

Abstract
Calcineurin is an important signal transduction mediator in T cells, neurons, the heart, and kidneys. Recent evidence points to unique actions of the two main isoforms of the catalytic subunit. Although the β isoform is required for T-cell development, α is important in the brain and kidney. In addition, mice lacking α but not β suffer from failure to thrive and early mortality. The purpose of this study was to identify the cause of postnatal death of calcineurin α null (CnAα−/−) mice and to determine the mechanism of α activity that contributes to the phenotype. CnAα−/− mice and wild-type littermate controls were fed a modified diet and then salivary gland function and histology were examined. In vitro studies were performed to identify the mechanism of α action. Data show that calcineurin is required for normal submandibular gland function and secretion of digestive enzymes. Loss of α does not impair nuclear factor of activated T-cell activity or expression but results in impaired protein trafficking downstream of the inositol trisphosphate receptor. These findings show a novel function of calcineurin in digestion and protein trafficking. Significantly, these data also provide a mechanism to rescue to adulthood a valuable animal model of calcineurin inhibitor-mediated neuronal and renal toxicities.
Calcineurin is most familiar as the target of immunosuppression drugs cyclosporine and tacrolimus. However, calcineurin is also a key signal transduction molecule in a variety of cell types. Genetic knockout of the two main, closely related isoforms of the catalytic subunit has led to a number of new observations that have added to our knowledge of calcineurin action in the immune system,1,2 brain,3 muscle,4,5 heart,6 and kidney.7–9 Importantly, calcineurin α null (CnAα−/−) and calcineurin β null (CnAβ−/−) mice have significant phenotypic distinctions, suggesting that the isoforms have unique functions. For example, the majority of CnAα−/− mice die 3 to 4 weeks after birth10 whereas CnAβ−/− mice reach maturity and are fertile.2 CnAβ−/− mice, however, are immunocompromised and graft-tolerant6 whereas Rag−/− mice with CnAα−/−-reconstituted immune systems can still be immunosuppressed with cyclosporine.1 These findings highlight an important distinction between the action of the isoforms and suggest that although CnAβ is the predominant isoform in the immune system, CnAα may be important in nonimmune tissues. Data from our laboratory and other investigators have shown that CnAβ acts through nuclear factor of activated T cell (NFAT)c whereas CnAα does not.9,11 As such, the mechanism of CnAα action still is unknown.
Previously, we reported that loss of CnAα but not CnAβ results in mislocalization of the water channel aquaporin 2 in the kidney collecting duct.8 Cameron et al12 showed that calcineurin can be immunoprecipitated with the inositol-3 phosphate receptor (IP3R) and the ryanodine receptor, whereas Guo et al13 reported that overexpression of constitutively active CnAα in the heart rescued embryonic lethality of calreticulin null mice. Together, these findings led us to develop the hypothesis that CnAα plays a novel role as a downstream target of calcium release from endoplasmic reticulum (ER) calcium channels. Consistent with this model, IP3R type II−/−/III−/− mice were reported to share features with CnAα null pups including failure to thrive (FTT) and early lethality.14 Futatsugi et al14 identified a defect in the salivary gland that led to nutritional deficiencies in the double null pups. The mice could be rescued to adulthood by feeding the pups “predigested” chow. We reasoned that if our model of CnAα action downstream of ER calcium release was correct, a similar strategy also might rescue CnAα−/− pups.
In this study we report the rescue of CnAα−/− mice to adulthood. Our data show that calcineurin is required for normal salivary gland function and early digestion. Moreover, we report a unique role for the α isoform downstream of the IP3R. Loss of this action results in altered vesicle trafficking in vivo and in vitro. Interestingly, this action of calcineurin is independent from NFATc regulation and provides yet another example of a nonimmune function of calcineurin that may be relevant to toxicities associated with clinical inhibition of calcineurin.
Materials and Methods
Materials
Antibodies were obtained as follows: calcineurin Aα, calcineurin Aβ, Rab5B, Rab8A, NFATc1, NFATc2, NFATc3, and NFATc4 were from Santa Cruz Biotechnology (Santa Cruz, CA); and calnexin and GM130 were from BD Transduction Laboratories (San Diego, CA). Cyclosporine, brefeldin A, acetylcholine, and xestospongin were purchased from Sigma-Aldrich (St. Louis, MO).
Animal Models
CnAα−/−
Mice were generated by Johnathan Seidman (Howard Hughes Medical Institute, Harvard Medical School, Boston, MA) as previously described1 and have been maintained in our laboratory. Mice were maintained and bred at the Atlanta Veterans Administration Medical Center and all procedures were performed in accordance with an approved Institutional Animal Care and Use Committee protocol. Genotypes of offspring from heterozygous breeding pairs were determined by polymerase chain reaction using the following primers: 5′-GGCAGGAGAGTAAATTCTTGC-3′, 5′-GTGGAATTCTCTGGAGACAAACCACC-3′, and neo-5′-TCTTGATTCCCACTTTGTGGTTCTA-3′. CnAα−/− mice were created on a mixed genetic background.1 Therefore, all experiments were performed with littermate controls. Likewise, littermate controls also were fed the modified gel diet for all experiments involving rescued CnAα−/− mice.
