

Abstract
Duchenne muscular dystrophy (DMD) is a severe neuromuscular disorder, and one of the most frequently encountered, but one for which there is as yet no treatment. Laminin-111 protein therapy was recently shown to be a promising approach to prevent muscle disease in the mdx mouse model of DMD. The present study demonstrated that transgenic expression of laminin α1 chain in mdx animals, resulting in laminin-111 heterotrimer formation in mdx muscle, does not improve the dystrophic phenotype. The mdx mice overexpressing laminin-111 (mdxLMα1) display features of mdx littermates: dystrophic pattern of muscle biopsy, elevated creatine kinase levels, reduced muscle strength, and decreased sarcolemmal integrity. Increased expression of integrin α7 is not beneficial for mdxLMα1 muscle, and components of the dystrophin-glycoprotein complex are not restored at the sarcolemma on laminin-111 overexpression. In summary, further studies are needed to verify the functionality of laminin-111 protein therapy in DMD and to describe the molecular events resulting from this approach.
Duchenne muscular dystrophy (DMD) is a severe, inherited neuromuscular disorder and the most prevalent form of muscular dystrophy, occurring in 1 of 3500 male births.1 DMD patients experience progressive muscle wasting, with clinical onset at 2 to 5 years of age; they lose the ability to walk between ages 7 to 13, and die in their 20s because of cardiopulmonary failure.2 DMD is caused by deletions and mutations in the dystrophin gene (DMD)3 that lead to absence of the sarcolemma-docked cytoskeletal protein and reduction of the dystrophin-glycoprotein complex (DGC), which together provide a mechanical link between the cytoskeleton and extracellular matrix.4,5 The mdx dystrophin-deficient mouse is the best characterized mouse model for DMD,6,7 although the progressive muscle wasting presents itself in a much milder form than in humans.7
Although the genetic defects underlying DMD were identified more than 20 years ago,3 there is still no effective treatment of this devastating neuromuscular disease. Recently, however, a remarkable protein therapy strategy in the mdx mouse was undertaken by Rooney et al,8 who demonstrated that systemic injections of laminin-111 derived from the Engelbreth-Holm-Swarm (EHS) tumor could ameliorate dystrophic symptoms in the mdx mouse. Additionally, such an approach facilitated myoblast transplantation in this mouse model.9
Laminin-211, an extracellular matrix protein consisting of α2, β1, and γ1 chains, is the major laminin isoform in skeletal muscle. The α2 subunit binds to α-dystroglycan (a DGC component) and to integrin α7β1.10 Laminin-111 (consisting of α1, β1, and γ1 chains) is not expressed in skeletal muscle, but it has been shown to functionally replace laminin-211 in laminin α2 chain-deficient muscular dystrophy upon transgenic overexpression in the neuromuscular system.11–13 Similarly to the laminin α2 subunit, the laminin α1 chain binds to α-dystroglycan14 and to integrin α7β1.15 Of note, integrin α7 is upregulated in the skeletal muscle in both DMD patients and mdx mice.16 Furthermore, integrin α7 has been proposed to be an important modifier of dystrophic symptoms in mice and to have roles complementary to those of the DGC.8,17–20
Data reported by Rooney et al8 indicate that laminin-111 is a highly effective therapeutic agent in the mdx mouse model of DMD and could have applications in human disease. These remarkable results show promise for patients, and therefore the potency of laminin protein therapy needs to be tested in additional preclinical tests involving different molecular approaches.
In the present study, we tested by transgenic means the ability of murine laminin-111 to prevent dystrophin-deficient muscular dystrophy in mice. We generated mdxLMα1 mice, dystrophin-deficient animals that significantly overexpress laminin α1 chain (ie, the α subunit of the laminin-111 heterotrimer) in skeletal muscle. Transgenic expression of laminin α1 chain resulted in laminin-111 formation in mdxLMα1 skeletal muscle basement membranes. Despite the substantial deposition of laminin-111, mdxLMα1 animals exhibited the same dystrophic features as mdx mice, with fiber size variability, central nucleation, necrosis, sarcolemmal damage, decreased grip strength, and elevated creatine kinase (CK) levels. The expression of integrin α7 was increased in mdxLMα1 muscle, but the components of the DGC were not restored at the sarcolemma. Taken together, the present results contradict those of Rooney et al8 and suggest that laminin protein therapy in mdx mice requires further verification.
Material and Methods
Transgenic Animals
C57BL/10ScSn-Dmdmdx/J (mdx) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). The transgenic animals overexpressing laminin α1 chain under the control of β-actin promoter (LMα1TG) have been described previously.11 The mdx females were bred with LMα1TG males. All males born were mdx. These were further genotyped for the presence of the transgene (mdxLMα1 mice), as described previously.11 Mice were maintained in animal facilities according to animal care guidelines. All mouse experimentation was approved by the local (Lund district) ethics committee.
Histology and Immunofluorescence Microscopy
Cryosections (8 μm thick) of skeletal muscle (quadriceps femoris, gastrocnemius, soleus, tibialis anterior, triceps brachii, and diaphragm) from 8- to 10-week-old wild-type, mdx, mdxLMα1, and dy3KLMα1TG mice (n = 3 per group) were either stained with hematoxylin and eosin or subjected to immunofluorescence analysis using the following antibodies: rat monoclonal mAb200 against LMα1LG4,11 rabbit polyclonal 15277 against dystrophin (Abcam, Cambridge, UK), rat monoclonal 4H8-2 against laminin α2 chain (Alexis Biochemicals; Enzo Life Sciences, Plymouth Meeting, PA), rat monoclonal MTn15 against tenascin-C,11 rat monoclonal LT3 against laminin β1 chain (Chemicon; Millipore, Temecula, CA), rabbit polyclonal 1083+ against laminin γ1 chain (kindly provided by Dr. T. Sasaki), rabbit polyclonal U31 against integrin α7B subunit (kindly provided by Dr. U. Mayer),21,22 mouse monoclonal IIH6 against α-dystroglycan (Upstate Biotechnology, Lake Placid, NY), rabbit polyclonal against β-dystroglycan,11 mouse monoclonal 5B1 against β-sarcoglycan (Novocastra, Newcastle upon Tyne, UK), and mouse monoclonal DRP3/20C5 against utrophin (Novocastra). Stainings were performed as described previously.11,22,23 In addition, muscles from 5-week-old mdx and mdxLMα1 animals were subjected to hematoxylin and eosin staining.
