

Abstract
The Kv1 family voltage-dependent K+ channels assemble with cytosolic β subunits (Kvβ), which are composed of a flexible N terminus followed by a structured core domain. The N terminus of certain Kvβs inactivates the channel by blocking the ion conduction pore, and the core domain is a functional enzyme that uses NADPH as a cofactor. Oxidation of the Kvβ-bound NADPH inhibits inactivation and potentiates channel current, but the mechanism behind this effect is unknown. Here we show that after oxidation, the core domain binds to part of the N terminus, thus restraining it from blocking the channel. The interaction is partially mediated by two negatively charged residues on the core domain and three positively charged ones on the N terminus. These results provide a molecular basis for the coupling between the cellular redox state and channel activity, and establish Kvβ as a target for pharmacological control of Kv1 channels.
Keywords: aldo-keto reductase, double-mutant cycle analysis, electrostatic interactions, voltage-dependent potassium channel
Voltage-dependent K+ channels open in response to membrane depolarization, and many of them quickly close, i.e., inactivate, even when the depolarization is maintained. Inactivating K+ current regulates firing frequencies of a neuron and is essential for integration of synaptic inputs (1–3). There are several known mechanisms of channel inactivation, and one of them, N-type inactivation, was first elucidated by Aldrich et al. (4–7) in the drosophila Shaker K+ channel. They showed that the distal N terminus of the Shaker K+ channel blocks the ion conduction pathway and thus causes inactivation, and the N terminus has since been referred to as an inactivation gate. This mechanism was nicknamed “ball-and-chain” type inactivation, because the blocker, the “ball,” is tethered to the channel by a stretch of unstructured amino acids, the “chain.” A similar mechanism was also reported in voltage-dependent Na+ channels (8). In mammalian Kv1 family channels, which are closely related to the Shaker K+ channel, an inactivation gate can come from the channel itself, as in Kv1.4 channels (9, 10), or from an associated β subunit (Kvβ), such as Kvβ1 and Kvβ3 (11–14).
Kvβ is a cytosolic protein that associates with the tetramerization domains (T1) of Kv1 channels to form a stable complex (15–22). There are three Kvβ genes in mammals, Kvβ1–3, which all encode a highly conserved core domain ~330 residues long (23). Studies in Kvβ1 and Kvβ2 have shown that the conserved cores are functional aldo-keto reductases (AKR) that use NADPH as a cofactor (24–26). In addition to the core domain, Kvβ1 has an unstructured N-terminal segment, ~70 amino acids long, which closes the channel by the N-type inactivation mechanism (13, 27, 28). Perfusion of a synthetic peptide corresponding to just the first 24 amino acids of Kvβ1 is sufficient to induce channel inactivation, indicating that the inactivation gate is contained within that region (13). Further structural and functional studies have shown that binding of the inactivation gate to the channel is largely determined by the first few residues and that the receptor site for the inactivation gate is the hydrophobic central cavity located approximately midway through the ion conduction pathway (27, 29).
The AKR activity of the Kvβ1 core and the ability of the Kvβ1 N terminus to induce channel inactivation are linked. Conversion of the bound NADPH to NADP+, whether enzymatically by a substrate, nonenzymatically by an oxidant such as H2O2, or by exchange with NADP+, leads to an increase in current (24, 25). Although it has been shown that the potentiation of current is mainly due to a partial elimination of channel inactivation (24), how this change in inactivation is coupled to oxidation of NADPH remains unknown. In this study we therefore set out to determine the molecular mechanism by which N-type inactivation is modulated by the redox state of the cofactor on Kvβ1.
Results
Part of the Kvβ1 N Terminus Affects Redox Modulation.
To examine if residues on the N terminus of Kvβ1 are involved in redox modulation, residues 2–70 of Kvβ1 were mutated to alanine one or two at a time. The wild-type (WT) and mutant Kvβs were then coexpressed with Kv1.1, and channel modulation by the known Kvβ substrate 4-cyanobenzaldehyde (4-CY) was measured (Fig. 1). When an inside-out patch expressing Kv1 and Kvβ1 was perfused with 4-CY, a large increase in current was observed (Fig. 1 A and B and Fig. S1). Most of the mutant Kvβ1s had a similar response to that of the WT: 4-CY induced a large increase in current, and the ΔIss, which is defined as change in the steady-state current normalized by the initial peak current, was not significantly different from that of the WT Kvβ1 (Fig. 1 C and E and Fig. S1). However, alanine mutations to residues from 33 to 40 and 43 to 50 resulted in a significantly smaller ΔIss (Fig. 1 D and E and Fig. S1). We define these two segments collectively as the redox regulation sequence (RRS).