NFATc-Luciferase Transgenic Mice
Transgenic NFATc-luciferase reporter mice were obtained from Jeffery Molkentin (Cincinnati Children's Hospital, Cincinnati, OH) as described previously6,15 and were crossed with CnAα+/− mice in our laboratory. Dissected salivary glands were homogenized and NFATc-mediated luciferase activity was measured using a commercial kit (Promega, Madison, WI). Briefly, tissue sections were homogenized with 1 μL lysis buffer/mg tissue and particulate matter separated by centrifugation. Luciferase assay reagent (100 μL) was added to 20 μL of supernatant and luminescence was measured for 10 seconds using an OptoComp luminometer (MGM Instruments, Hamden, CT). Results were normalized by subtracting values obtained from identically processed samples from NFATc-luciferase–negative littermate mice.
IP3RII+/−/III+/− Mice
Double heterozygous mice were obtained from Dr. Ju Chen (University of California, San Diego, San Diego, CA). Double heterozygous mice were bred to generate double-null and wild-type littermates. Salivary glands then were obtained for examination of calcineurin activity.
Histology
Tissue Preparation
Salivary glands were harvested and then either fixed in 10% formalin for paraffin embedding or 4 parts paraformalydehyde:1 part glutaraldehyde for plastic embedding. Paraffin tissues were sectioned at 4 μm and then mounted on glass slides for H&E staining or immunohistochemistry. Plastic-embedded tissues were cut ultra thin, mounted on glass slides, and then stained with toluidine blue.
Immunohistochemistry
Paraffin-embedded tissue sections (4-μm thick) were prepared by dewaxing followed by quenching of endogenous peroxidases by incubation in 0.3% H2O2 in methanol for 30 minutes. Antigens were unmasked by incubation in 10 mmol/L citrate buffer at 80°C for 10 minutes. Sections then were rehydrated in PBS–0.1% bovine serum albumin for 15 minutes before addition of appropriate blocking serum for 15 minutes. Sections were incubated with primary antibodies (dilutions, 1:100 to 250) overnight in a humidified chamber at 4°C. The following day, sections were washed 3 times in PBS–0.1% bovine serum albumin and then incubated with biotinylated secondary antibodies (dilutions, 1:100) for 1 hour at room temperature. Bound antibody was identified by immunoperoxidase staining following the manufacturer's instructions (Vector Laboratories, Burlingame, CA). In addition, sections were counterstained with hematoxylin. Finally, coverslips were mounted with Permount (Sigma-Aldrich) and sections were viewed by bright-field microscopy.
Salivary Gland Assays
Saliva was collected using capillary tubes after injection of pilocarpine (1 mg/kg body weight) administered intramuscularly under xylazine and ketamine anesthesia. Serum and salivary amylase and blood urea nitrogen levels were measured using Reflotron test strips in a Reflotron benchtop analyzer (Roche, Indianapolis, IN). Peroxidase and lysozyme activities were measured using commercially available kits from Molecular Probes (Eugene, OR). Sialic acid was measured using a sialic acid quantitation kit (Sigma-Aldrich). Electrolytes (sodium, potassium, chloride, and bicarbonate) were measured using a clinical analyzer (Emory University Hospital, Atlanta, GA). Osmolality was measured using a WESCOR osmometer (Logan, UT). Total protein was measured using a bicinchoninic acid reagent (Bio-Rad, Hercules, CA).
Calcineurin Activity
Calcineurin phosphatase activity was determined as previously described.16 Briefly, the calcineurin substrate peptide RII was synthesized with a phospho-serine at residue 15 and an amino-terminus TAMRA fluorescent tag. In a 96-well plate, the labeled substrate was mixed in equal parts with reaction buffer and sample and allowed to incubate at 30°C for 10 minutes. Each well then was transferred to a 96-well plate coated with titanium-oxide (Glygen, Baltimore, MD), followed by gentle shaking to allow binding of phosphorylated substrate. Finally, supernatants containing unbound peptide then were moved to a new 96-well plate and the amount of dephosphorylated peptide was determined by fluorimetry at 485 nm excitation and 528 nm emission. Calcineurin activity then was determined by extrapolating fluorescence of experimental samples from a standard curve of purified calcineurin (Sigma-Aldrich).
Subcellular Fractionation
Salivary gland tissues or cells were harvested, mechanically disrupted, and then suspended in buffer containing 0.25 mol/L sucrose, 0.02 mol/L HEPES, and 1 mmol/L EDTA. Tissues also were disrupted using a Dounce homogenizer and centrifuged at 1000 × g for 10 minutes to pellet debris. Supernatants were layered on a sucrose gradient (OptiPrep; Sigma-Aldrich) and then centrifuged at 100,000 × g for 60 minutes until subcellular layers separated. Seven or 8 fractions from low density to high density were collected and protein concentrations were determined using the Bradford Method (Sigma-Aldrich). Fractions were characterized by Western blotting for protein markers of the ER, Golgi, trans-Golgi network, and vesicles as indicated. Antibodies were obtained from Santa Cruz Biotechnology or Chemicon (Billerica, MA).
Cell Culture
ParC5 cells were obtained from David Quissell and maintained as previously reported17 and were transfected with empty vector or constitutively active calcineurin Aα18 by electroporation. A total of 10 μg DNA was electroporated in 10 × 106 cells using a Bio-Rad GenePulser electroporater according to the manufacturer's instructions. Cells were re-plated, allowed to recover, and then expanded for subcellular fractionation.