The area corresponding to tenascin-C labeling was quantified from stitched photos of the entire area of the diaphragm cross-section, using ImageJ software, version 143u (NIH, Bethesda, MD); five mdx and six mdxLMα1 animals were analyzed. Unpaired t-test was used for statistical analysis. P < 0.05 was considered statistically different. GraphPad Prism software, version 2.01 (La Jolla, CA) was used for all statistical analyses.
Quantification of Fiber Size Distribution and Central Nucleation
Diaphragm and limb muscles from three mdx and three mdxLMα1 animals (8 to 10 weeks old) were analyzed. For central nucleation, the tibialis anterior, triceps brachii, and diaphragm muscles were examined. The entire area of each muscle cross-section was considered (which corresponds to at least 1746, 1389, and 1889 fibers for each muscle type, respectively). For limbs, muscles from both collateral limbs were used. Unpaired t-test was used for statistical analysis, with significance set at P < 0.05. Minimal Feret's diameter of muscle fibers24 was measured for at least 1993 and 1330 fibers in diaphragm and triceps brachii muscle, respectively. ImageJ software, version 143u (NIH) was used for measurements. The χ2 test was calculated for fiber distribution comparison, with significance set at P < 0.05. All distributions of fiber size compared by pairs were related to the genotype.
Treadmill Exercise and Evans Blue Dye Uptake
Eight- to 11-week-old mdx (n = 3) and mdxLMα1 mice (n = 4) were exercised for 30 minutes on a treadmill Exer 6M (Columbus Instruments, Columbus, OH) at a downhill angle of 15 degrees. During the first 2 minutes, the speed was gradually increased from 7 m/min to 14–16 m/min. Within 30 minutes after completed exercise, the mice were injected intraperitoneally with Evans Blue dye (EBD; Sigma-Aldrich, St. Louis, MO) dissolved in sterile saline (0.5 mg EBD/0.05 mL saline; 50 μL/10 g body weight). After approximately 16 hours, muscles (quadriceps femoris, tibialis anterior, posterior compartment of calf, and triceps brachii) were collected and quickly frozen in liquid nitrogen. Unexercised mice were injected with EBD and used as controls. Cryosections (8 μm) of the muscles were fixed in ice-cold acetone at −20°C for 10 minutes and then were stained with laminin γ1 antibody. Under fluorescence microscopy analysis, the muscle fiber EBD uptake was visualized by red emission. Total muscle area and area of EBD-positive fibers (red staining) in each muscle were quantified using ImageJ software, version 143u (NIH). Muscles from both collateral limbs were used for quantification. Mann-Whitney U-test was used for statistical analysis, with significance set at P < 0.05.
Grip Strength Analyses
Forelimb grip strength was measured on a grip strength meter (Columbus Instruments). Wild-type (n = 8), mdx (n = 13), and mdxLMα1 (n = 15) males, 2 to 3 months old, were analyzed. Mice were pulling the metal bar five times. The two lowest values were rejected, and the mean of the three remaining values was counted. One-way analysis of variance followed by Bonferroni's multiple comparison test was used for statistical analysis, with significance set at P < 0.05.
Creatine Kinase Quantification
Blood was collected from the tail vein of 2- to 3-month-old control (mdx/+), mdx, and mdxLMα1 mice (n = 8; n = 16 and n = 16, respectively) into EDTA tubes and was centrifuged two times for 5 minutes at 1100 x g. Plasma was sent to the Clinical Chemistry Laboratory at Skåne University Hospital. The CK_P_S Cobas method was used to quantify enzyme activity. Kruskal–Wallis one-way analysis of variance followed by Dunn's test was used for statistical analysis, with significance set at P < 0.05.
Immunoblotting
For laminin and integrin detection, proteins were isolated from 100 mg of wild-type, mdx, mdxLMα1, and dy3KLMα1 muscles (n = 3 per group) as described previously.23 The supernatants were collected and the protein concentration was determined using a Pierce BCA assay (Thermo Fisher Scientific, Rockford, IL). The SDS-polyacrylamide gel electrophoresis and immunoblotting were performed as described previously.23 Integrin-containing samples were run under nonreducing conditions, and laminin-containing samples were run under both reducing and nonreducing conditions. Membranes were incubated overnight at 4°C with rabbit polyclonal antibody detecting laminin α1 chain, LG1-3 domains (1:500)23 (kindly provided by Dr. T. Sasaki); rabbit polyclonal antibody recognizing both laminin β1 and γ1 chain (1:1000; Sigma-Aldrich); and rabbit polyclonal antibody against integrin α7B (1:1500) (kindly provided by Dr. U. Mayer). Laminin α1 and β1/γ1 chain expression was normalized to α-actinin expression (detected with mouse monoclonal antibody DM1A, 1:3000; Sigma-Aldrich). One-way analysis of variance followed by Bonferroni's multiple comparison test was used for statistical analysis, except that for laminin α1 chain immunoblotting the Mann-Whitney U-test was applied, with significance set at P < 0.05.
Results
Generation of mdx Mice Overexpressing Laminin α1 Chain
Transgenic mice overexpressing laminin α1 chain (LMα1TG) and producing laminin-111 in the neuromuscular system were shown to substantially alleviate congenital muscular dystrophy in laminin α2 chain-deficient mice.11–13 To test the ability of transgenically expressed laminin α1 chain to rescue the phenotype of mdx mice, we crossed both strains to obtain dystrophin-deficient mdx animals overexpressing laminin α1 subunit; these progeny are hereafter referred to as mdxLMα1 mice) (Figure 1, A and B). We next determined the expression of laminin α1 chain in mdxLMα1 skeletal muscle (Figure 1B). Laminin α1 chain is absent from basement membranes in mdx muscle tissue; however, immunofluorescence staining in mdxLMα1 muscle showed continuous deposition of laminin α1 subunit alongside sarcolemma, in a similar manner as in laminin α2 chain-deficient mice overexpressing laminin α1 chain (dy3KLMα1). Importantly, transgenic expression of laminin α1 in skeletal muscle is secured already at postnatal day 1 (Figure 1C) and even at embryonic stages (data not shown). We further investigated the expression of laminin α1 subunit by Western blot analyses. Similarly to dy3KLMα1 muscle and EHS laminin extracts, laminin α1 chain is assembled into a laminin-111 heterotrimer with laminin β1 and γ1 subunits (900 kDa) in mdxLMα1 skeletal muscle (analysis run under nonreduced conditions and probed with a laminin α1 chain antibody) (Figure 1D).
