

Abstract
We found a spontaneous autosomal mutation in a mouse leading to neutrophil infiltration with ulceration in the upper dermis of homozygous offspring. These animals had increased neutrophil numbers, associated with normal lymphocyte count, in peripheral blood and bone marrow, suggesting a myeloproliferative disorder; however, granulocyte precursor proliferation in bone marrow was actually reduced (because circulating neutrophils were less susceptible to apoptosis). Neutrophil infiltration of the skin and other organs and high serum levels of immunoglobulins and autoantibodies, cytokines, and acute-phase proteins were additional abnormalities, all of which could be reduced by high-dose corticosteroid treatment or neutrophil depletion by antibodies. Use of genome-wide screening localized the mutation within an 0.4-Mbp region on mouse chromosome 6. We identified insertion of a B2 element in exon 6 of the Ptpn6 gene (protein tyrosine phosphatase, non-receptor type 6; also known as Shp-1). This insertion involves amino acid substitutions that significantly reduced the enzyme activity in mice homozygous for the mutation. Disease onset was delayed, and the clinical phenotype was milder than the phenotypes of other Ptpn6-mutants described in motheaten (me, mev) mice; we designated this new genotype as Ptpn6meB2/meB2 and the phenotype as meB2. This new phenotype encompasses an autoinflammatory disease showing similarities to many aspects of the so-called neutrophilic dermatoses, a heterogeneous group of skin diseases with unknown etiology in humans.
The motheaten (me) mouse phenotype with patchy absence of hair and pigment in the skin was first identified in 1965 in a C57BL/6 mouse at the Jackson Laboratory and was published in 1975.1 The me mutation is a deletion of a cytosine residue in the Ptpn6 gene (protein tyrosine phosphatase, non-receptor type 6; also known as Shp-1) that generates a frame shift and a premature stop codon, whereby the Ptpn6 protein is truncated and becomes functionally inactive.2,3 Since the motheaten phenotype was first described,1,4,5 additional me forms have been discovered,6,7 all associated with different mutations in the mouse Ptpn6 gene. However, both the type of mutation in the Ptpn6 gene and the environmental conditions can dramatically affect the clinical (inflammatory) symptoms.7
Protein tyrosine phosphatases (PTPs) comprise a large family of 107 proteins, both receptor and nonreceptor types, and both human and murine. For reviews, see Alonso et al8 and Mustelin et al.8,9 The PTPN6 human and Ptpn6 murine proteins are nonreceptor PTPs and possess a relatively unique structure, with two SH2 domains at the N-terminus and a catalytic domain at the C-terminus of the enzyme (see Supplemental Figure S1B at http://ajp.amjpathol.org). Aliases and synonyms in the literature include Ptp1C, SH-PTP1, and Hcph.
Apparently, both SH2 domains [exons 2 to 3 (SH2N) and exons 4 to 5 (SH2C)] are required for binding to the phosphorylated tyrosine residues of the target protein, and the catalytic (phosphatase) domain (exons 7 to 13), which has a highly conserved crystal structure,10 is responsible for tyrosine dephosphorylation.11 Ptpn6 is thought to down-regulate signaling pathways of a number of cytokine receptors, T-cell and B-cell receptors (TCR and BCR), and chemokine and integrin receptors via tyrosine dephosphorylation of the cytoplasmic domains of these receptors.12–14 The SH2 tandem repeat occurs in only one other nonreceptor phosphatase (PTPN11; also known as SHP-2), otherwise, the dual SH2 binding (docking) site is typical for the Syk family of protein tyrosine kinases.15 The other special feature of the Ptpn6 gene is that it has two promoter regions. The two translation start sites (ATG) are 7 kbp apart, and, whereas the longer form (IA) is expressed mostly in epithelial cell types, the slightly shorter (IB) transcript of Ptpn6 is expressed mostly in hematopoietic cells.16,17
Consistent with the importance of Ptpn6, mice harboring the me/me mutation have a short lifespan (2 to 3 weeks). The viable me mutation (me-v) has two forms of alternative splicing, both of which affect the phosphatase catalytic domain3; however, the mutant Ptpn6 protein maintains approximately 10% to 20% activity of the wild-type enzyme.2,18 This phosphatase activity is, however, insufficient for normal intracellular regulation, and the lifespan of me-v/me-v mice is only slightly longer (9 to 10 weeks) than that of me/me mice.6 The third and most recently described motheaten phenotype, spin [ie, mice with spontaneous inflammation (Ptpn6m1Btlr)], is associated with a longer lifespan, compared with previously described me phenotypes: male and female mice are fertile and survive past 1 year of age.7
Here, we describe a new spontaneous mutation, the insertion of a B2 element into exon 6 of the mouse Ptpn6 gene leading to alteration in the normal function of Ptpn6. Homozygous Ptpn6meB2/meB2 (meB2) mice develop early skin inflammation of the paws and later an autoinflammatory disease involving other organs. The meB2 mice identified in our colony had an intermediate lifespan (between that of the me and the spin mice), and the homozygous mice could reach 6 to 8 months of age without treatment. We characterized this novel meB2 mutation, and intercrossed the me and me-v lines to generate combined Ptpn6 heterozygosity. We concluded that altered phosphatase function due to the defects in Ptpn6 drives this autoinflammatory disease. Many of the clinical symptoms and laboratory parameters associated with the meB2 phenotype bear a resemblance to those described in human neutrophilic dermatoses (Sweet's syndrome and pyoderma gangrenosum).19–21
Materials and Methods
Materials and Animals
All chemicals, unless indicated otherwise, were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Chicago, IL). Mouse recombinant cytokines and enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems (Minneapolis, MN) or BD Biosciences (San Diego, CA). Inbred BALB/c, C57BL/6, 129SV, CAST/Ei, and heterozygous motheaten C57BL/6J-Ptpn6me/J, C57BL/6J-Ptpn6me-v/J, and C3FeLe.B6 a/a-Ptpn6me/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal procedures were conducted under a protocol approved by the Institutional Animal Care and Use Committee of Rush University Medical Center (Chicago, IL).
A hereditary ulcerative autoinflammatory skin disease caused by massive polymorphonuclear neutrophil infiltration in the upper dermis was found in our breeding colonies. Based on the pedigree of four generations (n = 92), we identified the heterozygous founder male. Because of the clinical signs and histopathology resembling those that might occur in the human neutrophilic dermatoses (a heterogeneous group of diseases with unknown etiology), which are uniformly associated with sterile neutrophil infiltration of the skin, we initially designated this autoinflammatory skin disorder as neutrophilic dermatosis-like disease, or NDLD.
Healthy littermates (wild type or heterozygous) were maintained and bred under standard pathogen-free condition in the Comparative Research Center at Rush University Medical Center (Chicago, IL). Clinically sick mice were euthanized, or separated as soon as possible after the onset of the disease and maintained under different treatments. After identification of the Ptpn6 meB2 locus on chromosome 6, heterozygous males were mated with inbred wild-type females of various strains to identify additional recombinations and to reduce the size of the locus. Later, when the mutation was found in the Ptpn6 gene, all commercially available motheaten heterozygous mice (as listed above) were intercrossed with our meB2 mice to generate combined heterozygosity for the various mutations of the Ptpn6 gene.
Mice were assessed for paw inflammation and skin lesions three times a week, and body weight was measured weekly. In the absence of other treatments or during experiments, the mice received 10 mg hydrocodone bitartrate and 650 mg acetaminophen in 100 mL drinking water changed twice a week. Animals were euthanized before reaching the moribund stage. Blood or serum was collected by cardiac exsanguination under anesthesia. Organs and tissues were collected, fixed in 10% neutralized formalin, and embedded in paraffin. Sections (5 μm to 6 μm thick) were stained with hematoxylin and eosin or were deparaffinized, rehydrated in phosphate-buffered saline (PBS, pH 7.4), and stained with monoclonal antibody (mAb) specific for Gr-1/Ly-6G granulocyte marker (clone RB6-8C5; eBioscience, San Diego, CA).
Genome-Wide Screening
A set of 72 simple sequence length polymorphism (SSLP, microsatellites) markers that cover all mouse chromosomes was used for initial genome-wide screening of the mouse genome. DNA samples from 21 sick and 71 healthy mice (males and females) were tested. This population represented all parents of the first sick mice detected (generation 0) and the generations of three subsequent intercrosses.
The association of the disease with each genomic marker was estimated using χ2 statistics. The sick phenotype was scored as a binary trait, either 0 (not sick) or 1 (sick), and the genotype of each marker was either heterozygous or homozygous for the two parental genomes. As a result of the genome-wide scan, marker D6Mit254 on chromosome 6 demonstrated a particularly high association with the disease, with a χ2 value of 18.7 (P < 0.000015). An additional eight markers on mouse chromosome 6 were used to confirm locus association (see Supplemental Figure S1A at http://ajp.amjpathol.org). Based on the pattern of inheritance for a single recessive gene causing the disease, we further mapped the locus on chromosome 6 using an additional 16 markers and narrowed the expected locus to a region of 5.1 Mbp (see Supplemental Figure S1A at http://ajp.amjpathol.org). Additional animals with additional recombinations and the use of higher marker densities allowed us to narrow the locus to 0.4 Mbp between markers D6Abn176 (124.5 Mbp) and D6Abn123 (124.9 Mbp) (see Supplemental Figure S1A at http://ajp.amjpathol.org).
Twenty genes were found within this 0.4-Mbp NDLD locus (Ensemble release 52, December 11, 2008; http://www.ensembl.org/Mus_musculus). To identify any disease-causing gene or genes, we used real-time PCR with TaqMan probes (Applied Biosystems, Foster City, CA) for all 20 genes and found no significant differences at the level of transcription (data not shown). Gene expression profiles (bone marrow and spleen cells; GEO accession code GSE17161) were also tested using Affymetrix microarray chip hybridization (unpublished data) (Mouse Genome 430 2.0 gene chips) as described previously,22,23 but all 20 genes within the 0.4-Mbp locus demonstrated 1.01- to 1.48-fold changes.
Based on analyses of clinical phenotypes, histology sections and laboratory results, and publications related to any of the 20 genes present in the 0.4-Mbp locus, we selected a few candidate genes and sequenced their cDNA products. One of the candidate genes was protein tyrosine phosphatase, non-receptor type 6 (Ptpn6), in which we discovered the insertion of a B2 element in exon 6. A 16-nucleotide-long sequence of exon 6 was duplicated, and a 246-bp sequence (called B2 repeat or short interspersed element, SINE)24–28 was inserted between the duplicated exon 6 sequences (see Supplemental Figure S1C at http://ajp.amjpathol.org). This rearrangement resulted in the loss of 36 nucleotides of the wild-type coding sequence (see Supplemental Figure S1E at http://ajp.amjpathol.org), which was replaced with 21 nucleotides of a B2 repeat within the Ptpn6 cDNA sequence between exons 6 and 7 (see Supplemental Figure S1, C–E, at http://ajp.amjpathol.org). Overall, exon 6 was shortened by five amino acids (see Supplemental Figure S1E at http://ajp.amjpathol.org), but the insertion did not cause a frame shift. This deletion and insertion evidently affected the tertiary structure of the Ptpn6 protein,10 but the primary structure (amino acid sequences of both SH2 and the phosphatase domains) remained intact in meB2 mice (see Supplemental Figure S1B at http://ajp.amjpathol.org). A repeated genome-wide screening of NDLD sick mice and their healthy homozygous wild-type littermates in the BALB/c background (after 12 backcrosses) did not reveal any linkage to any other chromosomal region, nor did the cDNA sequences of the other 19 genes carry any mutation that would change the protein sequence (data not shown).
