

Abstract
The cellular and molecular mechanisms of phagocytic clearance of apoptotic cells and debris have been intensely studied in invertebrate model organisms and in the mammalian immune system. This evolutionarily conserved process serves multiple purposes. Uncleared debris from dying cells or aggregated proteins can be toxic and may trigger exaggerated inflammatory responses. Even though apoptotic cell death and debris accumulation are key features of neurodegenerative diseases, relatively little attention has been paid to this important homeostatic function in the central nervous system (CNS). This review attempts to summarize our knowledge of phagocytic clearance in the CNS, with a focus on retinal degeneration, forms of which are caused by mutations in genes within known phagocytic pathways, and on Alzheimer's disease (AD). Interest in phagocytic clearance mechanisms in AD was stimulated by the discovery that immunization could promote phagocytic clearance of amyloid-β; however, much less is known about clearance of neuronal and synaptic corpses in AD and other neurodegenerative diseases. Because the regulation of phagocytic activity is intertwined with cytokine signaling, this review also addresses the relationships among CNS inflammation, glial responses, and phagocytic clearance.
Two decades of work in both Caenorhabditis elegans and Drosophila melanogaster models, as well as in mammalian non-neural cells, has revealed numerous receptors and intracellular effector molecules involved in the recognition and engulfment of apoptotic cells (Figure 1).1 However, to what extent and in which cell types these molecules function in the context of specific neurodegenerative diseases is largely unstudied.
Figure 1.
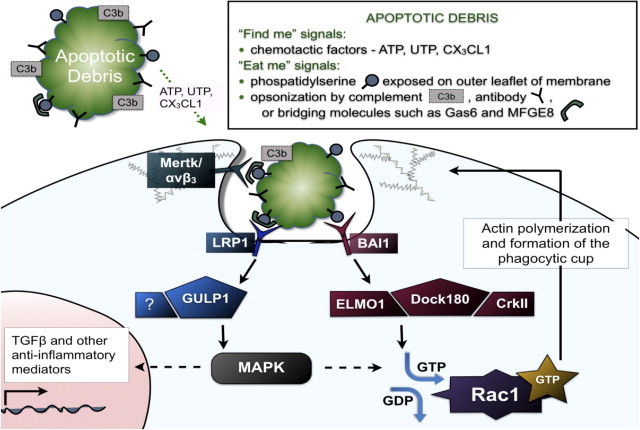
Steps involved in phagocytosis of apoptotic debris. Apoptotic cells release diffusible find-me factors, such as ATP and chemokines, and express cell-surface eat-me signals, including phosphatidylserine. Complement and antibody molecules serve as opsonins for debris, and bridging molecules such as milk fat globule EGF factor 8 protein (MFGE8, also known as lactadherin) or growth arrest-specific protein 6 (GAS-6) bind to phosphatidylserine, targeting apoptotic debris for recognition. The phagocyte can thereby identify debris either by direct recognition of phosphatidylserine through receptors such as brain-specific angiogenesis inhibitor 1 (BAI1) or indirectly through recognition of opsonins/bridging molecules via lipoprotein receptor proteins (LRP), complement receptors, Fc receptors, αvβ3/5 integrin, and the receptor tyrosine kinase Mer (MerTK), among others. Signaling via these receptors is presumed to induce phagocytosis, but the precise players for all of these pathways have not yet been described. The small GTPase Rac1 has been implicated as a key downstream player responsible for regulating cytoskeletal alterations that are necessary for formation of the phagocytic cup and subsequent engulfment, and at least two signaling pathways upstream of Rac1 activation have been elucidated. First, a complex of ELMO-Dock180-CrkII acts downstream of the phosphatidylserine receptor, BAI1, and functions as a guanine exchange factor for Rac1. Second, LRP1 interacts with GULP, an adaptor protein that has been linked to Rac1 activation, possibly via mitogen-activated protein kinase (MAPK). Depending on the context, signaling may lead not only to phagocytosis, but also to changes in cell morphology, induction of migration, and secretion of cytokines. For example, engulfment of apoptotic debris stimulates production of anti-inflammatory mediators, such as transforming growth factor β (TGF-β).
Phagocytes in the CNS: Macrophages, Microglia, Astrocytes, and Neurons
Although infiltrating macrophages and their CNS-resident counterparts, microglia, are considered the professional phagocytes in the brain, there are other populations of potential phagocytes in the CNS, including astrocytes, neural stem cells, and possibly even neurons. These cell types derive from different lineages, exhibit different characteristics, and are likely to have distinct roles in phagocytic clearance.
Microglia derive from the hematopoietic lineage, and express typical pattern recognition receptors, including the Toll-like receptors, Fc and complement receptors, cytokine receptors, CD40 and MHC molecules (Table 1). Microglia perform typical immune cell functions, including phagocytosis and antigen presentation, as well as production of inflammatory mediators and modulation of the general immune response.2 Microglia are well known for clearing dead and dying neurons after injury and therefore are a prime candidate for playing a role in phagocytic clearance in neurodegeneration.
Table 1.
Expression of Potential Phagocytic Receptors in Central Nervous System Cells
| Receptor | Ligand | Relevance | Expression detected |
References | ||
|---|---|---|---|---|---|---|
| Microglia | Astrocytes | Neurons | ||||
| Phosphatidylserine receptors | ||||||
| BAI1 | PS | Uptake of apoptotic cells | × | × | × | 1 |
| TIM4 | PS | Uptake of apoptotic cells | 1 | |||
| Scavenger family receptors | PS | Uptake of apoptotic cells | 1, 89 | |||
| Opsonin receptors | ||||||
| MerTK | Gas6 (binds PS) | Uptake of POS, apoptotic cells | × | × | 3, 26 | |
| Gas 6 production⁎ | × | × | ||||
| αvβ3/5 integrin | Mfge8 (binds PS) | Uptake of apoptotic cells, uptake of Aβ | × | × | × | 3, 79 |
| MFGE8 production⁎ | × | |||||
| Complement receptor | Complement | Uptake of apoptotic cells, uptake of Aβ | × | × | × | 22, 55 |
| Complement production⁎ | × | × | × | |||
| FcR | Immunoglobulins | Uptake of apoptotic cells, uptake of Aβ | × | × | × | 64, 90 |
| Immunoglobulin production⁎ | ||||||
| Pattern recognition receptors | ||||||
| TLRs/CD14 | Aβ | Uptake of apoptotic cells, uptake of Aβ | × | × | × | 3, 91, 92, 93 |
| FPRL1 | Aβ | Uptake of Aβ | × | × | × | 94, 95 |
| RAGE | Aβ | Uptake of Aβ, BBB-associated Aβ receptor | × | × | × | 5, 95, 96, 97 |
| CD36 scavenger receptor | Aβ, α-synuclein | Uptake of Aβ | × | × | 42, 95, 98, 99 | |
| Lipoprotein receptors | ||||||
| LDLR | ApoE | Uptake of Aβ-ApoE complexes, mediate Aβ-induced changes in ApoE production | × | × | 3, 95 | |
| ApoE production⁎ | × | |||||
| LRP1 | ApoE | Uptake of Aβ-ApoE complexes, BBB-associated Aβ receptor | × | × | 3, 95 | |
| Uptake of Aβ-ApoJ complexes | × | |||||
| LRP2/megalin | Clusterin (ApoJ) | Clusterin production⁎ | × | × | × | 55, 75, 95 |
Aβ, amyloid-β; Apo, apolipoprotein; BAI1, brain-specific angiogenesis inhibitor-1; BBB, blood-brain barrier; FcR, Fc receptor; FPRL1, formyl peptide receptor-like 1; Gas6, growth-arrest specific 6; LDLR, low density lipoprotein receptor; LRP, low density lipoprotein receptor-related; MerTK, Mer receptor tyrosine kinase; Mfge8, milk fat globule-EGF factor 8 (also known as lactadherin); POS, photoreceptor outer segments; PS, phosphatidylserine; RAGE, receptor for advanced glycation endproducts; TIM4, T-cell immunoglobulin and mucin-domain-containing molecule 4; TLR, Toll-like receptor.