Figure 1.
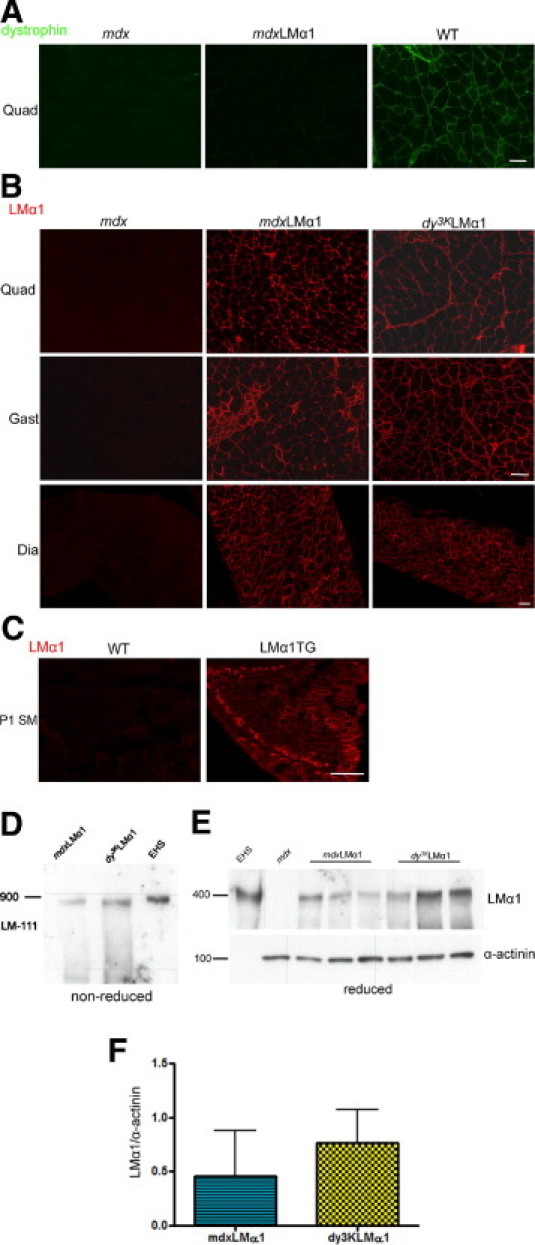
Laminin-111 expression in mdxLMα1 mice. A: Dystrophin immunostaining in mdx, mdxLMα1, and wild-type (WT) skeletal muscle confirms dystrophin absence from mdxLMα1 muscle. B: Laminin α1 chain immunostaining demonstrates uniform expression of laminin α1 subunit in basement membranes of mdxLMα1 skeletal muscle. It is expressed in a similar manner as in laminin α2 chain-deficient mice overexpressing laminin α1 chain (dy3KLMα1). As expected, it is absent from mdx muscle. Quadriceps (Quad), gastrocnemius (Gast), and diaphragm (Dia) muscles are shown. C: Laminin α1 chain is not expressed in wild-type muscle of newborn mice at postnatal day 1 (P1), but is present in muscle from littermates overexpressing transgenic laminin α1 chain. Scale bars = 50 μm (A–C, all images). D: Immunoblotting of skeletal muscle tissue extracts from mdxLMα1 and dy3KLMα1 mice and EHS laminin extract with a rabbit polyclonal antibody against laminin α1 LG3 domain under nonreducing conditions. The laminin-111 (LM-111) heterotrimer is present in mdxLMα1 muscle (900-kDa band). E: Immunoblotting with the same antibody against laminin α1 chain and α-actinin under reducing conditions. A 400-kDa band corresponding to laminin α1 chain is absent from mdx skeletal muscle extract, but is present in mdxLMα1 (n = 3) and dy3KLMα1 (n = 3) muscles. EHS laminin was used as a positive control. F: Quantification of laminin α1 chain signals revealed no significant difference in expression between mdxLMα1 and dy3KLMα1 muscles (P = 0.4000). Laminin α1 chain expression was normalized to α-actinin expression. Mann-Whitney U-test was used for statistical analysis, with significance set at P < 0.05. Results are reported as means ± SD.
We also compared expression levels of laminin α1 subunit between mdxLMα1 and dy3KLMα1 muscle (Figure 1, E and F). Laminin α1 chain expression levels were not significantly different between two mouse strains, but there was a trend toward decreased production of laminin α1 subunit in mdxLMα1 muscle, compared with dy3KLMα1 muscle (Figure 1, E and F). It is possible, however, that production of laminin α1 chain could be influenced by the normal expression of laminin α2 chain in mdxLMα1 muscle (Figure 2A). Production of laminin α2 subunit also seemed unchanged in mdx muscle (Figure 2A). Because laminin α chains can be secreted independently as monomers,25 we also analyzed whether sufficient amounts of laminin β1 and γ1 chains were available for the two major laminin α chains present in mdxLMα1 muscle basement membranes. Western blot analysis revealed substantial (5-fold) upregulation of laminin β1 and γ1 subunits in mdxLMα1 muscle, compared with wild-type and mdx muscle (Figure 2, B and C). Immunofluorescent labeling with antibodies against laminin β1 and γ1 chains showed stronger signals in mdxLMα1 muscle and revealed the deposition of β1 and γ1 subunits in muscle basement membranes (Figure 2D), to which they are secreted together with laminin α2 and α1 chains (forming laminin-211 and laminin-111 heterotrimers, respectively).
Figure 2.
