

Abstract
The Raf/MEK/extracellular signal-regulated kinase (ERK) pathway participates in many processes altered in development and progression of cancer in human beings such as proliferation, transformation, differentiation, and apoptosis. Kinase suppressor of Ras 1 (KSR1) can interact with various kinases of the Raf/MEK/extracellular signal-regulated kinase pathway to enhance its activation. The role of KSR1 in endometrial carcinogenesis was investigated. cDNA and tissue microarrays demonstrated that expression of KSR1 was up-regulated in endometrial carcinoma. Furthermore, inhibition of KSR1 expression by specific small hairpin RNA resulted in reduction of both proliferation and anchorage-independent cell growth properties of endometrial cancer cells. Because inhibition of apoptosis has a pivotal role in endometrial carcinogenesis, the effects of KSR1 in regulation of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)–induced apoptosis were investigated. KSR1 knock-down sensitized resistant endometrial cell lines to both TRAIL- and Fas-induced apoptosis. Sensitization to TRAIL and agonistic anti-Fas antibody was caused by down-regulation of FLIP (FLICE-inhibitory protein). Also investigated was the molecular mechanism by which KSR1 regulates FLIP protein levels. It was demonstrated that KSR1 small hairpin RNA did not affect FLIP transcription or degradation. Rather, FLIP down-regulation was caused by Fas-associated death domain protein–dependent inhibition of FLIP translation triggered after TRAIL stimulation in KSR1-silenced cells. Re-expression of heterologous KSR1 in cells with down-regulated endogenous KSR1 restored FLIP protein levels and TRAIL resistance. In conclusion, KSR1 regulates endometrial sensitivity to TRAIL by regulating FLIP levels.
The Raf/MEK/extracellular signal-regulated kinase (ERK) pathway participates in many processes altered in development and progression of cancer in human beings such as proliferation, transformation, differentiation, and apoptosis.1 Constitutive or enhanced activation of ERK signaling is a common downstream event in many types of malignant diseases.
The kinase suppressor of Ras 1 (KSR1) is considered a scaffold protein that interacts and regulates the intensity and duration of mitogen-activated protein kinase (MAPK) pathway activation.2 KSR1 was first identified as a suppressor of Ras-induced tumor phenotype in Caenorhabditis elegans and Drosophila melanogaster genetic screens.3–5 KSR1 can interact with various kinases of the Raf/MEK/ERK signaling pathway to enhance its activation.6–8 Strong evidence of KSR1 scaffolding function was provided in experiments in which overexpression of KSR1 in deficient mouse embryonic fibroblasts resulted in maximal assembly of the Raf-1/MEK/ERK/MAPK complex and optimal ERK/MAPK signaling.9 KSR1 contains a kinase domain in which the lysine typical of the ATP binding site of other serine/threonine kinases is substituted by an arginine. Whether KSR1 contains intrinsic kinase activity is controversial.10
KSR1 is critical for Ras-induced transformation by active forms of Ras both in vitro9,11,12 and in vivo.13 Thus, KSR1 is a suitable target to block Ras-induced tumorigenesis.14
Ras/Raf/MEK/ERK signaling is involved in regulation of apoptotic cell death triggered by antineoplastic drugs.15 KSR1 regulation of Ras/Raf/MEK/ERK has also been involved in modulation of apoptotic response to death receptors. Colonic epithelial cells from KSR1-deficient mice are more sensitive to tumor necrosis factor-α than are wild-type cells,16,17 which suggests that KSR1 may participate in control of apoptosis triggered by death receptors.
It has been previously demonstrated that endometrial cancer cells (ECCs) exhibit resistance to tumor necrosis factor family members such as Fas ligand and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL).18,19 TRAIL is capable of inducing apoptosis in tumor cells while sparing normal cells.20,21 Thus, TRAIL is a promising therapeutic agent against cancer, and is currently under clinical trials.22,23 However, there is growing evidence that some tumor populations are resistant to apoptosis triggered by TRAIL, which makes the intracellular signaling pathways that modulate apoptotic responses to TRAIL an important unresolved issue.
Binding of TRAIL to its functional receptors TRAILR1(DR4) and TRAILR2(DR5) initiate an intracellular response.24–26 Both TRAIL and Fas ligand have been reported to trigger similar intracellular signaling pathways, and their engagement to TRAIL and Fas receptors leads to formation of a death-inducing signaling complex that is composed primarily by the TRAIL/Fas receptor, the adaptor protein FADD (Fas-associated death domain), and the inactive proform of caspase 8. On formation of the death-inducing signaling complex, procaspase 8 is processed into its active form, caspase 8, through autoproteolytic cleavage, which initiates intracellular apoptotic signaling.27–29 One of the critical regulators of apoptosis triggered by Fas ligand and TRAIL is the FLICE (FADD-like interleukin-1β–converting enzyme)–inhibitory protein (FLIP).30,31 It has been previously reported that high levels of FLIP found in ECCs have a pivotal role in mediating resistance to TRAIL-induced apoptosis.18 Intracellular FLIP protein levels should be tightly regulated. Such regulation can be achieved by regulating FLIP transcription, protein translation, or its degradation via the ubiquitin-proteasome system.
Expression of KSR1 is increased in endometrial carcinoma, which suggests a possible role in endometrial carcinogenesis. Inhibition of KSR1 expression by lentiviral-delivered small hairpin RNA (shRNA) in ECCs resulted in marked reduction of both proliferation and anchorage-independent cell growth properties of ECCs. Because suppression of apoptosis is an important step in endometrial carcinogenesis, the effects of KSR1 inhibition on apoptotic response of ECCs was studied. Inhibition of KSR1 expression sensitized resistant ECC lines to both TRAIL- and Fas-induced apoptosis via a mechanism dependent on down-regulation of FLIP. Also assessed were the molecular mechanisms underlying such FLIP down-regulation. KSR1 shRNA blocks FLIP translation through regulation of TRAIL-induced phosphorylation of the eukaryotic initiation factor 4E (eIF4E), which suggests inhibition of FLIP protein translation.
Materials and Methods
Reagents, Antibodies, and Plasmids
MTT (3-[4,5-dimethylthiazol-2-yl]−2,5-diphenyl tetrazolium bromide) assay, cycloheximide, and monoclonal antibody to tubulin and Anti-FLAG M2 were obtained from Sigma-Aldrich Corp. (St Louis, MO). Caspase inhibitor BAF, proteasome inhibitors MG-132 and ALLN, antibody to caspase 8, and recombinant TRAIL were from Calbiochem (La Jolla, CA). Antibodies to caspase 9 and cleaved caspase 3 were obtained from Cell Signaling Technology, Inc. (Beverly, MA). Bortezomib (Millennium Pharmaceuticals, Cambridge, MA) was obtained from the Department of Pharmacy (Hospital Arnau de Vilanova, Lleida, Spain). Monoclonal antibodies to FLIP (NF6) and Fas agonistic antibody (CH11) were purchased from Alexis Corp. (Lausen, Switzerland). Antibodies to ITCH and FADD were from BD Biosciences (Franklin Lakes, NJ). Antibodies to KSR1 (sc-25416 for immunohistochemistry and sc-9317 for Western blot analysis) and ubiquitin were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phospho-eIF4E serine 209 was from Millipore Corp. (Bedford, MA). Peroxidase-conjugated anti-mouse, anti-rat, and anti-rabbit antibodies were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Lentiviral vector containing FLAG-tagged mouse FLIP cDNA was a gift from Dr. Joan Comella (Universitat Autònoma de Barcelona, Barcelona, Spain). FLAG-tagged pcDNA3-KSR1 has been previously described.11
Collection of Tissue Samples for cDNA Array Studies
For cDNA studies, two independent series of samples were used. The first series (series 1) was used for cDNA hybridization. Series 1 contained 24 ECCs that had been previously characterized pathologically and molecularly,32 and eight normal endometrial samples including four each in the late proliferative phase (histologic dating, 10 to 14 days) and early secretory phase (histologic dating, 15 to 19 days). In brief, the series included 10 grade 1, five grade 2, and nine grade 3 ECCs. Eighteen tumors were classified as stage I, and the remainder as stages II to IV. Normal cycling endometria were obtained from patients ranging in age from 26 to 45 years who were undergoing hysterectomy for treatment of small intramural leiomyomas. Samples were obtained from the mid-fundus. All women had a clinical history of normal cycles [mean (SD), 28 (2) days], and none had received hormone therapy in the previous year. Endometria were dated according to morphologic criteria, and in all patients the structure of the endometrium was normal and correlated well with the days of the clinical cycle. Sections of frozen samples were split for RNA isolation and confirmatory histology via H&E staining. The study was approved by the ethics committee, and specific informed consent was obtained from all patients.
The second series (series 2) was used to cross-validate data obtained from cDNA arrays. In series 2, mRNA data were obtained from eight normal endometria (four each in the proliferative and secretory phases) and 26 ECCs comprising 17 grade 1, six grade 2, and three grade 3 tumors. Twenty-four tumors were classified as stage I, two as stage II, and one as stage III. The use of two independent series in cDNA studies provided an independent validation of cDNA array data.
RNA Extraction, Probe Synthesis, Hybridization on cDNA Arrays, and Data Analysis
Procedures were performed as previously reported.32 In brief, total RNA was isolated using TRIZOL reagent (Life Technologies, Inc., Gaithersburg, MD). Three milligrams of total RNA and T7-(deoxythymidine)-24 oligo primer were used to synthesize the double-strand cDNA using the Superscript Choice System (Life Technologies, Inc.). In vitro transcription was performed using Megascript T7 (Ambion, Inc., Austin, TX). Three microliters of antisense RNA was used to generate fluorescence cDNA by transcriptional synthesis using the SuperScript enzyme protocol (Life Technologies, Inc.). All endometrial samples were labeled with Cy5-deoxyuridine triphosphate fluorochrome (Amersham Biosciences AB, Uppsala, Sweden), and the reference sample (Universal Human Reference RNA; Stratagene Corp., La Jolla, CA) was labeled with Cy3-deoxyuridine triphosphate fluorochrome (Amersham Biosciences AB) as previously described.32 The cDNA array chips used contained 9726 clones corresponding to 6386 different genes. The chips included 2489 clones that were printed in duplicate to assess reproducibility [Bioinformatics Unit, Centro Nacional de Investigaciones Oncológicas (CNIO), Madrid, Spain].
After hybridization, slides were scanned (ScanArray 5000 XL scanner; GSI Lumonics, Inc., Kanata, ON, Canada) at wavelengths of 635 and 532 nm for Cy5 and Cy3 dyes, respectively, to obtain 10-μm resolution images, which were quantified using the GenePix Pro 6.0 program (Axon Instruments, Inc., Union City, CA). Fluorescence intensity measurements from each array element were compared with the median of the local background in each channel. Elements with values less than this median were excluded. In addition, spots smaller than 25 μm were manually deleted. The expression ratios of the duplicated spots on the array were averaged. For statistical analysis, genes were selected in which expression differed by a factor of at least twofold with respect to the reference pool in a minimum of 10% of the samples.