Asterisk indicates that the ligand is an opsonin (bridging molecule), rather than the direct target of the phagocytic receptor.
Astrocytes originate from neural stem cells, sharing common precursors with oligodendrocytes and neurons. A recent microarray study on acutely isolated mouse brain astrocytes unexpectedly revealed that these cells express many components of evolutionarily conserved phagocytic pathways and numerous receptors involved in innate immunity, including Toll-like receptors, scavenger receptors, and mannose receptors, as well as components of the complement system (Table 1).3 Activation of these receptors prompts not only phagocytosis, but also production of cytokines that lead to amplification and/or suppression of the immune response.4
A few studies suggest that even neurons are capable of engulfment, although in many cases the phenomenon is probably better characterized as endocytosis or pinocytosis. Neurons and neuronal cell lines are able to take up aggregated extracellular amyloid-β (Aβ) in vitro, and both low-density lipoprotein receptor-related protein 1 (LRP1) and receptor for advanced glycation end products (RAGE) have been implicated in this process5–7 In addition, it has been hypothesized that neurons participate in pruning of their neighbors' synapses during development. In a mouse model of prion disease, ultrastructural evidence suggested that dendritic spines enwrap degenerating presynaptic boutons.8 Whether neurons actually completely phagocytose and process the degenerating material is not clear. Although neurons express at least some of the relevant receptors and intracellular engulfment machinery (Table 1), signaling through these receptors may have different downstream effects than in immune cells, with expression of adaptor proteins and coreceptors that are specific to neurons.9
Although microglia are considered by some to be the macrophages of the brain, it has been suggested that peripherally derived macrophages have different properties than resident microglia and could contribute to clearance of Aβ plaques.10 This has been difficult to study, however. When peripheral monocytes incorporate into the parenchyma, it is usually impossible to distinguish them from resident microglia morphologically, because there are no specific markers that definitively differentiate them. In addition, there is debate about whether peripheral monocytes infiltrate into the parenchyma to a significant degree under nontraumatic circumstances.11,12 Some studies using bone marrow transplant with green fluorescent protein-expressing myeloid cells suggest that, in Alzheimer's disease models, monocytes are recruited to the brain, evolve morphologically and functionally into microglia, and localize to sites of Aβ deposition. Critics of this theory, however, have shown that the irradiation involved in bone marrow transplantation increases the leakiness of the blood-brain barrier and that the previous model has not unequivocally demonstrated that infiltration occurs to a significant degree without radiation.12
Activated Phagocytes: A Phenotypic Spectrum
Activation of a glial cell in response to debris not only stimulates phagocytosis, but also results in other downstream effects, such as secretion of cytokines and production of reactive oxygen species.13 An issue that complicates analysis of the literature is that researchers refer to activation of glia without acknowledging or accounting for the ambiguity of that label. This is problematic, because there is a continuum of activation for microglia that entails a range of phenotypes, and the expression of activation markers can vary.14 For example, activation markers such as p38, CD45, and FcR have been shown to be differentially regulated over time in amyloid precursor protein (APP) transgenic mice exposed to anti-Aβ immunotherapy.14 Using one marker is clearly insufficient to fully characterize the activation phenotype of a glial cell. Macrophage activation has been described in terms of classical (M1) and alternative (M2) activation, largely based on cytokine and receptor expression profiles.15 Classical activation has as a hallmark the production of pro-inflammatory cytokines and free radicals; alternative activation is a less well defined anti-inflammatory phenotype. Expression profiling offers some utility, and use of delineations that have been established for macrophages serves as a starting point. Nonetheless, the immunomodulatory milieu in the CNS differs, and activation phenotypes in the CNS may not mimic those in the periphery.16 Quantitative real-time PCR examining expression of genes associated with inflammation indicated that, in AD brain and in AD mouse models, innate immune cells exhibit a hybrid activation state characteristic of both classical and alternative activation.17 Further studies are needed to generate functional delineations, so that researchers can better categorize the spectrum of glial activation phenotypes.
Regulation of Phagocytosis
Cell-cell interactions and the cytokine environment determine whether phagocytes are ready for clearance. Several studies have shown that astrocytes modulate microglial phagocytic activity in vitro.4,18 Astrocyte-conditioned medium has a general inhibitory effect,4 and a relatively unexplored concept is that interactions of CD40 and CD40 ligand (CD40L) could be involved.19 CD40-CD40L binding plays a role in peripheral immune activation and results in a range of outcomes, including up-regulation of costimulatory molecules, activation of antigen-presenting cells, and stimulation of cytokine production.18
Neuronal signals also appear to influence the activation of glia. For example, in culture, neuronal activity modulates interferon-γ-induced major histocompatibility complex class II (MHCII) expression on astrocytes and microglia, ultimately dampening inflammatory activity.20 Thus, it seems likely that neurons influence the overall activation phenotype and phagocytic properties of surrounding glia. In the context of neurodegeneration, neurons may lose their ability to dampen glial proinflammatory activity.
T cells are able to modulate microglial activities in vitro, and studies have shown that Treg cells can be neuroprotective.21 Although T cells have the machinery for direct communication (eg, through CD40-CD40L interactions), their relatively limited access to CNS parenchyma implies that indirect mechanisms such as cytokine signaling could be involved. Further work is needed to evaluate the complex integration of proinflammatory and anti-inflammatory cytokine signaling in glial cells and their combined effects on phagocytic activity.22,23
Clearance of Neuronal and Synaptic Corpses
Even though one of the key features of neurodegeneration is cell death and synapse loss, we know little about the ultimate fate of cell corpses and debris in disease states. Growing evidence implicates autophagy as a key process in several neurodegenerative diseases.24 In theory, however, a neuron undergoing autophagic death must still ultimately be cleared by phagocytosis. Although culture studies have convincingly shown that microglia can ingest apoptotic neurons and neuritic blebs,13 the majority of the literature provides only anecdotal descriptions of the fate of degenerating neurons, either in disease or in normal aging. Direct perturbations of glial phagocytic behaviors, perhaps using transgenic and conditional knockout mouse approaches, will be necessary to test cellular and molecular mechanisms of apoptotic neuronal clearance.
Retinal Degeneration
Phagocytic clearance plays a critical role in the maintenance of the retinal photoreceptors, and retinal degeneration can occur as a result of defects in modification, recognition, engulfment, or degradation of photoreceptor debris. Rod photoreceptors shed their outer segment tips diurnally in response to light, and the retinal pigment epithelium (RPE) is responsible for removal of this cellular debris.25 The RPE uses the same phagocytic mechanisms as are involved in the recognition and engulfment of apoptotic cells elsewhere in the body (Figure 1). In particular, opsonins such as milk fat globule EGF factor 8 protein (Mfge8; also known as lactadherin) and growth arrest-specific protein 6 (Gas6) bind phosphatidylserine and target the photoreceptor outer segments (POS) for recognition by the RPE,26,27 acting as ligands for receptors such as the lipid scavenger receptor CD36, an integrin adhesion receptor (αvβ5), and MerTK (Figure 2).28,29
Figure 2.