Fig. 1.

Residues 33–50 of the Kvβ1 N terminus mediate redox modulation. (A–D) K+ current recorded on inside-out patches expressing Kv1.1 with (A, B) Kvβ1 WT, (C) Kvβ1 P55A-Q56A, or (D) Kvβ1 I45A-I46A. In each case, the black trace was recorded before 4-CY or vehicle (1% ethanol) perfusion. After perfusion, a patch was held at −100 mV and its current level was monitored every 30 s with a 200-ms pulse to +60 mV until the current level reached steady state. The solution was then exchanged to the normal inside buffer and the red trace was recorded. This protocol was used for current traces shown in all figures. Scale bars represent 300 pA and 10 ms. Only the first 40 ms of each trace is shown, and the full 200-ms traces are shown in Fig. S1. (E) Bar graph of the 4-CY effect (ΔIss) on alanine mutations to residues 2–70 of the Kvβ1 N terminus. The error bars are SEM from 5 to 15 independent patches. **P < 0.01, ***P < 0.001 vs. Kvβ1 WT by 4-CY; unpaired Student's t test.
The effect of each mutation on channel inactivation, i.e., rates of onset of and recovery from inactivation, was also measured. Many of the mutations to residues 2–11 had a large effect on the recovery rate, consistent with their known function as the inactivation gate (27). Mutations to residues in the RRS had a less than twofold effect on the rates of onset of and recovery from inactivation (Fig. S2), suggesting that residues in the RRS are not directly involved in interactions with the inactivation gate receptor site.
To verify that the RRS is indeed required for redox modulation, we did two experiments. First, we produced a mutant, Kvβ1 (Δ33–50), by deleting residues 33–50 of Kvβ1. When Kvβ1 (Δ33–50) was coexpressed with Kv1.1, inactivating current was still observed, indicating that the shortened N terminus can still access and bind the inactivation gate receptor site on the channel. However, 4-CY induced only a small change in channel current, not significantly different from that of the vehicle control but significantly lower than that of the WT (Fig. 2 A and D).
Fig. 2.

The RRS is required for redox modulation. (A–C) K+ current recorded from inside-out patches coexpressing (A) Kv1.1 and Kvβ1 (Δ33–50); (B) Kv1.1 and Kv4.2N-Kvβ1 chimera, of which the first 32 residues of Kvβ1 were replaced by the first 40 residues of Kv4.2; and (C) Kv1.1 and Kv4.2N-Kvβ1 (18gly), of which the RRS of the chimera was replaced by 18 glycines. The black and red traces were recorded before and after perfusion of 4-CY. (D) Bar graph of ΔIss by 4-CY. The error bars are SEM from 6 to 15 independent patches. *** indicates P < 0.001 and n. s. indicates not significantly different (P > 0.05); ANOVA followed by Tukey's post hoc test.
Second, to further demonstrate that the RRS is important for redox modulation and that the inactivation gate itself is not involved, we constructed a chimera, Kv4.2N-Kvβ1, in which the first 32 residues of Kvβ1 containing the inactivation gate were replaced by the N-terminal inactivation gate from the Kv4.2 channel. The Kv4.2N-Kvβ1 chimera induced N-type inactivation when coexpressed with Kv1.1, and, more importantly, when NADPH was oxidized the inactivation was inhibited with a ΔIss of 0.62 ± 0.01 (n = 8; Fig. 2 B and D). The same chimera with residues 33–50 of Kvβ1 substituted with 18 glycines, Kv4.2N-Kvβ1 (18gly), was also capable of inactivation, but in contrast the current was not potentiated by substrate perfusion (ΔIss = 0.08 ± 0.003; n = 9; Fig. 2 C and D). These experiments confirmed the necessity of the RRS for mediating redox modulation.