To identify the set of genes that were differentially expressed in ECCs and normal endometria, the Welch t-test was used, which does not require equal within-group variance. To avert an excessive number of false-positive results, the step-down maxT method was used. Because the sample size was small, the adjusted P values were obtained from 50,000 random permutations. Statistical comparison was performed using the Pomelo II program (http://pomelo2.bioinfo.cnio.es). The SOTA and TreeView programs (Bioinformatics Unit, CNIO) were used for clustering analysis, assuming Euclidean distances between genes.
Semiquantitative RT-PCR and Real-Time PCR
The expression of KSR1 in ECCs and normal endometrium was evaluated using semiquantitative RT-PCR performed using the Superscript II system for reverse transcription (Life Technologies, Inc.), beginning with 0.5 μg total RNA and random primers in a 20-μL reaction volume. Reverse transcription product and 0.4 to 1.0 μg oligonucleotide were heated at 95°C for 5 minutes, followed by 30 cycles of PCR amplification: 94°C denaturing for 1 minute, 55° to 58°C annealing for 1 minute, and 72°C extension for 1 minute, followed by final extension at 72°C for 10 minutes. All reactions included two pairs of primers, one corresponding to GADPH (glyceraldehyde-3-phosphate dehydrogenase), which served as a reference control gene, and the other for the gene being tested. Primer sequences were as follows: KSR1 F, 5′-GCACTCGTTTGTGAGGGA-3′, and R, 5′-ACAGCCCGAAGTCTGTGA-3′. PCR products were visualized on 3% agarose gels stained with ethidium bromide. Images were captured and analyzed using the Quantity One 4.1.1 analyzer and software (Gel Doc 2000, Bio-Rad Laboratories, Inc., Hercules, CA). Statistical comparison (t-test) was performed using a commercially available software package (SPSS for Windows, version 10.1; SPSS, Inc., Chicago, IL).
FLIP quantitative real-time PCR was performed as previously described.33 FLIP and β-glucuronidase were obtained (Assay-on-Demand Gene Expression Products ABI P/N 4331182:Hs00236002_m1 and ABI P/N 4326320E; Applied Biosystems, Inc., Foster City, CA).
Tissue Microarrays and Immunohistochemistry
Two tissue microarrays (TMAs) were constructed and processed as previously described34 using tissues from the surgical pathology files from Hospital Universitari Arnau de Vilanova de Lleida (Lleida, Spain) and Hospital de Sant Pau (Barcelona, Spain). The first TMA was constructed from 80 paraffin-embedded samples of normal endometrium in different phases of the menstrual cycle: 20 proliferative, 50 secretory, and 10 menstrual. The second TMA was constructed from 157 endometrial cancers including 47 grade 1, 58 grade 2, and 26 grade 3 ECCs, 15 serous carcinomas, four clear-cell carcinomas, and seven mixed müllerian malignant tumors. One hundred eight tumors were classified as stage I, 15 were stage II, 22 were stage III, and one was stage IV. Staging information was incomplete in 11 cases. Ninety-five tumors had been previously evaluated for common alterations of endometrial carcinoma including microsatellite instability and mutations in KRAS, CTNNB-1, or PTEN. A tissue array device (Beecher Instruments, Silver Spring, MD) was used. Two selected cylinders (0.6 mm in largest diameter) from two different areas were included in each case. Control normal tissues from the same endometrial carcinoma specimens were also included. Immunohistochemistry was performed using a KSR1 antibody (SC-25416; Santa Cruz Biotechnology, Inc.; 1:400 dilution), and the reaction was visualized using the EnVision Detection Kit (DAKO Corp., Carpinteria, CA) using diaminobenzidine chromogen as a substrate. Immunohistochemical results were evaluated by two pathologists (X.M.-G. and J.P.) by considering the percentage and intensity of the staining. A histologic score was obtained from each sample, and ranged from 0 (no immunoreaction) to 300 (maximum immunoreactivity). The score was obtained by applying the following formula: Histoscore = (1 × percentage of cells with light staining) + (2 × percentage of cells with moderate staining) + (3 × percentage of cells with strong staining). Because each TMA included two different tumor cylinders from each case, immunohistochemical evaluation was performed after examining both samples. The reliability of such scoring for interpretation of immunohistochemical staining in endometrial carcinoma TMAs, and the reproducibility of TMA immunostaining, with comparison of TMA results with those obtained in sections from the corresponding paraffin blocks of 37 randomly selected cases, has been described previously.18,34,35
Statistical analysis was performed on a database (SPSS for Windows version 11.5; SPSS, Inc.). The H score results were compared using the Mann-Whitney U-test, the Student t-test, the Kruskal-Wallis test, and analysis of variance when applicable. Correlations between quantitative variables were established using the Pearson and Spearman Rho tests. P < 0.05 was considered statistically significant.
Cell Lines, Culture Conditions, and Transfection
The Ishikawa 3-H-12 (IK) cell line was purchased from the European Collection of Cell Cultures. KLE cells were a gift from Dr. Palacios (CNIO). RL-95 and HEC-1-A cells were a gift from Dr. Reventos (Hospital Vall d'Hebron, Barcelona, Spain). The IK cell line was grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich Corp.) supplemented with 10% fetal bovine serum (Invitrogen Corp.) and 1 mmol/L HEPES, 1 mmol/L sodium pyruvate, 2 mmol/L L-glutamine, and 1% penicillin or streptomycin (all from Sigma-Aldrich Corp.) at 37°C with saturating humidity and 5% CO2. When indicated, transfection plasmid constructs were performed using lipofectamine 2000 reagent (Invitrogen Corp.) following the manufacturer's instructions.
Lentiviral Production and Infection
Oligonucleotides to produce plasmid-based shRNA were cloned into the FSV vector using AgeI-BamHI restriction sites. shRNA target sequences were as follows: KSR1 1.3, 5′-GCCTCCTTATTGCAGAAAGTT-3′; KSR1 2.6, 5′-GCACTGACAAAGAAGGCTCTT-3′; FADD, 5′-CATGGAACTCAGACGCATCT-3′; caspase 8, 5′-GAATCACAGACTTTGGACAA-3′; FLIP, 5′-CCTCACCTTGTTTCGGACTA-3′; CK2β, 5′-TGGTTTCCCTCACATGCTCT-3′; and ITCH, 5′-CCAGAAGTCAAGGTCAATTA-3′. Lentiviral production was performed as previously described.36
Colony Formation Assay and Assessment of Apoptosis
Three days after infection, cells transduced with shRNA for FSV or KSR1 were trypsinized and resuspended in 0.3% agar diluted in medium at a concentration of 3000 cells/mL. Cell colonies were evaluated after 15 days in culture as previously described.34
BrdU Incorporation
For determination of proliferation, cells were incubated with 3 ng/mL 5-bromodeoxyuridine (BrdU; Sigma-Aldrich Corp.) for 20 minutes and fixed with 4% paraformaldehyde. After DNA denaturing with 2 mol/L HCl for 30 minutes and neutralization with 0.1 mol/L Na2B4O7 (pH 8.5) for 2 minutes, cells were blocked in PBS solution containing 5% horse serum, 5% fetal bovine serum, 0.2% glycine, and 0.1% Triton X-100 for 1 hour. Subsequently, cells were subjected to indirect immunofluorescence with a mouse anti-BrdU monoclonal antibody (DAKO Corp.) and fluorescein isothiocyanate–conjugated anti-mouse secondary antibody (Molecular Probes, Inc., Eugene, OR). Nuclei were counterstained with 5 μg/mL Hoechst 33258, and cells were visualized using an epifluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Western Blot Analysis
Western blot analysis was performed as previously described.36 Densitometric quantification of Western blot bands was performed using Quantity One software (Bio-Rad Laboratories, Inc.).
Statistical Analysis
All data are given as mean (SE) of at least three independent experiments. Differences between groups were determined using the Student t-test. P < 0.05 was considered statistically significant.
Results
KSR1 Is Overexpressed in Endometrial Carcinoma
In a cDNA array study, normal endometrium and ECC cDNA expression profiles were compared. Supervised analysis of a cDNA array revealed statistically significant differences (unadjusted P value < 0.001, and adjusted P value < 0.07) in expression of 155 genes between ECCs and normal endometrium, including 62 up-regulated and 93 down-regulated genes in ECCs (Moreno-Bueno et al; unpublished results). One of the up-regulated genes in ECCs compared with normal tissue was KSR1 (accession No. H88143). KSR1 was up-regulated 2.1-fold over nontumor samples. This result was validated using semiquantitative RT-PCR in the same series analyzed using cDNA arrays and an independent series (series 1 or series 2) (Figure 1A). Statistically significant differences were observed in KSR1 expression in both studied series.
Figure 1.
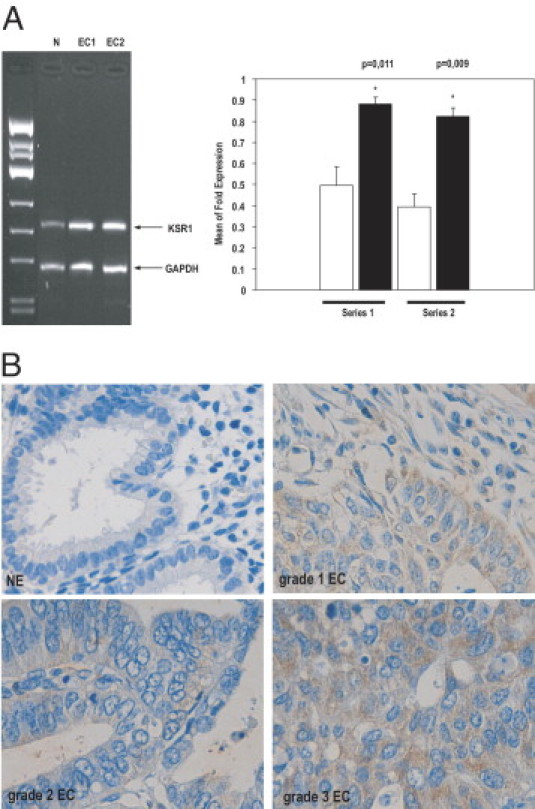
KSR1 expression is increased in endometrial carcinoma. A: Representative gel (left) and quantification (right) of KSR1 mRNA expression in two different series of normal endometrium (N; white bars) versus two different series of endometrial carcinoma (EC1 and EC2; black bars) elicits up-regulation of KSR1 mRNA in endometrial tumor samples. B: Endometrial carcinoma TMA was constructed and immunostained (see Materials and Methods). Representative images of KSR1 immunostaining of normal endometrium (NE) and grade 1, 2, and 3 endometrial carcinomas (EC).