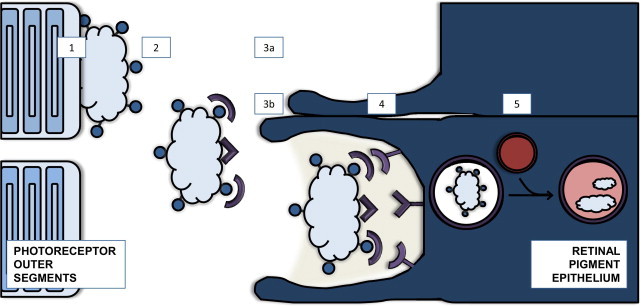
Sequence of events and factors involved in clearance of photoreceptor outer segments (POS) in the retina. Photoreceptors shed their tips diurnally in response to light, and the retinal pigment epithelium (RPE) is responsible for clearing this cellular debris.25 Retinal degeneration can occur as a result of defects in modification, recognition, engulfment, or degradation of photoreceptor debris. 1: Membrane modification. ABCA4 functions in removal of unmetabolizable byproducts such as all-trans retinal and N-retinylidene-PE on the POS membranes. 2: Segments are shed and expose phosphatidylserine. 3: Opsonization. 3a: GAS-6, protein S, and MFGE8 opsonize the shed POS by binding phosphatidylserine. 3b: Tubby and tubby-like protein 1 are ligands for receptor tyrosine kinase MerTK and promote phagocytosis via an unknown target, independent of PS. 4: Receptor recognition and engulfment. Receptors such as MerTK, lipid scavenger receptor CD36, integrin adhesion receptor (αvβ5), and TYRO3 recognize the shed POS and promote engulfment. 5: Intracellular trafficking of phagosomes. Phagosome-lysosome fusion leads to degradation of engulfed material. Mutations in genes involved in these pathways that have been linked to human disease include ABCA4 (Stargardt's macular degeneration), MERTK (retinitis pigmentosa), and MYO7A (Usher's syndrome).
Defects in clearance of POS lead to retinal degeneration characterized by photoreceptor death and progressive loss of vision. Due to a lack of clearance, material from the shed segments accumulates in the outer retina, and subsequently the photoreceptors and the RPE lose contact. The interaction between photoreceptors and the RPE is critical for photoreceptor survival; on accumulation of debris and disruption of contact, therefore, photoreceptor cells degenerate.25
There is evidence that POS membranes must be modified before they are engulfed. For example, in Stargardt macular degeneration, mutation of ABCA4 results in processing defects that lead to accumulation of all-trans retinal and N-retinylidene-PE on the POS membrane. Subsequent to engulfment by the RPE cells, these molecules react and create an unmetabolizable byproduct, A2E, which accrues as lipofuscin deposits. In addition, this byproduct can be converted to epoxides that are toxic to the RPE. The inability to degrade these products and the subsequent damage renders RPE cells unable to support photoreceptor cells, and thus leads to retinal degeneration.25
Retinitis pigmentosa is another retinal disorder that has been associated with defects in phagocytic clearance. The connection between clearance and retinitis pigmentosa was shown in an animal model, the Royal College of Surgeons (RCS) rat.30 This strain has a naturally occurring mutation in the gene encoding MerTK, the receptor responsible for recognizing Gas6-opsonized POS and inducing engulfment. Subsequent analyses identified cohorts of patients with retinitis pigmentosa who have mutations in the MERTK gene.31 Of note, studies in the RCS rat have shown that lentiviral-mediated gene therapy with MerTK can correct the phagocytic defect, slow photoreceptor loss, and preserve retinal function for up to 7 months.32 This suggests engulfment receptor defects as a possible target for therapy of some forms of retinitis pigmentosa.
To degrade debris from the shed segments, the RPE must engulf and then also mediate intracellular trafficking of the material to promote phagosome-lysosome fusion. Trafficking of the vesicles containing engulfed material is critical for clearance of POS debris. Usher type 1 syndrome has been associated with defects in myosin VIIa, and studies in knockout animals suggest that this retinal degeneration is due to disturbed vesicular trafficking and fusion between phagosomes and lysosomes.25 The term phagocytic clearance refers not only to the uptake of debris, but also to the degradation of engulfed material. This retinal disorder highlights the fact that these two processes are distinct but intimately tied, and dysfunction of either can result in retinal pathology.
These examples illuminate the fact that there are multiple steps involved in phagocytic clearance of cellular debris, and that secondary degeneration can arise due to genetic defects in any of these processes. It will be of great interest to determine whether similar genetic defects could be causative for any neurodegenerative or neurodevelopmental diseases affecting the brain and spinal cord.
Alzheimer's Disease
The amyloid-β cascade hypothesis posits that accumulation of Aβ protein is the fundamental initiator of Alzheimer's disease (AD).33 It is known that Aβ is neurotoxic in itself, but in addition the glial response to Aβ can contribute to its toxicity (Figure 3). The fact that pathology involves accumulation of extracellular Aβ led some researchers to hypothesize that plaque formation is due to defects in phagocytic clearance of the peptide and its aggregates. Studies have therefore been initiated to examine the role that phagocytic cells play in the development of Aβ plaques and AD pathology. Although it is known that reactive glia localize to plaques, possess receptors that recognize Aβ, and are able to phagocytose it, ultimately many questions remain about their contributions to AD.
Figure 3.
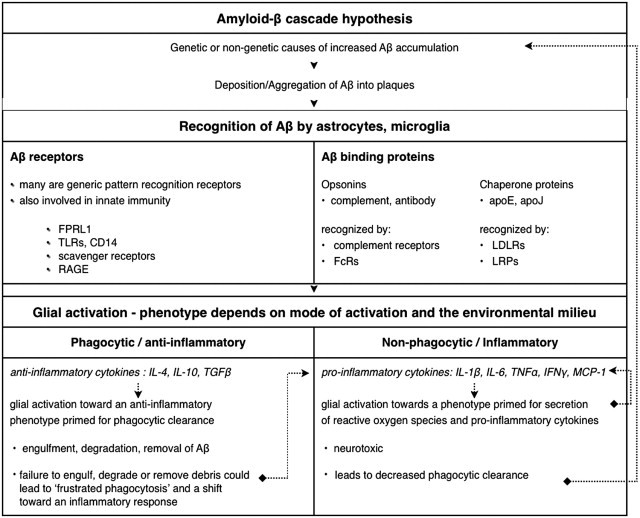
Sequence of events connecting the amyloid cascade hypothesis to putative glial cell responses. Receptors that stimulate phagocytosis are also involved in inducing inflammatory pathways, and clearly the mechanism of activation determines the activation phenotype of the cell.14,18 It has been posited that frustrated phagocytosis may occur when glia are unable to clear debris such as Aβ.35 Persistence of extracellular debris leads to continual stimulation and production of degrading enzymes and reactive oxygen species, as well as proinflammatory signals. This chronic inflammation creates an environment that is toxic to neurons and promotes neurodegeneration. In theory, tipping the balance to improve phagocytic clearance and decrease secretion of proinflammatory mediators may ameliorate Aβ pathology. The dotted lines illustrate the concept that pathology may be amplified due to a pro-inflammatory milieu and/or defects in phagocytic clearance.
Evidence for Phagocytosis of Aβ
Astrocytosis and microgliosis are invariant features of severe AD,34,35 and the reactive phenotype may be a sign that astrocytes and microglia are attempting to clear Aβ, recognized as foreign. In fact, microglia and astrocytes have been shown to contain Aβ in AD brain. This accumulation is presumed to be a result of engulfment, and not due to endogenous production of Aβ by glia, although both microglia and astrocytes do express the amyloid precursor protein.36 Astrocytes distant from plaques do not contain significant amounts of Aβ, and astroglial Aβ deposits are clearly spatially associated with plaques.34 Evidence from electron microscopy shows that microglia surround amyloid fibril deposits in AD brain tissue, suggesting that microglia are involved in their phagocytosis.37 Finally, postmortem immunohistochemistry and light microscopy have shown significant amounts of Aβ within microglia of AD patients treated with immunization therapy.38
Although there is evidence that glia are capable of phagocytosing Aβ, questions remain as to why clearance is inefficient in the AD brain, and researchers have attempted to use in vitro and ex vivo studies to understand these processes. These studies have yielded contradictory results with regard to microglial and astrocytic responses to Aβ. In culture, both microglia and astrocytes appear to be able to phagocytose Aβ,39,40 at least when in the soluble form. Nonetheless, the glial response to Aβ in tissue isolated from AD brains appears to vary, and there have been conflicting reports as to whether astrocytes and microglia can clear Aβ plaques ex vivo.4,39,40 The relative roles of astrocytes and microglia in Aβ clearance in vivo remain to be definitively clarified.