Mutations in the Ptpn6 gene have been previously described in association with the motheaten phenotype. Therefore, to follow standard nomenclature associated with Ptpn6 deficiency in mice, we changed the original NDLD designation to meB2 phenotype and Ptpn6meB2/meB2 homozygous genotype.
Cytokine Measurements and Laboratory Tests
Measurements of IL-1β, IL-4, IL-6, IL-10, IFN-γ, and TNF-α were determined in the sera and 4-day supernatants of spleen cell cultures of wild-type, heterozygous, and homozygous meB2 sick mice by ELISA. Spleen cell cultures of the three genotypes were also stimulated with 20 μg/mL lipopolysaccharide or 2.5 μg/mL concanavalin-A, and the secreted cytokines were measured in 72-hour supernatants. Secreted cytokine concentrations were normalized and expressed as picograms per million cells.29
Total peripheral blood leukocyte counts in wild-type, heterozygous, and meB2 sick mice were determined in Turk-stained cell suspensions, and differential leukocyte cell counts were assessed in Wright-Giemsa-stained smears. Monoclonal antibodies specific for CD45 (a ubiquitous leukocyte marker), Gr-1 (neutrophil), CD3 (T lymphocyte), and B220 (B lymphocyte markers) were used for flow cytometry (described below). In addition, antibodies specific for histone, C-reactive protein, serum amyloid A, single-stranded and double-stranded DNA, and anti-nuclear antibodies (all from Alpha Diagnostic International, San Antonio, TX) were measured in the sera. Antibodies against citrullinated cyclic peptides (Quanta Lite kit with anti-mouse IgGAM; Inova Diagnostics, San Diego, CA) and anti-mouse IgG and IgM rheumatoid factors (BioVendor/Shibayagi Co., Shibukawa, Gumma, Japan) were also determined in the sera of sick, heterozygous, and healthy littermates. Of note, from this list only anti-citrullinated antibodies were not detected in meB2 sick mice older than 3 to 4 months of age.
The neutrophil accumulation in various tissues was investigated by monitoring myeloperoxidase activity as described previously.30 Briefly, different organs were homogenized in 0.5% hexadecyltrimethilammonium bromide in PBS and ultrasonicated at 3 × 50 W ultrasonic power (Virsonic Digital 50; VirTis, Gardiner, NY) for 90 seconds on ice. Myeloperoxidase activity was measured in the supernatants based on the H2O2 oxidation of 3,3′,5,5′-tetramethylbenzidine.30 The results were normalized to myeloperoxidase activity measured in purified neutrophils (1 × 106) from the peripheral blood of normal mice. Enzyme activity was measured using the human/mouse/rat active DuoSet IC kit from R&D Systems.
Monoclonal Antibodies, Fluorescent Cell Surface Labeling, and Flow Cytometry
The following fluorochrome-labeled or biotinylated mAbs were purchased from BD Biosciences: fluorescein isothiocyanate-conjugated Armenian hamster anti-mouse CD69 (clone H1.2F3), phycoerythrin (PE)-conjugated rat anti-mouse CD8 (clone 53-6.7), PerCP-Cy5.5-conjugated rat anti-mouse CD4 (clone RM4-5), PE-Cy7-conjugated rat anti-mouse B220 (clone RA3-6B2) or PE-Cy7-conjugated rat anti-mouse CD19 (clone 1D3), allophycocyanin (APC)-conjugated rat anti-mouse CD25 (clone PC61), APC-Cy7-conjugated, Alexa Fluor 488-conjugated or PE-conjugated Armenian hamster anti-mouse CD3 (clone 145-2C11), fluorescein isothiocyanate (FITC) conjugated rat anti-mouse IgD (clone 11-26c.2a), biotinylated or PE-conjugated Gr-1/Ly-6G, PerCP-Cy5.5-conjugated rat anti-mouse IgM (clone R6-60.2), APC-conjugated rat anti-mouse CD5 (clone 53-7.3), biotin-conjugated rat anti-mouse CD23 (clone B3B4), and Alexa Fluor 488-conjugated rat anti-mouse CD44 (clone IM7). Biotinylated mAbs were detected with APC-Cy7-conjugated streptavidin. FITC-annexin V and propidium iodide (both from BD Biosciences) staining were used to distinguish early and late apoptotic cells.31 Labeling was performed according to the manufacturer's instructions, and cells were immediately analyzed by flow cytometry. Live cells are negative for both annexin V and propidium iodide; early apoptotic cells are positive for annexin V and negative for propidium iodide; late apoptotic (death) cells are positive for both annexin V and propidium iodide.
We used a three- to six-color labeling technique for the simultaneous detection of cell surface and intracellular molecules in cells harvested from heparinized peripheral blood, spleen, and occasionally bone marrow or thymus. Briefly, 1 × 106 cells (after erythrocyte lysis) were seeded in 96-well U-bottom assay plates (Falcon; BD Biosciences). Fc receptors were blocked with anti-CD16/32 (Fc Block; BD Biosciences) for 15 minutes at 4°C, and the cells were then incubated with mAb cocktails in 100 μL flow cytometry staining buffer for 30 minutes at 4°C in the dark. Cells were washed twice and finally resuspended in 200 μL 0.1% formaldehyde in PBS.
Samples were measured and analyzed using a FACSCanto II flow cytometer and FACSDiva software version 5.0.3 (BD Biosciences). Acquisition was performed by gating on leukocytes (CD45+), neutrophils (Gr-1+), and lymphoid cells in the forward/side scatter dot plots. Cell surface marker-defined populations were as follows: CD3+: total T cells; CD3+ CD4+: CD4+ T cells; CD3+ CD8+: CD8+ T cells; B220+: total B cells; IgDlow IgMhigh CD19+ CD23− CD5+/−: B1 cells (B1a/b); and IgDhigh IgMlow CD19+ CD23+: conventional B2 cells.32 Specific cell proportions were expressed as percentage of total cells unless otherwise stated. Data were collected from 10,000 cells within the lymphoid gate in each sample. Fluorescent histogram plots were used to compare the mean fluorescence intensities of different samples and to calculate the proportions of positively stained cells.
Determination of in Vivo Proliferation of Neutrophils and Their Lineage Distribution in Bone Marrow and Peripheral Blood
Mice in each group were injected intraperitoneally with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) at a dose of 250 μg and were sacrificed 3 to 5 days later. Blood was collected into heparinized tubes, and bone marrow was harvested by flushing the marrow cavity of femurs and tibias with cold PBS. Red blood cells were eliminated by hypotonic lysis. The nucleated cells were dispensed into flow cytometry tubes or 96-well microplates and were fixed and permeabilized using a commercially available kit (BD Biosciences). Alexa Fluor 488-labeled azide was added to the cells, and a click reaction33 between EdU and the azide was performed using a Click-iT cell reaction buffer kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Cells were then washed and surface-stained simultaneously with APC-conjugated anti-CD45 antibody (common leukocyte antigen) and PE-labeled Gr-1 mAb specific for the myeloid lineage (both antibodies from eBioscience). Flow cytometry was performed using a BD FACSCanto II instrument and FACSDiva software version 5.0.3. Phycoerythrin fluorescence was measured after gating on APC-CD45+ leukocytes and was expressed as percent Gr-1+ (myeloid) or Gr-1− (nonmyeloid) populations of all gated CD45+ cells. Alexa Fluor 488 fluorescence (EdU/thymidine incorporation) inside the cells was also measured and expressed as percent Gr-1+EdU+ or Gr-1−EdU+ of all CD45+ cells.
Treatments
In addition to the pain-killer and any necessary antibiotics (cefazolin, sulfamethoxazole, or trimethoprim), which did not affect the onset, severity, or cellular reactions of affected animals, both sick and healthy homozygous wild-type and heterozygous meB2 littermates were also treated with i) 15 mg/kg i.m. daily dose of methylprednisolone (Solu-Medrol; Pfizer, New York, NY), ii) purified rat neutralizing mAb against mouse TNF-α (eBioscience), iii) rat anti-CD44 mAb IM7,34,35 or iv) rat anti-mouse granulocyte Gr-1 mAb. Purified rat IgG (from naïve animals) was administered to age-matched controls (littermates, if possible). Anti-CD44 mAb (clone IM7l; ATCC, Manassas, VA) induces shedding of CD44-bearing cells, and so CD44+ leukocytes are unable to leave the circulation.34–37 Anti-Gr-1 rat mAb (clone RB6-8C5) is specific for mouse neutrophils and cytotoxic to these cells in vivo.35,38 The initial dose was 200 μg mAb or normal rat IgG injected intravenously, and then 100 μg i.p. was injected every second day. Antibodies were diluted in PBS for in vivo treatment and were sterilized with 0.22 μm pore-size syringe filters. As a fifth treatment, gliotoxin (Biomol, Plymouth Meeting, PA), a strong inhibitor of IκBα degradation and thus an inhibitor of NF-κB activation,39–42 was dissolved in dimethylsulfoxide at a concentration of 25 mg/mL and then was further diluted with sterile PBS. Mice were injected i.p. with 10 μg gliotoxin (or no gliotoxin) in 200 μL sterile PBS once a day. For a long-term and combined treatment, mice were injected once a week with 100 μg of anti-CD44 mAb IM7 and 5 mg prednisone or with 100 μg to 200 μg IM7 mAb and 10 μg gliotoxin (i.p.) with ad libitum 10 mg hydrocodone and 650 mg acetaminophen in 100 mL of drinking water (approximately 1 mL/day). These two treatment regimes (anti-CD44 and steroid or anti-CD44 and gliotoxin) had the same results, and homozygous Ptpn6meB2/meB2 mice remained symptom-free for 6 months posttreatment.
The efficacy of treatment was measured using a digital microcaliper (World Precision Instruments, Sarasota, FL) before the treatment and then every second day, immediately before subsequent injections. The results are expressed as percent changes in cumulative (sagittal and frontal) joint thickness (mm) relative to the initially measured (before the treatment) value. Measurements were performed early on the same area (digits, paw, joint diameter, etc), selected before the treatments (Figure 1D).
Figure 1.