Statistics
All statistical analyses were completed using GraphPad Prism software (La Jolla, CA). A P value of less than 0.05 was considered statistically significant. Unless otherwise stated, all comparisons were a two-tailed Student's t-test.
Results
CnAα−/− pups were placed on a modified diet consisting of powdered standard rodent chow suspended in 1.5% gelatin beginning at approximately 14 days of age. Wild-type littermate mice were treated identically. Figure 1 shows that the modified diet produced an immediate reversal of FTT in the null pups. Once on the diet, CnAα−/− mice showed a period of rapid weight gain and then maintained a normal body weight through 8 weeks of age. Adult mice then were grossly indistinguishable from littermates; male mice were found to be fertile (data not shown). Therefore, the next goals were to determine the reason CnAα−/− mice suffer FTT and the mechanism of CnAα action.
Figure 1.

Rescue of early lethality of CnAα−/− mice. Supplemental feeding of a modified diet (MD) consisting of powdered chow in 1.5% gelatin was initiated at 2 weeks of age as indicated. Data shown are the mean ± SEM of body weight in grams of 12 CnAα−/− littermates on a conventional diet (open squares) and 12 WT (closed circle) and 12 CnAα−/− mice supplied with a modified diet (open circles).
Rescue of weight gain and body mass with supplemental feeding suggested that, similar to IP3R II−/−/III−/− mice, CnAα−/− mice suffer from a nutritional deficiency. First, CnAα−/− mice were observed to have normal swallowing ability (as evidenced by ingestion of milk by newborn pups), adequate mobility to access food, and normal-appearing dentition and tooth formation (data not shown). Next, saliva was obtained from CnAα−/− mice after stimulation with the reversible cholinesterase inhibitor pilocarpine. There was no difference in the amount or rate of saliva produced by wild-type (WT) and null mice (Figure 2, A and B) and there was no difference between male and female mice (data not shown). Saliva then was analyzed. First, salivary osmolality was increased in null mice, however, electrolyte composition and protein content were normal. Salivary urea was increased along with blood urea nitrogen and serum osmolality, consistent with renal insufficiency as previously reported (Table 1).10 Next, amylase, peroxidase, and lysozyme activities were measured and found to be decreased (changes in lysozyme activity did not reach significance). Finally, activity of the main mucosal enzyme sialic acid was significantly lower in null mice (Figure 2, C–F).
Figure 2.
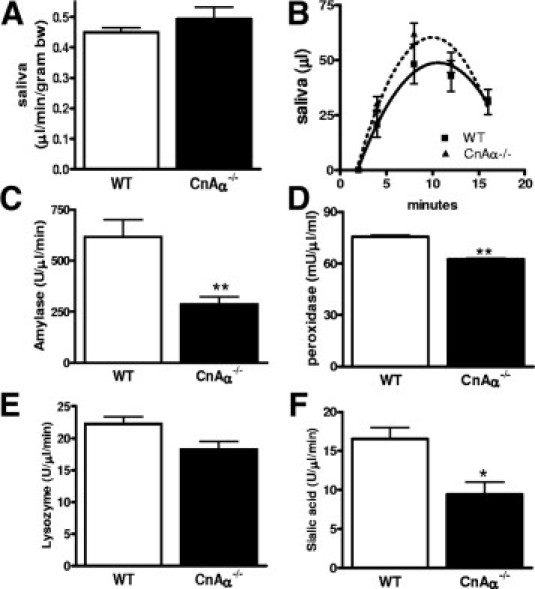
Loss of calcineurin alters salivary gland function. A and B: Volume and rate of saliva collected over a 20-minute time period from WT (N = 6) and CnAα−/− mice (N = 6) after injection with pilocarpine. C–F: Amylase, peroxidase, lysozyme, and sialic acid measured in saliva obtained from WT (N = 6) and CnAα−/− (N = 7) littermates maintained on the modified diet. *P < 0.05; **P < 0.01.
Table 1.
Characterization of Salivary Composition
| Wild-type | CnAα−/− | |
|---|---|---|
| Osmolality (mg/dL) | 194 ± 10 | 227 ± 6⁎ |
| Sodium level (mg/mL) | 73.7 ± 6.8 | 72.3 ± 4.1 |
| Potassium level (mEq/L) | 29.7 ± 0.8 | 29.8 ± 0.8 |
| Chloride level (mEq/L) | 95.3 ± 3.2 | 92.1 ± 3.0 |
| Bicarbonate level (mEq/L) | 25.3 ± 2.5 | 25.8 ± 1.5 |
| Total protein level (mg/mL) | 0.58 ± 0.06 | 0.42 ± 0.05 |
| Salivary BUN level (mg/dL) | 13.6 ± 0.8 | 26.3 ± 4.8⁎ |
| Serum amylase level (U/L) | 6.8 ± 0.4 | 6.5 ± 0.2 |
| Serum osmolality (mg/dL) | 312 ± 1 | 335 ± 3⁎ |
| Serum BUN level (mg/dL) | 27.3 ± 1.1 | 77.7 ± 7.5† |
BUN, blood urea nitrogen.
P < 0.05 compared with wild-type;
P < 0.001 compared with WT.