One factor to consider when deciphering the meaning of studies is that the aggregation state of Aβ influences the glial response, and different forms of Aβ appear to be taken up by different mechanisms. Culture studies have suggested that soluble Aβ is taken up by microglial pinocytosis,41 whereas fibrillar Aβ is usually described as being phagocytosed.42 Of note, oligomeric and fibrillar Aβ stimulate differential activation of microglia,43 and have different effects on phagocytosis.15 In particular, plaque composition is variable in the AD brain,44 and microglia preferentially associate with dense core plaques, as opposed to diffuse deposits.35
Recruitment and Activation of Glia
Multiple mediators play a role in recruitment and activation of glial cells in AD, including Aβ itself,18 the complement system,45 chemokines and other cytokines, and immune signaling pathways.46 These signals may arise from neurons, microglia, or astrocytes in the vicinity of plaques. Importantly, the characteristics of these signals influence whether glia are phagocytic or secrete proinflammatory cytokines.
Amyloid-β triggers microglial production of proinflammatory mediators, and these mediators have been shown to inhibit phagocytosis. Real-time PCR measurements show that Aβ treatment of microglia isolated from AD brains induces expression of proinflammatory genes, and not genes involved in phagocytosis.47 In culture, the addition of proinflammatory cytokines inhibits both β1 integrin-dependent and opsonized zymosan-stimulated phagocytosis; however, anti-inflammatory cytokines are able to restore phagocytic activity of microglia residing in a proinflammatory milieu.22 Transforming growth factor-β (TGF-β) increased Aβ clearance in culture, and reduced plaque burden in hAPP transgenic mice overexpressing TGF-β.48
A recent study found that knockout of the cytokine receptor CX3CR1 in microglia prevents neuron loss in a mouse model of AD, further supporting the idea that microglial response to certain cytokines has a negative impact in AD.38 Mechanistically, it appears that proinflammatory cytokines induce production of prostaglandins, which act on E prostanoid receptors to inhibit phagocytosis. This is supported by the fact that antagonists of prostaglandin E2 (PGE2) rescue phagocytosis,22 and by studies showing that knockout of the E prostanoid receptor subtype 2 increases phagocytosis of Aβ.39
Findings from one study suggest that norepinephrine influences microglial activation toward a beneficial response. Stimulation of microglia with norepinephrine suppressed Aβ-induced cytokine production and increased microglial migration and phagocytosis of Aβ.49
Interactions between astrocytes and microglia may also be involved in regulation of phagocytosis in the AD brain. When microglia are exposed to isolated Aβ plaques, they phagocytose and clear the cores; however, coculture with astrocytes or astrocyte-conditioned medium suppresses microglial phagocytosis.4 The mechanisms underlying this interaction are not completely clear, but it is known that exposing astrocytes to Aβ results in up-regulation and secretion of cytokines and reactive species such as tumor necrosis factor α (TNFα), IL-1β, and nitrite,50 and these may be the key factors negatively regulating microglial phagocytosis. CD40-CD40 ligand interactions merit further investigation in the context of plaque clearance. Microglia and astrocytes express CD40 and CD40L, respectively, and expression of both of these factors is increased in AD.19 In addition, there is increased soluble CD40L in plasma of AD patients, and these levels may be predictive of disease progression.51 In vitro studies have shown that CD40 interaction with soluble CD40L in the presence of Aβ results in inhibition of microglial phagocytosis and a shift toward secretion of proinflammatory cytokines.22,52 This cytokine profile has been shown to be associated with decreased phagocytic activity (Figure 3).22 The relevance of astrocytic-microglial communication via CD40L has yet to be demonstrated in vivo.
Recognition of Aβ
Recognition of Aβ may occur via direct receptor binding or through opsonization by complement and immunoglobulin. Additionally, chaperone molecules such as apolipoproteins can bind Aβ and have been implicated in engulfment. These have respective receptors on glia, and binding of Aβ-containing complexes leads to uptake. Some of these components have been associated with AD through genome-wide association studies, including complement receptor 1, apolipoproteins E and J, and phosphatidylinositol-binding clathrin assembly lymphoid myeloid leukemia protein (PICALM).53,54
Aβ Receptors
A number of receptors have been implicated in recognition and uptake of Aβ by glia (Table 1), and many are also more generally involved in innate immune responses, as well as in lipoprotein recognition and uptake. It is probable that receptors that directly recognize Aβ may promote its internalization, as well as relaying signals to modulate the inflammatory response.
Complement System
The complement pathway may be involved in regulating inflammatory signaling and/or targeting material for engulfment in AD.45 In vitro experiments have suggested that complement-dependent phagocytic clearance is inhibited by proinflammatory cytokines,22 and this may explain why complement is not sufficient to stimulate clearance of plaques in the inflamed AD brain.
Neurons, astrocytes, and microglia have all been reported to express components of the complement system,55 and complement factors have been detected in the vicinity of Aβ plaques.56,57 In vitro studies have shown that Aβ directly binds both complement C3 and C1q, and that, when co-incubated in cell-free suspension, Aβ can be isolated along with C3 activation fragments.58 This suggests that Aβ is sufficient to activate the complement pathway and may augment the inflammatory reaction or promote engulfment. On complement activation, subsequent cascades of events not only lead to glial recruitment, activation, and engulfment, but also result in formation of the membrane attack complex, which is presumed to lead to enhanced neurotoxicity.56 The detrimental consequences of complement activation seem to suggest that it could be a potential target for therapeutics; however, eliminating complement C3 appears to worsen outcome, at least in murine models of AD.
In hAPP transgenic mice, complement C3 deficiency caused by overexpression of a complement inhibitor, soluble complement receptor-related protein y (Scrry), or by knockout, results in increased Aβ plaque deposition.59,60 This suggests that complement may help prevent plaque formation, perhaps acting as an opsonin for Aβ and targeting it for clearance by glia. Also, complement C3 inactivation results in increased neuronal loss and accumulation of degenerating neurons.59,60 It remains to be determined whether the accumulation of degenerating neurons is secondary to persistence of neurotoxic Aβ (as the authors suggested) or, alternatively, could be due to loss of a direct complement-mediated engulfment of degenerating neurons.
Genome-wide association studies of late-onset sporadic AD found significant associations with genetic variants of a C3 receptor, CR1,54 but there are conflicting reports regarding whether astrocytes and/or microglia express CR1.55,61
Antibodies
Natural antibodies to Aβ appear to exist in the bloodstream,62 and immunization with Aβ or infusion of antibodies against Aβ appears to promote clearance.63 Experimentally, antibodies have been shown to localize to plaques in vivo and to opsonize Aβ in vitro, presumably targeting Aβ for phagocytosis by glia.64 It is unclear why, in AD, natural antibodies fail to facilitate clearance of Aβ plaques. Studies on samples from individual patients have shown that the array of anti-Aβ antibodies in different patients varies in its ability to inhibit neurotoxicity induced by Aβ in vitro. This is likely due to the fact that they have different avidities and epitope specificity,62 and it suggests that individual variation in antibody repertoires could play a role in susceptibility to the disease.