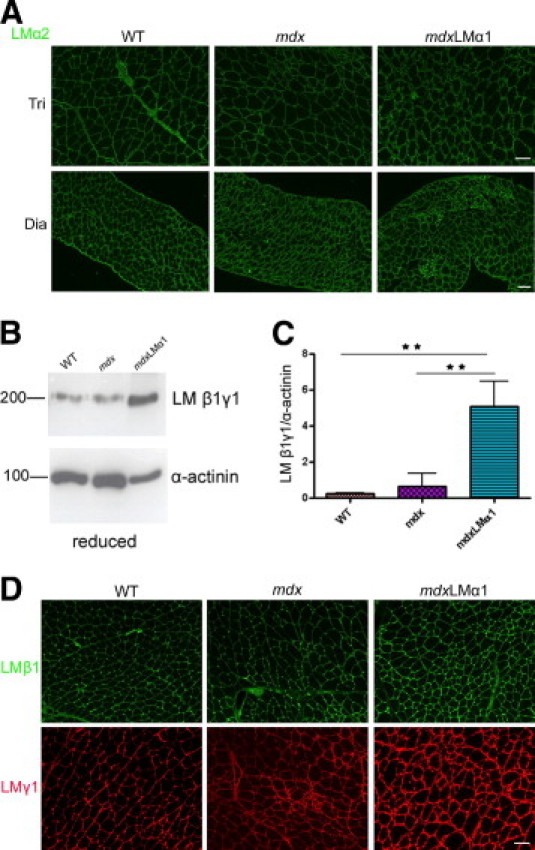
Expression of other laminin chains in mdxLMα1 and mdx mice. A: Immunostaining with laminin α2 chain antibody reveals no reduction in laminin α2 chain expression in mdx and mdxLMα1 muscles, compared with the wild type. B and C: Immunoblotting of skeletal muscle tissue extracts from wild-type (n = 3), mdx (n = 3), and mdxLMα1 mice (n = 3) reveals significant 5-fold upregulation of laminin β1 and γ1 subunits (both at approximately 200 kDa) in mdxLMα1 muscles, compared with both wild-type muscle (**P < 0001) and mdx muscle (**P < 0001). One-way analysis of variance followed by Bonferroni's multiple comparison test was used for statistical analysis. Results are reported as means ± SD. D: Increased immunofluorescent signals for laminin β1 and γ1 chains in mdxLMα1 muscle basement membranes, compared with wild-type and mdx basement membranes. They form heterotrimers with α subunits. Scale bars: 50 μm (A, all images in the same row, and D, all images).
The mdxLMα1 Mice Are as Dystrophic as mdx Mice
Despite high expression of laminin-111 in basement membranes of mdx muscles, none of the dystrophic features were improved in mdxLMα1 animals. The mdx limb muscles undergo an early phase of degeneration with widespread necrosis, followed by a regenerative phase that is initiated at approximately week 6.26 Diaphragm and limb muscles (quadriceps femoris, tibialis anterior, gastrocnemius, and triceps brachii) from 5-week-old mdx and mdxLMα1 mice displayed similar extensive areas of acute necrosis (Figure 3) (tibialis anterior least affected). Analyses of 8- to 10-week-old mdx and mdxLMα1 littermates revealed degenerating/regenerating and necrotic regions with mononuclear cell infiltrates in all muscles (quadriceps, triceps brachii, soleus, and diaphragm are shown in Figure 4A; gastrocnemius and tibialis anterior are not shown). Fiber splitting and endomysial fibrosis (Figure 4A) were also evident, especially in diaphragm muscle. Detailed comparative analyses of central nucleation between mdx and mdxLMα1 mice showed no significant difference in the amount of regenerating fibers (tibialis anterior, triceps brachii, and diaphragm), revealing robust muscle damage and degeneration/regeneration cycles in both genotypes (Figure 4B). Likewise, fiber size distribution was not significantly different between mdx and mdxLMα1 diaphragm and triceps brachii muscles (Figure 4C).
Figure 3.
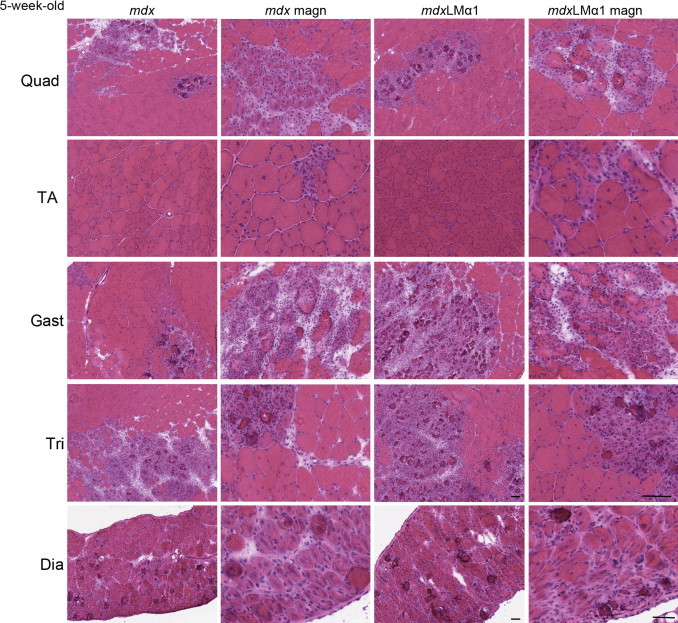
Histological analyses of 5-week-old mdx and mdxLMα1 muscles. Hematoxylin and eosin staining of quadriceps femoris (Quad), tibialis anterior (TA), gastrocnemius (Gast), triceps brachii (Tri), and diaphragm (Dia) muscles reveals dramatic focal necrosis in both mdx and mdxLMα1 muscles, often covering very large areas of muscle. The TA muscle was least affected by necrosis; only single, small necrotic patches were found. Scale bars: 50 μm.
Figure 4.