Examination of pancreas from CnAα−/− mice revealed a normal appearance including islet formation, number, and size, and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining revealed no increase in cell death (data not shown). Serum amylase and insulin levels also were unchanged in null mice and fecal trypsin-like enzyme activity was normal (data not shown). Together, these data suggest that changes in the salivary gland including vesicle formation and enzyme secretion are the most likely explanation for the nutritional deficiency and failure to thrive of CnAα−/− mice.
As a target of immunosuppressant drugs including cyclosporine, the finding that loss of calcineurin can alter normal salivary gland function may have important clinical implications. For example, cyclosporine has long been noted to cause anorexia in animal models and in human beings, although no mechanism has been described previously. Figure 3A shows that rats treated with cyclosporine for 2 weeks gained significantly less weight than vehicle-treated rats. We therefore examined salivary gland function in WT mice treated with cyclosporine or vehicle (10% ethanol) and found that, similar to loss of CnAα, cyclosporine did not alter pilocarpine-stimulated saliva production (Figure 3B). However, cyclosporine treatment significantly reduced amylase and lysozyme activities compared with vehicle treatment. In addition, sialic acid levels were significantly lower with cyclosporine treatment (Figure 3, C–F).
Figure 3.
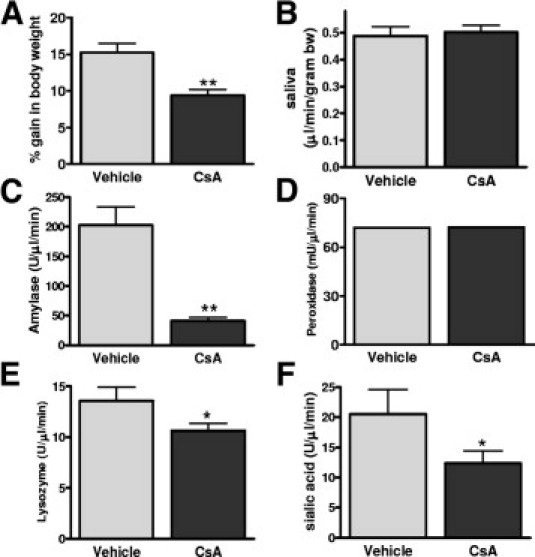
Cyclosporine treatment alters salivary gland function. A: Weight gain of rats treated with vehicle alone (10% ethanol) (N = 6) or cyclosporine (CsA) (10 mg/kg) (N = 6) for 2 weeks. **P < 0.01. B: Volume of saliva produced by pilocarpine injected–mice after 4 days of cyclosporine (CsA) (20 mg/kg) (N = 6) or vehicle treatment (N = 6). C–F: Amylase, peroxidase, lysozyme, and sialic acid measured from vehicle-treated (N = 6) and cyclosporine-treated (N = 6) WT saliva samples. *P < 0.05; **P < 0.01.
Next, salivary glands were obtained from WT and CnAα−/− littermates, stained with H&E, and examined by light microscopy. Figure 4 shows that although sublingual and parotid glands lacked obvious histologic defects (Figure 4, A and D vs B and E), submandibular glands from CnAα−/− mice were altered significantly (Figure 4, C vs F). Plastic-embedded, toluidine blue–stained sections further illustrated a defect decrease in secretory vesicle (Figure 4, G and H). Quantitation of these changes revealed significant decreases in vesicle number, mucosal acini cell size, and protein content of serosal acini (Figure 4, I and J). NFAT transcription factors are well-characterized substrates of calcineurin. To determine whether loss of CnAα altered NFAT, expression of all four calcium-responsive isoforms (NFATc1–c4) was examined by immunohistochemistry. Figure 5 shows that NFATc1 and NFATc4 are the predominant isoforms expressed in the submandibular gland (Figure 5, A and G). Although mucosal acini size is altered, expression of the two isoforms is maintained in CnAα−/− tissues (Figure 5, B and H). Likewise, activity of the ubiquitously expressed NFATc-driven luciferase reporter construct4,5 was unchanged in null salivary gland tissue (Figure 5I). In contrast, treatment of WT mice with cyclosporine for 3 days resulted in a decrease in NFAT activity (Figure 5J). These results suggest that the primary role of CnAα in the salivary gland is NFATc-independent.
Figure 4.

CnAα is required for vesicle formation in the salivary gland. Representative histology of H&E-stained sublingual (A and D), parotid (B and E), and submandibular glands (C and F) from WT (top row) and CnAα−/− (bottom row) mice. Original magnification, ×200. Representative submandibular gland sections from WT (G) and CnAα−/− (H) mice that have been thin-sectioned, paraffin-embedded, and stained with toluidine blue. Original magnification, ×600. I–J: Quantitation of vesicle number and cell size in mucosal cells, and estimation of protein content in serosal acini from 4 to 6 WT and CnAα−/− mice (**P < 0.01).
Figure 5.

Expression of NFAT isoforms and activity is unchanged in CnAα−/− salivary glands. NFATc isoform expression in WT (left) and CnAα−/− (right) submandibular tissue. NFATc1 (A and B), NFATc2 (C and D), NFATc3 (E and F), and NFATc4 (G and H) was determined by immunohistochemistry using specific antibodies. Data shown are representative of at least 2 independent experiments using four to six mice per group. Original magnification, ×200. I: Luciferase production in salivary glands from three WT and three CnAα−/− mice expressing a transgenic NFATc-responsive luciferase promoter construct.4J: Luciferase production in salivary glands from three WT mice treated with vehicle alone (10% ethanol) or cyclosporine (20 mg/kg) daily for 3 days. *P < 0.05.