Electrostatic Interaction Between the RRS and AKR Core.
Because NADPH oxidation happens on the core domain of Kvβ1, and because neither the N-terminal (Fig. S3) nor the C-terminal (24) cytoplasmic region of the channel is necessary for redox modulation, we reasoned that the RRS may mediate redox modulation by interacting with the core domain. In particular, the presence of three positively charged residues on the RRS suggests that there are electrostatic interactions between the RRS and the core.
This hypothesis was examined in three experiments. First, we mutated the three positively charged residues on the RRS one at a time to glutamate. Reversing the charge on R48 abolishes redox modulation; the ΔIss is 0.10 ± 0.01 (n = 8; Fig. 3A), which is not significantly different from the vehicle control. The ΔIss values of R37E and K39E are 0.18 ± 0.01 (n = 4) and 0.33 ± 0.01 (n = 4; Fig. 3A), respectively, significantly smaller than that of the WT but also significantly larger than the vehicle control, suggesting that these individual mutations remain partially redox sensitive. Because the two residues are close to each other, we mutated both to glutamate at the same time, and the double mutant, R37E-K39E, had a ΔIss of 0.09 ± 0.01 (n = 6; Fig. 3A and Fig. S4), not significantly different from that of the vehicle control. In addition, each of the three single charge-reversal mutants had a smaller ΔIss than the charge-neutral double alanine mutants shown in Fig. 1. Combined, these results suggest that electrostatic interactions may play a role in redox modulation.
Fig. 3.

Charged residues on the RRS and Kvβ1 core mediate redox modulation. (A) The 4-CY effect (ΔIss) measured on charge reversal mutations on the RRS and the core domain of Kvβ1. The error bars are SEM from 3 to 15 independent patches. ***P < 0.001 vs. Kvβ1 WT by 4-CY; unpaired Student's t test. (B) The tetrameric core domain of Kvβ2 as viewed looking down the fourfold axis, which is marked with a “+” symbol, from the membrane-facing side. The two glutamate residues, equivalent to E265 and E349 in Kvβ1, are shown as red space-fill. The N terminus of the Kvβ2 core is labeled with an N for each subunit. (C) A monomer of Kvβ2 core, with NADP shown in cyan and the N terminus of the Kvβ2 core marked with a blue sphere. Representative currents for three mutations are shown in Fig. S4.
We then looked for negatively charged residues on the Kvβ1 core involved in redox modulation. The crystal structure of the Kvβ2 core (30), whose amino acid sequence is 85% identical to that of Kvβ1, was used as a guide to identify glutamate and aspartate residues located on the surface. There are a total of 34 negatively charged residues on the core of Kvβ1 and 27 of them are predicted to reside on the solvent-accessible surface. All of the surface-exposed negatively charged residues were mutated one at a time to an arginine, except for E314 and E315, which were simultaneously mutated due to their proximity, and the effect on redox modulation was measured. Only two mutations, E265R and E349R, have significantly smaller ΔIss values, whereas all others had a ΔIss not significantly different from that of the WT (Fig. 3A and Fig. S4). To eliminate concerns that the two mutations may reduce ΔIss by simply reducing the enzymatic activity, we expressed and purified the two mutant proteins and examined their enzymatic activity. Both mutants had a similar rate of NADPH oxidation to that of the WT (Fig. S5).
Finally, we investigated if these charged residues on the RRS and the core interact with each other. In the Kvβ2 crystal structure, which contains an NADP+ (30), the two glutamate residues equivalent to E265 and E349 are located roughly 14 Å apart on the channel-facing side of the tetramer (Fig. 3 B and C). This distance could be easily covered by the 10 residues between R37 and R48, suggesting that the oppositely charged residues interact with each other. We reasoned that if the loss of redox modulation by a charge reversal mutation on the RRS, e.g., R48E, can be rescued by a charge reversal mutation on the core, e.g., either E265R or E349R, then the pair likely interact. This is based on the concept of double-mutant cycle analysis, which has previously been used successfully to pinpoint interacting residues between a peptide and a protein (27, 31). Indeed, a rescue was observed with the double mutation R48E-E349R, which has a ΔIss (0.57 ± 0.03; n = 6; Fig. 4 A and B) almost as large as that of the WT. In contrast, the double mutation R48E-E265R has a ΔIss (0.11 ± 0.01; n = 5; Fig. 4A) not substantially different from the two single mutations. The rescue of redox modulation by a charge swap, combined with its specificity, strongly suggests that R48 and E349 physically interact with each other. Similarly, when the R37E-K39E double mutant was paired with either E265R or E349R, a rescue was observed for the triple mutations R37E-K39E-E265R, with a ΔIss of 0.57 ± 0.02 (n = 6), but not for the R37E-K39E-E349R mutation, with a ΔIss of 0.10 ± 0.01 (n = 5; Fig. 4A). Thus, R37 and K39 may both interact with E265, whereas R48 may interact with E349.
