

Abstract
Chronic heart failure often results in catabolic muscle wasting, exercise intolerance, and death. Oxidative muscles, which have greater expression of the metabolic master gene peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α) and its target genes, are more resistant to catabolic wasting than are glycolytic muscles; however, the underlying mechanism is unknown. To determine the functional role of PGC-1α in oxidative phenotype–associated protection, skeletal muscle–specific PGC-1α transgenic mice were crossbred with cardiac-specific calsequestrin transgenic mice, a genetic model of chronic heart failure. PGC-1α overexpression in glycolytic muscles significantly attenuated catabolic muscle wasting induced by chronic heart failure. In addition to inactivation of forkhead transcription factor signaling through enhanced Akt/protein kinase B expression, in glycolytic muscles, PGC-1α overexpression led to enhanced expression of inducible nitric oxide synthase and endothelial nitric oxide synthase, production of nitric oxide, and expression of antioxidant enzyme including superoxide dismutases (SOD1, SOD2, and SOD3) and catalase, and reduced oxidative stress. These findings suggest that PGC-1α protects muscle from catabolic wasting in chronic heart failure through enhanced nitric oxide antioxidant defenses and inhibition of the forkhead transcription factor signaling pathways.
Chronic heart failure (CHF) is a major cause of death, affecting more than two million persons in the United States annually.1 CHF often leads to skeletal muscle wasting, or cardiac cachexia, characterized by muscle atrophy and reduced endurance, strength, and power as a result of abnormalities in skeletal muscles.2–4 Patients with CHF often exhibit the clinical symptom of exercise intolerance, manifested as early activation of the glycolytic pathways and fatigue during work. The symptom of exercise intolerance is strongly correlated with death.2,3 The underlying pathophysiologic mechanisms are poorly understood, hindering ability to develop effective therapies.
Increased production of reactive oxygen species (ROS)5–7 and reduced antioxidant gene expression8 have been functionally linked to muscle catabolism. ROS activates proteasome-dependent protein degradation by activating expression of muscle-specific E3 ligases, muscle atrophy F-box protein (MAFbx/atrogin-1), and muscle ring-finger protein 1 (MuRF1).9 ROS also causes muscle atrophy through activation of the autophagy-lysosome pathway.10,11 Activation of these degradation processes is mediated through the p38 mitogen-activated protein kinase signaling pathway.12 These studies demonstrate the importance of ROS in catabolic muscle wasting.
The resistance to catabolic muscle wasting of oxidative muscles, rich in type I and/or type IIa myosin heavy-chain protein, mitochondria, and capillaries,13–18 provides an excellent opportunity to decipher the mechanism underlying the “built-in” defenses. Overexpression of calsequestrin (CSQ) under control of the α-myosin heavy-chain promoter in transgenic mice leads to severe cardiac hypertrophy and eventually to CHF as a result of impairment of physiologic Ca2+ regulation and β-adrenergic receptor signaling.19,20 Using this genetic model of CHF, evidence was recently obtained to support the oxidative phenotype-associated protection as at least partly due to the presence and function of inducible antioxidant defenses.17 In response to cachectic stimuli, oxidative muscles demonstrated significantly less oxidative stress and more increased inducible nitric oxide synthase (iNOS) expression, nitric oxide (NO) production, and NO-dependent antioxidant gene expression.17,21 These findings suggest that NO-dependent antioxidant defenses in skeletal muscle are involved in protecting oxidative myofibers from cachexia. However, the regulatory factor or factors that control the built-in antioxidant defenses remains to be determined, which is the focus of the present study.
Several lines of evidence support a role of peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α) in maintaining skeletal muscle oxidative phenotype and in protection against oxidative stress. First, oxidative muscles demonstrate significantly more PGC-1α expression and less oxidative stress in response to cachectic stimuli than do glycolytic muscles, primarily composed of type IId/x and type IIb myofibers.21 Second, skeletal muscle catabolic wasting is associated with decreased PGC-1α expression in atrophying glycolytic myofibers.17 Third, endurance exercise training, known to promote oxidative phenotype, stimulates PGC-1α expression in skeletal muscle,22,23 and overexpression of PGC-1α promotes oxidative phenotype24 and prevents denervation- or fasting-induced muscle atrophy through suppression of FOXO3 signaling and atrogene transcription.25 What is not known is whether PGC-1α protects against CHF-induced muscle wasting and whether similar and additional mechanisms underlie the protection.
In the present study, it was hypothesized that PGC-1α has a critical role in protection against CHF through regulation of the Akt/protein kinase B–forkhead transcription factor (FOXO) signaling and/or the NO-dependent antioxidant defenses. To test this hypothesis, skeletal muscle–specific PGC-1α and cardiac muscle–specific CSQ was generated in double-transgenic mice (PGC-1α × CSQ) by crossbreeding. Consistent with this hypothesis, muscle-specific overexpression of PGC-1α significantly attenuated muscle atrophy induced by CHF in CSQ mice. In addition to enhanced Akt expression and inactivation of FOXO3a, overexpression of PGC-1α resulted in enhanced iNOS and endothelial nitric oxide synthase (eNOS) expression and NO production, and profound enhancement of antioxidant gene expression and reduction of oxidative stress in glycolytic muscles. These findings provide novel insights into how oxidative myofibers are protected from cardiac cachexia and highlight the importance of PGC-1α in NO-dependent antioxidant defense in skeletal muscle.
Materials and Methods
Experimental Animals
Transgenic mice (DBA) with cardiac-specific overexpression of CSQ under the promoter of α-myosin heavy chain19,20 were crossbred with transgenic mice (C57BL/6) with muscle-specific overexpression of PGC-1α under the promoter of muscle creatine kinase.24 Single- and double-transgenic mice and their wild-type littermates were housed in temperature-controlled quarters (21°C) with 12-hour light-dark cycles, with free access to water and a normal chow diet (Purina Mills, Inc., Richmond, IN). The CSQ breeder mice were treated with the β-blocker metoprolol (350 mg/kg of body weight per day in drinking water) before and during breeding, as previously described.26 At 8 weeks, when CSQ mice normally developed cachexia,17,20 all of the mice were humanely euthanized using isoflurane-induced anesthesia and cervical dislocation, and muscles were harvested for analysis. All animal protocols were approved by the Duke University Institutional Animal Care and Use Committee.