To determine the mechanism of CnAα action, salivary glands were obtained from null and WT littermate mice for in vitro analyses. First, total calcineurin activity was examined in lysates from WT and CnAα−/− salivary glands. Activity was reduced by approximately one third in CnAα−/− samples, suggesting that CnAβ contributes the majority of total calcineurin activity in salivary glands (Figure 6A). Next, seven high-density to low-density fractions were collected from salivary glands pooled from three mice of each genotype. Expression and activity of calcineurin isoforms was examined. Figure 6B shows that the α isoform is expressed in the highest density fractions 1 to 3. In contrast, expression of the β isoform is highest in fractions 5 and 6. In CnAα−/− tissue, expression of CnAβ was shifted toward fraction 7 whereas CnAα was undetectable, as expected. Calcineurin activity also was examined in the same subcellular fractions; activity in fractions 1 and 2 was reduced significantly in CnAα−/− tissue compared with WT. Consistent with changes in β expression, calcineurin activity was increased in fraction 7 in null tissues compared with WT (Figure 6C). Finally, to determine the intracellular compartment where α is expressed, subcellular fractions were characterized using makers of the ER, Golgi apparatus, trans-Golgi network, and intracellular vesicles (Figure 6D). In both wild-type and null samples, factions 1 to 3 showed significant expression of calnexin supporting the ER as the site of α localization. Interestingly, in null samples, distribution of proteins identifying post-ER structures is altered compared with wild-type samples. Specifically, peak GM130 expression is in fraction 2 rather than fraction 4 and Rab8 is decreased in fractions 1 and 2. More striking, Rab5 expression is observed in fractions 1 to 3 in null tissues but absent in these fractions in the wild-type samples.
Figure 6.

Alterations in vesicle trafficking with loss of calcineurin. A: Calcineurin activity determined in salivary gland lysates from three wild-type and three CnAα−/− mice. Data shown are the mean ± SEM. *P < 0.05. B: Expression of CnAα and CnAβ in salivary gland lysates from wild-type and CnAα−/− mice detected by Western blotting. Actin also was detected as a loading control. C: Calcineurin activity in the same fractions shown in panel A. *P < 0.05; **P < 0.01. Two-way analysis of variance. D: Subcellular fractions prepared from WT and CnAα−/− salivary gland tissues pooled from at least two mice per genotype. Subcellular compartments were characterized by Western blotting with markers of the ER, Golgi apparatus, trans-Golgi network, and vesicles as indicated. Asterisks indicate changes when compared with WT.
To determine whether calcineurin activity is required for normal vesicle trafficking, cultured fibroblasts were treated with cyclosporine or vehicle alone for 4 hours and then Rab5 localization was determined in fractionated lysates. Figure 7A shows that inhibition of calcineurin is sufficient to prompt redistribution of Rab5 expression to higher-density vesicles that also express calnexin. This was examined further in Par5C salivary gland cells. Immunofluorescence for Rab5 shows that vesicle localization is altered with cyclosporine A treatment (Figure 7, B vs C). Moreover, the pattern of vesicle distribution is comparable with ER localization induced by brefeldin A treatment (Figure 7D) and distinct from Golgi localization induced by room temperature incubation (Figure 7E). Finally, Rab5 distribution was examined in CnAα−/− and CnAβ−/− fibroblasts. Figure 7F shows that Rab5 localization is decreased in the highest-density fractions and shifted toward fractions that also express calnexin. In contrast, Rab5 expression in CnAβ−/− fractions remains highest in fraction 8. Inhibition of remaining α activity with cyclosporine A produced a shift of Rab5 expression comparable with that of CnAα−/− cells. Taken together, these data indicate that α action is required for normal post-ER processing of intracellular vesicles.
Figure 7.

CnAα is required for vesicle trafficking. A: Rab5 expression in subcellular fractions from cultured fibroblasts treated with vehicle alone or cyclosporine for 4 hours. Calnexin also was detected for comparison. B–E: Rab5 localization in ParC5 cells determined by immunofluorescence after treatment with vehicle alone (B), cyclosporine A (C), brefeldin A (D), or incubation at room temperature (E). Nuclei were identified with DAPI staining for comparison. F: Calnexin (CNX) and Rab5 expression in subcellular fractions generated from WT, CnAα−/−, and CnAβ−/− cells. CnAβ−/− cells also were treated with vehicle or cyclosporine A for 4 hours. Asterisks indicate changes when compared with wild-type or vehicle control.