The antibody response in AD patients may be enhanced through immunization with Aβ or with passive infusion of antibodies. A study in human patients showed that intravenous immunoglobulin infusion resulted in decreases in Aβ concentration in cerebrospinal fluid, with a consequent increase in the bloodstream, perhaps suggesting clearance from the cerebrospinal fluid and redistribution of Aβ to the bloodstream; this study also showed that treatment resulted in cognitive improvement.65 Importantly, immunization therapies appear to increase clearance of Aβ from the parenchyma. Postmortem immunohistochemistry and light microscopy on brains of AD patients immunized with Aβ1–42 showed significant clearance of plaques in response to treatment and revealed that Aβ was taken up by microglia.38 This indicates that immunization can improve pathological indices of AD, and it suggests that opsonization of Aβ with antibodies increases phagocytosis and clearance of plaques.
Chronic administration of anti-Aβ antibody to hAPP transgenic mice resulted in localization of antibodies to plaques, increased FcR expression on microglia, and led to a reduction in Aβ deposits due to phagocytic clearance by microglia.64 Phagocytosis may not be the only mechanism by which antibodies promote clearance. Studies have indicated that antibody-mediated clearance occurs even in FcR knockout animals and when antibody lacking the Fc region is administered.66,67 Furthermore, antibody therapy has been shown in some studies to mediate cognitive improvement without any effect on plaque clearance.68 This could be due to antibody-mediated neutralization of the toxic effect of Aβ,62 which could have a subsequent effect on inflammation and cognitive function.
At least in vitro, FcR-mediated phagocytic clearance of Aβ is not affected by the presence of proinflammatory cytokines, unlike complement-mediated clearance.22 Perhaps immunization therapy has been so effective because it works despite a general inhibition of phagocytosis created by proinflammatory cytokines in the AD brain.
Chaperone Molecules
Apolipoproteins are Aβ-binding proteins with particular relevance to phagocytic clearance. They are generally known for their role in lipid and cholesterol homeostasis, but, in addition, apolipoprotein E (ApoE)-containing lipoprotein particles may modulate cellular uptake of Aβ through formation of Aβ-ApoE complexes. Apoliprotein E binds Aβ, and these complexes are recognized by receptors such as low-density lipoprotein receptor-related protein (LRP) and low-density lipoprotein receptor (LDLR); ligand binding stimulates clathrin-mediated endocytosis. There is reason to believe that defects in these mechanisms could be involved in AD, especially given that genome-wide association studies have implicated variants of both ApoE and the clathrin assembly protein PICALM as AD risk factors.53,69 In support of this notion, studies in transgenic mice showed that knockout of ApoE reduces uptake and degradation of Aβ within astrocytes in vitro,70 and overexpression of LDLR prevents amyloid deposition and increases Aβ clearance.71 Of note, presenilin, a component of the gamma secretase complex with a genetic variant linked to early-onset AD, has been linked to regulation of LDLR endocytosis.72 Thus, there are multiple lines of evidence connecting ApoE and LDLR to Aβ clearance in AD.
In particular, the APOE allele encoding the apolipoprotein ε4 isoform has been linked to an increased risk of sporadic AD.69 Although some studies have demonstrated that there are isoform-dependent differences in Aβ uptake, there are contradictory findings and no overall consensus or trend. Of note, apolipoproteins may also regulate the inflammatory state of the brain. Some studies suggest that ApoE provides an overall anti-inflammatory effect; for example, transgenic mice expressing the ApoE4 isoform exhibit greater production of inflammatory mediators than mice expressing ApoE3.73
The lipidation state of ApoE may also modulate its function. It was found that the ATP binding cassette transporter A1 (ABCA1) mediates lipidation of ApoE and modulates amyloid plaque formation, and genomic studies have suggested that polymorphisms in ABCA1 are associated with increased risk of AD.74 This is noteworthy, interesting considering the roles of ABCA family members in processing of material that undergoes phagocytic clearance in the retina, and specifically because mutations in the ABCA4 gene cause material to be toxic to the engulfing cells, leading to a form of retinal degeneration. In theory, the effects of ABCA1 mutations associated with AD could be mediated through a similar mechanism.
A role for clusterin or apolipoprotein J (ApoJ) in AD pathogenesis has been suggested,75 and it has recently been linked to AD in two independent genome-wide association studies.53,54 Clusterin has been shown to be secreted by astrocytes and increased in regions associated with AD pathology.75 Culture studies suggest that clusterin binds Aβ and enhances endocytosis within glial cells via the megalin receptor, also known as LRP-2,76 which is a member of the LDLR family. In addition, clusterin is a complement inhibitor (also known as complement-associated protein SP-40,40 and as complement lysis inhibitor) and may suppress activity of the cytolytic membrane attack complex.75 These findings generally suggest that clusterin plays a protective role, and future studies should examine whether the polymorphisms associated with AD have altered function that may contribute to pathogenesis (as has been done for the APOE alleles).
α2-Macroglobulin (α2M) is another extracellular chaperone protein implicated in the binding and clearance of Aβ; in vitro, the neurotoxicity of Aβ is abrogated by co-incubation with α2M.77 Like Aβ-ApoE complexes, the Aβ-α2M complex acts as a ligand for LRP, and LRP mediates internalization. One study showed that LRP expression is required for the beneficial effect of α2M, which suggests that α2M exerts its protective effects through mediating clearance of Aβ from the extracellular space.77 It is possible, however, that the receptor is protective through downstream anti-inflammatory effects, and the mechanisms of neuroprotection have not been determined.
Ultimately, the effects of chaperone proteins on phagocytic clearance may be direct, through binding Aβ and promoting its uptake, or indirect, exerting effects on phagocytic clearance through interactions with inflammatory pathways.
Degradation and Clearance of Aβ
The mechanisms by which Aβ is normally degraded and cleared are still under intensive study. There is in vitro evidence that both microglia and astrocytes are capable of degrading Aβ,40,41 but this ability appears to be affected by the inflammatory environment; for example, interferon-γ inhibits degradation of Aβ, whereas IL-4, IL-10, and TGF-β enhance degradation.23
Even if a cell engulfs debris, phagocytosis does not guarantee degradation, and in fact Aβ may be trafficked to the bloodstream instead of being degraded in phagocytes. Amyloid immunotherapy is associated with a reduction in Aβ deposits in the parenchyma and with increases in serum Aβ levels, suggesting that transport from the parenchyma to the bloodstream occurs.64
Neuronal Death and Synapse Loss in AD
The number of apoptotic cells is increased in AD brain,78 and although this is primarily ascribed to increased neuronal death, it could also reflect deficiencies in apoptotic clearance. Levels of the phosphatidylserine-binding opsonin Mfge8 are decreased in AD,79 and could result in persistence of apoptotic corpses that are not properly marked for engulfment. Synapse loss is also a well-documented feature of AD.80 It remains to be determined whether the molecular machinery of apoptotic cell recognition and engulfment plays a significant role in synapse elimination (important in both development and degeneration).
Does Clearance Matter? Clinical Implications
A recent report described a flow cytometric analysis to measure Aβ phagocytosis in peripheral blood mononuclear cells.81 The AD mononuclear cells had significantly lower phagocytic activity than in age-matched controls, making this a potentially useful clinical test. It will be of great interest to test also the phagocytic activity of CNS-resident cells in AD (both microglia and astrocytes).