To demonstrate that the increased KSR1 mRNA expression was associated with increased protein expression, immunohistochemistry for KSR1 was performed in two TMAs constructed as described (see Materials and Methods). To test the specificity of KSR1 antibody used for immunohistochemistry, IK cells were infected with lentiviruses carrying the empty vector (FSV) or an FSV vector containing two shRNAs targeting human KSR1 (KSR1 1.3 and KSR1 2.6). Three days later, the ability of KSR1 shRNAs to cause its down-regulation was assessed using Western blot analysis. KSR1 2.6 but not KSR1 1.3 down-regulated KSR1 protein levels (see Supplemental Figure S1A at http://ajp.amjpathol.org). To validate the specificity of antibody used in TMAs, immunohistochemistry was performed on IK cells infected with lentiviruses carrying shRNA 2.6 or the control vector using the same protocol used for TMA staining. The antibody did not stain IK cells infected with shRNA 2.6, but stained IK cells infected with control vector (Figure S1B).
To investigate whether KSR1 was expressed in the endometrium, expression of KSR1 was analyzed in a TMA constructed from normal endometrium samples. In normal endometrium, KSR1 was evaluated in all of the 80 cases included in the first TMA. KSR1 immunostaining varied according to the phase of the menstrual cycle (proliferative or secretory). KSR1 immunoexpression was significantly higher in the secretory phase [mean (range) histoscore, 3.11 (0 to 15)] than in the proliferative endometrium [0.95 (0 to 5)] (P = 0.0157). Staining was cytoplasmic. Afterward, expression of KSR1 was analyzed in a TMA constructed from ECCs. Because endometrial carcinomas arise from epithelial endometrial cells, only expression of KSR1 in epithelial fraction was evaluated, in normal and neoplastic tissue. However, stromal fraction demonstrated low levels of KSR1 expression. Because of cylinder losses caused by the immunostaining procedure, KSR1 was evaluated in 134 of 157 cases. Overall, KSR1 immunoexpression was significantly increased in endometrial carcinoma compared with normal endometrium (Figure 1B). KSR1 was expressed in 122 cases (91%). The KSR1 histoscore was 46.49 (0 to 182) in endometrial carcinoma, and 2.22 in normal endometrium (P < 0.00001).
KSR1 expression was significantly increased in nonendometrial carcinoma compared with endometrial carcinoma (P = 0.01204). KSR1 expression was significantly higher in grade 3 tumors than in grade 1 (P = 0.01802) and grade 2 (P = 0.01057) tumors. No statistically significant differences were observed when comparing grade 1 and grade 2 endometrial carcinomas (P = 0.9011). However, no statistical significance was observed in pathologic stage (P = 0.7235). Moreover, no significant relationship was observed between KSR1 expression and microsatellite instability (P = 0.5599) or mutations in KRAS (P = 0.2967), CTNNB-1 (P = 0.6457), or PTEN (P = 0.8390). Statistical analysis did not demonstrate any significant correlation between KSR1 and FLIP (P = 0.15) in the tissue samples. The lack of correlation between one protein involved in signaling pathways and a putative target is a common observation in TMA studies. Expression of FLIP, as of many other proteins, is controlled by multiple signaling molecules or pathways such as NF-κB, PI3K/Akt, or CK2. These pathways are also altered in endometrial carcinoma. Therefore, the total FLIP levels in vivo may be the result of different signaling pathways. Thus, it is difficult to correlate FLIP expression with the data obtained in a single pathway or molecule.
Inhibition of KSR1 Expression Reduces Tumor Phenotype of ECCs
Increased proliferation and anchorage-independent cell growth are two hallmarks of tumor phenotype. To ascertain the role of KSR1 in endometrial carcinogenesis, two endometrial cancer cell lines (IK and KLE) were infected with viruses carrying KSR1 shRNA. The effects of KSR1 knock-down on cell proliferation and anchorage-independent cell growth were evaluated. Compared with cells infected with the empty vector, KSR1 down-regulation caused a marked decrease in BrdU incorporation, which suggests that KSR1 knock-down reduces cell proliferation (Figure 2A). As a negative control, IK cells transduced with shRNA targeting the CK2β subunit of the protein casein kinase 2 (CK2) were used. CK2 is a ubiquitous, highly conserved, tetrameric serine/threonine kinase composed of two regulatory subunits called CK2β and two catalytic subunits, which can be two of either the subunits CK2α or CK2α′.37–39 Increased CK2 activity is associated with cell growth and proliferation, and many types of tumors exhibit aberrant or increased CK2 activity.40–42 Recently, it was demonstrated that CK2β knock-down reduces proliferation of ECCs34 Matched cultures were lysed and analyzed at Western blotting with antibodies to cyclin D1 and phosphorylated ERK. The reduction of cell proliferation correlated with a marked reduction of cyclin D1. ERK phosphorylation was also decreased, indicating that KSR1 shRNA was efficiently decreasing MAPK signaling (Figure 2B).
Figure 2.
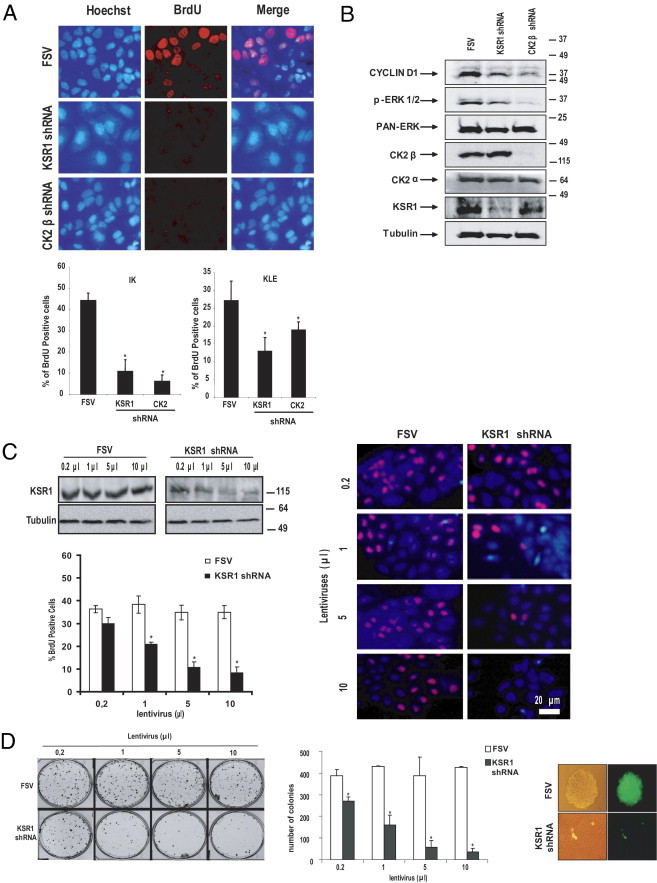
KSR1 knock-down inhibits proliferation and anchorage-independent cell growth. A: IK and KLE cells were infected with lentiviruses carrying the empty vector (FSV), KSR1 shRNA or CK2β shRNAs, and after 3 days, were analyzed using BrdU incorporation. Quantification of BrdU-positive IK and KLE cells (bottom) revealed that KSR1 shRNA cause a marked decrease in proliferating cells. Representative images of BrdU immunofluorescence in IK cells (top). B: IK cells were infected with lentiviruses carrying the empty vector (FSV), KSR1 shRNA or CK2β shRNAs, and after 3 days, lysates were collected. Western blot analysis demonstrated that both KSR1 shRNA and CK2 shRNA lead to marked down-regulation of CyclinD1, p-ERK. Membrane was reprobed using anti-KSR1 and anti-CK2β antibody to ensure specific inhibition of KSR1 and CK2β and anti-tubulin and anti–pan-ERK to ensure equal protein loading. C: KSR1 shRNA causes a dosage-dependent decrease in proliferating cells. IK cells were infected with increasing amounts of lentiviruses carrying the empty vector (FSV) or KSR1 shRNA. Western blot analysis demonstrated dosage-dependent inhibition of KSR1 expression (left). Proliferating cells of matched IK cultures were analyzed using BrdU incorporation. Quantification of BrdU-positive IK cells (left) and representative images of BrdU-stained cells (right). D: Soft agar experiment of IK cells infected with increasing amounts of lentiviruses carrying the empty vector (FSV) or KSR1 shRNA caused a dosage-dependent colony formation. Representative image of colonies in soft agar (left) and quantification of the number of colonies (middle) in FSV- or KSR1 shRNA–infected cells. Right, Higher magnification image demonstrates that KSR1-infected cells are alive, although they do not form colonies. Error bars represent SE from three independent experiments. *P < 0.05.
The intracellular amount of KSR1 is determinant for its function on proliferation and oncogenic potential.9 To demonstrate that high intracellular amounts of KSR1 are determinant to promote proliferation of ECCs, a titration experiment was performed in which IK cells were infected with increasing dosages of lentiviruses carrying KSR1 shRNA or the empty vector. Lentiviral titration resulted in graded inhibition of endogenous KSR1 expression that consistently correlated with a decrease in BrdU incorporation and mitotic index (Figure 2C). In contrast, no effects were observed with increasing dosages of lentiviruses carrying the empty vector.
Likewise, to investigate the role of KSR1 protein levels on anchorage-independent growth properties, IK cells were infected with increasing dosages of lentiviruses carrying KSR1 shRNAs, and after 3 days, cell cultures were trypsinized and cultured in soft agar for 15 days, as described previously. As shown in Figure 2D, KSR1 shRNA caused dosage-dependent inhibition of colonies, which indicates that KSR1 levels regulate anchorage-independent cell growth.
KSR1 Controls Sensitivity to TRAIL- and aFas-Induced Cell Death in ECCs
Deregulation of apoptotic cell death is also a critical event in development and progression of cancer. After it was ascertained that KSR1 regulates both proliferation and anchorage-independent cell growth, its potential role in regulation of apoptosis was investigated. For this purpose, IK cells were infected with virus carrying shRNAs to KSR1, and after 3 days, cells were treated with TRAIL or aFas, and the apoptotic nuclei were visualized at Hoechst staining. Treatment of IK cells with shRNA KSR1 2.6 alone resulted in a moderate increase in apoptotic nuclei; however, addition of aFas or TRAIL resulted in a dramatic increase in nuclei (Figure 3A). In contrast, shRNA 1.3, which did not cause down-regulation of KSR1, failed to sensitize IK cells to TRAIL-induced apoptosis. Moreover, TRAIL and aFas treatment caused activation of caspase 8 and caspase 3 in cells infected with lentiviruses carrying KSR1 shRNA (Figure 3B). Similar results were obtained with other endometrial carcinoma cell lines (see Supplemental Figure S2 at http://ajp.amjpathol.org). These data demonstrate that KSR1 inhibition sensitizes ECCs to apoptosis triggered by either TRAIL or aFas.