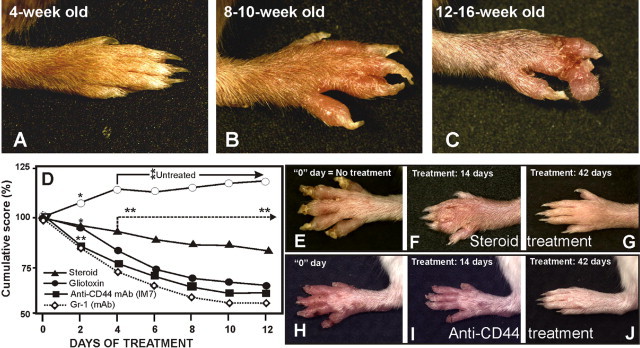
Progression of paw inflammation and therapeutic effects of various agents. Untreated homozygous Ptpn6meB2/meB2 (meB2 sick) animals (A) develop severe paw inflammation, which starts with edema and redness of the digits and carpal/tarsal areas (B), resembling arthritis. Inflammation leads to superficial scars and, if infected, abscesses (C). If these animals were not treated, they died of interstitial pneumonia within a few months (Figure 2, E and F). If the treatment was started in time (8 to 12 weeks of age; E and H), even without antibiotics, the healing process was fast and impressive, and only mild inflammatory reactions could be observed after 2 to 3 weeks of therapy (F and I). Clinically complete recovery (with or without mild scars, depending on the efficacy of the therapy) could be achieved by 6 to 7 weeks of therapy (G and J) and could be maintained for more than 1 year with a low dose of oral prednisone (2 × 5 mg/kg per week) with 100 μg anti-CD44 monoclonal antibody (once a week) or 10 μg of gliotoxin (twice a week). If, however, the mice were weaned off the treatment, a dramatic recurrence of the condition occurred within 1 to 2 weeks, with even more severe symptoms and a diminished response to resumed therapy. (D) Cumulative results of treatment. Each treatment group comprised 9 to 12 mice; only 1 to 3 mice were simultaneously treated in each group. Sick mice were selected for treatment 10 to 12 days after disease onset. *P < 0.01; **P < 0.001 relative to day 0 of treatment. Each group displayed massive healing or progression that was highly significant after day 4 (**P < 0.01). Macroscopic images show the same paw of a representative animal.
Combined Heterozygosity
Heterozygous C57BL/6J-Ptpn6me/J (Jackson Laboratory stock no. 000810;), C57BL/6J-Ptpn6me-v/J (stock no. 000811) and C3FeLe.B6 a/a-Ptpn6me/J (stock no. 000225; same allelic variant as stock 000810 but in a mixed genetic background) males were intercrossed with our heterozygous BALB/c-Ptpn6meB2+/− females, and combined heterozygosity (heterozygous for two different mutant alleles of the Ptpn6 gene) was assessed by the evaluation of clinical phenotypes. Any combination of mutant Ptpn6 alleles resulted in the motheaten phenotype, and disease onset, disease severity, and maximum lifespan all reflected the combination of the original phenotypes. For example, combined heterozygosity of Ptpn6me/meB2 exhibited early paw inflammation and a short lifespan (∼1.5 to 2 months), which is shorter than the homozygous meB2 but longer than the me/me homozygous littermates. The Ptpn6me-v/meB2 genotype developed the first symptoms of the disease at 2 to 4 weeks of age and survived up to 3 to 4 months (data not shown).
Bone Marrow Chimeras
Wild-type female BALB/c mice (5 to 6 weeks old) were lethally irradiated twice with 4.5 Gy (Gammacell, Cs-source; Atomic Energy of Canada, Mississauga, ON, Canada)35 4 hours apart and then injected intravenously with 2.5 × 107 bone marrow cells of the Ptpn6meB2/meB2 genotype or their wild-type littermates. Chimera mice (n = 5 and n = 6 in two experiments) that received bone marrow from Ptpn6meB2/meB2 donor mice that were not yet sick developed the disease 3 to 5 weeks after the bone marrow transplantation. The clinical phenotypes were very similar to those described for homozygous meB2 sick mice, with the gradual development of leukocytosis. However, the disease-affected chimera recipients (at the time of disease onset) exhibited almost 50% fewer lymphocytes (both B and T cells) and 20% fewer neutrophils in the peripheral blood, compared with mice that underwent normal control bone marrow transplantation. The wild-type (littermate) bone marrow donor cells did not transfer the disease phenotype into wild-type recipients (n = 2 × 5 recipients).
DNA Binding Studies
Transcription factor binding activity was determined using the electrophoretic mobility shift assay (EMSA, or super gel-shift assay) as described previously.43,44 Briefly, nuclear protein extracts (3 μg to 10 μg protein) prepared from spleen or bone marrow cells of 5- to 6-week-old mice were incubated with 50,000 cpm of 32P end-labeled oligonucleotide probes for 20 to 30 minutes at room temperature in 10- or 20-μL reaction volumes containing 12% glycerol, 12 mmol/L HEPES-NaOH (pH 7.9), 60 mmol/L KCl, 5 mmol/L MgCl2, 4 mmol/L Tris-Cl (pH 7.9), 0.6 mmol/L EDTA (pH 7.9), 0.6 mmol/L dithiothreitol, and 0.25 μg of poly(dI-dC). The 32P-labeled consensus double-stranded NF-κB binding oligonucleotide sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′; the recognition site is underlined) was used to detect NF-κB subunits. To demonstrate binding specificity, a 100-fold molar excess (10 ng) of nonlabeled specific (see above) or nonspecific (5′-GCAGAGCATATAAGGTGAGGTAGGA-3′) oligonucleotides was included in the binding reaction. For the super gel-shift assays, 2 μg each of anti-Rel A (p65), anti-NF-κB1 (p50), anti-c-Rel, or anti-RelB antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were included in the binding reaction. Protein-DNA and protein-DNA-antibody complexes were resolved in 5% polyacrylamide gels (PAGE) that had been pre-electrophoresed for 30 minutes at room temperature in 0.25 × TBE gel-running buffer (22.5 mmol/L Tris borate and 0.5 mmol/L EDTA, pH 8.3). Gels were dried and exposed to radiographic film at −70°C for 8 to 16 hours, or the radioactivity of the samples was measured and analyzed by scanning densitometry on a STORM PhosphoImager (Molecular Dynamics, Sunnyvale, CA). NF-κB subunits were measured by ELISA (Active Motif, Carlsbad, CA).
Western Blot Analysis
Spleen cells (after erythrolysis) or bone marrow samples were homogenized for 30 seconds (10,000 rpm) on ice and then lysed (100 mg tissue or 1 × 106 cells in 1 mL) in cold radioimmunoprecipitation (RIPA) lysis buffer containing Halt protease inhibitor cocktail (Pierce; Thermo Fisher Scientific, Rockford, IL) and 0.1% NaN3 for 1 hour at 4°C. Cell lysates were sonicated as described above. The protein content of the lysates was measured using Pierce's bicinchoninic acid assay and analyzed by Western blotting as described previously.43,44 Briefly, proteins were separated in a sodium dodecyl sulfate containing 10% to 12% polyacrylamide gel (SDS-PAGE) and electrophoretically transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). The membrane was stained with anti-I-κBα, anti-I-κBε, or anti-NF-κB antibodies (Santa Cruz Biotechnology), rabbit anti-mouse Ptpn6 mAb (Abcam, Cambridge, MA), or anti-mouse β-actin (Santa Cruz Biotechnology). The reactions were visualized using the enhanced chemiluminescence (ECL) detection system (Amersham International, Arlington Heights, IL).
Statistical Analysis
In vitro and in vivo results were analyzed using the Student's t-test to compare means and the one-way analysis of variance method using least significant difference post hoc tests for multiple comparisons. The group means and SEM (mean ± SEM) was applied. All statistical analyses were performed using the SPSS statistical software package (version 16.0; SPSS, Chicago, IL). A value of P < 0.05 was considered statistically significant.
Results
Macroscopic Features of the Ptpn6meB2/meB2 (meB2) Phenotype
Sterile neutrophil invasion of the skin, especially in the paws, dominated the clinical phenotype observed in our mouse colony. As described under Materials and Methods, an extensive genome-wide screening narrowed the genomic region of interest (see Supplemental Figure S1A at http://ajp.amjpathol.org), and sequencing of the Ptpn6 gene revealed a novel mutation (see Supplemental Figure S1C at http://ajp.amjpathol.org). The earliest inflammatory symptoms of paws of disease-affected animals appeared as soon as 3 to 5 weeks of age, a time point equivalent to the entire lifespan of the me mice.1,4,5 The earliest spontaneous skin lesions appeared on the trunk, most likely because of the microinjuries of intracage movement by the mother, which became less evident as soon as fur covered the body (data not shown). In contrast, inflammation of the paws was noted approximately 2 weeks later, and paw inflammation never healed spontaneously thereafter, because of the constant cage activity and microdamages of the pups (Figure 1, A–C). The disease was progressive, both macroscopically (Figure 1, A–C) and histologically (Figure 2). The animals were maintained under pathogen-free conditions and received 0.1 g/L hydrocodone bitartarate and 6.5 g/L acetaminophen in drinking water. Homozygous meB2 mice could survive up to 1 year, the longest period tested to date.
Figure 2.
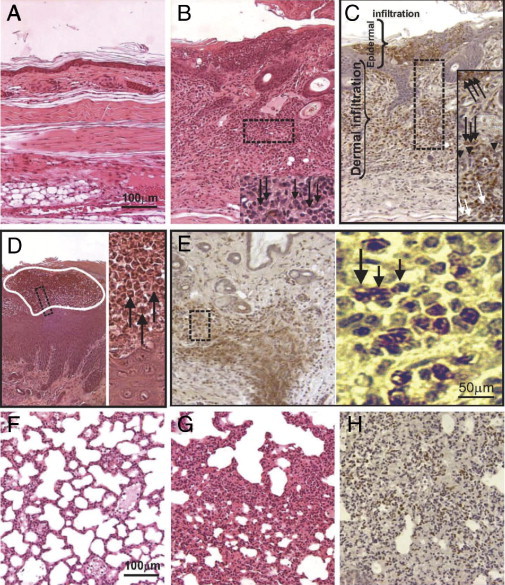
Skin (A–E) and lung (F–H) histology and immunohistochemistry for polymorphonuclear (neutrophil) leukocytes in normal sections from control wild-type littermates of 5-week-old animals (A and F) and from age-matched sick mice (B–E, G, and H). B-E: Inflamed skin sections with neutrophilic infiltration in an untreated animal. G and H: Lung (serial sections) with interstitial pneumonia in an untreated animal. Sections were stained with hematoxylin and eosin (A, B, D, F, and G) or paraffin-embedded and rehydrated sections (C, E, and H) were stained for neutrophils using biotinylated Gr-1 monoclonal antibody and peroxidase-labeled streptavidin (the brown cells are neutrophils). B–E: Boxed areas are shown in higher magnification in the inset (B and C) or in the adjacent full-height image (D and E). In panel D, the area outlined in white is a microabscess in the epidermis, a portion of which appears in the inset. In the high-magnification images, black arrows indicate neutrophils, based on cell and nuclear shapes and staining with mAb Gr-1 (B, C, D, and E), arrowheads indicate macrophages/histiocytes (C), and white arrows indicate fibroblast-like cells (C). Original magnification ×20 (A–H, main image; scale bar = 100 μm); ×40 (B–E, high-magnification image; scale bar = 50 μm).
Before any paw inflammation started, up to 3 to 5 weeks of age, a gentle but continuous irritation (eg, 5 to 10 minutes massage) or relatively brief hypoxia (induced, for example, with strangulation above the wrist or ankle for 20 minutes) triggered paw inflammation within 24 to 48 hours. Similar exogenous manipulation is used for differential diagnosis to challenge symptom-free pyoderma gangrenosum (pathergy). Moreover, the warming (39°C to 40°C for 20 to 30 minutes) of a symptom-free, seemingly normal paw of a homozygous Ptpn6meB2/meB2 mouse was an even greater stimulant and initiated paw inflammation within a few hours. Notably, whenever swelling and inflammation of the paw started, either spontaneously or induced by exogenous factors, it did not heal spontaneously (data not shown).