Since the inception of the Aβ hypothesis, many have suggested that stimulating clearance of Aβ could ameliorate AD. Immunization therapies have shown some potential, and both passive transfer of immunoglobulins and immunization with Aβ peptide or peptide fragments show promise. Initial studies in transgenic APP mice found that immunization with Aβ42 in young animals prevented the development of plaques, neuritic dystrophy, and astrogliosis and that treatment of older animals reduced the extent and progression of pathology.63 This led to further studies aimed at determining whether cognitive function is improved as a result of immunization and Aβ clearance; the results have been conflicting. In an APP transgenic mouse model, Tg2576, it was found that, although treatment with a monoclonal antibody targeted against Aβ improved cognitive measures, there was no significant reduction in parenchymal Aβ.68 In contrast, another study using the same mouse model found that immunization with anti-Aβ resulted in reduction in Aβ deposits, in addition to improved memory as measured by a Y maze task.64 Human studies have also yielded conflicting results, but at least one study has shown cognitive improvement on administration of immunoglobulin.65
Discussion
Type of Debris and Glial Activation
Engulfment of apoptotic debris induces anti-inflammatory responses that result in a quiet death, free from inflammation and cytotoxicity to neighboring cells,1 similar to what is seen in clearance of POS in the retina. In contrast, materials associated with Alzheimer's disease, Parkinson's disease, and prion disease, including Aβ, α-synuclein, neuromelanin, and prion protein appear to induce a proinflammatory glial response that is neurotoxic.47,50,82–84 Why this difference? Many receptors involved in recognition of these materials are pattern recognition receptors that may have evolved to recognize and respond to pathogens. A response to pathogens induces phagocytes to produce cytotoxic elements aimed at killing the invader.85 Ultimately, the cell's inflammatory response to these materials appears to parallel activation that would occur in response to pathogens; this reaction could be overkill and maladaptive in the case of neurodegenerative diseases.
Alternatively, glia may not possess the machinery to effectively engulf and degrade these materials, resulting in an overload of toxic debris. Failure to clear efficiently may lead to frustrated phagocytosis,35 and the phagocyte may ramp up the inflammatory response in the face of continual stimulation caused by persistence of debris.
Clearance: For Better or for Worse?
Phagocytic clearance may be considered beneficial if it removes toxic or inflammatory stimuli from the extracellular environment and prevents consequent detrimental responses. Clearance of apoptotic cells serves as a paradigm: phagocytic removal of dying cells helps avoid an exaggerated immune response after cell death. One might expect that a parallel model would exist in the case of Aβ clearance; however, we still do not have definitive answers regarding i) whether phagocytic clearance is required to eliminate toxicity of Aβ and ii) whether engulfment prevents or promotes detrimental effects of Aβ. These distinctions are important in contemplating treatment strategies. In the meantime, there are many features to consider when evaluating studies that attempt to address these questions.
First, the form of Aβ (soluble, fibrillar, or complexed with chaperone proteins) influences its effects, in terms of i) toxicity, ii) capacity for phagocytic clearance, and iii) effect on inflammation.15,22,41,43 The soluble form of Aβ is more toxic to neurons than the aggregated forms,43 and some researchers have concluded that aggregation is therefore protective. Formation of the aggregated or fibrillar form may be preferable to the soluble form in this sense; however, it is important to note that the fibrillar form has the propensity to cause glial inflammatory responses, which can then cause secondary neurotoxicity. Alternatively, some studies have shown that Aβ-chaperone protein complexes can be less toxic,77 and if they do not adversely activate glia, they may be the optimal way to sequester Aβ. Defects in chaperone systems could be involved in the pathogenesis of AD, especially considering that association studies have linked polymorphisms of chaperone proteins and their receptors to risk of AD. Further studies are needed to evaluate whether formation of Aβ-chaperone complexes avoids the toxicity/inflammatory response observed to occur with soluble and fibrillar Aβ and, more important, whether phagocytosis is required to mediate these beneficial effects.
Second, it is unclear which makes inflammation/toxicity worse: i) persistence of extracellular debris and continued stimulation of glia or ii) internalization and subsequent glial activation. Either of these could lead to inflammation and secondary neurotoxicity. Stimulation by extracellular debris appears sufficient to induce glial activation and inflammation, suggesting that persistence of plaques is undesirable. On the other hand, it is possible that internalization perpetuates the inflammatory reaction to a worse degree. As an example, exposure of astrocytes to α-synuclein leads to up-regulation of inflammatory mediators, and the response appears to be related to the dose of α-synuclein internalized.86 This supports the notion that glia sense intracellular accumulation on phagocytosis. If this mimics activity in vivo, it suggests that glial phagocytosis may augment inflammation.
Relevance of Clearance and Therapies to Promote Phagocytosis
Although substantial evidence indicates that defects in clearance are causative for retinal degeneration, the importance of apoptotic clearance in neurodegenerative diseases is much less clear. Promoting certain types of phagocytic responses may be the key; for example, mimicking the anti-inflammatory effects that occur during clearance of apoptotic cells may be beneficial. Immunization studies are generally limited to models of AD, and they have yielded conflicting results with regard to efficacy. Nonetheless, a study in human patients showed that intravenous immunoglobulin infusions promoted Aβ clearance from the cerebrospinal fluid, and this was associated with cognitive improvement.65
Epidemiological studies have suggested that chronic use of nonsteroidal anti-inflammatory drugs (NSAIDS) may help treat or prevent progression of Alzheimer's disease and Parkinson's disease.87,88 NSAIDs have multiple effects, and the mechanism mediating the beneficial outcomes for these diseases has not been defined. NSAIDs modulate microglial cytokine expression, and could affect neurodegenerative diseases through their effects on microglial phagocytosis.22
Finally, gene therapy has shown promise in animal models of retinal degeneration. Studies have shown that gene therapy can correct the phagocytic defect associated with mutations in the MerTK and Myo7a genes, leading to slowed photoreceptor loss and preservation of retinal function.25,32 This suggests that other engulfment pathway defects could be corrected by gene therapy. Although this may be relevant only for conditions with monogenetic causes, it is possible that its application could be more widespread, and gene therapy approaches could be used to increase phagocytic clearance in a variety of neurodegenerative diseases.
Conclusion
Studies on phagocytic clearance in the context of retinal degeneration illuminate the fact that numerous steps in the pathway are critical for normal homeostasis. Important steps include modification of debris before internalization, recognition by appropriate receptors, internalization, and trafficking through degradative pathways. Although these steps are well studied in the retina, a comprehensive story has yet to emerge for clearance of aggregates, neuronal corpses, or dysfunctional synapses in the context of specific neurodegenerative diseases. The use of cell type-specific and inducible knockout mice to eliminate critical components of defined engulfment pathways in relevant models of neurodegenerative disease can begin to answer some of these important questions.
Acknowledgments
We are grateful to Dr. Kodi Ravichandran (Department of Microbiology, University of Virginia) for stimulating our interest in apoptotic clearance and for his generous ongoing collaboration.
Footnotes
Supported in part by the University of Virginia NIH-funded Medical Scientist Training Program (J.D.S.) and the National Institutes of Health (NIH NS065447 to J.W.M.).