Figure 3.
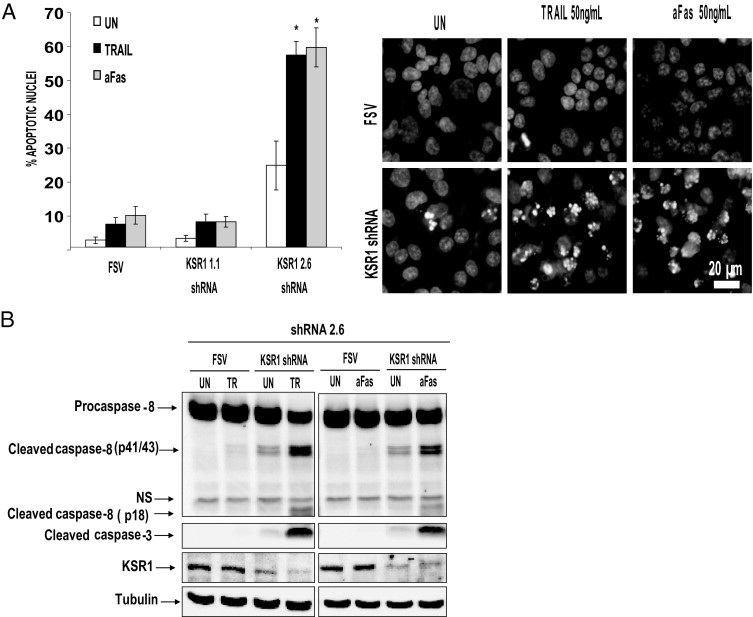
Down-regulation of KSR1 sensitizes IK to TRAIL- and aFas-induced apoptosis. A: Treatment of IK with shRNA to KSR1 plus either TRAIL or aFas increases the number of apoptotic nuclei and caspase activation. IK cells were infected with lentiviruses carrying two different shRNAs targeting KSR1 (KSR1 1.3 and KSR1 2.6). After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL for 8 hours, and nuclei were visualized at Hoechst staining. Results are given as percentage of apoptotic nuclei compared with the total number of nuclei (left). Representative images of Hoechst-stained IK cells (right). Error bars represent SE from three independent experiments. *P < 0.05. B: KSR1 shRNA plus 50 ng/mL TRAIL or aFas activate caspases 8 and 3. IK cells were infected with lentiviruses carrying KSR1 shRNA 2.6. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL or aFas. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with tubulin to ensure equal amounts of protein, and antibodies to KSR1 to check inhibition by the specific shRNA. Blots are representative of three experiments.
KSR1 Sensitization to TRAIL and aFas Requires FLIP Down-Regulation and Expression of Caspase 8 and FADD
High levels of FLIP are responsible for resistance to TRAIL-induced apoptosis in ECCs.18 Because of the critical role of FLIP in death receptor apoptosis resistance in ECCs, whether KSR1-mediated resistance to TRAIL and aFas was caused by regulation of FLIP protein levels was examined. IK cells were infected with lentiviruses carrying KSR1 shRNA, and after 3 days, they were exposed to TRAIL and aFas. Addition of either TRAIL or aFas to IK cells infected with shRNA to KSR1 resulted in a dramatic decrease in FLIP protein levels (Figure 4A). Similar results were obtained with other endometrial carcinoma cell lines (see Supplemental Figure S3 at http://ajp.amjpathol.org).
Figure 4.
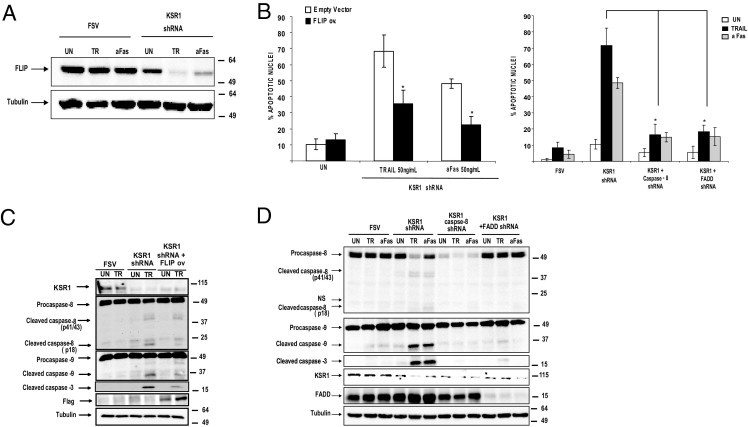
KSR1 sensitization to TRAIL and aFas requires FLIP down-regulation and caspase 8 and FADD expression. A: FLIP protein levels are decreased after addition of TRAIL or aFas in cells infected with KSR1 shRNA, but not in FSV-infected cells. IK cells were infected with lentiviruses carrying shRNA KSR1. After 3 days to enable down-regulation of the protein, cells were left untreated (UN) or were treated with 50 ng/mL TRAIL or aFas. After 8 hours, cells were lysed. Cell lysates were analyzed at Western blotting with antibodies to FLIP. Membranes were reprobed with tubulin to ensure equal amounts of protein. B: FLIP overexpression or silencing of FADD and caspase 8 reduce the number of apoptotic nuclei caused by KSR1 shRNA plus TRAIL or aFas treatment. IK cells were infected with lentiviruses carrying KSR1 shRNA alone or with a FLAG-tagged FLIP expression plasmid (left graph) or either FADD or caspase 8 shRNAs (right graph). After 3 days, cells treated with 50 ng/mL TRAIL or aFas were added to the culture. Nuclei exhibiting apoptotic features were visualized using Hoechst staining, and counted after 8 hours. Results of apoptotic nuclei quantification are given as the percentage of apoptotic nuclei compared with the total number of nuclei. C: FLIP overexpression inhibits activation of caspases 8, 9, and 3. IK cells were infected with lentiviruses carrying a FLAG-tagged FLIP expression plasmid. After 3 days, cells were stimulated with 50 ng/mL TRAIL or aFas. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with anti-KSR1 antibodies to ensure protein inhibition, and anti-FLAG to check heterologous FLIP expression. Membranes were reprobed again with tubulin to ensure equal amounts of protein. NS, nonspecific bands. D: Down-regulation of caspase 8 or FADD inhibits activation of caspases 8, 9, and 3. IK cells were infected with lentiviruses carrying KSR1 alone or together with FADD or caspase 8 shRNAs. After 3 days, 50 ng/mL TRAIL or aFas was added to the culture. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with anti-FADD, anti–caspase 8, and anti-KSR1 antibodies to ensure protein inhibition by the specific shRNAs. Membranes were reprobed again with tubulin to ensure equal amounts of protein. NS, nonspecific bands.
To ascertain whether such FLIP down-regulation was the cause of sensitization to TRAIL in KSR1-silenced ECCs, IK cells were infected with a plasmid coding for a FLAG-tagged mouse FLIP. After 3 or 4 days to enable FLIP expression, cells were treated with aFas. Apoptotic nuclei were then visualized at Hoechst staining, and caspase processing at Western blot analysis. Overexpression of FLIP resulted in a dramatic reduction of apoptotic nuclei caused by KSR1 shRNA plus either TRAIL or aFas (Figure 4B). Consistent with this observation, FLIP overexpression inhibited processing of caspases 8, 9, and 3 (Figure 4C).
To test whether KSR1 sensitization to TRAIL- and aFas-induced apoptosis required a functional death-inducing signaling complex, IK cells were co-infected with viruses carrying KSR1, plus either caspase 8 or FADD shRNAs. After 3 days, cells were treated with TRAIL. Hoechst staining revealed a decrease in the number of apoptotic nuclei in cells infected with KSR1 shRNA plus either FADD or caspase 8 shRNAs and exposed to TRAIL or aFas (Figure 4B). Accordingly, knock-down of FADD and caspase 8 blocked activation of all caspases analyzed at Western blotting.
These results indicate that TRAIL sensitization to apoptosis after KSR1 down-regulation requires adaptor protein FADD and initiator caspase 8 in the death-inducing signaling complex to trigger an apoptotic response (Figure 4D).
KSR1 shRNA Leads to a FADD-Dependent Inhibition of FLIP Translation
Next investigated were the mechanisms by which KSR1 regulates FLIP levels. First, whether FLIP levels were transcriptionally regulated was assessed. For this purpose, real-time PCR was performed on mRNA extracted from IK cells infected with lentiviruses carrying KSR1 shRNA. Compared with cells carrying the empty vector, cells carrying KSR1 shRNA did not demonstrate a decrease in FLIP mRNA levels, even in IK cells treated with TRAIL (Figure 5A). As a positive control of FLIP down-regulation, FLIP mRNA from IK cells infected with lentiviruses carrying FLIP shRNA or CK2β shRNA was analyzed.
Figure 5.
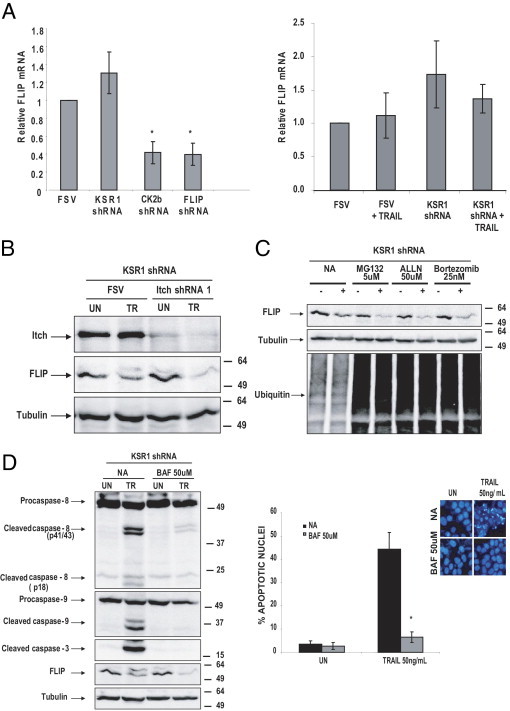
KSR1 shRNA does not alter FLIP transcription and degradation. A: KSR1 shRNA did not change the levels of FLIP mRNA. IK cells were infected with lentiviruses carrying KSR1. After 3 days, cells were stimulated with 50 ng/mL TRAIL or aFas. mRNA was extracted and subjected to reverse transcription. mRNA relative levels were analyzed using real-time PCR. Results are given as relative mRNA levels compared with untreated (UN) cells. B: The ubiquitin ligase ITCH did not prevent FLIP down-regulation. IK cells were infected with lentiviruses carrying KSR1 alone or together with ITCH shRNA. After 3 days, 50 ng/mL TRAIL was added to the culture. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to FLIP. Membranes were reprobed with anti-ITCH antibodies to ensure protein inhibition by the specific shRNA. Membranes were reprobed again with tubulin to ensure equal amounts of protein. C: The ubiquitin-proteasome system is not involved in FLIP down-regulation. IK cells were infected with lentiviruses carrying KSR1. After 3 days, cells were treated with 50 ng/mL TRAIL plus the indicated dosages of the proteasome inhibitors MG-132, ALLN, or bortezomib. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to FLIP. Membranes were reprobed with anti-ubiquitin antibodies to ensure effectiveness of the proteasome inhibitors. Membranes were reprobed again with tubulin to ensure equal amounts of protein. D: Caspases are not responsible for FLIP down-regulation. Right, IK cells were infected with lentiviruses carrying KSR1. After 3 days, cells were treated with 50 ng/mL TRAIL plus the caspase inhibitor BAF. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to FLIP. Membranes were reprobed with anti-caspase antibodies to check caspase inhibition by BAF. Membranes were reprobed again with tubulin to ensure equal amounts of protein. Left, Parallel cultures were Hoechst stained, and the number of nuclei exhibiting apoptotic features was scored. Results of apoptotic nuclei quantification are given as the percentage of apoptotic nuclei compared with the total number of nuclei. Error bars represent SE from three independent experiments. *P < 0.05.
