J Biol Chem. 2010;285:32954–32966. doi: 10.1074/jbc.M110.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiema T, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21:337–347. doi: 10.1016/j.molcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 55.Millon-Fremillon A, et al. Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent beta1-integrin affinity. J Cell Biol. 2008;180:427–441. doi: 10.1083/jcb.200707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oxley CL, et al. An integrin phosphorylation switch: the effect of beta3 integrin tail phosphorylation on Dok1 and talin binding. J Biol Chem. 2008;283:5420–5426. doi: 10.1074/jbc.M709435200. [DOI] [PubMed] [Google Scholar]
- 57.Anthis NJ, et al. Beta integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. J Biol Chem. 2009;284:36700–36710. doi: 10.1074/jbc.M109.061275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bledzka K, et al. Tyrosine phosphorylation of integrin beta3 regulates kindlin-2 binding and integrin activation. J Biol Chem. 2010;285:30370–30374. doi: 10.1074/jbc.C110.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deshmukh L, et al. Integrin {beta}3 phosphorylation dictates its complex with the Shc phosphotyrosine-binding (PTB) domain. J Biol Chem. 2010;285:34875–34884. doi: 10.1074/jbc.M110.159087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takala H, et al. Beta2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood. 2008;112:1853–1862. doi: 10.1182/blood-2007-12-127795. [DOI] [PubMed] [Google Scholar]
- 61.Kopp PM, et al. Studies on the morphology and spreading of human endothelial cells define key inter- and intramolecular interactions for talin1. Eur J Cell Biol. 2010;89:661–673. doi: 10.1016/j.ejcb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patla I, et al. Dissecting the molecular architecture of integrin adhesion sites by cryo-electron tomography. Nat Cell Biol. 2010;12:909–915. doi: 10.1038/ncb2095. [DOI] [PubMed] [Google Scholar]
- 63.Etzioni A. Genetic etiologies of leukocyte adhesion defects. Curr Opin Immunol. 2009;21:481–486. doi: 10.1016/j.coi.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Wang S, Basson MD. Integrin-linked kinase: a multi-functional regulator modulating extracellular pressure-stimulated cancer cell adhesion through focal adhesion kinase and AKT. Cell Oncol. 2009;31:273–289. doi: 10.3233/CLO-2009-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cox D, et al. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 66.Auzzas L, et al. Targeting alphavbeta3 integrin: design and applications of mono- and multifunctional RGD-based peptides and semipeptides. Curr Med Chem. 2010;17:1255–1299. doi: 10.2174/092986710790936301. [DOI] [PubMed] [Google Scholar]
- 67.Cantor JM, et al. Integrin-associated proteins as potential therapeutic targets. Immunol Rev. 2008;223:236–251. doi: 10.1111/j.1600-065X.2008.00640.x. [DOI] [PubMed] [Google Scholar]
- 68.Xiong JP, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arnaout MA, et al. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19:495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Askari JA, et al. Linking integrin conformation to function. J Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie C, et al. Structure of an integrin with an alphaI domain, complement receptor type 4. EMBO J. 2010;29:666–679. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiong JP, et al. Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. J Cell Biol. 2009;186:589–600. doi: 10.1083/jcb.200905085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adair BD, et al. Three-dimensional EM structure of the ectodomain of integrin {alpha}V{beta}3 in a complex with fibronectin. J Cell Biol. 2005;168:1109–1118. doi: 10.1083/jcb.200410068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao T, et al. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senetar MA, et al. Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motil Cytoskeleton. 2007;64:157–173. doi: 10.1002/cm.20173. [DOI] [PubMed] [Google Scholar]
- 76.Fehon RG, et al. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calderwood DA, et al. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci U S A. 2003;100:2272–2277. doi: 10.1073/pnas.262791999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wegener KL, et al. Structural basis for the interaction between the cytoplasmic domain of the hyaluronate receptor layilin and the talin F3 subdomain. J Mol Biol. 2008;382:112–126. doi: 10.1016/j.jmb.2008.06.087. [DOI] [PubMed] [Google Scholar]
- 79.Goksoy E, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell. 2008;31:124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goult BT, et al. The structure of an interdomain complex that regulates talin activity. J Biol Chem. 2009;284:15097–15106. doi: 10.1074/jbc.M900078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 82.Czuchra A, et al. Genetic analysis of beta1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hodivala-Dilke KM, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Law DA, et al. Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature. 1999;401:808–811. doi: 10.1038/44599. [DOI] [PubMed] [Google Scholar]
- 85.Calderwood DA, et al. Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol. 2001;3:1060–1068. doi: 10.1038/ncb1201-1060. [DOI] [PubMed] [Google Scholar]