Fig. 4.

Charge-swap double-mutant rescue of redox modulation. (A) 4-CY effects (ΔIss) measured on double-mutations of Kvβ1. The error bars are SEM from 5 to 15 independent patches. ***P < 0.001 vs. Kvβ1 WT; unpaired Student's t test. (B) K+ current recorded on inside-out patches expressing Kv1.1 with Kvβ1 WT, R48E, E349R, or the double mutant R48E-E349R, before (black) and after (red) perfusion of 4-CY. Scale bars represent 300 pA and 10 ms.
Further Test of the Redox Modulation Mechanism.
Because the affinity of the receptor site for the inactivation gate itself is unaffected by the redox state of the Kvβ cofactor (Fig. S6), we hypothesized that the interaction between the RRS and the core physically restrains the N terminus from reaching its receptor site. If the RRS interacts with the enzymatic core of Kvβ1, then only the first 32 residues of the 70-residue N terminus are unrestrained. Although we do not know precisely which residue on the Kvβ1 core interacts with the first residue (V33) of the RRS, the linear distance on Kvβ between residue 265, which interacts with residue R37, and the receptor site of the inactivation gate is ~70 Å (15). This distance is likely too long to be covered by a 32 amino acid unstructured peptide, which is predicted to have a mean end-to-end distance of 38–61 Å if modeled as a self-avoiding random walk (32, 33).
This model allows us to make two testable predictions. First, if enough amino acid residues are inserted before the RRS on Kvβ1, then the N terminus will still have sufficient length to reach the inactivation gate even after the RRS binds to the core, rendering the channel insensitive to redox modulation. However, if the same number of amino acid residues is inserted after the last residue on the RRS, then redox modulation will not be affected. To test this, glycine residues were inserted between amino acid 32 and 33 of Kvβ1. Whereas inserting 10 glycine residues did not significantly alter redox modulation of channel inactivation with a ΔIss of 0.56 ± 0.01 (n = 5; Fig. 5 A and D), inserting 20 glycine residues dramatically decreased ΔIss to 0.13 ± 0.004 (n = 6; Fig. 5 B and D). Furthermore, redox modulation was preserved when the 20 glycine residues were inserted after residue L50 (Fig. 5 C and D). These results are in full agreement with the hypothesized redox modulation mechanism.
Fig. 5.

Further test of the model for redox modulation. (A–C) K+ current recorded from inside-out patches coexpressing (A) Kv1.1 with Kvβ1 (10gly32), of which 10 glycines were inserted after V32; (B) Kv1.1 with Kvβ1 (20gly32), of which 20 glycines were inserted after V32; or (C) Kv1.1 with Kvβ1 (20gly50), of which 20 glycines were inserted after L50. The black and red traces were recorded before and after perfusion of 4-CY. Scale bars represent 300 pA and 10 ms. (D) ΔIss after 4-CY modulation. The error bars are SEM from 5 to 15 independent patches. *** indicates P < 0.001 and n. s. indicates not significantly different (P > 0.05); ANOVA followed by Tukey's post hoc test.
Second, if electrostatic interactions contribute significantly to binding of the RRS to the core, redox modulation should be sensitive to the ionic strength of the medium. Redox modulation of N-type inactivation was therefore measured in an intracellular solution containing 450 mM K+ instead of the regular 150 mM. As shown in Fig. S7, 4-CY induced a modest change in channel current with a ΔIss of 0.13 ± 0.01 (n = 8). The smaller ΔIss was not due to a compromised enzymatic activity in high K+ concentration, because after the reaction was complete, perfusion of the normal intracellular solution restored the ΔIss to 0.67 ± 0.07 (n = 3). These results are consistent with the hypothesis that electrostatic interactions contribute to redox modulation.