There is evidence to suggest that CnAα may act in a pathway downstream of calreticulin and ER calcium channels. Loss of this function could produce altered processing and/or trafficking of salivary gland vesicles. Therefore, in the final set of experiments, we investigated a functional role for CnAα downstream of the IP3R. First, in vitro suspensions of salivary gland tissue from WT and CnAα−/− mice were pretreated with cyclosporine or xestospongin to block the IP3R channel and then stimulated with acetylcholine. Inhibition of calcineurin blocked acetylcholine-induced amylase secretion comparably with xestospongin (Figure 8A). Likewise, xestospongin significantly reduced calcineurin activity in salivary gland suspensions (Figure 8B). Similarly, Figure 8C shows that calcineurin activity is reduced significantly in IP3R II+/−/III+/− and IP3R II−/−/III−/− salivary gland tissue. Finally, Par5C salivary acinar cells were transfected with either empty vector or constitutively active CnAα,18 and then the effect of inhibiting the IP3R was examined on vesicle trafficking. In vector-transfected cells, xestospongin resulted in redistribution of Rab5 in a pattern similar to cyclosporine treatment in Figure 7A. Overexpression of CnAα, however, blocked this effect (Figure 8D). Taken together, these data show a requirement for CnAα downstream of the IP3R.
Figure 8.
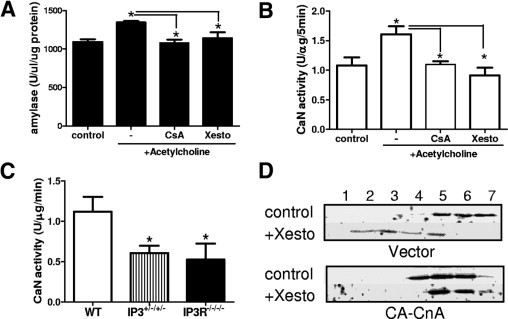
CnAα is activated downstream of the IP3R. A: Amylase secretion from WT salivary gland suspensions pretreated for 30 minutes with cyclosporine (100 μmol/L) or xestospongin followed by acetylcholine (100 μmol/L) for 15 minutes. *P < 0.05, analysis of variance. Data are representative of at least three independent experiments. B: Calcineurin activity in the salivary gland suspensions described in panel A. *P < 0.05, analysis of variance. C: Calcineurin activity in salivary gland tissue obtained from wild-type (N = 4), IP3R II+/−/III+/− (N = 4), or IP3R II/III double-null mice (N = 4). *P < 0.05, analysis of variance. D: Par5C acinar cells were first transfected with either empty vector or constitutively active CnAα (CA-CnAα) by electroporation. Cells were allowed to recover for 48 hours and then were treated with vehicle control or xestospongin (10 μmol/L, 1 hour). Subcellular fractions were obtained and Rab5 expression was determined by Western blotting. Data are representative of at least three independent experiments.
Discussion
In this study, we report the rescue of an important animal model, provide evidence that CnAα is dispensable for regulation of NFAT expression and activity, and show a requirement role for the α isoform downstream of the IP3R in the salivary gland. CnAα−/− mice were first reported by Seidman's group in 1996. Early studies focused on the neuronal changes3,19 and the effects of CnAα−/− immune reconstitution in Rag−/− mice.1 Changes in the brain were consistent with the predicted role of calcineurin as a tau phosphatase.19 Build-up of hyperphosphorylated tau has been linked to the formation of neurofibrillary plaques such as those observed in the brains of individuals with Alzheimer's disease.20–22 Similar lesions were found in CnAα−/− mice along with memory impairment and changes in the hippocampus.19 In addition, the investigators noted that CnAα−/− mice had stunted growth, were infertile, and died early of unknown causes. Since that time, dozens of publications have used CnAα−/− mice but none have addressed the cause of FTT and early lethality. Our report identifies a cause of both and, more importantly, shows that the mice can be rescued to adulthood. This breakthrough is particularly significant given our previous work showing that CnAα−/− kidneys recapitulate many aspects of cyclosporine nephrotoxicity. An adult model will enable us to further explore the mechanisms of one of the most common and serious side effects of calcineurin inhibitor therapy in transplant patients.
The lack of a significant immune defect in CnAα−/− mice and the finding that Rag−/− mice with CnAα−/− immune systems could still be immunosuppressed with cyclosporine1 strongly suggested that an isoform other than α predominated in T cells. This was confirmed in 2002 when Molkentin's group published the phenotype of CnAβ−/− mice. Bueno et al2 reported that CnAβ−/− mice had significant T-cell defects and were immune-compromised. Calcineurin action in the T cell is well characterized to involve the NFAT transcription factors.23,24 Previously, we reported that cells lacking the α isoform retained nuclear translocation of NFATc in response to calcium.9 In contrast, NFATc remained in the cytoplasm in β−/− cells. This experiment suggested that, similar to T-cell development, β is required for regulation of NFATc whereas α is dispensable. Data in the present study further expand this observation. In salivary glands, we find that NFATc activity is unchanged in CnAα−/− tissues and that NFATc1 and NFATc4, the predominant NFATc isoforms expressed in WT tissues, are still expressed in nulls. Taken together, these data support a model of CnAβ/NFAT signaling that can be distinct from CnAα.
Further supporting a separation of α and β function is our finding that the two isoforms are expressed in different subcellular fractions. Studying isoform-specific differences and actions is challenging given the close amino acid identity, interaction with common intracellular binding partners, and ubiquitous co-expression of the two isoforms. Current assay methods do not distinguish between α and β activity. The finding that α and β segregate to distinct subcellular compartments and, moreover, that isoform-specific activity within those fractions can be observed, is the first potential mechanism to study α- and β-specific actions in vivo. Importantly, localization of the α but not β isoform to fractions containing the ER is consistent with a functional role downstream of the IP3R that is specific to the α isoform.