Genotyping
Mouse genomic DNA was isolated from tail tissue using a phenol-chloroform–based DNA extraction protocol for PCR with primers for the CSQ allele, 5′-CTCTGACAGAGAAGCAGGCACTTTAC-3′ and 5′-GATGAACAGGTGTGTTCTCTTCAT-3′, and the Pgc-1α allele, 5′-GCAGGATCACATAGGCAG GATGTGGCC-3′ and 5′-GGAAGATCTGGGCAAAGAGGCTGGTCC-3′. The PCR reaction for the CSQ allele was initial denaturation at 96°C for 4 minutes, 36 cycles of denaturation at 96°C for 30 seconds, annealing at 60°C for 30 seconds, extension at 72°C for 45 seconds, and a final extension at 72°C for 7 minutes. The PCR reaction for the Pgc-1α allele was initial denaturation at 96°C for 4 minutes, 30 cycles of denaturation at 96°C for 30 seconds, annealing at 60°C for 30 seconds, extension at 72°C for 45 seconds, and a final extension at 72°C for 7 minutes. The PCR products were resolved using electrophoresis followed by image acquisition.
Real-Time PCR
Total RNA was extracted from gastrocnemius muscle using Trizol reagent (Invitrogen Corp, Carlsbad, CA), and was reverse-transcribed into cDNA. Real-time PCR was performed using a sequence detection system (ABI Prism 7000; Applied Biosystems, Inc, Foster City, CA) with an initial hold of 50°C for 2 minutes, then 95°C for 10 minutes, followed by cyclic reactions of 95°C for 15 seconds and 60°C for 1 minute. The reagents used in real-time PCR including primers, probes, and TaqMan PCR Universal Master Mix (all purchased from Applied Biosystems, Inc) were Pgc-1α (Mm00447183_m1), Pgc-1β (Mm00504720_m1), estrogen-related receptor-α (Esrra; Mm00433143_m1), vascular endothelial growth factor (Vegf; Mm00437306_m1), nuclear respiratory factor-1 (Nrf1; Mm00447996_m1), Nrsf2 (Gabpa; Mm00484598_m1), NADH dehydrogenase (ubiquinone) Fe-S protein 1 (Ndufs1; Mm00523631_m1), MAFbx/Atrogin1 (Mm00499518_m1), Murf1 (Mm00491305_m1), neuronal NOS (nNOS; Mm00435175_m1), iNOS (Mm00440485_m1), and eNOS (Mm00435217_m1). The cycle threshold was determined using the supplied software, and standard curves were constructed for quantification. 18S rRNA was used as reference.
Semiquantitative RT-PCR
Because of technical difficulties in using the SOD3 primers in real-time PCR, semiquantitative RT-PCR was performed for SOD3 mRNA. The primer sequences were 5′-TCTTCCTGTCCCCATAGCAC-3′ and 5′-AAGCCACACACATGCACATT-3′. The PCR reaction was initial denaturation at 94°C for 3 minutes, 27 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, extension at 72°C for 45 seconds, and a final extension at 72°C for 8 minutes. The PCR products were resolved using electrophoresis on 1% agarose gel containing 1 mg/mL of ethidium bromide followed by image acquisition under UV light. Quantification of the PCR products was performed using commercially available software (Scion Image; Scion Corp, Frederick, MD). Results were normalized using 18S rRNA, and are given as fold change to wild-type mice.
Immunoblot Analysis
Skeletal muscle sample preparation for immunoblot analysis has been described previously.17 Antibodies used for immunoblot analysis were PGC-1α (ab3242; Millipore Corp, Temecula, CA), cytochrome oxidase IV (ab14744; Novus Biologicals, Inc, Littleton, CO), ß-actin (sc-81178; Santa Cruz Biotechnology, Inc, Santa Cruz, CA), SOD1 (ab16831; Novus Biologicals, Inc), SOD2 (SOD-111; Assay Designs, Ann Arbor, MI), SOD3 (AF4817; R&D Systems, Inc, Minneapolis, MN), catalase (ab15834; Novus Biologicals, Inc); and phospho-Akt (9271), total Akt (9272), phospho-FOXO1/3a (9464), and total FOXO3a (9467) (all four from Cell Signaling Technology, Inc, Beverly, MA). Quantification of the proteins was performed using software (Scion Image; Scion Corp).
Protein Carbonylation Assay
To determine oxidative stress in skeletal muscle, protein carbonyl levels were determined in plantaris muscle lysates using the Millipore OxyBlot Protein Oxidation Detection Kit (S7150; Serologicals Corp, Norcross, GA). In brief, 40 μg of muscle lysate was resolved using 10% SDS-PAGE and transferred to nitrocellulose membrane (162-0097; BioRad Laboratories, Inc, Hercules, CA), followed by 1 hour of blocking with 5% milk in 1× PBS with Tween 20. The membrane was then washed twice with 1× PBS with Tween 20 for 5 minutes each time, and incubated with 1 × 2,4-dinitrophenylhydrazine (DNPH) for 15 minutes on a shaker. This was followed by washes with water for 1 minute. The membrane was treated with neutralization solution and incubated for 5 minutes. Subsequent incubations with primary and secondary antibodies were performed using Western blotting on an imaging system (Odyssey; LI-COR Biosciences, Lincoln, NE).
Nitrite Assay
Nitrite was assessed using the Griess Reagent System (G-7921; Molecular Probes, Inc, Eugene, OR). In brief, 30 mg of frozen gastrocnemius muscle powder was homogenized in 300 μL of lysis buffer (20 mmol/L Tris base, 200 mmol/L NaCl, 2 mmol/L EDTA disodium salt, 0.02% Triton X-100, pH7.5) in a Wheaton glass-on-glass homogenizer, and the homogenate was centrifuged at 16,000 × g for 5 minutes at 4°C. The protein concentration of the supernatants was determined using the RC DC protein assay (BioRad Laboratories, Inc). The amount of nitrite in each sample was then determined using the Griess Reagent System according to the manufacturer's instructions. Nitrite concentration was calculated after normalization by total protein.
Determination of Skeletal Muscle Fiber Size
Fiber type analysis was determined at immunofluorescence staining for myosin heavy-chain proteins in gastrocnemius muscles as described previously.27 After immunohistochemical staining, the size of type I fibers (rhodamine Red-X–labeled), type IIa fibers (cyanine Cy5–labeled), and type IIb fibers (fluorescein-labeled) were measured for gastrocnemius muscle using software (Scion Image; Scion Corp), and presented as pixels per fiber. To ensure consistent analysis, the region of gastrocnemius muscle section with the presence of all fiber types was chosen, that is, the deep region of the muscle. On average, 105 fibers (range, 83–115) for each fiber type per section were measured to calculate the mean fiber size for each genotype.
Statistical Analysis
Data are given as mean (SE). Comparisons of various treatments were analyzed for statistical significance using two-way analysis of variance followed by the Student-Newman-Kuels test, with P < 0.05 considered statistically significant.