Discussion
In conclusion, we have identified a stretch of residues on Kvβ1, the RRS, which mediates redox modulation, and we identified two negatively charged residues on the enzymatic core that likely interact with positively charged residues on the RRS. These results have led us to propose a model for redox modulation where the binding of the RRS to the core of Kvβ1 shortens the N terminus to a point that the inactivation gate can no longer block the pore (Fig. 6).
Fig. 6.

A cartoon model for redox modulation of N-type inactivation. When bound to NADPH (Left), the inactivation gate on the N terminus of Kvβ1 (green) was able to reach the receptor site on the Kv1 channel (shown with its membrane-spanning domain in purple and intracellular T1 domain in blue) and prevent K+ flux. When the cofactor was oxidized (Right), residues R37 and R48 (blue spheres) on the N terminus interacted with E265 and E349 (red spheres) on the Kvβ core, restraining the inactivation gate and permitting K+ flux. For clarity, only one Kvβ subunit is shown with an N terminus drawn as a black line.
Constraining access of the inactivation gate to the channel could be a general mechanism for modulation of channel activities. Ruppersberg et al. (34) found that oxidation of the seventh cysteine residue on Kvβ1 eliminated inactivation, likely due to formation of a disulfide bond, either between the inactivation gates or with other regions of the channel or Kvβ. N-type inactivation induced by Kvβ1 was compromised in the presence of phosphoinositides, likely by immobilization of the inactivation gate to the cell membrane (35). In addition, coexpression of leucine-rich glioma inactivated gene 1 (LGI1) with the Kv1 and Kvβ1 complex inhibits inactivation (36), and although the mechanism is less clear in this case, it may also involve restraining the N terminus of Kvβ by the LGI1 protein.
What prevents the RRS from interacting with the core domain when the core is bound to NADPH? One possibility is that the distance between residues E265 and E349 changes, likely by lengthening, when the Kvβ-bound cofactor is reduced, so that the two negatively charged residues become incapable of simultaneously interacting with all three positively charged residues on the RRS. Alternatively, one or both negatively charged residues could become solvent-inaccessible when the core domain is bound to NADPH. Further studies will be necessary to reveal the conformational change on Kvβ resulting from NADPH-to-NADP+ conversion. In addition, it should be possible to identify small-molecule compounds that target Kvβ and alter the interaction of the RRS and the core domain. These compounds should be able to modulate channel activity and have specificity to only the Kv1 family channels because of the exclusive assembly between Kvβ and Kv1 (18, 20, 21, 37).
Methods
Molecular Biology.
Full-length cDNA of rat Kv1.1 (accession no. NM_173095) or rat Kvβ1 (accession no. NM_017303) was cloned into a modified pBluescript vector (a gift from Dr. Mark Sonders, Columbia University, New York, NY) between the KpnI and EcoRI sites for in vitro transcription. The Kv4.2N-Kvβ1 chimera was generated by splicing the coding sequence for residues 1–40 of Kv4.2 (accession no. NM_031730) onto the coding sequence for residues 33–401 of Kvβ1 by the overlapping PCR method. This construct was then used as a template to generate Kv4.2N-Kvβ1 (18gly), of which residues 33–50 of Kvβ1 were replaced by 18 glycines. Kvβ1 (10gly32), Kvβ1 (20gly32), and Kvβ1 (20gly50) were constructed by inserting 10 or 20 glycines after V32 or L50 of Kvβ1, respectively. Kv1.1 (Δ1–34) was generated by amplifying the DNA sequence encoding residues 35–495-stop using the following two primers: CCGGTACCATGTGCTGCGAGCGCGTGG and CCGAATTCTTAAACATCGGTCAGG. Each PCR product was then inserted into the same modified pBluescript vector. Single or double site-directed mutations and the deletion mutation Kvβ1 (Δ33–50) were made using the QuikChange kit (Agilent Technologies). The sequences of all constructs were verified by DNA sequencing through the entire coding region. Kvβ1 has a cysteine residue at position 7 that can be oxidized on inside-out patches to affect channel inactivation (13). To eliminate concerns that the change in inactivation could be due to cysteine oxidation, we mutated the seventh position cysteine to an alanine for all of the constructs of Kvβ1 and used Kvβ1 (C7A) as the WT throughout this study. In separate experiments, 4-CY was found to induce similar changes on Kv1.1 coexpressed with the true WT Kvβ1 with the seventh position cysteine in the presence of 5 mM DTT (Fig. S8).