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Elliott M.R., Ravichandran K.S. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee C.Y., Landreth G.E. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117:949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A., Thompson W.J., Barres B.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeWitt D.A., Perry G., Cohen M., Doller C., Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Neurol. 1998;149:329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 5.Takuma K., Fang F., Zhang W., Yan S., Fukuzaki E., Du H., Sosunov A., McKhann G., Funatsu Y., Nakamichi N., Nagai T., Mizoguchi H., Ibi D., Hori O., Ogawa S., Stern D.M., Yamada K., Yan S.S. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X., Crick S.L., Bu G., Frieden C., Pappu R.V., Lee J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci USA. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuentealba R.A., Liu Q., Zhang J., Kanekiyo T., Hu X., Lee J.M., LaDu M.J., Bu G. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS One. 2010;5:e11884. doi: 10.1371/journal.pone.0011884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siskova Z., Page A., O'Connor V., Perry V.H. Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol. 2009;175:1610–1621. doi: 10.2353/ajpath.2009.090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Noort J.M., Bsibsi M. Toll-like receptors in the CNS: implications for neurodegeneration and repair. Prog Brain Res. 2009;175:139–148. doi: 10.1016/S0079-6123(09)17509-X. [DOI] [PubMed] [Google Scholar]
- 10.Simard A.R., Soulet D., Gowing G., Julien J.P., Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Tsuchiya T., Park K.C., Toyonaga S., Yamada S.M., Nakabayashi H., Nakai E., Ikawa N., Furuya M., Tominaga A., Shimizu K. Characterization of microglia induced from mouse embryonic stem cells and their migration into the brain parenchyma. J Neuroimmunol. 2005;160:210–218. doi: 10.1016/j.jneuroim.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 12.Mildner A., Schmidt H., Nitsche M., Merkler D., Hanisch U.K., Mack M., Heikenwalder M., Bruck W., Priller J., Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 13.Fraser D.A., Pisalyaput K., Tenner A.J. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan D., Gordon M.N., Tan J., Wilcock D., Rojiani A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- 15.Michelucci A., Heurtaux T., Grandbarbe L., Morga E., Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Carson M.J., Sutcliffe J.G. Balancing function vs. self defense: the CNS as an active regulator of immune responses. J Neurosci Res. 1999;55:1–8. doi: 10.1002/(SICI)1097-4547(19990101)55:1<1::AID-JNR1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Colton C.A., Mott R.T., Sharpe H., Xu Q., Van Nostrand W.E., Vitek M.P. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Town T., Nikolic V., Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calingasan N.Y., Erdely H.A., Altar C.A. Identification of CD40 ligand in Alzheimer's disease and in animal models of Alzheimer's disease and brain injury. Neurobiol Aging. 2002;23:31–39. doi: 10.1016/s0197-4580(01)00246-9. [DOI] [PubMed] [Google Scholar]
- 20.Biber K., Neumann H., Inoue K., Boddeke H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Reynolds A.D., Banerjee R., Liu J., Gendelman H.E., Mosley R.L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82:1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 22.Koenigsknecht-Talboo J., Landreth G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto M., Kiyota T., Walsh S.M., Liu J., Kipnis J., Ikezu T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J Immunol. 2008;181:3877–3886. doi: 10.4049/jimmunol.181.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohli L., Roth K.A. Autophagy: cerebral home cooking. Am J Pathol. 2010;176:1065–1071. doi: 10.2353/ajpath.2010.090850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 26.Hall M.O., Prieto A.L., Obin M.S., Abrams T.A., Burgess B.L., Heeb M.J., Agnew B.J. Outer segment phagocytosis by cultured retinal pigment epithelial cells requires Gas6. Exp Eye Res. 2001;73:509–520. doi: 10.1006/exer.2001.1062. [DOI] [PubMed] [Google Scholar]
- 27.Nandrot E.F., Anand M., Almeida D., Atabai K., Sheppard D., Finnemann S.C. Essential role for MFG-E8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc Natl Acad Sci USA. 2007;104:12005–12010. doi: 10.1073/pnas.0704756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryeom S.W., Silverstein R.L., Scotto A., Sparrow J.R. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J Biol Chem. 1996;271:20536–20539. doi: 10.1074/jbc.271.34.20536. [DOI] [PubMed] [Google Scholar]
- 29.Finnemann S.C., Nandrot E.F. MerTK activation during RPE phagocytosis in vivo requires alphaVbeta5 integrin. Adv Exp Med Biol. 2006;572:499–503. doi: 10.1007/0-387-32442-9_69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bok D., Hall M.O. The role of the pigment epithelium in the etiology of inherited retinal dystrophy in the rat. J Cell Biol. 1971;49:664–682. doi: 10.1083/jcb.49.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gal A., Li Y., Thompson D.A., Weir J., Orth U., Jacobson S.G., Apfelstedt-Sylla E., Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 32.Tschernutter M., Schlichtenbrede F.C., Howe S., Balaggan K.S., Munro P.M., Bainbridge J.W., Thrasher A.J., Smith A.J., Ali R.R. Long-term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther. 2005;12:694–701. doi: 10.1038/sj.gt.3302460. [DOI] [PubMed] [Google Scholar]
- 33.Selkoe D.J. Toward a comprehensive theory for Alzheimer's disease: Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 34.Nagele R.G., D'Andrea M.R., Lee H., Venkataraman V., Wang H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- 35.D'Andrea M.R., Cole G.M., Ard M.D. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 36.Haass C., Hung A.Y., Selkoe D.J. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itagaki S., McGeer P.L., Akiyama H., Zhu S., Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 38.Nicoll J.A., Barton E., Boche D., Neal J.W., Ferrer I., Thompson P., Vlachouli C., Wilkinson D., Bayer A., Games D., Seubert P., Schenk D., Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 39.Shie F.S., Breyer R.M., Montine T.J. Microglia lacking E prostanoid receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol. 2005;166:1163–1172. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyss-Coray T., Loike J.D., Brionne T.C., Lu E., Anankov R., Yan F., Silverstein S.C., Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 41.Mandrekar S., Jiang Q., Lee C.Y., Koenigsknecht-Talboo J., Holtzman D.M., Landreth G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koenigsknecht J., Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sondag C.M., Dhawan G., Combs C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pereson S., Wils H., Kleinberger G., McGowan E., Vandewoestyne M., Van Broeck B., Joris G., Cuijt I., Deforce D., Hutton M., Van Broeckhoven C., Kumar-Singh S. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J Pathol. 2009;219:173–181. doi: 10.1002/path.2580. [DOI] [PubMed] [Google Scholar]
- 45.Emmerling M.R., Watson M.D., Raby C.A., Spiegel K. The role of complement in Alzheimer's disease pathology. Biochim Biophys Acta. 2000;1502:158–171. doi: 10.1016/s0925-4439(00)00042-9. [DOI] [PubMed] [Google Scholar]
- 46.Heneka M.T., O'Banion M.K. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 47.Walker D.G., Link J., Lue L.F., Dalsing-Hernandez J.E., Boyes B.E. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes [Erratum appeared in J Leukoc Biol 2006, 80:448] J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 48.Wyss-Coray T., Lin C., Yan F., Yu G.Q., Rohde M., McConlogue L., Masliah E., Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 49.Jellinger K.A. Challenges in neuronal apoptosis. Curr Alzheimer Res. 2006;3:377–391. doi: 10.2174/156720506778249434. [DOI] [PubMed] [Google Scholar]
- 50.Johnstone M., Gearing A.J., Miller K.M. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- 51.Desideri G., Cipollone F., Necozione S., Marini C., Lechiara M.C., Taglieri G., Zuliani G., Fellin R., Mezzetti A., di Orio F., Ferri C. Enhanced soluble CD40 ligand and Alzheimer's disease: evidence of a possible pathogenetic role. Neurobiol Aging. 2008;29:348–356. doi: 10.1016/j.neurobiolaging.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 52.Nikolic W.V., Hou H., Town T., Zhu Y., Giunta B., Sanberg C.D., Zeng J., Luo D., Ehrhart J., Mori T., Sanberg P.R., Tan J. Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular beta-amyloid deposits in Alzheimer mice. Stem Cells Dev. 2008;17:423–439. doi: 10.1089/scd.2008.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease [Erratum appeared in Nat Genet 2009, 41:1156] Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 55.Barnum S.R. Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med. 1995;6:132–146. doi: 10.1177/10454411950060020301. [DOI] [PubMed] [Google Scholar]