Results
Skeletal Muscle–Specific Overexpression of PGC-1α Improves Cardiac Cachexia Without Affecting Development of CHF
Because oxidative myofibers are resistant to muscle atrophy,13–18 and PGC-1α overexpression promotes formation of oxidative myofibers,24 it was speculated that glycolytic skeletal muscles with PGC-1α overexpression would become resistant to CHF-induced muscle wasting. When MCK-PGC-1α mice were crossbred with α-MHC-CSQ mice,17,19,26 PGC-1α transgenic mice, whether in the CSQ background or not, demonstrated a six- to sevenfold increase in PGC-1α and cytochrome oxidase IV protein expression in glycolytic white vastus lateralis muscle (Figure 1, A and B). PGC-1α transgenic mice also exhibited a three- to fivefold increase in Pgc-1α mRNA expression in gastrocnemius muscle (Figure 1C). Consistent with previous findings,28 mRNA levels of Esrra, Nrf2/Gabpa, Ndufs-1, and Vegf were increased two- to fourfold in gastrocnemius muscle of PGC-1α transgenic mice (Figure 1C). The transcription of Pgc-1β, a coactivator in the same family as PGC-1α, was not significantly increased in PGC-1α transgenic mice (Figure 1C).
Figure 1.

Muscle-specific overexpression of PGC-1α improves cardiac cachexia. Littermate wild-type mice, MCK-PGC-1α (PGC-1α), α-MHC-calsequestrin (CSQ), and CSQ × PGC-1α double-transgenic mice (CSQ × PGC-1α) were euthanized at age 8 weeks for various tissue harvesting and measurement. A: Representative images of immunoblots for PGC-1α and COX IV proteins in glycolytic white vastus lateralis muscles from wild-type (WT) and PGC-1α transgenic mice using α-actin as loading control. B: Quantification of PGC-1α and COX IV protein expression (n = 5 to 8). C: Analysis of mRNA expression for PGC-1α, Pgc-1b, Esrra, Vegf, Nrf1, Nrf2, and Ndufs1 at real-time PCR using 18S ribosomal RNA as control of RNA quantity and quality in gastrocnemius muscle (n = 4 to 6). D–G: Heart, body, liver, and lung weights, respectively, of WT and PGC-1α mice with and without cardiac-specific overexpression of CSQ (n = 6 or 7). Values are given as mean (SE) *P < 0.05. ***P < 0.001. Con, control group of mice not in the CSQ background.
First measured was wet weight of various organs, and the data were normalized against tibia length to minimize the effect fluctuation of body weight due to the disease condition. CSQ mice developed severe cardiac hypertrophy with a profound increase (2.2-fold; P < 0.001) in biventricular heart weight (Figure 1D), and cardiac cachexia as indicated by loss of body weight (−39%; P < 0.001) and liver weight (−45%; P < 0.001) (Figure 1, E and F). The observed loss in body weight occurred after 4 weeks because significant differences in body weight were not detected at 4 weeks (data not shown). The lung weight in CSQ mice was similar to that observed in wild-type mice (Figure 1F) despite the loss of whole body weight and other organ weight such as liver (Figure 1F) and skeletal muscles (Table 1), which suggests a relative increase in lung weight due to impaired cardiac function. Overexpression of PGC-1α in skeletal muscle alone did not significantly change any of these parameters; however, although overexpression of PGC-1α in skeletal muscles in CSQ background did not have a significant effect on cardiac hypertrophy (Figure 1D) and heart failure (indicated by lung weight) (Figure 1G), it led to significantly attenuated body weight loss, from 39% in CSQ mice to 27% in CSQ × PGC-1α double-transgenic mice (P < 0.05) (Figure 1G). This protection seems to be specific to skeletal muscle because the reduction in liver weight was not significantly affected (Figure 1F), consistent with a protective function of PGC-1α expression in skeletal muscle. Taken together, muscle specific overexpression of PGC-1α significantly attenuates cardiac cachexia with no significant influences on development of CHF in CSQ mice.
Table 1.
Skeletal Muscle–Specific Expression of PGC-1α Attenuates CHF-Induced Atrophy in Glycolytic Muscle
| Genotype |
||||
|---|---|---|---|---|
| Muscle | Wild type (n = 6) | PGC-1α (n = 7) | CSQ (n = 6) | CSQ × PGC-1α (n = 6) |
| Soleus | ||||
| mg | 4.54 (0.17) | 5.19 (0.42) | 2.69 (0.40)† | 4.07 (0.25)⁎§ |
| mg/mm | 0.29 (0.01) | 0.32 (0.02) | 0.18 (0.02)† | 0.25 (0.01)⁎§ |
| mg/g | 0.24 (0.01) | 0.25 (0.02) | 0.26 (0.02) | 0.27 (0.02) |
| Plantaris | ||||
| mg | 10.64 (0.40) | 11.82 (0.66) | 4.71 (0.79)‡ | 7.47 (0.71)‡§ |
| mg/mm | 0.67 (0.02) | 0.72 (0.04) | 0.32 (0.04)‡ | 0.46 (0.03)‡§ |
| mg/g | 0.57 (0.01) | 0.57 (0.02) | 0.45 (0.03)‡ | 0.49 (0.02)⁎ |
| Gastrocnemius | ||||
| mg | 78.5 (3.8) | 86.8 (2.9) | 35.7 (6.7)‡ | 54.9 (6.5)‡§ |
| mg/mm | 4.97 (0.16) | 5.29 (0.15) | 2.41 (0.31)‡ | 3.41 (0.28)‡§ |
| mg/g | 4.17 (0.09) | 4.23 (0.06) | 3.43 (0.24)† | 3.69 (0.16)⁎ |
| Tibialis anterior | ||||
| mg | 30.8 (1.3) | 35.5 (0.9) | 15.1 (2.0)‡ | 23.5 (1.9)†¶ |
| mg/mm | 1.95 (0.06) | 2.16 (0.04) | 1.02 (0.10)‡ | 1.47 (0.08)†¶ |
| mg/g | 1.64 (0.03) | 1.73 (0.04) | 1.47 (0.07)⁎ | 1.61 (0.05) |
| Extensor digitorum longus | ||||
| mg | 5.90 (0.19) | 6.53 (0.27) | 3.23 (0.54)‡ | 4.81 (0.46)†§ |
| mg/mm | 0.37 (0.01) | 0.40 (0.01) | 0.22 (0.02)‡ | 0.30 (0.03)†§ |
| mg/g | 0.31 (0.01) | 0.32 (0.01) | 0.30 (0.02) | 0.32 (0.01) |
PGC-1α, MCK-PGC-1α transgenic; CSQ, α-MHC-CSQ transgenic.
Values are given as mean (SE) in absolute muscle weight (mg), normalized muscle weight by tibia length (mg/mm), and body weight (mg/g).
P < 0.05,
P < 0.01, and
P< 0.001 for comparison between WT and CSQ or between PGC-1α and CSQ × PGC-1α groups.
P < 0.05 and
P < 0.01 for comparison between PGC-1α and CSQ × PGC-1α groups.