The present study grew from the hypothesis that calcineurin may be a key signaling mediator downstream of the IP3R. This is based in part on the work of Guo et al,13 who showed that overexpression of constitutively activated calcineurin in the heart rescued embryonic lethality of mice lacking the ER chaperone protein calreticulin. These data suggested that calcineurin was a downstream mediator of calreticulin effects. However, calreticulin is an ER luminal protein whereas calcineurin, by all indications, resides in the cytoplasm. The link between calreticulin and calcineurin therefore could be the result of one of two important functions of calreticulin: ER calcium levels or protein folding. The IP3R was a logical choice of a mediator between the functions of calreticulin and activation of calcineurin. In 2005, Futatsugi et al14 reported that IP3R type II/III double null mice suffered from FTT and died early of nutritional deficiencies stemming from defects in the salivary gland and pancreas. The mice could be rescued by providing pups with what they termed pre-digested chow. We reasoned that if, similar to calreticulin, calcineurin was downstream of the IP3R, then CnAα−/− mice also might share the phenotype of nutritional deficiency. This was shown to be the case in our current study when a modified diet intervention also rescued CnAα−/− pups. Moreover, we show that CnAα−/− mice also have a salivary gland defect and, similar to IP3R II/III double null pups, likely suffered from a nutritional deficiency that led to their early death. Finally, data in this study indicate that one function of CnAα is to mediate trafficking of proteins.
This study showed a novel requirement for calcineurin in the salivary gland and identified a requirement for α isoform in IP3R-mediated protein trafficking. Both of these findings are likely to be relevant to clinical use of calcineurin inhibitors. First, the identification of the salivary gland as a tissue that requires calcineurin activity shows yet another nonimmune function of the enzyme. Moreover, the finding that the major features described in this study can be reproduced with cyclosporine argues for the development of α isoform–sparing calcineurin inhibitors. Interestingly, the role of CnAα in the salivary gland may shed light on anorexia and gingival hyperplasia, two common side effects of calcineurin inhibitor therapies. First, as we show in a rodent model, cyclosporine is associated with reduced weight gain. This is noted in many animal and human studies as anorexia, although no mechanism has been described previously. It will be interesting to carry these findings into patient populations and determine whether there are changes in salivary gland enzyme levels with calcineurin inhibitor therapies. Likewise, gingival hyperplasia is noted commonly in both animal models and human beings.25–27 Decreased lysozyme activity in the oral cavity may contribute to changes in antibacterial function leading to gingival inflammation and hyperplasia.
In conclusion, we report that FTT and early lethality of CnAα null mice can be rescued by feeding pups modified gelatin chow. Experimental data show that this function of CnAα is independent of the NFAT pathway, which is intact in CnAα−/− salivary glands. Rather, CnAα is required downstream of the IP3R and contributes to normal protein processing and trafficking of proteins including amylase. The decrease of digestive enzymes in the saliva of young mice, in turn, likely resulted in nutritional deficiency and early mortality. Finally, these findings show yet another nonimmune function of calcineurin and provide additional rationale for the development of α-sparing calcineurin inhibitor regimens.
Acknowledgments
We thank Jeffery Molkentin for CnAβ+/− mice and NFATc-luciferase mice, Johnathan Seidman for CnAα+/− mice, David Quissell for the ParC5 cells, and Janet Klein for discussion and review of the manuscript.
Footnotes
Funding for this work was provided by the Department of Veterans Affairs MERIT and PECASE awards (J.L.G.).
References
- 1.Zhang B.W., Zimmer G., Chen J., Ladd D., Li E., Alt F.W., Wiederrecht G., Cryan J., O'Neill E.A., Seidman C.E., Abbas A.K., Seidman J.G. T cell responses in calcineurin A alpha-deficient mice. J Exp Med. 1996;183:413–420. doi: 10.1084/jem.183.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bueno O.F., Brandt E.B., Rothenberg M.E., Molkentin J.D. Defective T cell development and function in calcineurin Abeta-deficient mice. Proc Natl Acad Sci U S A. 2002;99:9398–9403. doi: 10.1073/pnas.152665399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuo M., Zhang W., Son H., Mansuy I., Sobel R., Seidman J., Kandel E. A selective role of calcineurin Aalpha in synaptic depotentiation in hippocampus. Proc Natl Acad Sci U S A. 1999;96:4650–4655. doi: 10.1073/pnas.96.8.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons S.A., Wilkins B.J., Bueno O.F., Molkentin J.D. Altered skeletal muscle phenotypes in calcineurin Aalpha and Abeta gene-targeted mice. Mol Cell Biol. 2003;23:4331–4343. doi: 10.1128/MCB.23.12.4331-4343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parsons S.A., Millay D.P., Wilkins B.J., Bueno O.F., Tsika G.L., Neilson J.R., Liberatore C.M., Yutzey K.E., Crabtree G.R., Tsika R.W., Molkentin J.D. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber type switching but not hypertrophy. J Biol Chem. 2004;279:26192–26200. doi: 10.1074/jbc.M313800200. [DOI] [PubMed] [Google Scholar]