FLIP levels are also regulated by ubiquitin-proteasome–mediated degradation. The E3 ubiquitin ligase ITCH is responsible for ubiquitin conjugation and subsequent degradation of FLIP through the proteasome.43–45 These observations enabled analysis of whether the ubiquitin-proteasome system mediated FLIP down-regulation. For this purpose, IK cells were co-infected with KSR1 shRNA plus an shRNA targeting ubiquitin ligase ITCH expression and, thus, ubiquitin conjugation of FLIP. ITCH inhibition did not prevent down-regulation of FLIP (Figure 5B). To further exclude the role of the ubiquitin-proteasome system in FLIP down-regulation, IK cells were treated with the proteasome inhibitors MG-132, bortezomib, or ALLN. Addition of any of the proteasome inhibitors failed to inhibit reduction in FLIP protein caused by TRAIL in cells with down-regulated KSR1 (Figure 5C).
Another suggested mechanism for FLIP degradation is caspase processing. To investigate whether the decrease in FLIP levels was caused by caspase activation after TRAIL treatment, IK cells infected with FSV or KSR1 shRNA were treated with TRAIL or TRAIL plus the broad-caspase inhibitor BAF. Treatment of IK cells with BAF did not inhibit FLIP down-regulation, although it efficiently blocked processing of caspases 8, 9, and 3 and nuclei with apoptotic features (Figure 5D).
Forced expression of FLAG-tagged mouse FLIP overcame TRAIL-induced caspase processing and apoptosis in KSR1 shRNA-infected IK cells. Expression of endogenous FLIP levels was investigated in cells in which FLIP was overexpressed. The FLIP antibody used in the present study recognized human FLIP but not mouse FLIP, which enabled detection of endogenous FLIP protein. Western blot analysis of endogenous FLIP revealed that it was down-regulated even when heterologous mouse FLIP was overexpressed (Figure 6A). This indicates that TRAIL-induced FLIP down-regulation by KSR1 shRNA was not caused by a general or unspecific process of degradation or down-regulation of gene expression resulting from apoptotic cell death.
Figure 6.
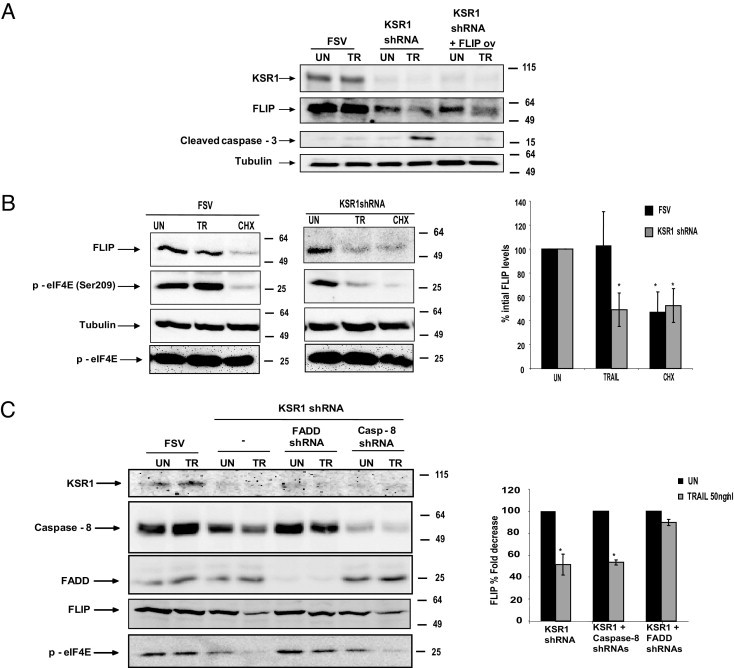
Reduction of FLIP levels is posttranscriptional and FADD-dependent. A: Endogenous FLIP is down-regulated even in cells with heterologous FLIP overexpression. IK cells were infected with lentiviruses carrying KSR1 shRNA plus a FLAG-tagged FLIP expression plasmid. After 3 days, cells were stimulated with 50 ng/mL TRAIL. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to detect endogenous FLIP expression, FLAG, and KSR1. Membranes were reprobed again with tubulin to ensure equal amounts of protein. B: CHX and TRAIL induce the same down-regulation of eIF4E phosphorylation and FLIP down-regulation in KSR1-silenced cells. IK cells were infected with lentiviruses carrying KSR1 (left panel) or the empty vector (middle panel). After 3 days, 50 ng/mL TRAIL or CHX was added to the culture. Cell lysates were analyzed at Western blotting with anti–phospho-eIF4E, anti-FLIP, or anti-tubulin. Densitometric quantification of FLIP expression in three different independent experiments (right panel). Results are given as the percentage of FLIP decrease. C: FADD shRNAs but not caspase 8 shRNAs prevent FLIP down-regulation and eIF4E dephosphorylation in KSR1-silenced cells. IK cells were infected with lentiviruses carrying KSR1 alone or together with FADD or caspase 8 shRNAs. After 3 days, 50 ng/mL TRAIL or aFas was added to the culture. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with anti-FLIP and anti–phospho-eIF4E antibodies. Membranes were reprobed again with FADD, caspase 8, and KSR-1 antibodies to check for protein knock-down. Membranes were also reprobed with tubulin to ensure equal amounts of protein (left). Densitometric quantification of FLIP expression in three different independent experiments (right). Results are given as the percentage of FLIP decrease. *P < 0.05.
Apart from transcription and degradation, FLIP protein levels may also be regulated by the mechanism of translation. To ascertain whether FLIP down-regulation was caused by inhibition of FLIP translation, IK cells were treated with TRAIL or with the inhibitor of translation cycloheximide (CHX) for 8 hours in cells infected with either KSR1 shRNA or FSV vector. Treatment with CHX led to a marked decrease in FLIP protein levels in FSV-transfected cells, which suggests that FLIP protein levels are maintained by continuous translation from its mRNA (Figure 6B). Addition of TRAIL to IK cells infected with KSR1 shRNA induced a level of decrease of eIF4E phoshorylation similar to that observed after CHX treatment, which suggests that TRAIL-induced inhibition of translation occurred in cells with silenced KSR1 (Figure 6B).
Also assessed were the effects of caspase 8 and FADD shRNAs on FLIP levels. Caspase 8 shRNA did not restore FLIP levels, although it completely inhibited caspase activation and apoptosis. Conversely, FADD shRNA inhibited FLIP down-regulation caused by TRAIL stimulation in KSR1-silenced IK cells (Figure 6C). Such a differential effect of caspase 8 and FADD shRNAs on FLIP protein levels strongly correlated with the phosphorylation status of eIF4E. FADD shRNA, but not caspase 8 shRNA, restored eIF4E phosphorylation in cells treated with KSR1 shRNA plus TRAIL. These data further support the inhibition of translation as a mechanism for FLIP down-regulation caused by TRAIL in KSR1-silenced cells.
Re-Introduction of Heterologous KSR1 Restores Apoptosis Resistance and Proliferation
To demonstrate the specific requirement of KSR1 in regulation of FLIP protein levels and subsequent apoptotic response to TRAIL, a heterologous mouse KSR1 cDNA that is not targeted by human KSR1 shRNA was re-expressed. For these experiments, IK cells were transduced with viruses carrying KSR1 shRNA, and the day after infection they were transfected with a plasmid-encoding mouse KSR1 cDNA. After 2 days, cells were treated with TRAIL.
Re-introduction of mouse KSR1 cDNA in IK cells caused marked inhibition of caspases 3, 8, and 9 processing, indicating that KSR1 is a key element in determination of life-death signal after TRAIL stimulation (Figure 7A). Proper overexpression of KSR1 in this experiment was monitored using antibodies to the FLAG tag because the antibody used in this study was unable to detect mouse KSR1.
Figure 7.