Skeletal Muscle–Specific Overexpression of PGC-1α Significantly Reduces CHF-Induced Muscle Atrophy
To determine whether PGC-1α overexpression renders resistance to CHF-induced muscle wasting, muscle mass was measured for soleus, plantaris, gastrocnemius, tibialis anterior, and extensor digitorum longus muscles in all four genetic backgrounds. Again, to minimize the effect of the reduction of body weight on the analysis, muscle weight was normalized against tibia length. CHF led to significant loss of muscle mass in the soleus (−38%; P < 0.01), plantaris (−52%; P < 0.001), gastrocnemius (−52%; P < 0.001), tibialis anterior (−48%; P < 0.001), and extensor digitorum longus (−41%) muscles compared with that in wild-type mice (Table 1). Moreover, the loss of muscle mass in these muscles was significantly attenuated by PGC-1α overexpression (Table 1). Specifically, compared with PGC-1α transgenic mice, loss of muscle mass in CSQ × PGC-1α double-transgenic mice was reduced to 22% for the soleus (P < 0.05), 36% for the plantaris (P < 0.05), 36% for the gastrocnemius (P < 0.05), 32% for the tibialis anterior (P < 0.05), and 25% for the extensor digitorum longus (P < 0.05) muscles. To determine whether PGC-1α–mediated protection depends on the fiber type specification, immunofluorescence-based fiber type analysis was performed in gastrocnemius muscle (Figure 2A). CHF resulted in significant reduction of types I (−28%; P < 0.01), IIa (−28%; P < 0.001), and IIb (−52%; P < 0.001) myofibers (Figure 2, B–D). PGC-1α overexpression alone resulted in a trend (not statistically significant) of decreased fiber size for all three fiber types but completely blocked the reduction in fiber size induced by CHF (Figure 2, B–D). These findings suggest that (1) skeletal muscles rich in oxidative myofibers such as soleus muscle are more resistant to catabolic wasting induced by CHF than are skeletal muscles of mixed fibers or predominantly glycolytic fibers; (2) oxidative myofibers, specifically types I and IIa fibers, are more resistant to catabolic wasting than are type IIb fibers; and (3) enhanced PGC-1α expression renders myofibers resistant to catabolic wasting independent of fiber type.
Figure 2.
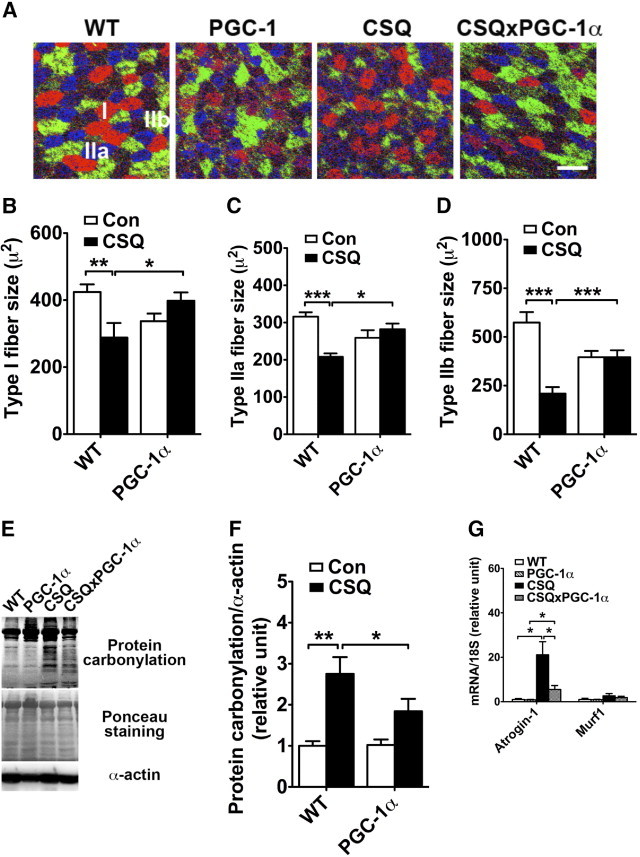
Muscle-specific overexpression of PGC-1α attenuates catabolic muscle wasting induced by chronic heart failure. A: Immunofluorescence analysis for fiber size in gastrocnemius muscle sections immunostained for myosin heavy chains I (red), IIa (blue), and IIb (green). Scale bar = 500 μm. B–D: Quantitative data of fiber size of various fiber types. Mean (SE) values of fiber size for each group of mice was calculated from muscle sections of 4 to 8 mice with 20 myofibers for each fiber type for each mouse. E: Analysis of protein carbonylation in plantaris muscle as an indication of the level of oxidative stress with ponceau staining and α-actin as loading controls. F: Quantification of protein carbonylation in plantaris muscle (n = 3). G: Analysis of mRNA expression for MAFbx/Atrogin-1 and Murf1 in gastrocnemius muscle at real-time PCR using 18S ribosomal RNA as control of RNA quantity and quality. Values are given as mean (SE) (n = 4 to 6). *P < 0.05. **P < 0.01. ***P < 0.001. Con, control group of mice not in the CSQ background; WT, wild type.
PGC-1α Overexpression in Skeletal Muscle Reduces Oxidative Stress and MAFbx/Atrogen-1 Expression Induced by CHF
Because ROS is an important mediator in muscle atrophy,5–7 whether the protection by PGC-1α overexpression is associated with reduced oxidative stress was tested by assessing the level of protein carbonylation.21 As expected, there was a significant increase in protein carbonylation in plantaris muscle (with mixed muscle fibers) in CSQ mice compared with their wild-type littermates (P < 0.01) (Figure 2, E and F). The increased protein carbonylation was significantly attenuated by PGC-1α overexpression (P < 0.05) (Figure 2, E and F). Consistent with the notion that ROS activates the proteasome-dependent protein degradation, it was observed that muscle-specific E3 ligases, MAFbx/Atrogin-1 mRNA was increased by 21-fold (P < 0.05) in CSQ transgenic mice compared with wild-type mice, with a trend toward increased Murf1 mRNA (Figure 2G). Induction of MAFbx/atrogin-1 mRNA was attenuated by overexpression of PGC-1α in skeletal muscles (Figure 2G). Taken together, muscle-specific overexpression of PGC-1α significantly attenuates muscle atrophy induced by CHF along with reduced oxidative stress and MAFbx/atrogen-1 E3 ligase mRNA expression.