- 6.Bueno O.F., Wilkins B.J., Tymitz K.M., Glascock B.J., Kimball T.F., Lorenz J.N., Molkentin J.D. Impaired cardiac hypertrophic response in calcineurin Abeta-deficient mice. Proc Natl Acad Sci U S A. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy R.N., Knotts T.L., Roberts B.R., Molkentin J.D., Price S.R., Gooch J.L. Calcineurin A-beta is required for hypertrophy but not matrix expansion in the diabetic kidney. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gooch J.L., Guler R.L., Barnes J.L., Toro J.J. Loss of calcineurin Aalpha results in altered trafficking of AQP2 and in nephrogenic diabetes insipidus. J Cell Sci. 2006;119:2468–2476. doi: 10.1242/jcs.02971. [DOI] [PubMed] [Google Scholar]
- 9.Gooch J.L., Roberts B.R., Cobbs S.L., Tumlin J.A. Loss of the alpha-isoform of calcineurin is sufficient to induce nephrotoxicity and altered expression of transforming growth factor-beta. Transplantation. 2007;83:439–447. doi: 10.1097/01.tp.0000251423.78124.51. [DOI] [PubMed] [Google Scholar]
- 10.Gooch J.L., Toro J.J., Guler R.L., Barnes J.L. Calcineurin A-alpha but not A-beta is required for normal kidney development and function. Am J Pathol. 2004;165:1755–1765. doi: 10.1016/s0002-9440(10)63430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilka S., Erdmann F., Migdoll A., Fischer G., Weiwad M. The proline-rich N-terminal sequence of calcineurin Abeta determines substrate binding. Biochemistry. 2009;48:1900–1910. doi: 10.1021/bi8019355. [DOI] [PubMed] [Google Scholar]
- 12.Cameron A.M., Steiner J.P., Roskams A.J., Ali S.M., Ronnett G.V., Snyder S.H. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 13.Guo L., Nakamura K., Lynch J., Opas M., Olson E.N., Agellon L.B., Michalak M. Cardiac-specific expression of calcineurin reverses embryonic lethality in calreticulin-deficient mouse. J Biol Chem. 2002;277:50776–50779. doi: 10.1074/jbc.M209900200. [DOI] [PubMed] [Google Scholar]
- 14.Futatsugi A., Nakamura T., Yamada M.K., Ebisui E., Nakamura K., Uchida K., Kitaguchi T., Takahashi-Iwanaga H., Noda T., Aruga J., Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science. 2005;309:2232–2234. doi: 10.1126/science.1114110. [DOI] [PubMed] [Google Scholar]
- 15.Wilkins B.J., Dai Y.S., Bueno O.F., Parsons S.A., Xu J., Plank D.M., Jones F., Kimball T.R., Molkentin J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 16.Roberts B., Pohl J., Gooch J.L. A fluorimetric method for determination of calcineurin activity. Cell Calcium. 2008;43:515–519. doi: 10.1016/j.ceca.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quissell D.O., Barzen K.A., Redman R.S., Camden J.M., Turner J.T. Development and characterization of SV40 immortalized rat parotid acinar cell lines. In Vitro Cell Dev Biol Anim. 1998;34:58–67. doi: 10.1007/s11626-998-0054-5. [DOI] [PubMed] [Google Scholar]
- 18.Cobbs S.L., Gooch J.L. NFATc is required for TGFbeta-mediated transcriptional regulation of fibronectin. Biochem Biophys Res Commun. 2007;362:288–294. doi: 10.1016/j.bbrc.2007.07.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kayyali U., Zhang W., Yee A., Seidman J., Potter H. Cytoskeletal changes in the brains of mice lacking calcineurin A alpha. J Neurochem. 1997;68:1668–1678. doi: 10.1046/j.1471-4159.1997.68041668.x. [DOI] [PubMed] [Google Scholar]
- 20.Ermak G., Morgan T.E., Davies K.J. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem. 2001;276:38787–38794. doi: 10.1074/jbc.M102829200. [DOI] [PubMed] [Google Scholar]
- 21.Harris C.D., Ermak G., Davies K.J. Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product calcipressin 1 (or RCAN1) in disease. Cell Mol Life Sci. 2005;62:2477–2486. doi: 10.1007/s00018-005-5085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ladner C., Czech J., Maurice J., Lorens S., Lee J. Reduction of calcineurin enzymatic activity in Alzheimer's disease: correlation with neuropathologic changes. J Neuropathol Exp Neurol. 1996;55:924–931. doi: 10.1097/00005072-199608000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Rao A., Luo C., Hogan P. Transcription factors of the NFAT family: regulation and function. Ann Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 24.Masuda E., Imamura R., Amasaki Y., Arai K., Arai N. Signalling into the T-cell nucleus: NFAT regulation. Cell Signal. 1998;10:599–611. doi: 10.1016/s0898-6568(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 25.Wright G., Welbury R.R., Hosey M.T. Cyclosporin-induced gingival overgrowth in children. Int J Paediatr Dent. 2005;15:403–411. doi: 10.1111/j.1365-263X.2005.00676.x. [DOI] [PubMed] [Google Scholar]
- 26.Faulds D., Goa K.L., Benfield P. Cyclosporin: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs. 1993;45:953–1040. doi: 10.2165/00003495-199345060-00007. [DOI] [PubMed] [Google Scholar]
- 27.Aalamian Z. Reducing adverse effects of immunosuppressive agents in kidney transplant recipients. Prog Transplant 11:271–282. 2001:283–284. doi: 10.1177/152692480101100409. [DOI] [PubMed] [Google Scholar]