Electrophysiology and Data Analysis.
Protocols for mRNA preparation, electrophysiological recordings, and data analysis have been described in detail in previous publications (24, 25). K+ currents were elicited by holding the patch at −100 mV for at least 30 s and stepping to +60 mV for 200 ms. ΔIss was calculated by the following equation:
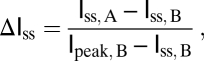 |
where Iss, A and Iss, B are the steady-state currents, measured at 200 ms after depolarization, after and before 4-CY oxidation, respectively. Ipeak, B is the peak current amplitude before 4-CY oxidation. The rates of onset of and recovery from inactivation were measured following the same protocols as in ref. (27).
For ionic strength manipulation of redox modulation, the regular intracellular solution contained (in mM): 100 KCl, 5 EGTA, and 50 KH2PO4 at pH 7.4. The pH was adjusted with KOH. The high ionic strength solution was identical to the regular solution except with 400 mM KCl.
Protein Expression and Fluorescence Measurement of the Single Turnover Enzymatic Reaction.
Expression of the Kvβ2 conserved core (accession no. CAA54142, residues 36–367) and the mutants has been described in ref. (38). The Kvβ2-bound NADPH fluorescence spectrum was recorded at room temperature (20–22 °C). The reaction mixture had 2 μM of Kvβ2 protein and 5 mM 4-CY in a final volume of 150 μL. The excitation wavelength was set at 360 nm with a slit size of 1-nm bandpass and the emission was measured at 454 nm with a slit size of 5-nm bandpass at various time points after the reaction was initiated with addition of 4-CY. The decrease of NADPH fluorescence was well-fit by a single exponential function, and the inverse of the time constant was defined as the rate of the oxidation reaction (24, 25).
Data Statistics.
The Origin 7.5 software package was used for statistical analysis of the data. The results are expressed as mean ± SEM. Student's t test and one-way ANOVA followed by Tukey's post hoc test were used to assess changes of a mean value. The statistical value of P < 0.05 is considered significant.
Supplementary Material
Acknowledgments
M.Z. is grateful to R. MacKinnon for encouragement and advice. This work was supported by the US National Institutes of Health (HL086392 to M.Z. and T32HL087745 to E.J.L.), the American Heart Association (0630148N to M.Z. and 0826067D to Y.P.), and the March of Dimes Birth Defects Foundation (research Grant 5-FY06-20 to M.Z.). M.Z. is a Pew Scholar in Biomedical Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1100316108/-/DCSupplemental.