Enzymatic Antioxidant System Is Enhanced by Muscle-Specific Overexpression of PGC-1α
Recent findings indicate that PGC-1α is a broad and powerful regulator of ROS metabolism because it can induce many ROS-detoxifying antioxidant enzymes.29–32 However, the relevance of this regulated antioxidant defense system in skeletal muscle, in particular in CHF-induced muscle wasting, is unknown. Thus, expression of a set of key antioxidant enzymes including SOD1 (CuZnSOD), SOD2 (MnSOD), SOD3 (EC-SOD), and catalase was examined in skeletal muscles. The assays would help determine whether the reduced oxidative stress was associated with enhanced expression of these antioxidant enzymes. It was observed that protein expression of SOD1, SOD2, SOD3, and catalase increased by 1.4-fold (P < 0.001), 3.2-fold (P < 0.001), 1.7-fold (P < 0.05), and 4.3-fold (P < 0.01), respectively, in glycolytic white vastus lateralis muscle with or without CHF (Figure 3,A and B). These findings indicate the possibility that PGC-1α–induced antioxidant enzyme expression in skeletal muscle provides protection against catabolic muscle wasting induced by CHF.
Figure 3.
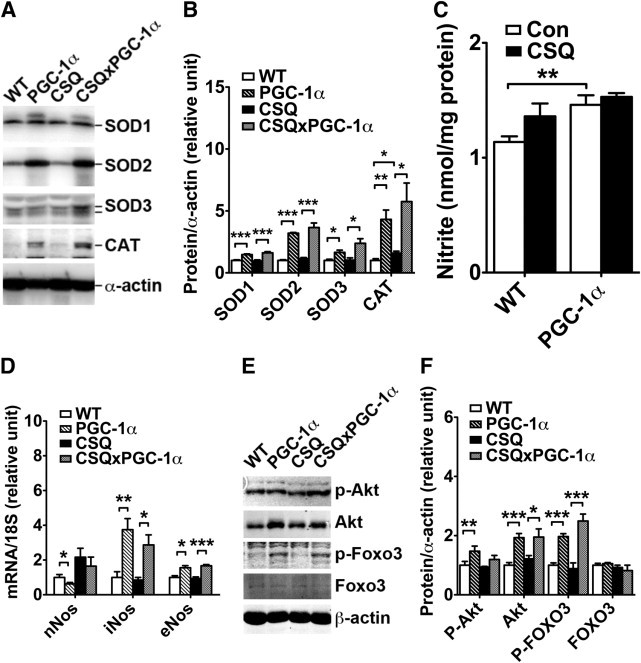
Muscle-specific overexpression of PGC-1α promotes NO and antioxidant enzyme expression and inhibits FOXO signaling. A: Representative immunoblots of SOD1, SOD2, SOD3, and catalase (CAT) in white vastus lateralis muscles using α-actin as loading control. B: Quantification of protein expression of SOD1, SOD2, SOD3, and CAT (n = 4 to 6). C: Nitrite concentrations in gastrocnemius muscles (n = 3 to 7). D: Analysis of mRNA expression of nNOS, iNOS, and eNOS in gastrocnemius muscle at real-time PCR using 18S ribosomal RNA as control of RNA quantity and quality (n = 4 to 6). E: Representative immunoblots of phosphorylated Akt (P-Akt), total Akt, phosphorylated FOXO3, and total FOXO3 in plantaris muscles using α-actin as loading control. F: Quantification of P-Akt, Akt, P-FOXO3, and FOXO3 in plantaris muscles (n = 5 to 7). Values are given as mean (SE). *P < 0.05. **P < 0.01. ***P < 0.001. Con, control group of mice not in the CSQ background; WT, wild type.
NOS Expression and NO Production Are Enhanced by Muscle-Specific Overexpression of PGC-1α
Recent findings indicate that NO promotes antioxidant enzyme expression under cachectic conditions and is sufficient to protect glycolytic muscles from catabolic wasting.21 Thus, it was speculated that the increased expression of these antioxidant enzymes was due to enhanced NO production by PGC-1α overexpression. Nitrite, a stable NO metabolite, in gastrocnemius muscle was significantly (P < 0.01) higher in PGC-1α transgenic mice than in wild-type mice (Figure 3C). CSQ transgenic mice also demonstrated a trend toward increased NO, which persisted in the PGC-1α-CSQ double-transgenic mice (Figure 3C). To determine which isoform of NOS might be responsible for the increased NO production due to PGC-1α overexpression, mRNA levels of nNOS, iNOS, and eNOS were measured using real-time PCR. mRNA levels of iNOS and eNOS were increased by 3.8-fold (P < 0.01) and 1.6-fold (P < 0.05), respectively, and nNOS mRNA was reduced (P < 0.05) by PGC-1α overexpression alone in gastrocnemius muscle (Figure 3D). CHF induced a 2.2-fold (P < 0.05) increase in nNOS mRNA and a 2.6-fold (P < 0.001) increase in the double-transgenic background. These findings support the notion that NO from different sources has different functions. Enhanced PGC-1α expression promotes NO production through enhanced iNOS and eNOS expression, whereas CHF promotes NO production through enhanced nNOS expression.
Enhanced Akt Expression and Suppressed FOXO Signaling in PGC-1α Transgenic Mice
FOXO activation–mediated transcription of ubiquitin ligases has a critical role in muscle wasting,33 and PGC-1α protects muscle from denervation-induced atrophy, possibly by blocking Murf1 E3 ligase transcription via suppression of the FOXO3 signaling.25 To determine whether PGC-1α overexpression may also protect muscle from cardiac cachexia through the same mechanism, the phosphorylation status of FOXO3 was measured. Compared with wild-type mice, PGC-1α transgenic mice demonstrated a 1.9-fold increase in phosphorylated FOXO3 in plantaris muscles, with no significant increases in total FOXO3 (Figure 3, E and F). The increased FOXO3 phosphorylation was concurrent with increased expression of Akt, the upstream kinase responsible for FOXO phosphorylation,33 but not the phosphorylation state of Akt. These findings are consistent with the notion that PGC-1α overexpression enhances FOXO phosphorylation (suppression of the activity) through enhanced Akt expression.
Discussion
It has long been known that glycolytic muscles are more vulnerable to catabolic muscle wasting than are oxidative muscles13–18; however, the underlying mechanism remains unknown. The present study confirms that PGC-1α overexpression is sufficient to prevent muscles, including muscles enriched in glycolytic fibers, from atrophy in an animal model of CHF, which suggests that PGC-1α has a key role in the oxidative muscle–specific resistance to catabolic wasting. Moreover, PGC-1α overexpression–mediated protection in CHF seems to be caused by increased expression of iNOS and eNOS, NO production, and antioxidant enzyme expression, leading to reduced oxidative stress in glycolytic muscles, in addition to enhanced Akt expression and suppressed FOXO signaling. These findings provide insights into the functional role of PGC-1α in skeletal muscle in protection against catabolic muscle wasting.