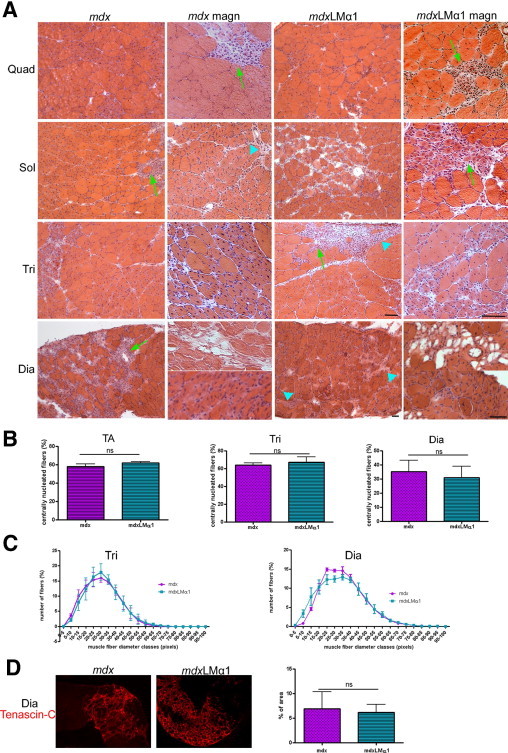
Analyses of mdxLMα1 and mdx muscle morphology. A: Hematoxylin and eosin staining of quadriceps femoris (Quad), soleus (Sol), triceps brachii (Tri), and diaphragm (Dia) muscles from 2-month-old mdxLMα1 (n = 3) and mdx (n = 3) mice reveals advanced muscular dystrophy in both genotypes. Images in columns 2 and 4 (magn) were taken at higher magnification than in columns 1 and 3, and are from a different individual. Robust muscle degeneration/regeneration is evident as fibers contain centrally located nucleus. Necrotic areas and fibrotic lesions are indicated by arrows and arrowheads, respectively. The two separate magnified images for mdx diaphragm muscle show presence of adipose tissue (top) and splitting fibers (bottom). B: Detailed analyses of central nucleation of muscle fibers show no significant differences between mdx (n = 3) and mdxLMα1 (n = 3) mice in all analyzed muscles (tibialis anterior, P = 0.0792; triceps brachii, P = 0.4470; and diaphragm, P = 0.5365). Unpaired t-test was used for statistical analysis. Results are reported as means ± SD. C: Fiber size distribution from mdxLMα1 and mdx triceps brachii (n = 3 for both genotypes) and diaphragm muscle (n = 3 for both genotypes). There is no shift toward bigger muscle fibers in mdxLMα1 muscles (triceps brachii, P = 0.9999; diaphragm, P = 0.9997). The χ2 test was used for statistical analysis. D: Tenascin-C labeling reveals fibrosis of diaphragm muscle from mdxLMα1 animals (n = 6). The area of fibrotic lesions is not reduced, compared with diaphragm muscle from mdx mice (n = 5) (P = 0.7527). Unpaired t-test was used for statistical analysis. Results are shown as means ± SD. Large patches of tenascin-C stained areas are shown in the micrographs, whereas the graphs represent the ratio of fibrotic lesion area to total area. The entire area of the diaphragm cross-section was used for quantification of tenascin-C staining. Scale bars: 50 μm.
Fibrosis is 10 times more pronounced in mdx diaphragm than in mdx hindlimb muscles.27 We observed the same trend using antibodies against collagen III (data not shown) and tenascin-C. Hence, we quantified the expression of tenascin-C in diaphragm muscle from mdx and mdxLMα1 mice (Figure 4D). Tenascin-C was deposited mostly focally in large patches in both genotypes, and fibrotic areas were not smaller in mdxLMα1 diaphragm muscle (Figure 4D).
The mdx mice are also characterized with contraction-induced damage of sarcolemma,28,29 decreased grip strength,30 and drastically elevated CK levels.31 Notably, sarcolemmal integrity was not increased in mdx mice upon laminin α1 chain overexpression. Nonexercised 2- to 3-month-old mdxLMα1 animals displayed substantial Evans Blue dye uptake in different muscles (Figure 5A). Treadmill exercise further enhanced muscle fiber damage in mdxLMα1, as well as in mdx littermates (Figure 5A), and EBD-uptake did not differ significantly between the genotypes (Figure 5B).
Figure 5.
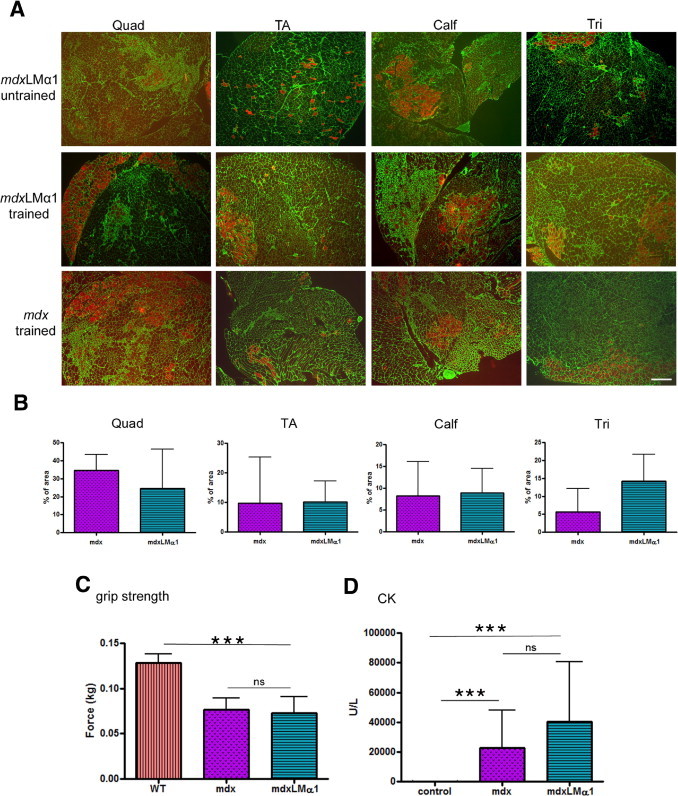
Examination of muscle function in mdxLMα1 mice. A: Analyses of sarcolemmal integrity. Untrained mdxLMα1 mice (n = 3) display damaged muscle fibers, as demonstrated by EBD uptake (fibers stained in red, top row). Treadmill exercise (trained) further enhances muscle injury in mdxLMα1 mice (n = 4) (middle row). The mdx mice subjected to training (n = 3) show a similar pattern of sarcolemmal disruption and EBD uptake as the mdxLMα1 mice (bottom row). Quadriceps femoris (Quad), tibialis anterior (TA), calf, and triceps brachii (Tri) muscles from both legs were analyzed. Laminin γ1 immunostaining (green) was used to covisualize muscle fibers. Moderately affected muscles were chosen for illustration. Scale bar = 100 μm for all images. B: Quantification of EBD uptake in exercised mdx and mdxLMα1 mice. Sarcolemmal damage is not reduced in mdxLMα1 muscles (quadriceps femoris, P = 0.4000; tibialis anterior P = 0.6286; calf, P = 0.7213; triceps brachii, P = 0.2286). There is considerable variability of EBD uptake in mdx and mdxLMα1 mice between animals from the same group and even between opposing limbs from the same individual. Mann-Whitney U-test was used for statistical analysis. Results are reported as means ± SD. C: Grip strength testing reveals no increase in forelimb muscle strength of mdxLMα1 mice (n = 6), compared with mdx animals (n = 7) (P > 0.05). Both mouse genotypes remained significantly weaker than age-matched wild-type mice (n = 5) (***P < 0.0001 for both mdx and mdxLMα1). One-way analysis of variance followed by Bonferroni's test was used for statistical analysis. Results are reported as means ± SD. D: Serum CK activity in mdx/+ control (n = 8), mdx (n = 10), and mdxLMα1 (n = 7) mice. There was no difference in CK activity between mdx and mdxLMα1 animals (P > 0.05), but for both genotypes the CK level was significantly elevated compared with the mdx/+ control (***P < 0.0001 for both mdx and mdxLMα1). Kruskal–Wallis one-way analysis of variance followed by Dunn's test was used for statistical analysis. Results are reported as means ± SD.