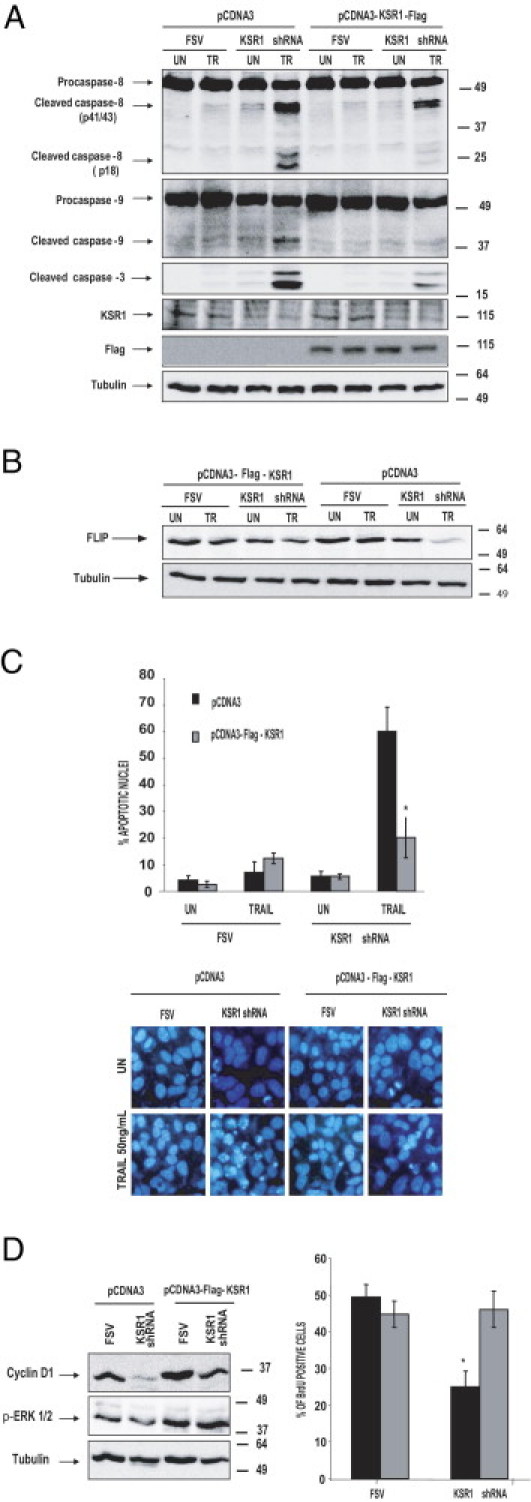
Re-introduction of heterologous KSR1 restores FLIP levels and TRAIL resistance. A: Western blot analysis of caspase activation reveals that re-introduction of KSR1 inhibits caspase activation. IK cells were infected with lentiviruses carrying the empty vector (FSV) or KSR1 shRNA, and the day after were transfected with pcDNA3 or pCDNA3-KSR1-FLAG. At 48 hours after transfection, cells were treated with 50 ng/mL TRAIL for 8 hours. Cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with anti-tubulin to ensure equal protein amounts, anti-KSR1 to check inhibition by the specific shRNA, and anti-FLAG to ensure heterologous KSR1 expression. B: Analysis of lysates from same cultures at Western blotting demonstrate that FLIP expression is re-sorted on re-introduction of KSR1. C: Hoechst staining of parallel cultures demonstrated marked reduction in the number of nuclei with apoptotic morphologic features. Results are given as the percentage of apoptotic nuclei compared with the total number of nuclei. Representative micrographs of Hoechst-stained cells. D: IK cells were infected with lentiviruses carrying the empty vector (FSV) or KSR1 shRNA, and the day after were transfected with pcDNA3 or pCDNA3-KSR1-FLAG. Cell lysates were collected and analyzed at Western blotting with anti-cyclinD1 or anti–p-ERK antibodies. Parallel cultures were analyzed for BrdU. Results are given as the percentage of BrdU-positive cells over the total number of cells. *P < 0.05.
More important, re-introduction of KSR1 caused marked re-expression of FLIP protein (Figure 7B). This result strongly supports the role of KSR1 in control of endogenous FLIP protein levels and, thus, death receptor–induced apoptosis.
Furthermore, overexpression of mouse KSR1 increased the levels of ERK phosphorylation and restored the levels of cyclinD1 observed in normal cells (Figure 7C), as well as BrdU incorporation (Figure 7D).
Discussion
To our knowledge, the present study provides the first demonstration that KSR1 expression is elevated in endometrial carcinoma. Moreover, it demonstrates that inhibition of KSR1 expression in ECCs causes a marked decrease in proliferation rates and anchorage-independent cell growth. Further, KSR1 has a critical role in control of sensitivity of ECCs to TRAIL-induced apoptosis and FLIP protein translation.
Previous studies have demonstrated that KSR1 is necessary to promote Ras-induced transformation in vivo and in vitro. Moreover, pharmacologic inhibition of KSR1 abrogated pancreatic cancer caused by Ras activation.14 This evidence supports an important role for KSR1 in development of cancer. Data from cDNA and tissue microarray studies demonstrated that KSR1 expression is elevated in endometrial carcinomas compared with normal endometrial tissue. These results provide the first evidence of up-regulated expression of KSR1 in cancer in human beings. Previous evidence demonstrated that Raf/MEK/ERK signaling is altered in endometrial carcinogenesis at different levels of the pathway such as mutations in KRAS or hypermethylation of RassF1A.46–48 Up-regulation of KSR1 as responsible for carcinogenesis is compatible with the scaffolding function of KSR1 in regulation of Raf/MEK/ERK signaling. The importance of KSR1 concentration was evidenced by experiments in which reintroduction of increasing amounts of KSR1 in KSR1−/− fibroblasts caused a dosage-dependent increase in ERK signaling and transformation.9 Therefore, it can be speculated that increased expression may have an important role in development and progression of endometrial carcinoma.
To determine the role of KSR1 protein levels in endometrial tumorigenesis, shRNA was used to inhibit endogenous KSR1 expression. The results showed that KSR1 inhibition blocked not only proliferation but also anchorage-independent cell growth. These results are in agreement with previous reports that demonstrated a role for KSR1 in control of cell cycle, proliferation, and oncogenic potential of tumor cells,9,12,49 and further demonstrate that KSR1 protein levels are important for the tumor phenotype of endometrial carcinoma.
Another hallmark of tumor cells is their ability to evade apoptosis. KSR1 regulates apoptosis triggered by anticancer agents such as cis-platin.50 KSR1 has also been involved in modulation of apoptotic response of B colonic epithelial cells to TNFα,16,17 which suggests a role for KSR1 in control of apoptosis triggered by death receptors.
Apoptosis is a critical event in regulation of endometrial homeostasis. Previous studies have demonstrated that proteins related to apoptosis regulation are deregulated in endometrial carcinoma.33–35,51 Insofar as extrinsic or death receptor apoptosis, the results demonstrated that ECCs exhibit high levels of FLIP, which confers apoptotic resistance to both TRAIL and Fas ligand.18,19 Among death receptors and ligands, the ability to selectively kill tumor cells has suggested that TRAIL may be a promising anti-cancer agent.52,53 Although agonistic TRAIL antibodies are under clinical trial for various types of malignant diseases, some cancer cell types escape apoptosis triggered by monotherapy with agonistic antibodies. The decisions between life and death on TRAIL stimulation can be regulated by several signaling pathways, depending on cellular context.54 Therefore, knowledge about molecules involved in regulation of TRAIL sensitivity to apoptosis may be helpful in development of specific combination treatments. In this regard, the present results represent the first evidence for a role of KSR1 in control of life/death decisions after TRAIL treatment. Inhibition of KSR1 not only caused sensitization to TRAIL-induced apoptosis, but also an increase in apoptosis even in the absence of TRAIL or Fas. This feature increases the potential of KSR1 inhibition as a tool for killing endometrial tumor cells.
The molecular mechanisms by which KSR1 controls FLIP protein levels were further elucidated. FLIP is constitutively or highly expressed in some tumors,55–59 and its expression can be regulated at both the transcriptional and posttranscriptional levels. It was recently demonstrated that CK2 regulates FLIP levels by modulating its transcription and proteasome-mediated degradation.19 In contrast, the present study demonstrated that KSR1 did not modify FLIP mRNA levels in either the presence or absence of TRAIL, which suggests posttranscriptional regulation. At the posttranscriptional level, FLIP can be regulated by both translation and degradation. An increasing number of reports demonstrate that FLIP protein is primarily degraded by the ubiquitin-proteasome system. The E3 ubiquitin ligase ITCH mediates proteasome degradation of FLIP.45 The present study demonstrated TRAIL-triggered FLIP down-regulation even in cells infected with ITCH shRNA. These data do not exclude the possibility that other ubiquitin ligases may function to ubiquitylate FLIP in endometrial cells. Nonetheless, proteasome inhibitors did not block FLIP reduction in KSR1-silenced cells, which suggests that KSR1 does not regulate proteasomal degradation of FLIP.
Another explanation for FLIP down-regulation is that it results from a general process of protein degradation associated with degeneration of a dying cell. However, this possibility was excluded after the observation that FLIP down-regulation occurred even in cells in which heterologous mouse FLIP was overexpressed. In this setting, activation of caspases and apoptosis was inhibited; however, endogenous FLIP protein was still down-regulated. This result demonstrates that endogenous FLIP protein levels are tightly regulated.
It was demonstrated that CHX, an inhibitor of translation, caused down-regulation of FLIP similar to that observed with treatment with TRAIL plus KSR1 shRNA. Increasing evidence points to regulation of translation as a mechanism involved in regulation of proteins of the apoptotic machinery. Recent findings have demonstrated that some chemotherapeutic drugs such as sorafenib inhibit Mcl-1 or FLIP protein translation, likely by regulating eIF4E phosphorylation60. Moreover, in other cell types, FLIP-S translation is critical in determination of TRAIL sensitivity.61–63 In line with these results, addition of TRAIL to ECCs with KSR1 shRNA caused inhibition of eIF4E phosphorylation to the same extent as that observed with CHX treatment.
Inhibition of the ERK pathway controls TRAIL-induced apoptosis.64–66 Because KSR1 is a critical regulator of ERK/MAPK signaling, it is logical to think that inhibition of KSR1 may sensitize ECCs to TRAIL by inhibition of ERK activation. However, previous studies have demonstrated that inhibition of ERK/MAPK signaling is not involved in sensitization of ECCS to apoptosis induced by TRAIL.67
TRAIL engagement of DR4 and DR5 receptors leads to recruitment of the adaptor protein FADD, which serves as a docking protein for both caspase 8 and FLIP. Therefore, it is logical that both FADD and caspase 8 shRNA overcame KSR1 sensitization to TRAIL. However, the effects of these two shRNAs on FLIP protein levels were completely different. Caspase 8 down-regulation did not prevent a decrease in FLIP, whereas FADD knock-down restored FLIP levels to those observed in untreated cells. Caspase 8 inhibits apoptosis because it impairs downstream caspase activation; however, it does not block binding of FADD on TRAIL stimulation. This result and those obtained with the caspase inhibitor BAF make it unlikely that down-regulation of FLIP is caused by caspase degradation, as observed in other cell types.30,68,69 In contrast, inhibition of FADD should inhibit all downstream signaling after TRAIL receptor signaling. Then, if it is postulated that KSR1 modulation of FLIP translation depends on ligand binding, eIF4E dephosphorylation and subsequent FLIP down-regulation should not occur in the absence of FADD. Addition of TRAIL in cells co-infected with KSR1 and FADD shRNAs did not cause dephosphorylation of eIF4E, and consistently, FLIP protein levels were maintained at normal levels. This result strongly supports TRAIL-induced inhibition of FLIP translation in the absence of KSR1.
Furthermore, it is believed that complementation experiments using mouse KSR1 cDNA provided strong evidence of the direct effects of KSR1 expression on proliferation and apoptosis of ECCs. Re-introduction of KSR1 overcame sensitization to TRAIL in cells with silenced endogenous KSR1 expression and, more important, restored FLIP levels to those observed in control cells, demonstrating a role for KSR1 in control of FLIP protein.