Skeletal muscle–specific overexpression of PGC-1α in the present study promoted transcription of its target genes functioning in mitochondrial biogenesis, mitochondrial respiration, and angiogenesis in skeletal muscle (Figure 1, A–C) but had no significant effect on development of CHF (Figure 1D, F, and G). The findings suggest that the improved cachectic condition, as demonstrated by the attenuated loss of body weight (Figure 1E), is not mediated by improved cardiac function. In light of the recent finding that cardiac-derived humoral factors are important in skeletal muscle catabolic wasting in CHF,34,35 the findings suggest that enhanced PGC-1α expression in skeletal muscle exerts a cell-autonomous protection rather than resulting from improved cardiac function. Exercise training and increased contractile activity have a profound positive effect on skeletal muscle in CHF.36–38 Inasmuch as exercise training induces PGC-1α gene expression and metabolic adaptations,22,23,39 the benefits of regular exercise in CHF could be mediated, at least in part, by induced PGC-1α expression. Future research will be necessary to ascertain the functional role of PGC-1α in exercise-induced protection against cardiac cachexia. It would also be intriguing to know whether improved cachectic condition by PGC-1α overexpression for a longer time (>8 weeks) would improve cardiac function and/or reduce mortality.
Skeletal muscle atrophy induced by CHF was clearly prevalent in glycolytic muscles when muscle weight was normalized against tibia length (Table 1), which could be significantly attenuated by overexpression of PGC-1α. Normalization of muscle weight by body weight significantly confounded the analyses because cachexia is inevitably associated with loss of body weight. Direct quantitative evidence was obtained that PGC-1α overexpression could render myofibers resistant to catabolic muscle wasting, even including glycolytic type IIb myofibers (Figure 2D). It seems that PGC-1α overexpression exerts more profound protection in glycolytic fibers than in oxidative fibers. First, types I and IIa fibers demonstrated approximately 30% reduction in fiber size in CSQ mice compared with wild-type littermates (Figure 2, B and C), in line with body weight reduction (−39%), whereas type IIb fiber demonstrated approximately 60% reduction in fiber size; that is, types I and IIa myofibers did not show preferential loss of mass compared with body weight. Type IIb fiber is clearly susceptible to CHF-induced atrophy. However, the absence of size reduction in types I and IIa fibers could be due to endogenous (already high) and/or exogenous PGC-1α expression, whereas the protection observed in type IIb fibers is clearly due to exogenous PGC-1α expression because these fibers demonstrated little PGC-1α expression in wild-type mice (Figure 1A).
The present findings provide proof-of-principle evidence to support targeting PGC-1α and its downstream Akt-FOXO signaling and NO-dependent antioxidant defenses in treatment and prevention of catabolic muscle wasting in CHF (Figure 4). It is possible that the built-in defenses in oxidative muscle depend on sustained PGC-1α expression. That endogenous PGC-1α protein expression does not increase significantly in oxidative muscles in cardiac cachexia17 suggests that the protective function of PGC-1α in oxidative muscle is not mediated by induced expression during the catabolic wasting process. Rather, the finding that PGC-1α overexpression leads to enhanced expression of iNOS and eNOS in glycolytic muscles is consistent with the notion that at least some of the protective functions of PGC-1α may be mediated by a priming effect on inducing basal level expression of NO synthases. It remains to be ascertained whether PGC-1α–induced antioxidant gene expression depends on iNOS and eNOS, although NO donors are sufficient to induce antioxidant gene expression in muscle cells both in vitro and in vivo.21
Figure 4.

Working model of the role of PGC-1α in protecting skeletal muscle from CHF-induced catabolic muscle wasting. PGC-1α inactivates FOXO by promoting Akt expression, leading to reduced atrogene expression. In contrast, PGC-1α promotes expression of antioxidant genes, possibly through enhanced expression of iNOS and eNOS. The antioxidant defenses in turn detoxify ROS induced by CHF, leading to less p38 activation and protein oxidation. These collectively attenuate atrophy or catabolic muscle wasting.
Members of the FOXO family have an important role in muscle atrophy through control of genes involved in proteasome- and lysosome-dependent proteolysis.40,41 The upstream kinase Akt inhibits FOXO activity through phosphorylation in atrophying muscle.33 Enhanced muscle expression of insulinlike growth factor 1 inhibits the atrophic process in models of cardiac cachexia,42,43 and PGC-1α overexpression is sufficient to attenuate muscle atrophy induced by expression of a constitutively active FOXO transgene.25 In the present study, PGC-1α overexpression did not lead to Akt activation (increased phosphorylation) but enhanced Akt expression (increased total Akt) and FOXO3 inactivation (increased phosphorylation). Inasmuch as enhanced antioxidant defenses and reduced oxidative stress are not likely due to, but may lead to, inhibition of FOXO signaling, it was speculated that enhanced NO and antioxidant defenses are an upstream or a parallel mechanism underlying PGC-1α overexpression-mediated protection against catabolic muscle wasting.
ROS has been considered an important mediator in muscle atrophy induced by many chronic diseases and syndromes.5–7 Its cause-and-effect role in muscle atrophy is supported by in vitro9,12,44 and in vivo21,44,45 studies that demonstrated that oxidative stress induced by oxidant supplement or proinflammatory cytokines leads to muscle atrophy. The causative effect of ROS is further strengthened by the finding that ROS scavengers or enhanced antioxidant genes attenuate muscle atrophy.21,46,47 In the present study, it was observed that CHF induced a significant increase in oxidative stress in glycolytic muscle, as indicated by the results of the carbonylation assay, and PGC-1α overexpression significantly reduced indices of oxidative stress (Figure 2E). Therefore, one of the important functions of PGC-1α might be promotion of antioxidant defenses in skeletal muscle in counteracting oxidative stress induced by various cachectic conditions.
ROS-detoxifying antioxidant enzymes have different functional localizations, with SOD2 in mitochondrial matrix, catalase in peroxisome, SOD3 in extracellular space, and SOD1 globally distributed in the cell. The different localizations of the antioxidant enzymes are likely related to their distinct ROS scavenging function. PGC-1α overexpression enhanced expression of all four antioxidant enzymes (Figure 3, A and B), consistent with its profound function against catabolic muscle wasting. This finding is consistent with previous studies that have linked overexpression of PGC-1α to suppression of oxidative stress in neural cells by promoting antioxidant gene transcriptions.48 More recently, PGC-1α has been shown to have an important role in regulating expression of myocardial mitochondrial antioxidants SOD2 and thioredoxin-2 and in protecting hearts against transverse aortic constriction-induced myocardial oxidative stress, hypertrophy, and dysfunction.31 Moreover, endurance exercise training, which promotes PGC-1α activity and expression, improves whole body peak oxygen consumption along with enhanced antioxidant gene expression in skeletal muscle in patients with CHF.8,49 These findings collectively support the view that PGC-1α has an important function in protecting skeletal muscle from catabolic wasting by promoting antioxidant enzyme expression.