Histopathological Features of the Skin and Other Organs
In addition to the epidermal thickening and spongiosis (Figure 2, A and B), the histopathology of sections prepared from the initial skin lesions revealed a predominantly neutrophilic dermal infiltrate with intraepidermal (occasionally subcorneal) neutrophil occupation forming pustules and ulcerations (Figure 2C). The blood vessels were dilated, but there was no evidence of leukocytoclastic vasculitis. Scattered macrophage-like cells were located primarily in the superficial dermis (Figure 2C). Immunohistochemical staining of the cell infiltrate with a granulocyte marker (Gr-1) and assessment of the nuclear shape under higher magnification showed that the infiltration was composed predominantly of neutrophils. Accumulations of immune deposits of IgG and IgM in kidney glomeruli were demonstrated several weeks after onset of the paw inflammation (data not shown). Although in the early stage the swelling of digits and paws mimicked arthritis (Figure 1B), the inflammation did not affect internal joint structure.
The most characteristic histological alterations in the early stage were detected in the skin, but more subtle changes could also be observed in the lungs, which consistently exhibited varying degrees of neutrophil and macrophage infiltration in the peri-alveolar interstitium, resembling interstitial pneumonia (Figure 2, G and H), even while the general condition and behavior of the animals appeared to be almost normal. The hypercellular (myeloid leukemia-like) bone marrow with massive bone resorption and osteoclast formation (data not shown), massive splenomegaly with myeloproliferative changes,3,45,46 and small neutrophil islands in the submucosal layer of the colon (data not shown) were evident histopathological abnormalities. Although the thymus was smaller in Ptpn6meB2/meB2 mice than in their wild-type or heterozygous littermates, its morphology was normal (data not shown). Homozygous Ptpn6meB2/meB2 females were infertile, their gonads were atrophic, and Graafian follicles could not be detected in the ovaries even after hormone-induced superovulation.47 In contrast, the testes histology was normal, and homozygous Ptpn6meB2/meB2 males were fertile if they had a late onset of the condition (6 to 8 weeks) or were being treated with early (preventive) suppressive therapy (Figure 1D). Thus, the meB2 homozygous phenotype was less severe than the phenotype of either me/me or me-v/me-v mice,2,3,6 but it appeared to be more extensive and progressive than that described in spin mice.7
Pathophysiological Abnormalities in meB2 Mice
The observed autoinflammatory skin disease, occasionally associated with mild neutrophilic infiltration of other organs, compelled us to test for serum biomarkers and abnormalities in the cellular composition. First, serum cytokines and various biomarkers were measured using a multiplex cytokine and growth factor panel (Figure 3A), and then serum cytokines, acute phase proteins (serum amyloid-A and C-reactive protein) (Figure 3B), serum immunoglobulins (Figure 3C), differential leukocyte counts in the peripheral blood (Figure 3D), and myeloperoxidase activities in different organs (Figure 3F) were determined in wild-type and homozygous meB2 mice.
Figure 3.
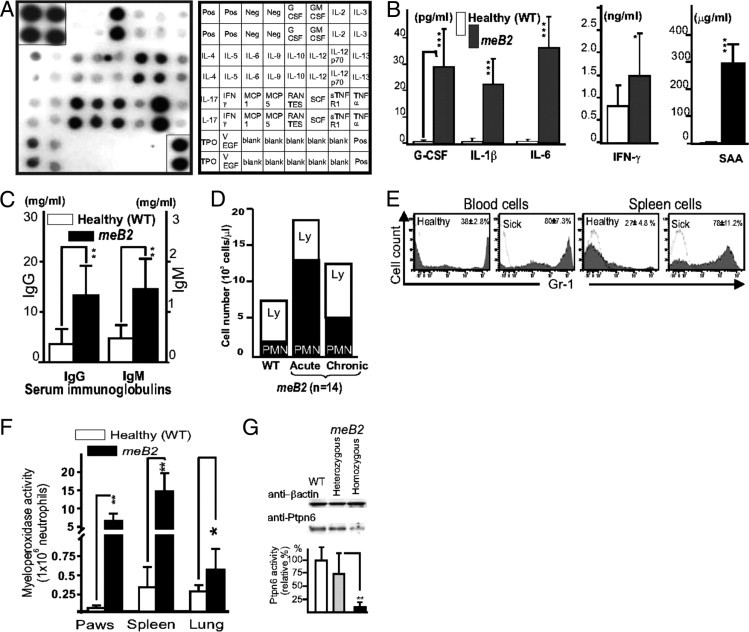
Serum bioassays and markers of normal [healthy wild-type (WT)] and age-matched Ptpn6meB2/meB2 (meB2 sick) mice. A: Multiplex cytokine assay showing the relative abundance of various cytokines in the sera of sick (Ptpn6meB2/meB2) mice (Chemicon ChemiArray mouse cytokine array I map; Millipore, Temecula, CA). The positive control dots (at the right top and left bottom corners) were cut out (after a 2-second exposure) and reincorporated into the panel to show the relative amounts of different cytokines, receptors, and growth factors detected after a 15-second exposure time. Standard abbreviations are used (including R, receptor; s, soluble; TPO, thrombopoietin). The sera of five homozygous meB2 sick animals were pooled, and 1:50 serum dilution was used. Only the positive controls were positive in membranes blotted with pooled sera of wild-type littermates (also 1:50 dilution) (data not shown). B: The serum levels of the measured cytokines and acute-phase protein serum amyloid A (SAA) (B) were similar to those observed on the capture immunodot membrane (A). C and D: Serum immunoglobulin levels (C) in healthy and sick mice (sick for >8 weeks) and ratios (and numbers) of lymphocytes (Ly) and neutrophils (PMN) in the peripheral blood (D) (n = 12 in each group). E and F: Gr-1+ (neutrophil) cell ratio in the peripheral blood and spleen (E) and myeloperoxidase activity in the paws, spleens and lungs (F) measured in 12 wild-type and 12 meB2 sick littermates 3 to 4 days after the onset of inflammatory paw disease. Data are reported as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. G: Ptpn6 protein expression in bone marrow cell lysates of wild-type, heterozygous, and homozygous (meB2 sick) mice, the β-actin expression in the same cell lysates, and the corresponding Ptpn6 activities measured in six bone marrow cell lysates (means ± SEM). **P < 0.01.
The high levels of anti-DNA (both single-stranded and double-stranded), anti-nuclear, and anti-histone antibodies (up to mg/mL levels) might have contributed to the massive hypergammaglobulinemia, including both IgG and IgM isotypes (Figure 3C). This hypergammaglobulinemia could be detected as early as 4 to 6 weeks after disease onset and increased with age (data not shown). Significant leukocytosis associated with relative lymphopenia was detected in meB2 sick mice (Figure 3D); however, absolute T and B cell numbers were normal or close to normal, although most of the CD4+ T and B cells were present in an activated state after the onset of the disease (Table 1). Only trace amounts of Ptpn6 protein could be detected in Western blot, associated with nearly zero enzyme activity (Figure 3G). A twofold to threefold greater percentage of CD4+/CD25+/CD69+ activated T cells and B220+/CD69+ activated B cells (Table 1), and an unusually high ratio of B1a/b cells, were the most prominent characteristic abnormalities associated with this relative lymphopenia (Figure 3D) observed in the peripheral blood and spleen of meB2 mice (Table 1). Large numbers of Gr-1+ polymorphonuclear (neutrophil) leukocytes were detected in the peripheral blood and spleen (Figure 3E). Thus, Ptpn6meB2/meB2 homozygous mice were not immunodeficient, as is frequently described for Ptpn6me/me or Ptpn6mev/mev mice.1,45,48,49 Instead, they possessed an autoinflammatory tendency early in life that eventuated into an autoimmune state over time. Nonetheless, the strength and level and especially the size of the window of autoimmune components characteristic of this inflammatory phenotype remain unclear, and more extensive studies are needed.
Table 1.
Cellular Composition of Blood and Spleen of Wild-Type and Homozygous meB2 Littermates Assessed by Flow Cytometry
| Cell type† | Peripheral blood |
Spleen |
||||||
|---|---|---|---|---|---|---|---|---|
| Healthy |
Ptpn6meB2/meB2 |
Healthy |
Ptpn6meB2/meB2 |
|||||
| % total | % sub | % total | % sub | % total | % sub | % total | % sub | |
| T cell (CD3) | 51.2 ± 3.2 | 22.2 ± 1.4⁎ | 47.8 ± 3.6 | 22.1 ± 0.3⁎ | ||||
| CD8+ T cell | 17.2 ± 4.1 | 15.6 ± 3.4 | 6.8 ± 0.5 | 4.4 ± 0.1 | ||||
| CD4+ T cell† | 30.0 ± 0.8 | 11.0 ± 2.5⁎ | 26.7 ± 0.6 | 17.1 ± 0.1⁎ | ||||
| Subpopulation | ||||||||
| CD4+CD69+ | 0.3 ± 0.1 | 1.0 | 1.2 ± 0.1⁎ | 10.9⁎⁎ | 6.2 ± 1.0 | 23.2 | 9.1 ± 2.3⁎ | 53.2⁎⁎ |
| CD4+CD44high | 15.9 ± 1.5 | 53.0 | 9.5 ± 2.2⁎ | 86.4⁎⁎ | 12.4 ± 1.0 | 46.4 | 5.8 ± 0.3⁎⁎ | 33.9 |
| B cell (B220+) | 27.3 ± 0.8 | 16.3 ± 1.0⁎ | 26.3 ± 4.7 | 14.2 ± 0.9⁎⁎ | ||||
| B cell (B220+) | 27.3 ± 0.8 | 16.3 ± 1.0⁎ | 26.3 ± 4.7 | 14.2 ± 0.9⁎⁎ | ||||
| Subpopulation | ||||||||
| B1a/b | 6.9 ± 1.1 | 25.3 | 7.2 ± 3.9 | 44.3⁎⁎ | 6.1 ± 1.6 | 23.2 | 8.5 ± 2.4 | 60.5⁎⁎ |
| B2 (conventional) | 17.3 ± 1.6 | 63.4 | 15.7 ± 5.4 | 52.3⁎ | 16.1 ± 2.5 | 61.3 | 4.1 ± 2.1 | 28.8⁎⁎ |
| CD69+B220+ | 0.3 ± 0.2 | 1.1 | 0.2 ± 0.1 | 1.5 | 0.8 ± 1.2 | 2.2 | 1.1 ± 0.2 | 7.8⁎ |
| Gr-1+ neutrophils | 36.2 ± 2.3 | 78.8 ± 2.3⁎⁎ | 27.0 ± 4.0 | 79.6 ± 2.8⁎⁎ | ||||
| Subpopulation‡ | ||||||||
| Live | 17.7 ± 4.3 | 66.5 | 63.4 ± 4.4 | 80.5⁎ | 8.8 ± 1.3 | 51.8 | 48.2 ± 4.3 | 60.6 |
| Pre-apoptotic | 7.4 ± 0.3 | 27.8 | 12.4 ± 2.4 | 15.7⁎⁎ | 2.9 ± 0.3 | 17.1 | 27.3 ± 5.6 | 34.9⁎⁎ |
| Late apoptotic | 2.5 ± 0.3 | 9.4 | 4.1 ± 0.9 | 5.2⁎⁎ | 5.3 ± 0.4 | 31.2 | 4.6 ± 1.2 | 5.8⁎⁎ |
Cells were analyzed (in all cases, n = 8 sick and n = 7 healthy littermates) at 2 days after disease onset together with their wild-type littermates.