Additionally, 2- to 3-month-old mdxLMα1 mice remained as weak as mdx animals, as revealed by grip strength testing of forelimbs (Figure 5C). Finally, CK levels were substantially elevated in 2- to 3-month-old mdxLMα1 mice, compared with control mice, and remained not significantly different from mdx animals (Figure 5D), emphasizing the poor overall condition of mdxLMα1 muscle and mirroring all of the dystrophic changes described here.
All these data confirm advanced muscular dystrophy in mdx mice producing laminin-111 in skeletal muscle. Their dystrophic phenotype was not improved in any way, compared with mdx littermates.
Laminin α1 Chain Overexpression Additionally Increases the Expression of Integrin α7 but Does Not Restore DGC in the mdx Muscle
It has been reported that expression of integrin α7 is increased in mdx mice8,16 and in mdx mice injected with laminin-111.8 In agreement with these reports, we detected an increase in expression of integrin α7B (the major cytoplasmic splice variant produced in muscle32) in mdx and mdxLMα1 muscle, compared with wild-type muscle (Figure 6A). This was especially evident in limb muscles, and to a lesser extent in diaphragm muscle. Western blot analyses confirmed the upregulation of integrin α7B in mdx animals (1.8 fold) and revealed further moderate increase in integrin α7B expression in mdxLMα1 limb muscle, compared with mdx limb muscle, to the same degree as detected by Rooney et al8 (1.4 fold) (Figure 6, B and C).
Figure 6.
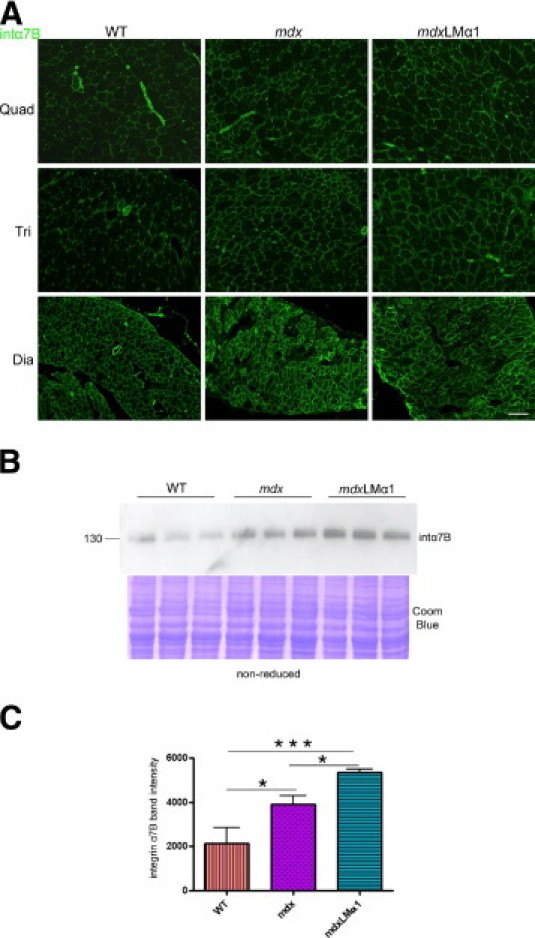
Analyses of integrin α7B expression in wild-type, mdx, and mdxLMα1 mice. A: Immunostaining reveals upregulation of integrin α7B in mdx and mdxLMα1 muscles. Quadriceps, triceps brachii, and diaphragm are shown. Scale bar = 100 μm for all images. B and C: Immunoblotting of skeletal muscle tissue extracts (nonreducing conditions) from wild-type (n = 3), mdx (n = 3), and mdxLMα1 mice (n = 3) using integrin α7B antibody. Densitometric analysis confirms significant upregulation of integrin α7B in mdx and mdxLMα1 vs wild-type mice (1.8 fold, *P < 0.05; 2.5-fold, ***P < 0.0001, respectively). Additionally, moderate increase of integrin α7B was noted in mdxLMα1 limb muscles, compared with mdx muscles (1.4 fold, *P < 0.05). Coomassie Blue staining was shown to demonstrate equal loading. One-way analysis of variance followed by Bonferroni's multiple comparison test was used for statistical analysis. Results are reported as means ± SD.
The injection of laminin-111 also resulted in enhanced expression of utrophin,8 which is the dystrophin homolog upregulated in mdx animals.33 Additionally, utrophin was shown to functionally replace dystrophin upon transgenic overexpression in this mouse model.34 We therefore analyzed the expression of utrophin in mdx mice overexpressing laminin α1 chain. Immunostaining revealed substantial upregulation of utrophin alongside sarcolemma in mdx and mdxLMα1 limb muscles, compared with wild-type muscles, where it is expressed only in the neuromuscular junctions (Figure 7, arrows). The immunofluorescent signal did not differ appreciably between mdx and mdxLMα1 muscle; that a moderate utrophin upregulation upon laminin α1 chain overexpression cannot be distinguished by immunohistochemical means.