Recently it was demonstrated that the protein kinase CK2 has an important role in mediating resistance of ECCs to TRAIL and aFas-induced cell death.19 In that study,19 pharmacologic inhibition of CK2 or knock-down of the CK2β regulatory subunit sensitized ECCs to apoptosis triggered by either TRAIL or aFas. In contrast, recent findings demonstrated that KSR1 interacts with CK2.70 Taking into account these results and the role of KSR1 and CK2 in apoptosis resistance, it is tempting to speculate that both proteins can interact and cooperate in control of FLIP. However, the mechanisms by which CK2 and KSR1 control FLIP levels are largely different. First, CK2 controlled transcription and degradation of FLIP, whereas KSR1 regulated its translation. Second, CK2β shRNA caused marked down-regulation of FLIP in the absence of TRAIL, whereas KSR1 shRNA mediated down-regulation of FLIP requires TRAIL treatment. Third, it has been suggested that KSR1 acts as a functional kinase.14,16,71,72 However, whether it possesses intrinsically kinase activity in mammals is still controversial, and further investigations will be required to address this point. Future work will examine whether CK2/KSR1 interactions and the putative kinase activity of KSR1 are required for maintenance of FLIP levels and, ultimately, TRAIL sensitivity.
Acknowledgments
Tumor samples were obtained from Xarxa Catalana de Bancs de Tumors and the Tumor Banc Platform of Red Temática de Investigación Cooperativa en Cáncer (RTICC). We thank Roger Dolcet, Eric Dolcet, and Laura Paya.
Footnotes
Supported by Fondo de Investigaciones Sanitarias (FIS) (grants FIS PI10/00604, FIS PI070304, FIS PI070276), from Ministerio de Ciencia e Innovación (SAF2002-10529-E, and SAF2004-05250), the National Cancer Institute, National Institutes of Health (grant CA90400), Marató de TV3 2005-47, RD06/0020/1034, RD06/0020/0013, 2009SGR794, and 2004XT00090, AECC, Catalunya Contra el Cancer and Programa de Intensificación de la Investigación, Instituto Carlos III. D.L. is supported by a predoctoral fellowship (FI05/00191) from FIS, Ministerio de Sanidad y Consumo. N.E. is supported by a fellowship (FI08/0012) from FIS. M.S. is supported by Red Temática de Investigación Cooperativa en Cáncer (RTICC; RD06/0020/1034). A.Y. is supported by a postdoctoral fellowship (JCI-2008-1969) from Programa Juan de la Cierva, Ministerio de Ciencia e Innovación. X.D. is supported by a Miguel Servet fellowship (CP05/00028) from FIS, Ministerio de Sanidad y Consumo.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.12.041.
Supplementary data
Specificity of KSR1 shRNA and antibodies. IK cells were infected with lentiviruses carrying two different shRNAs targeting KSR1 (KSR1 1.3 and KSR1 2.6). A: After 3 days to enable down-regulation of the protein, cells were lysed, and KSR1 was analyzed at Western blotting. B: Parallel cultures were infected, and after 3 days were trypsinized, embedded in agarose, and fixed. Immunohistochemistry of KSR1 was performed (see “Materials and Methods”).
KSR1 shRNA sensitizes HEC-1-A and KLE cells TRAIL-induced apoptosis. A: IK cells were infected with increasing amounts of lentiviruses carrying KSR1 shRNA or FSV empty vector. Western blot analysis demonstrated that progressive KSR1 down-regulation caused TRAIL-induced progressive decrease in FLIP levels and a consistent increase in caspase 3 activation. B: KLE and HEC-1-A endometrial carcinoma cell lines were infected with lentiviruses carrying shRNA targeting KSR1. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL for 8 hours, and nuclei were visualized via Hoechst staining. Results are given as the percentage of apoptotic nuclei compared with the total number of nuclei. Bottom: Representative images of Hoechst-stained IK cells. C: KSR1 shRNA plus 50 ng/mL TRAIL activated caspases 8, 9, and 3. KLE cells were infected with lentiviruses carrying shRNA to KSR1 2.6. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with tubulin to ensure equal amounts of protein and antibodies to KSR1 to check inhibition by the specific shRNA.
KSR1 shRNA triggers TRAIL-induced down-regulation of FLIP. KLE and HEC-1-A endometrial carcinoma cell lines were infected with lentiviruses carrying shRNA 2.6 targeting KSR1. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL for 8 hours and lysed. FLIP expression was analyzed at Western blotting.
References
- 1.Chang L., Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Morrison D.K. KSR: a MAPK scaffold of the Ras pathway? J Cell Sci. 2001;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 3.Kornfeld K., Hom D.B., Horvitz H.R. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- 4.Sundaram M., Han M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell. 1995;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- 5.Therrien M., Chang H.C., Solomon N.M., Karim F.D., Wassarman D.A., Rubin G.M. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 6.Therrien M., Michaud N.R., Rubin G.M., Morrison D.K. KSR modulates signal propagation within the MAPK cascade. Genes Dev. 1996;10:2684–2695. doi: 10.1101/gad.10.21.2684. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen A., Burack W.R., Stock J.L., Kortum R., Chaika O.V., Afkarian M., Muller W.J., Murphy K.M., Morrison D.K., Lewis R.E., McNeish J., Shaw A.S. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy F., Laberge G., Douziech M., Ferland-McCollough D., Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002;16:427–438. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortum R.L., Lewis R.E. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004;24:4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolch W. Coordinating ERK/MAPK signaling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 11.Joneson T., Fulton J.A., Volle D.J., Chaika O.V., Bar-Sagi D., Lewis R.E. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem. 1998;273:7743–7748. doi: 10.1074/jbc.273.13.7743. [DOI] [PubMed] [Google Scholar]
- 12.Razidlo G.L., Kortum R.L., Haferbier J.L., Lewis R.E. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J Biol Chem. 2004;279:47808–47814. doi: 10.1074/jbc.M406395200. [DOI] [PubMed] [Google Scholar]
- 13.Lozano J., Xing R., Cai Z., Jensen H.L., Trempus C., Mark W., Cannon R., Kolesnick R. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 2003;63:4232–4238. [PubMed] [Google Scholar]
- 14.Xing H.R., Cordon-Cardo C., Deng X.Z., Tong W., Campodonico L., Fuks Z., Kolesnick R. Pharmacologic inactivation of kinase suppressor of ras-1 abrogates Ras-mediated pancreatic cancer. Nat Med. 2003;9:1266–1268. doi: 10.1038/nm927. [DOI] [PubMed] [Google Scholar]
- 15.McCubrey J.A., Steelman L.S., Chappell W.H., Abrams S.L., Wong E.W., Chang F., Lehmann B., Terrian D.M., Milella M., Tafuri A., Stivala F., Libra M., Basecke J., Evangelisti C., Martelli A.M., Franklin R.A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolesnick R., Xing H.R. Inflammatory bowel disease reveals the kinase activity of KSR1. J Clin Invest. 2004;114:1233–1237. doi: 10.1172/JCI23441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan F., John S.K., Wilson G., Jones D.S., Washington M.K., Polk D.B. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004;114:1272–1280. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolcet X., Llobet D., Pallares J., Rue M., Comella J.X., Matias-Guiu X. FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Lab Invest. 2005;85:885–894. doi: 10.1038/labinvest.3700286. [DOI] [PubMed] [Google Scholar]
- 19.Llobet D., Eritja N., Encinas M., Llecha N., Yeramian A., Pallares J., Sorolla A., Gonzalez-Tallada F.J., Matias-Guiu X., Dolcet X. CK2 controls TRAIL and Fas sensitivity by regulating FLIP levels in endometrial carcinoma cells. Oncogene. 2008;27:2513–2524. doi: 10.1038/sj.onc.1210924. [DOI] [PubMed] [Google Scholar]
- 20.Ashkenazi A., Pai R.C., Fong S., Leung S., Lawrence D.A., Marsters S.A., Blackie C., Chang L., McMurtrey A.E., Hebert A., DeForge L., Koumenis I.L., Lewis D., Harris L., Bussiere J., Koeppen H., Shahrokh Z., Schwall R.H. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walczak H., Miller R.E., Ariail K., Gliniak B., Griffith T.S., Kubin M., Chin W., Jones J., Woodward A., Le T., Smith C., Smolak P., Goodwin R.G., Rauch C.T., Schuh J.C., Lynch D.H. Tumoricidal activity of tumor necrosis factor–related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 22.Koschny R., Walczak H., Ganten T.M. The promise of TRAIL: potential and risks of a novel anticancer therapy. J Mol Med. 2007;85:923–935. doi: 10.1007/s00109-007-0194-1. [DOI] [PubMed] [Google Scholar]
- 23.Johnstone R.W., Frew A.J., Smyth M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 24.LeBlanc H.N., Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 25.Walczak H., Degli-Esposti M.A., Johnson R.S., Smolak P.J., Waugh J.Y., Boiani N., Timour M.S., Gerhart M.J., Schooley K.A., Smith C.A., Goodwin R.G., Rauch C.T. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheridan J.P., Marsters S.A., Pitti R.M., Gurney A., Skubatch M., Baldwin D., Ramakrishnan L., Gray C.L., Baker K., Wood W.I., Goddard A.D., Godowski P., Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 27.Bodmer J.L., Holler N., Reynard S., Vinciguerra P., Schneider P., Juo P., Blenis J., Tschopp J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 28.Kischkel F.C., Lawrence D.A., Chuntharapai A., Schow P., Kim K.J., Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–620. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 29.Sprick M.R., Weigand M.A., Rieser E., Rauch C.T., Juo P., Blenis J., Krammer P.H., Walczak H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/s1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- 30.Micheau O., Thome M., Schneider P., Holler N., Tschopp J., Nicholson D.W., Briand C., Grutter M.G. The long form of FLIP is an activator of caspase-8 at the fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 31.Irmler M., Steiner V., Ruegg C., Wajant H., Tschopp J. Caspase-induced inactivation of the anti-apoptotic TRAF1 during Fas ligand–mediated apoptosis. FEBS Lett. 2000;468:129–133. doi: 10.1016/s0014-5793(00)01206-0. [DOI] [PubMed] [Google Scholar]
- 32.Moreno-Bueno G., Sanchez-Estevez C., Cassia R., Rodriguez-Perales S., Diaz-Uriarte R., Dominguez O., Hardisson D., Andujar M., Prat J., Matias-Guiu X., Cigudosa J.C., Palacios J. Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: sTK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res. 2003;63:5697–5702. [PubMed] [Google Scholar]
- 33.Llobet D., Pallares J., Yeramian A., Santacana M., Eritja N., Velasco A., Dolcet X., Matias-Guiu X. Molecular pathology of endometrial carcinoma: practical aspects from the diagnostic and therapeutic viewpoints. J Clin Pathol. 2000;62:777–785. doi: 10.1136/jcp.2008.056101. [DOI] [PubMed] [Google Scholar]