Data are reported as percentage of the mean ± SEM.
AnxV, annexin V; PI, propidium iodide; sub, subpopulation.
P < 0.05;
P < 0.01 compared with healthy mice.
Because of the relative lymphopenia, T-cell and B-cell numbers may be similar in healthy and sick (Ptpn6meB2/meB2) mice, but the ratio (activated cell types, or B1a/b cells) is significantly higher in the sick mice.
Live cells: AnxV−PI−. Pre-apoptotic cells: AnxV+PI−. Late apoptotic: AnxV+PI+.
Therapeutic Efficacy of Various Compounds on the meB2 Sick Phenotype
Cyclosporine (15 mg/kg daily dose for 4 weeks) and neutralizing antibody against TNF-α (clone TN3-19, eBioscience) had a relatively modest effect, mostly during the progressive or chronic phase of the disease (data not shown), compared with the other therapeutic regimens evaluated. A high daily dose of steroid (10 mg/kg), anti-CD44 mAb (clone IM7; 200 μg to 100 μg), Gr-1 mAb specific for mouse neutrophils (clone RB6-8C5), and gliotoxin, a strong inhibitor of IκBα degradation (10 μg/day), had a beneficial effect on paw inflammation, which was resolved almost completely during the treatment period (Figure 1, D–J). However, weaning from the effective therapy provoked a dramatic recurrence of multiorgan inflammation within 1 to 2 weeks, necessitating euthanization. On the other hand, maintenance of a relatively low-dose therapy (see Methods) protected the animals against disease flare-ups. Conventional anti-inflammatory drugs, such as ibuprofen (7.5 mg/kg per day), and acetaminophen with hydrocodone, uniformly failed to provide a therapeutic benefit.
Neutrophil Proliferation and Survival
The hyperproliferative-like bone marrow associated with massive leukocytosis, slightly increased numbers of immature neutrophils, and relative lymphopenia in the peripheral blood suggested an increase in neutrophil proliferation. Bone marrow neutrophil proliferation and survival were therefore measured using a thymidine analog, 5-ethynyl-2′-deoxyuridine (EdU), and were monitored using the click reaction (Figure 4, E–J).33 Although the numbers of Gr-1+ cells in wild-type and heterozygous meB2 mice were approximately the same, the Gr-1+ cell ratio relative to the total leukocyte cell number (CD45+) was significantly higher both in bone marrow (Figure 4A) and in peripheral blood (Figure 4B) in Ptpn6meB2/meB2 homozygous sick mice. Unexpectedly, the increased number of neutrophils was associated with reduced EdU incorporation into these cells, both in bone marrow and in peripheral blood of meB2 sick mice (Figure 4, C and D). Overall, these observations indicate an extended half-life of neutrophils, which resulted in leukocytosis and an increased number of neutrophils in blood (Figure 3D), potentially contributing to the accumulation of these cells in the spleen, skin, and lung (Figure 3F).
Figure 4.
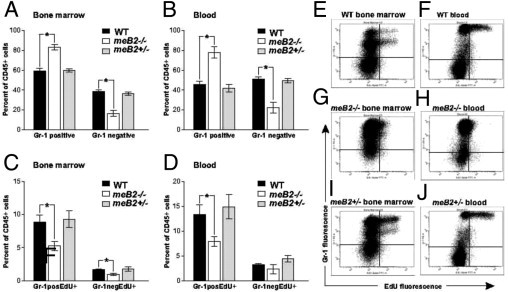
Distribution and EdU incorporation of myeloid (Gr-1+) and nonmyeloid (Gr-1−) leukocyte populations in the bone marrow and blood of wild-type (WT), heterozygous (meB2+/−) and homozygous meB2−/− (Ptpn6meB2/meB2) sick mice. Mice in each group were injected with 250 μg EdU i.p. and sacrificed 3 to 5 days later. Cells isolated from the bone marrow and peripheral blood were fixed, permeabilized, and reacted with Alexa Fluor 488 azide in vitro using the Click-iT cell reaction cocktail (Invitrogen). Cells were then surface-stained with an APC-conjugated antibody against CD45 (common leukocyte antigen) and PE-conjugated anti-Gr-1 (a myeloid lineage marker). The proportions of Gr-1+ and Gr-1− leukocytes (A, B) and EdU incorporation (C, D) by these two populations were determined by flow cytometry, and the results are expressed as % of CD45+ cells. E–J: Representative flow cytometry dot plots of bone marrow and blood samples (one of each group); data are reported as means ± SEM (n = 7 to 8 mice/group). *P < 0.05.
IκBα and IκBε Deficiencies Increase NF-κB Activity in Homozygous Ptpn6meB2/meB2 Mice
Neutrophils express a significant amount of the NF-κB inhibitor IκBα,50 and IκBα deficiency51 results in a sustained NF-κB response that constitutively maintains proinflammatory cytokine and acute-phase protein levels (as measured in the sera of Ptpn6meB2/meB2 mice) (Figure 3, C–D, and Table 2). IκBα deficiency may induce a severe widespread dermatitis and early death in mice,51 whereas IκBε-null mice present a constitutive up-regulation of IgM and IgG1,52 which was also detected in our homozygous Ptpn6meB2/meB2 mice (Figure 3C). Based on this information, we decided to test the expression levels of a few key regulatory components of the NF-κB pathway in our mice. First, we determined the levels of NF-κB components and found no intact c-Rel (p75) (Figure 5A) or RelA (p65) (Figure 5B) in the spleen cell lysates of homozygous meB2 mice. However, the low molecular weight forms of c-Rel and p65 could be detected in the total cell lysates, suggesting the presence of the free (non-IκB bound) forms of these proteins (Figure 5A). To test this hypothesis, we measured the levels of the activated (DNA binding) form of p65 in total spleen cell lysates by ELISA. Indeed, the spleen cells of sick animals contained a high level of the activated form of p65 (Rel A), whereas no active p65 could be detected in the spleen cells of healthy littermates (Figure 5C).
Table 2.
Cytokine Concentrations in the Supernatants of Spleen Cell Cultures with or without Stimulation with LPS or Con-A in Healthy Wild-Type and Asymptomatic Ptpn6meB2/meB2 Mice
| Cytokine | Cytokine level (pg produced by 1 × 106 cells) |
||
|---|---|---|---|
| No stimulation | LPS (10 μg/mL) | Con-A (2.5 μg/mL) | |
| IL-1β | |||
| Healthy | 2.79 ± 3.80 | 77.3 ± 55.2 | 3.67 ± 6.55 |
| Ptpn6meB2/meB2 | 18.2 ± 9.7⁎⁎ | 417 ± 197⁎ | 18.3 ± 10.1⁎⁎ |
| IL-6 | |||
| Healthy | 5.68 ± 18.8 | 387 ± 263 | 61.1 ± 62.1 |
| Ptpn6meB2/meB2 | 35.5 ± 20.2⁎⁎ | 2091 ± 837⁎⁎ | 339 ± 171⁎⁎ |
| TNF-α | |||
| Healthy | 7.80 ± 4.09 | 195 ± 65.1 | 149 ± 39.6 |
| Ptpn6meB2/meB2 | 142 ± 93.2⁎⁎⁎ | 1087 ± 238⁎⁎⁎ | 196 ± 81.7 |
| IL-10 | |||
| Healthy | 14.2 ± 18.2 | 822 ± 213 | 554 ± 603 |
| Ptpn6meB2/meB2 | 281 ± 56.5⁎⁎ | 4311 ± 215⁎⁎ | 599 ± 552 |
| IFNγ | |||
| Healthy | 69.9 ± 78.4 | 176 ± 1.3 | 2279 ± 426 |
| Ptpn6meB2/meB2 | 90.3 ± 90.7 | 81.1 ± 54.7⁎⁎ | 827 ± 416⁎⁎ |
Cytokines were measured by ELISA in 72 hours supernatants of lipopolysaccharide (LPS) or concanavalin-A (Con-A)-stimulated spleen cell cultures. No significant differences were observed for IL-4, IL-17, and GM-CSF expression (data not shown). [Spleen cells from heterozygous mice were also tested using the same conditions. Elevated cytokine levels were measured in the conditioned medium, but only IL-6 was significantly elevated, compared with wild-type littermates (data not shown).]
Data are reported as means ± SEM. Duplicate samples of 10 mice in each group.
P < 0.05;
P < 0.01;
P < 0. 001 compared with wild type.
Figure 5.
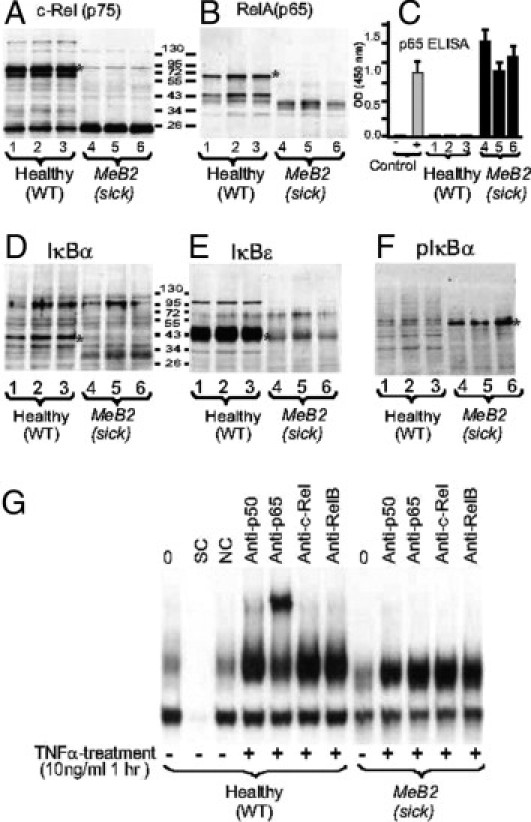
Western blots of the major components of NF-κB [c-Rel and p65 (RelA)], IκBα, and IκBε and phosphorylated IκBα (pIκBα) in spleen cell lysates from wild-type (healthy) and Ptpn6meB2/meB2 (sick) mice (A, B, and D–F), quantification of p65 (RelA) by ELISA (C), and a representative EMSA super gel-shift assay (G). Spleen cells were isolated and pooled from two wild-type and two sick littermates at 5 weeks (lanes 1 and 4), 7 weeks (lanes 2 and 5), and 9 weeks (lanes 3 and 6) of age. Erythrocytes were lysed, the cells washed twice with PBS and spleen cells were dissociated in lysis buffer. Cell lysates were normalized for protein content (10 μg/lane) and loaded for SDS-PAGE. After electrophoresis and transfer onto nitrocellulose, the same membrane was probed for c-Rel (A), then developed with ECL reagent, stripped, and restained with anti-RelA antibody (B). The other membrane (A) was stained, stripped, and restained with antibodies against IκBα (D), IκBε (E), and pIκBα (F). (C) Nuclear extracts were isolated from the same cells used for Western blotting, and 10 μg of nuclear protein was incubated with an NF-κB consensus-binding sequence in an ELISA plate and the results were quantified using a p65-specific antibody. Cell lysates (10 μg nuclear protein each) from the HeLa cell line without stimulation or after 1 hour of TNF-α (10 ng/mL) stimulation were used as negative (−) or positive (+) controls, respectively. G: EMSA super gel-shift assay using neutrophil lysates (one of the three cell types: T cells, B cells, and PMNs) from healthy and symptom-free Ptpn6meB2/meB2 mice. NC, nonspecific control; SC, specific control. (For details, see under Materials and Methods.) A, B, and D–F: Single asterisks indicate the appropriate protein bands. B: Healthy wild-type mice were significantly different from sick Ptpn6meB2/meB2 mice at P < 0.0001.