- 56.Tenner A.J., Fonseca M.I. The double-edged flower: roles of complement protein C1q in neurodegenerative diseases. Adv Exp Med Biol. 2006;586:153–176. doi: 10.1007/0-387-34134-X_11. [DOI] [PubMed] [Google Scholar]
- 57.Eikelenboom P., Hack C.E., Rozemuller J.M., Stam F.C. Complement activation in amyloid plaques in Alzheimer's dementia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;56:259–262. doi: 10.1007/BF02890024. [DOI] [PubMed] [Google Scholar]
- 58.Bradt B.M., Kolb W.P., Cooper N.R. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. 1998;188:431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wyss-Coray T., Yan F., Lin A.H., Lambris J.D., Alexander J.J., Quigg R.J., Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maier M., Peng Y., Jiang L., Seabrook T.J., Carroll M.C., Lemere C.A. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singhrao S.K., Neal J.W., Rushmere N.K., Morgan B.P., Gasque P. Differential expression of individual complement regulators in the brain and choroid plexus. Lab Invest. 1999;79:1247–1259. [PubMed] [Google Scholar]
- 62.Szabo P., Relkin N., Weksler M.E. Natural human antibodies to amyloid beta peptide. Autoimmun Rev. 2008;7:415–420. doi: 10.1016/j.autrev.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 63.Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 64.Wilcock D.M., Rojiani A., Rosenthal A., Levkowitz G., Subbarao S., Alamed J., Wilson D., Wilson N., Freeman M.J., Gordon M.N., Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Relkin N.R., Szabo P., Adamiak B., Burgut T., Monthe C., Lent R.W., Younkin S., Younkin L., Schiff R., Weksler M.E. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 66.Bacskai B.J., Kajdasz S.T., McLellan M.E., Games D., Seubert P., Schenk D., Hyman B.T. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das P., Howard V., Loosbrock N., Dickson D., Murphy M.P., Golde T.E. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma−/− knock-out mice. J Neurosci. 2003;23:8532–8538. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kotilinek L.A., Bacskai B., Westerman M., Kawarabayashi T., Younkin L., Hyman B.T., Younkin S., Ashe K.H. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strittmatter W.J., Saunders A.M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G.S., Roses A.D. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koistinaho M., Lin S., Wu X., Esterman M., Koger D., Hanson J., Higgs R., Liu F., Malkani S., Bales K.R., Paul S.M. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 71.Kim J., Castellano J.M., Jiang H., Basak J.M., Parsadanian M., Pham V., Mason S.M., Paul S.M., Holtzman D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tamboli I.Y., Prager K., Thal D.R., Thelen K.M., Dewachter I., Pietrzik C.U., St George-Hyslop P., Sisodia S.S., De Strooper B., Heneka M.T., Filippov M.A., Müller U., van Leuven F., Lutjohann D., Walter J. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci. 2008;28:12097–12106. doi: 10.1523/JNEUROSCI.2635-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Colton C.A., Needham L.K., Brown C., Cook D., Rasheed K., Burke J.R., Strittmatter W.J., Schmechel D.E., Vitek M.P. APOE genotype-specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004;147:62–67. doi: 10.1016/j.jneuroim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 74.Koldamova R., Fitz N.F., Lefterov I. The role of ATP-binding cassette transporter A1 in Alzheimer's disease and neurodegeneration. Biochim Biophys Acta. 2010;1801:824–830. doi: 10.1016/j.bbalip.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nuutinen T., Suuronen T., Kauppinen A., Salminen A. Clusterin: a forgotten player in Alzheimer's disease. Brain Res Rev. 2009;61:89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Zlokovic B.V., Martel C.L., Matsubara E., McComb J.G., Zheng G., McCluskey R.T., Frangione B., Ghiso J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. 1996;93:4229–4234. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fabrizi C., Businaro R., Lauro G.M., Fumagalli L. Role of alpha2-macroglobulin in regulating amyloid beta-protein neurotoxicity: protective or detrimental factor. J Neurochem. 2001;78:406–412. doi: 10.1046/j.1471-4159.2001.00419.x. [DOI] [PubMed] [Google Scholar]
- 78.Broe M., Shepherd C.E., Milward E.A., Halliday G.M. Relationship between DNA fragmentation, morphological changes and neuronal loss in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2001;101:616–624. doi: 10.1007/s004010000337. [DOI] [PubMed] [Google Scholar]
- 79.Boddaert J., Kinugawa K., Lambert J.C., Boukhtouche F., Zoll J., Merval R., Blanc-Brude O., Mann D., Berr C., Vilar J., Garabedian B., Journiac N., Charue D., Silvestre J.S., Duyckaerts C., Amouyel P., Mariani J., Tedgui A., Mallat Z. Evidence of a role for lactadherin in Alzheimer's disease. Am J Pathol. 2007;170:921–929. doi: 10.2353/ajpath.2007.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scheff S.W., Price D.A., Schmitt F.A., Mufson E.J. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 81.Avagyan H., Goldenson B., Tse E., Masoumi A., Porter V., Wiedau-Pazos M., Sayre J., Ong R., Mahanian M., Koo P., Bae S., Micic M., Liu P.T., Rosenthal M.J., Fiala M. Immune blood biomarkers of Alzheimer disease patients. J Neuroimmunol. 2009;210:67–72. doi: 10.1016/j.jneuroim.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 82.McGeer P.L., McGeer E.G. Glial reactions in Parkinson's disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 83.Wilms H., Rosenstiel P., Sievers J., Deuschl G., Zecca L., Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J. 2003;17:500–502. doi: 10.1096/fj.02-0314fje. [DOI] [PubMed] [Google Scholar]
- 84.Brown D.R., Schmidt B., Kretzschmar H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 85.Nagl M., Kacani L., Müllauer B., Lemberger E.M., Stoiber H., Sprinzl G.M., Schennach H., Dierich M.P. Phagocytosis and killing of bacteria by professional phagocytes and dendritic cells. Clin Diagn Lab Immunol. 2002;9:1165–1168. doi: 10.1128/CDLI.9.6.1165-1168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee H.J., Suk J.E., Patrick C., Bae E.J., Cho J.H., Rho S., Hwang D., Masliah E., Lee S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rogers J., Kirby L.C., Hempelman S.R., Berry D.L., McGeer P.L., Kaszniak A.W., Zalinski J., Cofield M., Mansukhani L., Willson P. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 88.Gagne J.J., Power M.C. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74:995–1002. doi: 10.1212/WNL.0b013e3181d5a4a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kinchen J.M., Ravichandran K.S. Journey to the grave: signaling events regulating removal of apoptotic cells. J Cell Sci. 2007;120:2143–2149. doi: 10.1242/jcs.03463. [DOI] [PubMed] [Google Scholar]
- 90.Okun E., Mattson M.P., Arumugam T.V. Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med. 2009;12:164–178. doi: 10.1007/s12017-009-8099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Landreth G.E., Reed-Geaghan E.G. Toll-like receptors in Alzheimer's disease. Curr Top Microbiol Immunol. 2009;336:137–153. doi: 10.1007/978-3-642-00549-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reed-Geaghan E.G., Savage J.C., Hise A.G., Landreth G.E. CD14 and Toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kielian T. Overview of Toll-like receptors in the CNS. Curr Top Microbiol Immunol. 2009;336:1–14. doi: 10.1007/978-3-642-00549-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iribarren P., Zhou Y., Hu J., Le Y., Wang J.M. Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol Res. 2005;31:165–176. doi: 10.1385/IR:31:3:165. [DOI] [PubMed] [Google Scholar]
- 95.Wilhelmus M.M., Otte-Höller I., van Triel J.J., Veerhuis R., Maat-Schieman M.L., Bu G., de Waal R.M., Verbeek M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Donahue J.E., Flaherty S.L., Johanson C.E., Duncan J.A., 3rd, Silverberg G.D., Miller M.C., Tavares R., Yang W., Wu Q., Sabo E., Hovanesian V., Stopa E.G. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 97.Deane R., Du Yan S., Submamaryan R.K., LaRue B., Jovanovic S., Hogg E., Welch D., Manness L., Lin C., Yu J., Zhu H., Ghiso J., Frangione B., Stern A., Schmidt A.M., Armstrong D.L., Arnold B., Liliensiek B., Nawroth P., Hofman F., Kindy M., Stern D., Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 98.Alarcón R., Fuenzalida C., Santibáñez M., von Bernhardi R. Expression of scavenger receptors in glial cells: Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem. 2005;280:30406–30415. doi: 10.1074/jbc.M414686200. [DOI] [PubMed] [Google Scholar]
- 99.Paresce D.M., Ghosh R.N., Maxfield F.R. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]