Previous findings suggest that enhanced NO signaling, specifically from iNOS, may protect muscle from atrophy.21 The underlying mechanism could be that NO promotes antioxidant gene expression through enhanced transcription of antioxidant response elements containing antioxidant genes.21 In the present study, nNOS was induced by CHF but not by PGC-1α overexpression, and iNOS and eNOS were induced by PGC-1α but not by CHF. These findings suggest that NO from different sources has distinct functions in catabolic muscle wasting. Although total NO production could only be estimated by measuring nitrite content, based on the apparent phenotypes associated with the four genetic backgrounds, it was speculated that CHF promotes NO production from nNOS, which is detrimental to development of catabolic muscle wasting, as has been reported in other muscular diseases.26,50 However, PGC-1α may promote NO production from iNOS and eNOS, facilitating antioxidant defenses against muscle wasting. The distinct functions of the NOS isoforms may be related to the subcellular locations of these enzymes as well.
Overall, the present study provides evidence that PGC-1α overexpression attenuates CHF-induced muscle atrophy through enhanced NO-dependent antioxidant defenses and reduced oxidative stress in addition to enhanced Akt expression and suppression of FOXO signaling. The findings support the view that PGC-1α has a key role in fiber type–specific susceptibility to cachectic muscle wasting. PGC-1α may mediate the protective function of endurance exercise training and is a potential target for developing effective therapies for skeletal muscle catabolic wasting induced by many chronic diseases including CHF.
Acknowledgments
We thank Bruce M. Spiegelman, Ph.D., for providing the PGC-1α mice; Howard A. Rockman, M.D., for providing the CSQ transgenic mice; and Avril V. Somlyo, Ph.D., and James W. Mandell, M.D., Ph.D., for critical review of the manuscript.
Footnotes
Supported by the National Institutes of Health (grant AR050429) and by the American Heart Association, Mid-Atlantic Affiliate (grant-in-aid 0555426U to Z.Y.)
References
- 1.Schocken D.D., Arrieta M.I., Leaverton P.E., Ross E.A. Prevalence and mortality rate of congestive heart failure in the United States. J Am Coll Cardiol. 1992;20:301–306. doi: 10.1016/0735-1097(92)90094-4. [DOI] [PubMed] [Google Scholar]
- 2.Drexler H., Coats A.J. Explaining fatigue in congestive heart failure. Annu Rev Med. 1996;47:241–256. doi: 10.1146/annurev.med.47.1.241. [DOI] [PubMed] [Google Scholar]
- 3.Massie B.M., Conway M., Rajagopalan B., Yonge R., Frostick S., Ledingham J., Sleight P., Radda G. Skeletal muscle metabolism during exercise under ischemic conditions in congestive heart failure: evidence for abnormalities unrelated to blood flow. Circulation. 1988;78:320–326. doi: 10.1161/01.cir.78.2.320. [DOI] [PubMed] [Google Scholar]
- 4.Wilson J.R., Mancini D.M., Dunkman W.B. Exertional fatigue due to skeletal muscle dysfunction in patients with heart failure. Circulation. 1993;87:470–475. doi: 10.1161/01.cir.87.2.470. [DOI] [PubMed] [Google Scholar]
- 5.Barreiro E., de la Puente B., Busquets S., Lopez-Soriano F., Gea J., Argiles J. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005;579:1646–1652. doi: 10.1016/j.febslet.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 6.Arias-Diaz J., Vara E., Torres-Melero J., Garcia C., Hernandez J., Balibrea J.L. Local production of oxygen free radicals and nitric oxide in rat diaphragm during sepsis: effects of pentoxifylline and somatostatin. Eur J Surg. 1997;163:619–625. [PubMed] [Google Scholar]
- 7.Fagan J.M., Ganguly M., Tiao G., Fischer J.E., Hasselgren P.O. Sepsis increases oxidatively damaged proteins in skeletal muscle. Arch Surg. 1996;131:1326–1331. doi: 10.1001/archsurg.1996.01430240080011. discussion 1331–1322. [DOI] [PubMed] [Google Scholar]
- 8.Linke A., Adams V., Schulze P., Erbs S., Gielen S., Fiehn E., Mobius-Winkler S., Schubert A., Schuler G., Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–1770. doi: 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- 9.Li Y., Chen Y., Li A., Reid M. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285:C806–C812. doi: 10.1152/ajpcell.00129.2003. [DOI] [PubMed] [Google Scholar]
- 10.Dobrowolny G., Aucello M., Rizzuto E., Beccafico S., Mammucari C., Bonconpagni S., Belia S., Wannenes F., Nicoletti C., Del Prete Z., Rosenthal N., Molinaro M., Protasi F., Fanò G., Sandri M., Musarò A. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–436. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Hernandez A., Cordero M.D., Salviati L., Artuch R., Pineda M., Briones P., Gomez Izquierdo L., Cotan D., Navas P., Sanchez-Alcazar J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- 12.McClung J.M., Judge A.R., Powers S.K., Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol. 2010;298:C542–C549. doi: 10.1152/ajpcell.00192.2009. [Epub ahead of press December 2, 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah A.J., Sahgal V., Quintanilla A.P., Subramani V., Singh H., Hughes R. Muscle in chronic uremia: a histochemical and morphometric study of human quadriceps muscle biopsies. Clin Neuropathol. 1983;2:83–89. [PubMed] [Google Scholar]
- 14.Butori C., Desnuelle C., Hofman P., Paquis V., Durant J., Carles M., Pesce A., Michiels J.F. Muscular involvement in the course of AIDS: anatomo-clinical study of 17 cases. Ann Pathol. 1995;15:424–430. [in French] [PubMed] [Google Scholar]
- 15.Minnaard R., Drost M.R., Wagenmakers A.J., van Kranenburg G.P., Kuipers H., Hesselink M.K. Skeletal muscle wasting and contractile performance in septic rats. Muscle Nerve. 2005;31:339–348. doi: 10.1002/mus.20268. [DOI] [PubMed] [Google Scholar]
- 16.Krolick K.A. Muscle-derived nitric oxide synthase expression, differences associated with muscle fiber-type, and disease susceptibility in a rat model of myasthenia gravis. Clin Immunol. 2006;121:286–293. doi: 10.1016/j.clim.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Li P., Waters R., Redfern S., Zhang M., Mao L., Annex B., Yan Z. Oxidative phenotype protects myofibers from pathological insults induced by chronic heart failure in mice. Am J Pathol. 2007;170:599–608. doi: 10.2353/ajpath.2007.060505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acharyya S., Butchbach M.E., Sahenk Z., Wang H., Saji M., Carathers M., Ringel M.D., Skipworth R.J., Fearon K.C., Hollingsworth M.A., Muscarella P., Burghes A.H., Rafael-Fortney J.A., Guttridge D.C. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–432. doi: 10.1016/j.ccr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Jones L., Suzuki Y., Wang W., Kobayashi Y., Ramesh V., Franzini-Armstrong C., Cleemann L., Morad M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest. 1998;101:1385–1393. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho M., Rapacciuolo A., Koch W., Kobayashi Y., Jones L., Rockman H. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J Biol Chem. 1999;274:22251–22256. doi: 10.1074/jbc.274.32.22251. [DOI] [PubMed] [Google Scholar]