IκBα and IκBε, the two major cytoplasmic inhibitors of NF-κB, were measured in spleen cell lysates of homozygous meB2 mice and their wild-type littermates. Both inhibitors were present at significantly lower levels in meB2 sick mice than in normal wild-type animals (Figure 5, D and F), which suggests that both factors have a role in the pathomechanisms responsible for the Ptpn6meB2/meB2 phenotype. The degradation of IκBs is tightly regulated via a multistep process that involves the site-directed phosphorylation and ubiquitination of these proteins.7,53,54 We therefore determined the phosphorylation status of these proteins. Because the level of phosphorylated IκBα was elevated in total spleen cell lysates from homozygous meB2 mice (Figure 5F), we concluded that the decreased levels of IκB proteins (Figure 5, D and E) were due to increased degradation, rather than decreased production (Figure 5D); furthermore, the phosphorylation processes seemed to be normal. Thus, it appears that the defect in Ptpn6 function affects one or more different inhibitory components that indirectly control the level of NF-κB (Figure 5E)—which indeed was extremely high, as demonstrated by super gel-shift assay. One of the best examples of the overactivated NF-κB system was obtained when spleen cells (either purified B, T, or polymorphonuclear cells from the spleen of symptom-free mice) were activated with TNF-α for a short period of time, and the nuclear extracts were tested by the EMSA super gel-shift assay (Figure 5G). Whereas all of the cells responded to TNF-α, the amounts of p50 and p65 were high in the nuclear extracts of wild-type cells but scarcely detectable or even undetectable in those isolated from Ptpn6meB2/meB2 mice.
To gain insight into the mechanisms by which NF-κB-controlled cytokine expression drives the autoinflammatory status in meB2 mice, spleen cells from wild-type, heterozygous (Ptpn6meB2/−), and homozygous Ptpn6meB2/meB2 mice, all 4 to 5 weeks old and asymptomatic (still healthy), were tested for cytokine production in response to the Toll-like receptor (TLR) ligands lipopolysaccharide and concanavalin-A or were left unstimulated for 72 hours. The most important results obtained are summarized in Table 2. Mice with the Ptpn6meB2/meB2 genotype spontaneously produced significantly more IL-1β, IL-6, IL-10, and TNF-α than their wild-type littermates, and all of these cytokine levels increased dramatically in response to stimulation with lipopolysaccharide (ligand for TLR4) and concanavalin-A (which induces the expression of various Toll-like receptors).55 Only IFN-γ was significantly suppressed (Table 2). The stimulation of spleen cells with concanavalin-A significantly increased IL-1β and IL-6 secretion in 3-day-old cultures, indicating that T cells also are affected by Ptpn6 deficiency. Taken together, our results show that homozygous Ptpn6meB2/meB2 mice still presenting a healthy phenotype (3 to 6 weeks of age) were strongly driven toward an autoinflammatory condition via Toll-like receptor activation. In sum, lymphocytes (adaptive immunity) and neutrophils and possible macrophages (innate immunity) appeared to be involved in and/or affected by the inflammatory mechanisms associated with the phenotype of Ptpn6meB2/meB2 mice.
Discussion
Diseased animals were discovered in our breeding colonies. The pedigree of the affected siblings identified one male that most likely carried a mutation in an unknown gene. Because approximately 25% (both males and females) of the selected breeding pairs spontaneously developed the condition (sterile inflammation), we concluded that the founder male had carried a recessive autosomal mutation in a single gene in the heterozygous state. Using genome-wide screening, we localized the genomic region on mouse chromosome 6 and narrowed the region to a size of 0.4 Mbp. This chromosome region contained 20 genes, including Ptpn6 (also known as Shp1), which carried a B2 insertion in exon 6. The insertion of the B2 element modified the function of Ptpn6, resulting in the development of an autoinflammatory disease characteristic of the motheaten (me) phenotype in mice. Although the clinical symptoms were delayed in onset and the pathophysiological abnormalities were milder in Ptpn6meB2/meB2 than in either me or me-v mice, the penetrance of the autoinflammatory phenotype was 100%, and the overall clinical features were closely similar to the me phenotypes described in all Ptpn6 mutant mice to date (see Supplemental Table S1 at http://ajp.amjpathol.org). We observed a severe inflammatory skin disease, occasionally accompanied with neutrophilic infiltrations in other organs (mostly in the lung), and the autoinflammatory phenotype was fully suppressible. Because of this altered but not completely abrogated Ptpn6 function, we were able to examine the pathophysiological and immunoregulatory abnormalities associated with this genotype and thus could assess the therapeutic effects of various treatments (Figure 1D) and compare the clinical features associated with this genotype for potentially unlimited periods of time.
Ptpn6 contains two tandem Src homology 2 (SH2) domains at the N-terminus, followed by a single phosphatase domain at the C-terminus that encompasses two tyrosine phosphorylation sites.56 Ptpn6 has been implicated in the negative regulation of a number of receptor tyrosine kinases (c-kit, CSF-1 receptor), cytokines (TNF-α, IL-1β, IL-3, IFN-αβ), Toll-like receptors,7,57–59 and NF-κB,60,61 as well as receptors of the immune system that contain a tyrosine-based inhibitory motif (eg, CD22, FcγRIIB, TCR, and BCR).8,9,12,53,62
The major etiological component was the limited amount of functionally active Ptpn6 enzyme (Figure 3G), which seemed to delay neutrophil apoptosis, leading to an extended lifetime of otherwise short-lived granulocytes. Although the hyperproliferative-like bone marrow suggested increased neutrophil proliferation, this was not the case; rather, the immature and mature neutrophils accumulated in both the bone marrow and peripheral blood (leukocytosis). In turn, neutrophils accumulated in the peripheral blood, and the retrograde effect of leukocytosis apparently created a myeloproliferative-like bone marrow hypercellularity and leukocyte deposition in the spleen (splenomegaly). Clearly, environmental injuries accelerated neutrophil invasion into the skin or other first-exposed organs, which may not occur in germ-free conditions, as described in spin mice.7 The reduction of apoptotic events in neutrophils was most likely the consequence of a defect in NF-κB regulation, due to the partial deficiency of Ptpn6,12,53,54 which affected mostly myeloid cell types63 but affected also lymphocytes64,65 and macrophages.66 The NF-κB machinery was affected at various levels in various hematopoietic cell types in which the myeloid form of Ptpn6 was preferentially expressed. c-Rel and RelA were present in free forms in neutrophils, most likely because of the absence or extremely low levels of IκBα and IκBε and possibly because of a defect in the degradation of the NF-κB/IκB complex, as demonstrated by the accumulation of nondegraded phosphorylated IκBα (Figure 5F). This seems to be especially important in the present case, because c-Rel dimers specifically promote the transcription of the anti-apoptotic Bcl-xL gene,67,68 and RelB deficiency results in the development of skin lesions.69 The apparent growth retardation observed for all me phenotypes, the osteoclastogenesis associated with TNFR-associated factor 6 (TNFRAF-6),70,71 and the inhibition of new bone formation are also secondary consequences of diminished NF-κB regulation.72
Although the diminished Ptpn6 activity affected both lymphoid and myeloid cell types, the autoinflammatory symptoms were due to altered neutrophil function (and possibly some macrophage function). Dramatically altered function of innate immunity dominated the clinical symptoms, and a less pronounced autoimmune phenotype could be detected weeks after disease onset, most likely as a secondary phenomenon. The accumulation of neutrophils in skin (or other organs) does not require the presence of T and B cells, but certain chemotactic factors or bacterial endotoxins are required for neutrophil extravasation.73 Ptpn6 is a negative regulator of both BCR and TCR signaling,2,3,13,18,74 and thus a diminished expression and activity of Ptpn6 may activate B or T cells, as observed in the sick mice, which demonstrated high levels of anti-nuclear antibodies, significantly higher ratios of activated T and B cells, and a high ratio of B1a/b cells.62 Thus, the lymphopenia was mild and only relative, and indeed, although lymphopenia is a critical component of the me phenotype, the autoimmune reactions were overlapped by overwhelming inflammatory reactions caused by innate immune responses.73
Many cytokine-receptor signaling pathways in myeloid cells are activated by tyrosine phosphorylation, and the dephosphorylation caused by Ptpn6 mediates a regulatory signal leading to the deactivation of these signaling pathways. Overall, the loss or partial loss of the negative regulatory function of Ptpn6 in me phenotypes may lead to both inflammatory and autoimmune phenotypes, as evidenced by the increased amounts of a wide range of autoantibodies against single-stranded and double-stranded DNAs, histones, and chromatins, potentially mimicking a number of autoimmune diseases. In turn, depending on the type of mutation and possibly because of the heterogeneity of environmental factors, the me phenotype appears to result from the combination of these two states (ie, both autoinflammatory and autoimmune), and the exposure to exogenous injury (pathergy, as seen in pyoderma gangrenosum) may dominate the clinical phenotype in more severe forms of the condition.
The mutation in the Ptpn6 gene did not affect either the two docking sites (SH2N and SH2C) or the catalytic domain of the enzyme, and resulted in the transcription of a slightly shorter mRNA. We therefore expected the mutation in the Ptpn6 gene to result in an almost full-length protein with some functional deficiency. However, only trace amounts of Ptpn6 protein (Figure 3G) and eventually only a trace amount of phosphatase activity could be detected in the bone marrow or spleen cell lysates using the human/mouse/rat active SHP-1 kit from R&D (Figure 3G).
In addition, the autoinflammatory symptoms were even milder than those observed in me or me-v mice and resembled some features found in the human neutrophilic dermatoses, a heterogeneous group of human diseases with an unknown etiology that share the presentation of inflammatory or autoinflammatory symptoms driven by neutrophil extravasation. We have discovered numerous functionally inactive splice variants in a number of patients with Sweet's syndrome and pyoderma gangrenosum. The partial deletion of exon 4 with the complete lack of exon 5 of the PTPN6 gene in all patients with pyoderma gangrenosum may have a diagnostic value, and might indicate PTPN6 involvement in this disease. These preliminary results in human patients suggest an association of PTPN6 gene expression with neutrophilic dermatoses.75 It is clear that PTPN6 alone does not drive dominant phenotypes in pyoderma gangrenosum or Sweet's syndrome, but most likely is involved in the pathomechanisms of these diseases, either in a downstream signaling pathway or in association with a defect in another phosphatase gene. For example, in PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne)76 mutations in both PTPN12 and proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1; alias CD2BP1) might be involved in this autoinflammatory condition (unpublished data). Another disease-modifying alteration appears to be a correlation between altered PTPN6 function and NF-κB regulation,60,61 which may significantly affect neutrophil survival. Regardless, the me phenotype in mice and neutrophilic dermatoses in humans are fully suppressible,77 and the treatment repertoire may be not restricted to steroids (Figure 1D), TNFα blockade (etanercept or infliximab),78,79 or IL1R antagonist protein (anakinra).80
Acknowledgments
The authors thank many colleagues for assistance with the animal breeding and with the numerous assays performed over many years, the sample collection, and the histology and immunohistochemistry work. Biotin labeling of RNA samples and Affymetrix hybridization were performed by staff of the Genomic Core Facility of the University of Illinois at Chicago.