Figure 7.
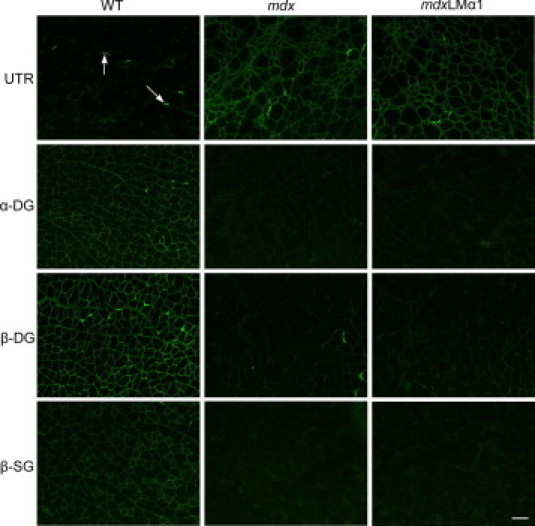
Expression of dystrophin-glycoprotein complex components in wild-type, mdx, and mdxLMα1 limb muscles. In wild-type muscle, utrophin (UTR) is expressed at the neuromuscular junction (arrows). In mdx and mdxLMα1 muscle, it is abundantly present along the sarcolemma. On the other hand, α-dystroglycan and β-dystroglycan (DG) and β-sarcoglycan (SG) are severely reduced in both mdx and mdxLMα1 muscle cell membranes. Scale bars: 100 μm for all images.
The effect of laminin-111 injections on expression of dystrophin-associated proteins is unknown, but laminin α1 chain overexpression could influence the expression of DGC components. We therefore analyzed the expression of α-dystroglycan, β-dystroglycan, and β-sarcoglycan in wild-type, mdx, and mdxLMα1 muscles. All three DGC components are severely reduced in mdx muscle5 (Figure 7). Laminin-111 is a strong ligand for α-dystroglycan,14 but none of the dystroglycan subunits (which link dystrophin/utrophin to the cell membrane and extracellular matrix) were restored at the sarcolemma upon laminin α1 chain overexpression in mdx mice (Figure 7). Similarly, β-sarcoglycan expression was not normalized either (Figure 7). Lack of DGC restoration might be associated with failure of improvement of the dystrophic phenotype in mdxLMα1 mice, which further indicates that integrin α7 alone is not sufficient for amelioration of disease symptoms.
Discussion
To date, the laminin-111 protein therapy approach seems to bypass several major obstacles that hinder various genetic strategies for curing DMD. The results presented by Rooney et al8 are striking, considering that the injected 900-kDa protein must traverse many barriers before it reaches skeletal muscle. It is debatable, however, whether laminin-111 would be beneficial for dystrophin-deficient muscle, which already expresses normal levels of laminin-211, as well as elevated levels of integrin α7 and utrophin. Furthermore, it is remarkable that laminin-111 could functionally replace dystrophin, a protein that displays completely different structure, function, and subcellular localization. Hence, additional preclinical studies are needed to verify its functionality and safety and to describe the molecular events resulting from such intervention. In the present study, we tested by transgenic means the efficacy of laminin-111 in preventing dystrophin-deficient muscular dystrophy in mice. Although laminin α1 chain overexpression resulted in laminin-111 deposition in muscle basement membranes, its expression did not prevent the muscular dystrophy in mdx animals.
Even allowing that our transgenic method is obviously different from the protein therapy introduced by Rooney et al,8 there is an evident discrepancy in the results obtained. One possibility is that injected EHS laminin undergoes fragmentation, which might facilitate the interaction of some laminin epitopes with their receptors. Hence, it is not excluded that smaller laminin molecules could be more effective in preventing the development of muscular dystrophy than a full-length laminin particle.
Another possible explanation for the discrepancies observed is that EHS laminin might be slightly different from endogenous35 or transgenic laminin-111, although it has also been shown that EHS laminin-111 is chemically, structurally, and immunologically identical to that obtained from nontumorigenic tissues.36 Additionally, commercial EHS laminin might contain other components that influence the outcome of laminin-111 protein therapy. Finally, although laminin α1 chain cDNA stays under the control of the β-actin promoter and in theory should be ubiquitously expressed, it is overexpressed primarily in the neuromuscular system.11,12 Injected EHS laminin might have a more systemic effect, and muscle phenotype improvement might result from global molecular events triggered by EHS laminin in different tissues.
Integrin α7 has been suggested to be one of the key molecules modifying disease progression in mdx mice treated with EHS laminin.8 In our mdxLMα1 mouse model, we also detected significant upregulation of integrin α7B, but this event might not be sufficient to secure the reduction of dystrophic symptoms by laminin-111 in mdx mice. We have previously shown that laminin α1 chain overexpression regulates the expression of integrin α7 in laminin α2 chain-deficient mice, and that this positive regulation is beneficial for muscle tissue.22 In those animals, however, the entire DGC complex remains intact and available for interaction with laminin-111, certainly contributing to the rescue of dystrophic phenotype. This is further confirmed by our more recent data: when laminin-111 binding to integrin α7 is maintained, but simultaneously the link between laminin-111 and DGC is disrupted, limb muscles are not spared from laminin α2 chain-deficient muscular dystrophy.23 Thus, upregulation of integrin α7 alone, without concomitant complete restoration of DGC, might not lead to alleviation of the muscle phenotype in mdx mice.
In summary, further steps are needed to verify the efficacy of laminin-111 injections into mice. Additionally, the molecular mechanisms triggered by systemic introduction of laminin-111 in muscle and other tissues must be characterized in more detail before bringing laminin-111 protein therapy to clinical trials.
Acknowledgments
We thank Drs. Takako Sasaki and Ulrike Mayer for providing antibodies and Dr. Virginie Carmignac for help with χ2 testing.
Footnotes
Supported in part by the Muscular Dystrophy Association, the Anna-Greta Crafoord Foundation for Rheumatological Research, the Swedish National Research Council (Vetenskapsrådet), and the Alfred Österlund Foundation.