- 21.Yu Z., Li P., Zhang M., Hannink M., Stamler J., Yan Z. Fiber type-specific nitric oxide protects oxidative myofibers against cachectic stimuli. PLoS One. 2008;3:e2086. doi: 10.1371/journal.pone.0002086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baar K., Wende A., Jones T., Marison M., Nolte L., Chen M., Kelly D., Holloszy J. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 23.Terada S., Goto M., Kato M., Kawanaka K., Shimokawa T., Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun. 2002;296:350–354. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 24.Lin J., Wu H., Tarr P., Zhang C., Wu Z., Boss O., Michael L., Puigserver P., Isotani E., Olson E., Lowell B., Bassel-Duby R., Spiegelman B. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 25.Sandri M., Lin J., Handschin C., Yang W., Arany Z.P., Lecker S.H., Goldberg A.L., Spiegelman B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki M., Carlson K.M., Marchuk D.A., Rockman H.A. Genetic modifier loci affecting survival and cardiac function in murine dilated cardiomyopathy. Circulation. 2002;105:1824–1829. doi: 10.1161/01.cir.0000014926.32463.89. [DOI] [PubMed] [Google Scholar]
- 27.Waters R.E., Rotevatn S., Li P., Annex B.H., Yan Z. Voluntary running induces fiber type-specific angiogenesis in mouse skeletal muscle. Am J Physiol Cell Physiol. 2004;287:C1342–C1348. doi: 10.1152/ajpcell.00247.2004. [DOI] [PubMed] [Google Scholar]
- 28.Arany Z., Foo S., Ma Y., Ruas J., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S., Baek K., Rosenzweig A., Spiegelman B. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 29.Valle I., Alvarez-Barrientos A., Arza E., Lamas S., Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 30.Chen S.-D., Lin T.-K., Yang D.-I., Lee S.-Y., Shaw F.-Z., Liou C.-W., Chuang Y.-C. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res. 2010;88:605–613. doi: 10.1002/jnr.22225. [DOI] [PubMed] [Google Scholar]
- 31.Lu Z., Xu X., Hu X., Fassett J.T., Zhu G., Tao Y., Li J., Zhang P., Zhao B., Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carreira R.S., Lee Y., Ghochani M., Gustafsson A.B., Gottlieb R.A. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6:462–472. doi: 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stitt T., Drujan D., Clarke B., Panaro F., Timofeyva Y., Kline W., Gonzalez M., Yancopoulos G., Glass D. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy–induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 34.Shyu K.G., Lu M.J., Wang B.W., Sun H.Y., Chang H. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur J Clin Invest. 2006;36:713–719. doi: 10.1111/j.1365-2362.2006.01718.x. [DOI] [PubMed] [Google Scholar]
- 35.Heineke J., Auger-Messier M., Xu J., Sargent M., York A., Welle S., Molkentin J.D. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–425. doi: 10.1161/CIRCULATIONAHA.109.882068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenk K., Schur R., Linke A., Erbs S., Matsumoto Y., Adams V., Schuler G. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur J Heart Failure. 2009;11:342–348. doi: 10.1093/eurjhf/hfp020. [DOI] [PubMed] [Google Scholar]
- 37.Hambrecht R., Schulze P.C., Gielen S., Linke A., Möbius-Winkler S., Erbs S., Kratzsch J., Schubert A., Adams V., Schuler G. Effects of exercise training on insulin-like growth factor-I expression in the skeletal muscle of non-cachectic patients with chronic heart failure. Eur J Cardiovasc Prev Rehab. 2005;12:401–406. doi: 10.1097/01.hjr.0000173106.68485.b7. [DOI] [PubMed] [Google Scholar]
- 38.Otis J.S., Lees S.J., Williams J.H. Functional overload attenuates plantaris atrophy in tumor-bearing rats. BMC Cancer. 2007;7:146. doi: 10.1186/1471-2407-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geng T., Li P., Okutsu M., Yin X., Kwek J., Zhang M., Yan Z. PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C572–C579. doi: 10.1152/ajpcell.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S., Goldberg A. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao J., Brault J.J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S.H., Goldberg A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 42.Schulze P., Fang J., Kassik K., Gannon J., Cupesi M., MacGillivray C., Lee R., Rosenthal N. Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ Res. 2005;97:418–426. doi: 10.1161/01.RES.0000179580.72375.c2. [DOI] [PubMed] [Google Scholar]
- 43.Song Y., Li Y., Du J., Mitch W., Rosenthal N., Delafontaine P. Muscle-specific expression of IGF-1 blocks angiotensin II–induced skeletal muscle wasting. J Clin Invest. 2005;115:451–458. doi: 10.1172/JCI22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y., Chen Y., John J., Moylan J., Jin B., Mann D., Reid M. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haddad F., Zaldivar F., Cooper D., Adams G. IL-6-induced skeletal muscle atrophy. J Appl Physiol. 2005;98:911–917. doi: 10.1152/japplphysiol.01026.2004. [DOI] [PubMed] [Google Scholar]
- 46.Dalla Libera L., Ravara B., Gobbo V., Danieli Betto D., Germinario E., Angelini A., Vescovo G. Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol. 2005;38:803–807. doi: 10.1016/j.yjmcc.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 47.Appell H., Duarte J., Soares J. Supplementation of vitamin E may attenuate skeletal muscle immobilization atrophy. Int J Sports Med. 1997;18:157–160. doi: 10.1055/s-2007-972612. [DOI] [PubMed] [Google Scholar]
- 48.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D.K., Bachoo R., Spiegelman B.M. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 49.Ennezat P.V., Malendowicz S.L., Testa M., Colombo P.C., Cohen-Solal A., Evans T., LeJemtel T.H. Physical training in patients with chronic heart failure enhances the expression of genes encoding antioxidative enzymes. J Am Coll Cardiol. 2001;38:194–198. doi: 10.1016/s0735-1097(01)01321-3. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki N., Motohashi N., Uezumi A., Fukada S.-I., Yoshimura T., Itoyama Y., Aoki M., Miyagoe-Suzuki Y., Takeda S.I. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–2476. doi: 10.1172/JCI30654. [DOI] [PMC free article] [PubMed] [Google Scholar]