Footnotes
Supported in part by philanthropic support from the J.O. Galante Endowment Chair at Rush University Medical Center, Chicago (T.T.G.), the Grainger Foundation, Forest Park, IL (T.T.G., K.M.), and the Clark W. Finnerud MD Endowment Chair (M.D.T.).
A.B.N. and S.S. contributed equally to the present work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at DOI: 10.1016/j.ajpath.2010.12.053.
Supplementary data
References
- 1.Green M.C., Shultz L.D. Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J Hered. 1975;66:250–258. doi: 10.1093/oxfordjournals.jhered.a108625. [DOI] [PubMed] [Google Scholar]
- 2.Shultz L.D., Schweitzer P.A., Rajan T.V., Yi T., Ihle J.N., Matthews R.J., Thomas M.L., Beier D.R. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 3.Tsui H.W., Siminovitch K.A., de Souza L., Tsui F.W. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 4.Sidman C.L., Shultz L.D., Unanue E.R. The mouse mutant “motheaten” I. Development of lymphocyte populations. J Immunol. 1978;121:2392–2398. [PubMed] [Google Scholar]
- 5.Sidman C.L., Shultz L.D., Unanue E.R. The mouse mutant “motheaten” II. Functional studies of the immune system. J Immunol. 1978;121:2399–2404. [PubMed] [Google Scholar]
- 6.Shultz L.D., Coman D.R., Bailey C.L., Beamer W.G., Sidman C.L. “Viable motheaten,” a new allele at the motheaten locus. I. Pathology. Am J Pathol. 1984;116:179–192. [PMC free article] [PubMed] [Google Scholar]
- 7.Croker B.A., Lawson B.R., Rutschmann S., Berger M., Eidenschenk C., Blasius A.L., Moresco E.M., Sovath S., Cengia L., Shultz L.D., Theofilopoulos A.N., Pettersson S., Beutler B.A. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1, MyD88, and a microbial trigger. Proc Natl Acad Sci USA. 2008;105:15028–15033. doi: 10.1073/pnas.0806619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Mustelin T., Vang T., Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. 2005;5:43–57. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- 10.Yang J., Liang X., Niu T., Meng W., Zhao Z., Zhou G.W. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:28199–28207. doi: 10.1074/jbc.273.43.28199. [DOI] [PubMed] [Google Scholar]
- 11.Banville D., Stocco R., Shen S.H. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics. 1995;27:165–173. doi: 10.1006/geno.1995.1020. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–359. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasler P., Zouali M. B cell receptor signaling and autoimmunity. FASEB J. 2001;15:2085–2098. doi: 10.1096/fj.00-0860rev. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Somani A.K., Siminovitch K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signaling. Semin Immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 15.Andersen J.N., Jansen P.G., Echwald S.M., Mortensen O.H., Fukada T., Del Vecchio R., Tonks N.K., Moller N.P. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 16.Yi T.L., Cleveland J.L., Ihle J.N. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol Cell Biol. 1992;12:836–846. doi: 10.1128/mcb.12.2.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wlodarski P., Zhang Q., Liu X., Kasprzycka M., Marzec M., Wasik M.A. PU. 1 activates transcription of SHP-1 gene in hematopoietic cells. J Biol Chem. 2007;282:6316–6323. doi: 10.1074/jbc.M607526200. [DOI] [PubMed] [Google Scholar]
- 18.Kozlowski M., Mlinaric-Rascan I., Feng G.-S., Shen R., Pawson T., Siminovitch K.A. Expression and catalytic activity of the tyrosine phosphatase PTP1C is severely impaired in motheaten and viable motheaten mice. J Exp Med. 1993;178:2157–2163. doi: 10.1084/jem.178.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brydges S., Kastner D.L. The systemic autoinflammatory diseases: inborn errors of the innate immune system. Curr Top Microbiol Immunol. 2006;305:127–160. doi: 10.1007/3-540-29714-6_7. [DOI] [PubMed] [Google Scholar]
- 20.Kanazawa N., Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601–618. doi: 10.1111/j.1346-8138.2007.00342.x. [DOI] [PubMed] [Google Scholar]
- 21.Farasat S., Aksentijevich I., Toro J.R. Autoinflammatory diseases: clinical and genetic advances. Arch Dermatol. 2008;144:392–402. doi: 10.1001/archderm.144.3.392. [DOI] [PubMed] [Google Scholar]
- 22.Adarichev V.A., Vermes C., Hanyecz A., Mikecz K., Bremer E., Glant T.T. Gene expression profiling in murine autoimmune arthritis during the initiation and progression of joint inflammation. Arthritis Res Ther. 2005;7:R196–R207. doi: 10.1186/ar1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Underhill G.H., George D., Bremer E.G., Kansas G.S. Gene expression profiling reveals a highly specialized genetic program of plasma cells. Blood. 2003;101:4013–4021. doi: 10.1182/blood-2002-08-2673. [DOI] [PubMed] [Google Scholar]
- 24.Krayev A.S., Markusheva T.V., Kramerov D.A., Ryskov A.P., Skryabin K.G., Bayev A.A., Georgiev G.P. Ubiquitous transposon-like repeats B1 and B2 of the mouse genome: b2 sequencing. Nucleic Acids Res. 1982;10:7461–7475. doi: 10.1093/nar/10.23.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kramerov D.A., Tillib S.V., Ryskov A.P., Georgiev G.P. Nucleotide sequence of small polyadenylated B2 RNA. Nucleic Acids Res. 1985;13:6423–6437. doi: 10.1093/nar/13.18.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pattanakitsakul S., Zheng J.H., Natsuume-Sakai S., Takahashi M., Nonaka M. Aberrant splicing caused by the insertion of the B2 sequence into an intron of the complement C4 gene is the basis for low C4 production in H-2k mice. J Biol Chem. 1992;267:7814–7820. [PubMed] [Google Scholar]
- 27.Blumenstein M., Hossfeld D.K., Duhrsen U. Indirect radiation leukemogenesis in DBA/2 mice: increased expression of B2 repeats in FDC-P1 cells transformed by intracisternal A-particle transposition. Ann Hematol. 1998;76:53–60. doi: 10.1007/s002770050363. [DOI] [PubMed] [Google Scholar]
- 28.Kroutter E.N., Belancio V.P., Wagstaff B.J., Roy-Engel A.M. The RNA polymerase dictates ORF1 requirement and timing of LINE and SINE retrotransposition. PLoS Genet. 2009;5:e1000458. doi: 10.1371/journal.pgen.1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Végvári A., Szabó Z., Szántó S., Nesterovitch A.B., Mikecz K., Glant T.T., Adarichev V.A. Two major interacting chromosome loci control disease susceptibility in murine model of spondyloarthropathy. J Immunol. 2005;175:2475–2483. doi: 10.4049/jimmunol.175.4.2475. [DOI] [PubMed] [Google Scholar]
- 30.Cao T., Pinter E., Al Rashed S., Gerard N., Hoult J.R., Brain S.D. Neurokinin-1 receptor agonists are involved in mediating neutrophil accumulation in the inflamed, but not normal, cutaneous microvasculature: an in vivo study using neurokinin-1 receptor knockout mice. J Immunol. 2000;164:5424–5429. doi: 10.4049/jimmunol.164.10.5424. [DOI] [PubMed] [Google Scholar]
- 31.Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. A novel assay for apoptosis: Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 32.Wells S.M., Kantor A.B., Stall A.M. CD43 (S7) expression identifies peripheral B cell subsets. J Immunol. 1994;153:5503–5515. [PubMed] [Google Scholar]
- 33.Hoffmann M.H., Tuncel J., Skriner K., Tohidast-Akrad M., Turk B., Pinol-Roma S., Serre G., Schett G., Smolen J.S., Holmdahl R., Steiner G. The rheumatoid arthritis-associated autoantigen hnRNP-A2 (RA33) is a major stimulator of autoimmunity in rats with pristane-induced arthritis. J Immunol. 2007;179:7568–7576. doi: 10.4049/jimmunol.179.11.7568. [DOI] [PubMed] [Google Scholar]
- 34.Mikecz K., Brennan F.R., Kim J.H., Glant T.T. Anti-CD44 treatment abrogates tissue edema and leukocyte infiltration in murine arthritis. Nat Med. 1995;1:558–563. doi: 10.1038/nm0695-558. [DOI] [PubMed] [Google Scholar]
- 35.Hutás G., Bajnok E., Gál I., Finnegan A., Glant T.T., Mikecz K. CD44-specific antibody treatment and CD44 deficiency exert distinct effects on leukocyte recruitment in experimental arthritis. Blood. 2008;112:4999–5006. doi: 10.1182/blood-2008-04-150383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi M., Dennis K., Peschon J.J., Chandrasekaran R., Mikecz K. Antibody-induced shedding of CD44 from adherent cells is linked to the assembly of the cytoskeleton. J Immunol. 2001;167:123–131. doi: 10.4049/jimmunol.167.1.123. [DOI] [PubMed] [Google Scholar]
- 37.Sarraj B., Ludányi K., Glant T.T., Finnegan A., Mikecz K. Expression of CD44 and L-selectin in the innate immune system is required for severe joint inflammation in the proteoglycan-induced murine model of rheumatoid arthritis. J Immunol. 2006;177:1932–1940. doi: 10.4049/jimmunol.177.3.1932. [DOI] [PubMed] [Google Scholar]
- 38.Angyal A., Egelston C., Kobezda T., Olasz K., László A., Glant T.T., Mikecz K. Development of proteoglycan-induced arthritis depends on T cell-supported autoantibody production, but does not involve significant influx of T cells into the joints. Arthritis Res Ther. 2010;12:R44. doi: 10.1186/ar2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pahl H.L., Krauss B., Schulze-Osthoff K., Decker T., Traenckner E.B., Vogt M., Myers C., Parks T., Warring P., Mühlbacher A., Czernilofsky A.P., Baeuerle P.A. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med. 1996;183:1829–1840. doi: 10.1084/jem.183.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Umezawa K., Ariga A., Matsumoto N. Naturally occurring and synthetic inhibitors of NF-kappaB functions. Anticancer Drug Des. 2000;15:239–244. [PubMed] [Google Scholar]