- 34.Pallares J., Llobet D., Santacana M., Eritja N., Velasco A., Cuevas D., Lopez S., Palomar-Asenjo V., Yeramian A., Dolcet X., Matias-Guiu X. CK2beta is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am J Pathol. 2009;174:287–296. doi: 10.2353/ajpath.2009.080552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pallares J., Martinez-Guitarte J.L., Dolcet X., Llobet D., Rue M., Palacios J., Prat J., Matias-Guiu X. Survivin expression in endometrial carcinoma: a tissue microarray study with correlation with PTEN and STAT-3. Int J Gynecol Pathol. 2005;24:247–253. doi: 10.1097/01.pgp.0000163849.37129.d4. [DOI] [PubMed] [Google Scholar]
- 36.Dolcet X., Llobet D., Encinas M., Pallares J., Cabero A., Schoenenberger J.A., Comella J.X., Matias-Guiu X. Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281:22118–22130. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- 37.Padmanabha R., Chen-Wu J.L., Hanna D.E., Glover C.V. Isolation, sequencing, and disruption of the yeast CKA2 gene: casein kinase II is essential for viability in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:4089–4099. doi: 10.1128/mcb.10.8.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikkawa U., Mann S.K., Firtel R.A., Hunter T. Molecular cloning of casein kinase II alpha subunit from Dictyostelium discoideum and its expression in the life cycle. Mol Cell Biol. 1992;12:5711–5723. doi: 10.1128/mcb.12.12.5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allende J.E., Allende C.C. Protein kinases: 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 1995;9:313–323. doi: 10.1096/fasebj.9.5.7896000. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed K., Gerber D.A., Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226–230. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- 41.Litchfield D.W. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tawfic S., Yu S., Wang H., Faust R., Davis A., Ahmed K. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16:573–582. doi: 10.14670/HH-16.573. [DOI] [PubMed] [Google Scholar]
- 43.Kim Y., Suh N., Sporn M., Reed J.C. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002;277:22320–22329. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- 44.Poukkula M., Kaunisto A., Hietakangas V., Denessiouk K., Katajamaki T., Johnson M.S., Sistonen L., Eriksson J.E. Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail. J Biol Chem. 2005;280:27345–27355. doi: 10.1074/jbc.M504019200. [DOI] [PubMed] [Google Scholar]
- 45.Chang L., Kamata H., Solinas G., Luo J.L., Maeda S., Venuprasad K., Liu Y.C., Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 46.Velasco A., Bussaglia E., Pallares J., Dolcet X., Llobet D., Encinas M., Llecha N., Palacios J., Prat J., Matias-Guiu X. PIK3CA gene mutations in endometrial carcinoma: correlation with PTEN and K-RAS alterations. Hum Pathol. 2006;37:1465–1472. doi: 10.1016/j.humpath.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Pallares J., Velasco A., Eritja N., Santacana M., Dolcet X., Cuatrecasas M., Palomar-Asenjo V., Catasus L., Prat J., Matias-Guiu X. Promoter hypermethylation and reduced expression of RASSF1A are frequent molecular alterations of endometrial carcinoma. Mod Pathol. 2008;21:691–699. doi: 10.1038/modpathol.2008.38. [DOI] [PubMed] [Google Scholar]
- 48.Lagarda H., Catasus L., Arguelles R., Matias-Guiu X., Prat J. K-ras mutations in endometrial carcinomas with microsatellite instability. J Pathol. 2001;193:193–199. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH769>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 49.Razidlo G.L., Johnson H.J., Stoeger S.M., Cowan K.H., Bessho T., Lewis R.E. KSR1 is required for cell cycle reinitiation following DNA damage. J Biol Chem. 2009;284:6705–6715. doi: 10.1074/jbc.M806457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim M., Yan Y., Kortum R.L., Stoeger S.M., Sgagias M.K., Lee K., Lewis R.E., Cowan K.H. Expression of kinase suppressor of Ras1 enhances cisplatin-induced extracellular signal-regulated kinase activation and cisplatin sensitivity. Cancer Res. 2005;65:3986–3992. doi: 10.1158/0008-5472.CAN-03-2334. [DOI] [PubMed] [Google Scholar]
- 51.Dolcet X., Llobet D., Pallares J., Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 52.Duiker E.W., Mom C.H., de Jong S., Willemse P.H., Gietema J.A., van der Zee A.G., de Vries E.G. The clinical trail of TRAIL. Eur J Cancer. 2006;42:2233–2240. doi: 10.1016/j.ejca.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 53.Carlo-Stella C., Lavazza C., Locatelli A., Vigano L., Gianni A.M., Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res. 2007;13:2313–2317. doi: 10.1158/1078-0432.CCR-06-2774. [DOI] [PubMed] [Google Scholar]
- 54.Falschlehner C., Emmerich C.H., Gerlach B., Walczak H. TRAIL signaling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–1475. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Rippo M.R., Moretti S., Vescovi S., Tomasetti M., Orecchia S., Amici G., Catalano A., Procopio A. FLIP overexpression inhibits death receptor–induced apoptosis in malignant mesothelial cells. Oncogene. 2004;23:7753–7760. doi: 10.1038/sj.onc.1208051. [DOI] [PubMed] [Google Scholar]
- 56.Lee S.H., Kim H.S., Kim S.Y., Lee Y.S., Park W.S., Kim S.H., Lee J.Y., Yoo N.J. Increased expression of FLIP, an inhibitor of Fas-mediated apoptosis, in stomach cancer. Apmis. 2003;111:309–314. doi: 10.1034/j.1600-0463.2003.1110203.x. [DOI] [PubMed] [Google Scholar]
- 57.Korkolopoulou P., Goudopoulou A., Voutsinas G., Thomas-Tsagli E., Kapralos P., Patsouris E., Saetta A.A. c-FLIP expression in bladder urothelial carcinomas: its role in resistance to Fas-mediated apoptosis and clinicopathologic correlations. Urology. 2004;63:1198–1204. doi: 10.1016/j.urology.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Dutton A., O'Neil J.D., Milner A.E., Reynolds G.M., Starczynski J., Crocker J., Young L.S., Murray P.G. Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin's lymphoma cells from autonomous Fas-mediated death. Proc Natl Acad Sci USA. 2004;101:6611–6616. doi: 10.1073/pnas.0400765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X., Jin T.G., Yang H., DeWolf W.C., Khosravi-Far R., Olumi A.F. Persistent c-FLIP(L) expression is necessary and sufficient to maintain resistance to tumor necrosis factor–related apoptosis-inducing ligand-mediated apoptosis in prostate cancer. Cancer Res. 2004;64:7086–7091. doi: 10.1158/0008-5472.CAN-04-1498. [DOI] [PubMed] [Google Scholar]
- 60.Rosato R.R., Almenara J.A., Coe S., Grant S. The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007;67:9490–9500. doi: 10.1158/0008-5472.CAN-07-0598. [DOI] [PubMed] [Google Scholar]
- 61.Panner A., Parsa A.T., Pieper R.O. Translational regulation of TRAIL sensitivity. Cell Cycle. 2006;5:147–150. doi: 10.4161/cc.5.2.2359. [DOI] [PubMed] [Google Scholar]
- 62.Panner A., Nakamura J.L., Parsa A.T., Rodriguez-Viciana P., Berger M.S., Stokoe D., Pieper R.O. mTOR-independent translational control of the extrinsic cell death pathway by RalA. Mol Cell Biol. 2006;26:7345–7357. doi: 10.1128/MCB.00126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panner A., James C.D., Berger M.S., Pieper R.O. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol Cell Biol. 2005;25:8809–8823. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ley R., Balmanno K., Hadfield K., Weston C., Cook S.J. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 65.Corazza N., Jakob S., Schaer C., Frese S., Keogh A., Stroka D., Kassahn D., Torgler R., Mueller C., Schneider P., Brunner T. TRAIL receptor-mediated JNK activation and Bim phosphorylation critically regulate Fas-mediated liver damage and lethality. J Clin Invest. 2006;116:2493–2499. doi: 10.1172/JCI27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han J., Goldstein L.A., Gastman B.R., Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 67.Llobet D, Eritja N, Yeramian A, Pallares J, Sorolla A, Domingo M, Santacana M, Gonzalez-Tallada FJ, Matias-Guiu X, Dolcet X: The multikinase inhibitor sorafenib induces apoptosis and sensitises endometrial cancer cells to TRAIL by different mechanisms. Eur J Cancer 46:836-850 [DOI] [PubMed]
- 68.Krueger A., Schmitz I., Baumann S., Krammer P.H., Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 69.Kataoka T., Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappa B signaling pathway. Mol Cell Biol. 2004;24:2627–2636. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ritt D.A., Zhou M., Conrads T.P., Veenstra T.D., Copeland T.D., Morrison D.K. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007;17:179–184. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 71.Xing H.R., Campodonico L., Kolesnick R. The kinase activity of kinase suppressor of Ras1 (KSR1) is independent of bound MEK. J Biol Chem. 2004;279:26210–26214. doi: 10.1074/jbc.M401323200. [DOI] [PubMed] [Google Scholar]
- 72.Xing H.R., Kolesnick R. Kinase suppressor of Ras signals through Thr269 of c-Raf-1. J Biol Chem. 2001;276:9733–9741. doi: 10.1074/jbc.M008096200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specificity of KSR1 shRNA and antibodies. IK cells were infected with lentiviruses carrying two different shRNAs targeting KSR1 (KSR1 1.3 and KSR1 2.6). A: After 3 days to enable down-regulation of the protein, cells were lysed, and KSR1 was analyzed at Western blotting. B: Parallel cultures were infected, and after 3 days were trypsinized, embedded in agarose, and fixed. Immunohistochemistry of KSR1 was performed (see “Materials and Methods”).
KSR1 shRNA sensitizes HEC-1-A and KLE cells TRAIL-induced apoptosis. A: IK cells were infected with increasing amounts of lentiviruses carrying KSR1 shRNA or FSV empty vector. Western blot analysis demonstrated that progressive KSR1 down-regulation caused TRAIL-induced progressive decrease in FLIP levels and a consistent increase in caspase 3 activation. B: KLE and HEC-1-A endometrial carcinoma cell lines were infected with lentiviruses carrying shRNA targeting KSR1. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL for 8 hours, and nuclei were visualized via Hoechst staining. Results are given as the percentage of apoptotic nuclei compared with the total number of nuclei. Bottom: Representative images of Hoechst-stained IK cells. C: KSR1 shRNA plus 50 ng/mL TRAIL activated caspases 8, 9, and 3. KLE cells were infected with lentiviruses carrying shRNA to KSR1 2.6. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL. After 8 hours, cells were lysed, and protein extracts were analyzed at Western blotting with antibodies to the indicated caspases. Membranes were reprobed with tubulin to ensure equal amounts of protein and antibodies to KSR1 to check inhibition by the specific shRNA.
KSR1 shRNA triggers TRAIL-induced down-regulation of FLIP. KLE and HEC-1-A endometrial carcinoma cell lines were infected with lentiviruses carrying shRNA 2.6 targeting KSR1. After 3 days to enable down-regulation of the protein, cells were treated with 50 ng/mL TRAIL for 8 hours and lysed. FLIP expression was analyzed at Western blotting.