References
- 1.Connor JA, Stevens CF. Prediction of repetitive firing behaviour from voltage clamp data on an isolated neurone soma. J Physiol. 1971;213:31–53. doi: 10.1113/jphysiol.1971.sp009366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dodson PD, Forsythe ID. Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004;27:210–217. doi: 10.1016/j.tins.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Roeper J, Pongs O. Presynaptic potassium channels. Curr Opin Neurobiol. 1996;6:338–341. doi: 10.1016/s0959-4388(96)80117-6. [DOI] [PubMed] [Google Scholar]
- 4.Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 5.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 6.Murrell-Lagnado RD, Aldrich RW. Energetics of Shaker K channels block by inactivation peptides. J Gen Physiol. 1993;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murrell-Lagnado RD, Aldrich RW. Interactions of amino terminal domains of Shaker K channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong CM, Bezanilla F, Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;62:375–391. doi: 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stühmer W, et al. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989;8:3235–3244. doi: 10.1002/j.1460-2075.1989.tb08483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comer MB, et al. Cloning and characterization of an Ito-like potassium channel from ferret ventricle. Am J Physiol. 1994;267:H1383–H1395. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- 11.Heinemann SH, Rettig J, Graack HR, Pongs O. Functional characterization of Kv channel beta-subunits from rat brain. J Physiol. 1996;493:625–633. doi: 10.1113/jphysiol.1996.sp021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinemann SH, Rettig J, Wunder F, Pongs O. Molecular and functional characterization of a rat brain Kv beta 3 potassium channel subunit. FEBS Lett. 1995;377:383–389. doi: 10.1016/0014-5793(95)01377-6. [DOI] [PubMed] [Google Scholar]
- 13.Rettig J, et al. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 14.Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- 15.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 16.Parcej DN, Eckhardt-Strelau L. Structural characterisation of neuronal voltage-sensitive K+ channels heterologously expressed in Pichia pastoris. J Mol Biol. 2003;333:103–116. doi: 10.1016/j.jmb.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Sokolova O, et al. Conformational changes in the C terminus of Shaker K+ channel bound to the rat Kvbeta2-subunit. Proc Natl Acad Sci USA. 2003;100:12607–12612. doi: 10.1073/pnas.2235650100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- 20.Parcej DN, Scott VE, Dolly JO. Oligomeric properties of alpha-dendrotoxin-sensitive potassium ion channels purified from bovine brain. Biochemistry. 1992;31:11084–11088. doi: 10.1021/bi00160a018. [DOI] [PubMed] [Google Scholar]
- 21.Sewing S, Roeper J, Pongs O. Kv beta 1 subunit binding specific for shaker-related potassium channel alpha subunits. Neuron. 1996;16:455–463. doi: 10.1016/s0896-6273(00)80063-x. [DOI] [PubMed] [Google Scholar]
- 22.Shen NV, Chen X, Boyer MM, Pfaffinger PJ. Deletion analysis of K+ channel assembly. Neuron. 1993;11:67–76. doi: 10.1016/0896-6273(93)90271-r. [DOI] [PubMed] [Google Scholar]
- 23.Pongs O, et al. Functional and molecular aspects of voltage-gated K+ channel beta subunits. Ann N Y Acad Sci. 1999;868:344–355. doi: 10.1111/j.1749-6632.1999.tb11296.x. [DOI] [PubMed] [Google Scholar]
- 24.Pan Y, Weng J, Cao Y, Bhosle RC, Zhou M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J Biol Chem. 2008;283:8634–8642. doi: 10.1074/jbc.M709304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage-dependent Shaker family potassium channels by an aldo-keto reductase. J Biol Chem. 2006;281:15194–15200. doi: 10.1074/jbc.M513809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tipparaju SM, Barski OA, Srivastava S, Bhatnagar A. Catalytic mechanism and substrate specificity of the beta-subunit of the voltage-gated potassium channel. Biochemistry. 2008;47:8840–8854. doi: 10.1021/bi800301b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 28.Morales MJ, Castellino RC, Crews AL, Rasmusson RL, Strauss HC. A novel beta subunit increases rate of inactivation of specific voltage-gated potassium channel alpha subunits. J Biol Chem. 1995;270:6272–6277. doi: 10.1074/jbc.270.11.6272. [DOI] [PubMed] [Google Scholar]
- 29.Decher N, et al. Structural determinants of Kvbeta1.3-induced channel inactivation: a hairpin modulated by PIP2. EMBO J. 2008;27:3164–3174. doi: 10.1038/emboj.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel beta subunit. Cell. 1999;97:943–952. doi: 10.1016/s0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- 31.Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- 32.Kohn JE, et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damaschun G, Damaschun H, Gast K, Zirwer D. Denatured states of yeast phosphoglycerate kinase. Biochemistry (Mosc) 1998;63:259–275. [PubMed] [Google Scholar]
- 34.Ruppersberg JP, et al. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- 35.Oliver D, et al. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 36.Schulte U, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 37.Rhodes KJ, et al. Voltage-gated K+ channel beta subunits: expression and distribution of Kv beta 1 and Kv beta 2 in adult rat brain. J Neurosci. 1996;16:4846–4860. doi: 10.1523/JNEUROSCI.16-16-04846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Y, et al. Cortisone dissociates the Shaker family K+ channels from their beta subunits. Nat Chem Biol. 2008;4:708–714. doi: 10.1038/nchembio.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.