References
- 1.van Deutekom J.C., van Ommen G.J. Advances in Duchenne muscular gene therapy. Nat Rev Genet. 2003;10:774–783. doi: 10.1038/nrg1180. [DOI] [PubMed] [Google Scholar]
- 2.Voit T., Tomé F.M.S. The congenital muscular dystrophies. In: Engel A.G., Franzini-Armstrong C., editors. Myology. ed 3. McGraw-Hill; New York: 2004. pp. pp 1203–1238. [Google Scholar]
- 3.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 4.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 5.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 7.Durbeej M., Campbell K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 8.Rooney J.E., Gurpur P.B., Burkin D.J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy [Erratum appeared in Proc Natl Acad Sci USA 2009, 106:15514] Proc Natl Acad Sci USA. 2009;106:7991–7996. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goudenege S., Lamarre Y., Dumont N., Rousseau J., Frenette J., Skuk D., Tremblay J.P. Laminin-111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol Ther. 2010;18:2155–2163. doi: 10.1038/mt.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciandra F., Gawlik K.I., Brancaccio A., Durbeej M. Dystroglycan, a possible mediator for reducing congenital muscular dystrophy? Trends Biotechnol. 2007;25:262–268. doi: 10.1016/j.tibtech.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Gawlik K., Miyagoe-Suzuki Y., Ekblom P., Takeda S., Durbeej M. Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum Mol Genet. 2004;13:1775–1784. doi: 10.1093/hmg/ddh190. [DOI] [PubMed] [Google Scholar]
- 12.Gawlik K.I., Li J.Y., Petersén Å.A., Durbeej M. Laminin alpha1 chain improves laminin alpha2 chain deficient peripheral neuropathy. Hum Mol Genet. 2006;15:2690–2700. doi: 10.1093/hmg/ddl201. [DOI] [PubMed] [Google Scholar]
- 13.Gawlik K., Durbeej M. Transgenic overexpression of laminin alpha1 chain in laminin alpha2 chain deficient mice rescues the disease throughout the lifespan. Muscle Nerve. 2010;42:30–37. doi: 10.1002/mus.21616. [DOI] [PubMed] [Google Scholar]
- 14.Talts J.F., Andac Z., Göhring W., Brancaccio A., Timpl R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999;18:863–870. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von der Mark H., Williams I., Wendler O., Sorokin L., von der Mark K., Pöschl E. Alternative splice variants of alpha7beta1 integrin selectively recognize different laminin isoforms. J Biol Chem. 2002;277:6012–6016. doi: 10.1074/jbc.M102188200. [DOI] [PubMed] [Google Scholar]
- 16.Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K., Kaufman S.J. Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J Cell Sci. 1997;110:2873–2881. doi: 10.1242/jcs.110.22.2873. [DOI] [PubMed] [Google Scholar]
- 17.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. Enhanced expression of the alpha7beta1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burkin D.J., Wallace G.Q., Milner D.J., Chaney E.J., Mulligan J.A., Kaufman S.J. Transgenic expression of alpha7beta1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am J Pathol. 2005;166:253–263. doi: 10.1016/s0002-9440(10)62249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo C., Willem M., Werner A., Raivich G., Emerson M., Neyses L., Mayer U. Absence of alpha7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum Mol Genet. 2006;15:989–998. doi: 10.1093/hmg/ddl018. [DOI] [PubMed] [Google Scholar]
- 20.Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J., Burkin D.J. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 21.Cohn R.D., Mayer U., Saher G., Herrmann R., van der Flier A., Sonnenberg A., Sorokin L., Voit T. Secondary reduction of alpha7B integrin in laminin alpha2 deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J Neurol Sci. 1999;163:140–152. doi: 10.1016/s0022-510x(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 22.Gawlik K.I., Mayer U., Blomberg K., Sonnenberg A., Ekblom P., Durbeej M. Laminin alpha1 chain mediated reduction of laminin alpha2 chain muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 2006;580:1759–1765. doi: 10.1016/j.febslet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 23.Gawlik K.I., Åkerlund M., Carmignac V., Elamaa H., Durbeej M. Distinct roles for laminin globular domains in laminin alpha1 chain mediated rescue of murine laminin alpha2 chain deficiency. PLoS One. 2010;5:e11549. doi: 10.1371/journal.pone.0011549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briguet A., Courdier-Fruh I., Foster M., Meier T., Magyar J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Yurchenco P.D., Quan Y., Colognato H., Mathus T., Harrison D., Yamada Y., O'Rear J.J. The alpha chain of laminin-1 is independently secreted and drives secretion of its beta- and gamma-chain partners. Proc Natl Acad Sci USA. 1997;94:10189–10194. doi: 10.1073/pnas.94.19.10189. [Erratum appeared in Proc Natl Acad Sci USA 1998, 95:1968] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coulton G.R., Morgan J.E., Partridge T.A., Sloper J.C. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol Appl Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 27.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 28.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connolly A.M., Keeling R.M., Mehta S., Pestronk A., Sanes J.R. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul Disord. 2001;11:703–712. doi: 10.1016/s0960-8966(01)00232-2. [DOI] [PubMed] [Google Scholar]
- 31.Glesby M.J., Rosenmann E., Nylen E.G., Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle Nerve. 1988;11:852–856. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- 32.Crawley S., Farrell E.M., Wang W., Gu M., Huang H.Y., Huynh V., Hodges B.L., Cooper D.N., Kaufman S.J. The alpha7beta1 integrin mediates adhesion and migration of skeletal myoblasts on laminin. Exp Cell Res. 1997;235:274–286. doi: 10.1006/excr.1997.3671. [DOI] [PubMed] [Google Scholar]
- 33.Matsumura K., Ervasti J.M., Ohlendieck K., Kahl S.D., Campbell K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 34.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;12:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 35.Zolkiewska A., Thompson W.C., Moss J. Interaction of integrin alpha7beta1 in C2C12 myotubes and in solution with laminin. Exp Cell Res. 1998;240:86–94. doi: 10.1006/excr.1998.4002. [DOI] [PubMed] [Google Scholar]
- 36.Kleinman H.K. Isolation of laminin-1 and type IV collagen from EHS sarcoma. J Tissue Cult Methods. 1994;16:231–233. [Google Scholar]