- 41.López-Franco O., Suzuki Y., Sanjuán G., Blanco J., Hernández-Vargas P., Yo Y., Kopp J., Egido J., Gómez-Guerrero C. Nuclear factor-kappa B inhibitors as potential novel anti-inflammatory agents for the treatment of immune glomerulonephritis. Am J Pathol. 2002;161:1497–1505. doi: 10.1016/s0002-9440(10)64425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q., Raghunath P.N., Vonderheid E., Odum N., Wasik M.A. Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promotor. Am J Pathol. 2000;157:1137–1146. doi: 10.1016/S0002-9440(10)64629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vermes C., Roebuck K.A., Chandrasekaran R., Dobai J.G., Jacobs J.J., Glant T.T. Particulate wear debris activates protein tyrosine kinases and nuclear factor-kappa B which downregulates type I collagen synthesis in human osteoblasts. J Bone Miner Res. 2000;15:1756–1765. doi: 10.1359/jbmr.2000.15.9.1756. [DOI] [PubMed] [Google Scholar]
- 44.Roebuck K.A., Vermes C., Carpenter L.R., Fritz E.A., Narayanan R., Glant T.T. Down-regulation of procollagen alpha1[I] messenger RNA by titanium particles correlates with nuclear factor kappaB (NF-kappaB) activation and increased Rel A and NF-kappaB1 binding to the collagen promoter. J Bone Miner Res. 2001;16:501–510. doi: 10.1359/jbmr.2001.16.3.501. [DOI] [PubMed] [Google Scholar]
- 45.Van Zant G., Shultz L. Hematologic abnormalities of the immunodeficient mouse mutant, viable motheaten (mev) Exp Hematol. 1989;17:81–87. [PubMed] [Google Scholar]
- 46.Shultz L.D., Bailey C.L., Coman D.R. Hematopoietic stem cell function in motheaten mice. Exp Hematol. 1983;11:667–680. [PubMed] [Google Scholar]
- 47.Fülöp C., Szántó S., Mukhopadhyay D., Bárdos T., Kamath R.V., Rugg M.S., Day A.J., Salustri A., Hascall V.C., Glant T.T., Mikecz K. Impaired cumulus mucification and female sterility in tumor necrosis factor-induced protein-6 deficient mice. Development. 2003;130:2253–2261. doi: 10.1242/dev.00422. [DOI] [PubMed] [Google Scholar]
- 48.Shultz L.D., Green M.C. Motheaten, an immunodeficient mutant of the mouse. II. Depressed immune competence and elevated serum immunoglobulins. J Immunol. 1976;116:936–943. [PubMed] [Google Scholar]
- 49.Lutzner M.A., Hansen C.T. Motheaten: an immunodeficient mouse with markedly less ability to survive than the nude mouse in a germfree environment. J Immunol. 1976;116:1496–1497. [PubMed] [Google Scholar]
- 50.Castro-Alcaraz S., Miskolci V., Kalasapudi B., Davidson D., Vancurova I. NF-kappa B regulation in human neutrophils by nuclear I kappa B alpha: correlation to apoptosis. J Immunol. 2002;169:3947–3953. doi: 10.4049/jimmunol.169.7.3947. [DOI] [PubMed] [Google Scholar]
- 51.Klement J.F., Rice N.R., Car B.D., Abbondanzo S.J., Powers G.D., Bhatt P.H., Chen C.H., Rosen C.A., Stewart C.L. IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mémet S., Laouini D., Epinat J.C., Whiteside S.T., Goudeau B., Philpott D., Kayal S., Sansonetti P.J., Berche P., Kanellopoulos J., Israël A. IkappaBepsilon-deficient mice: reduction of one T cell precursor subspecies and enhanced Ig isotype switching and cytokine synthesis. J Immunol. 1999;163:5994–6005. [PubMed] [Google Scholar]
- 53.Li Q., Verma I.M. NF-kappaB regulation in the immune system [Erratum appeared in Nat Rev Immunol 2002;2:975] Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 54.Renner F., Schmitz M.L. Autoregulatory feedback loops terminating the NF-kappaB response. Trends Biochem Sci. 2009;34:128–135. doi: 10.1016/j.tibs.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 55.Sodhi A., Tarang S., Kesherwani V. Concanavalin A induced expression of Toll-like receptors in murine peritoneal macrophages in vitro. Int Immunopharmacol. 2007;7:454–463. doi: 10.1016/j.intimp.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 56.Neel B.G., Tonks N.K. Protein tyrosine phosphatase in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 57.Zhao J., Brooks D.M., Lurie D.I. Lipopolysaccharide-activated SHP-1-deficient motheaten microglia release increased nitric oxide, TNF-alpha, and IL-1beta. Glia. 2006;53:304–312. doi: 10.1002/glia.20283. [DOI] [PubMed] [Google Scholar]
- 58.Sacre S.M., Andreakos E., Kiriakidis S., Amjadi P., Lundberg A., Giddins G., Feldmann M., Brennan F., Foxwell B.M. The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007;170:518–525. doi: 10.2353/ajpath.2007.060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyake A., Murata Y., Okazawa H., Ikeda H., Niwayama Y., Ohnishi H., Hirata Y., Matozaki T. Negative regulation by SHPS-1 of Toll-like receptor-dependent proinflammatory cytokine production in macrophages. Genes Cells. 2008;13:209–219. doi: 10.1111/j.1365-2443.2007.01161.x. [DOI] [PubMed] [Google Scholar]
- 60.Daigle I., Yousefi S., Colonna M., Green D.R., Simon H.U. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat Med. 2002;8:61–67. doi: 10.1038/nm0102-61. [DOI] [PubMed] [Google Scholar]
- 61.Neznanov N., Neznanova L., Kondratov R.V., O'Rourke D.M., Ullrich A., Gudkov A.V. The ability of protein tyrosine phosphatase SHP-1 to suppress NFkappaB can be inhibited by dominant negative mutant of SIRPalpha. DNA Cell Biol. 2004;23:175–182. doi: 10.1089/104454904322964779. [DOI] [PubMed] [Google Scholar]
- 62.Pao L.I., Lam K.P., Henderson J.M., Kutok J.L., Alimzhanov M., Nitschke L., Thomas M.L., Neel B.G., Rajewsky K. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 63.Kruger J., Butler J.R., Cherapanov V., Dong Q., Ginzberg H., Govindarajan A., Grinstein S., Siminovitch K.A., Downey G.P. Deficiency of Src homology 2-containing phosphatase 1 results in abnormalities in murine neutrophil function: studies in motheaten mice. J Immunol. 2000;165:5847–5859. doi: 10.4049/jimmunol.165.10.5847. [DOI] [PubMed] [Google Scholar]
- 64.Khaled A.R., Butfiloski E.J., Sobel E.S., Schiffenbauer J. Functional consequences of the SHP-1 defect in motheaten viable mice: role of NF-kappa B. Cell Immunol. 1998;185:49–58. doi: 10.1006/cimm.1998.1272. [DOI] [PubMed] [Google Scholar]
- 65.Khaled A.R., Butfiloski E.J., Villas B., Sobel E.S., Schiffenbauer J. Aberrant expression of the NF-kappaB and IkappaB proteins in B cells from viable motheaten mice. Autoimmunity. 1999;30:115–128. doi: 10.3109/08916939908994769. [DOI] [PubMed] [Google Scholar]
- 66.Wei G., Guo J., Doseff A.I., Kusewitt D.F., Man A.K., Oshima R.G., Ostrowski M.C. Activated Ets2 is required for persistent inflammatory responses in the motheaten viable model. J Immunol. 2004;173:1374–1379. doi: 10.4049/jimmunol.173.2.1374. [DOI] [PubMed] [Google Scholar]
- 67.Fu L., Lin-Lee Y.C., Pham L.V., Tamayo A.T., Yoshimura L.C., Ford R.J. BAFF-R promotes cell proliferation and survival through interaction with IKKbeta and NF-kappaB/c-Rel in the nucleus of normal and neoplastic B-lymphoid cells. Blood. 2009;113:4627–4636. doi: 10.1182/blood-2008-10-183467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarnico I., Lanzillotta A., Boroni F., Benarese M., Alghisi M., Schwaninger M., Inta I., Battistin L., Spano P., Pizzi M. NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem. 2009;108:475–485. doi: 10.1111/j.1471-4159.2008.05783.x. [DOI] [PubMed] [Google Scholar]
- 69.Barton D., HogenEsch H., Weih F. Mice lacking the transcription factor RelB develop T cell-dependent skin lesions similar to human atopic dermatitis. Eur J Immunol. 2000;30:2323–2332. doi: 10.1002/1521-4141(2000)30:8<2323::AID-IMMU2323>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Z., Jimi E., Bothwell A.L. Receptor activator of NF-kappa B ligand stimulates recruitment of SHP-1 to the complex containing TNFR-associated factor 6 that regulates osteoclastogenesis. J Immunol. 2003;171:3620–3626. doi: 10.4049/jimmunol.171.7.3620. [DOI] [PubMed] [Google Scholar]
- 71.Aoki K., Didomenico E., Sims N.A., Mukhopadhyay K., Neff L., Houghton A., Amling M., Levy J.B., Horne W.C., Baron R. The tyrosine phosphatase SHP-1 is a negative regulator of osteoclastogenesis and osteoclast resorbing activity: increased resorption and osteopenia in me(v)/me(v) mutant mice. Bone. 1999;25:261–267. doi: 10.1016/s8756-3282(99)00174-x. [DOI] [PubMed] [Google Scholar]
- 72.Iotsova V., Caamaño J., Loy J., Yang Y., Lewin A., Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 73.Yu C.C.K., Tsui H.W., Ngan B.Y., Shulman M.J., Wu G.E., Tsui F.W.L. B and T cells are not required for the viable motheaten phenotype. J Exp Med. 1996;183:371–380. doi: 10.1084/jem.183.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang J., Xu X., Liu Y. Activation-induced cell death in T cells and autoimmunity. Cell Mol Immunol. 2004 –192186. [PubMed] [Google Scholar]
- 75.Nesterovitch A.B., Gyorfy Z., Hoffman M.D., Moore E.C., Elbuluk N., Tryniszewska B., Rauch T.T., Simon M., Kang D., Fisher G.J., Mikecz K., Tharp M.D., Glant T.T. Alteration in the gene encoding protein tyrosine phosphatase non-receptor type-6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol. 2011;178:1434–1441. doi: 10.1016/j.ajpath.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wise C.A., Gillum J.D., Seidman C.E., Lindor N.M., Veile R., Bashiardes S., Lovett M. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–969. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- 77.Mazza J., Rossi A., Weinberg J.M. Innovative uses of tumor necrosis factor alpha inhibitors. Dermatol Clin. 2010;28:559–575. doi: 10.1016/j.det.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 78.McGowan J.W.4, Johnson C.A., Lynn A. Treatment of pyoderma gangrenosum with etanercept. J Drugs Dermatol. 2004;3:441–444. [PubMed] [Google Scholar]
- 79.Sapienza M.S., Cohen S., Dimarino A.J. Treatment of pyoderma gangrenosum with infliximab in Crohn's disease. Dig Dis Sci. 2004;49:1454–1457. doi: 10.1023/b:ddas.0000042245.20042.4f. [DOI] [PubMed] [Google Scholar]
- 80.Brenner M., Ruzicka T., Plewig G., Thomas P., Herzer P. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161:1199–1201. doi: 10.1111/j.1365-2133.2009.09404.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.