

Abstract
We have found a B2 repeat insertion in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6) in a mouse that developed a skin disorder with clinical and histopathological features resembling those seen in human neutrophilic dermatoses. Neutrophilic dermatoses are a group of complex heterogeneous autoinflammatory diseases that all demonstrate excessive neutrophil infiltration of the skin. Therefore, we tested the cDNA and genomic DNA sequences of PTPN6 from patients with Sweet's syndrome (SW) and pyoderma gangrenosum and found numerous novel splice variants in different combinations. Isoforms resulting from deletions of exons 2, 5, 11, and 15 and retention of intron 1 or 5 were the most common in a patients with a familial case of SW, who had a neonatal onset of an inflammatory disorder with skin lesions and a biopsy specimen consistent with SW. These isoforms were associated with a heterozygous E441G mutation and a heterozygous 1.7-kbp deletion in the promoter region of the PTPN6 gene. Although full-length PTPN6 was detected in all other patients with either pyoderma gangrenosum or SW, it was always associated with splice variants: a partial deletion of exon 4 with the complete deletion of exon 5, alterations that were not detected in healthy controls. The defect in transcriptional regulation of the hematopoietic PTPN6 appears to be involved in the pathogenesis of certain subsets of the heterogeneous group of neutrophilic dermatoses.
Among more than 100 genes of the large family of protein tyrosine phosphatases (PTPs), PTP nonreceptor type 6 (PTPN6, SHP1; OMIM 176883) has a critical function in the regulation of the myelopoietic system. PTPN6 is encoded by 17 exons and has a unique structure with two SH2 domains. Both SH2 domains (SH2N encoded by exons 2 and 3 and SH2C encoded by exons 4 and 5) are required for binding to the phosphorylated tyrosine residues of target proteins, whereas the catalytic (phosphatase) domain (exons 7 through 13) is responsible for enzyme activity.1 Another notable feature of the PTPN6 gene is that it has two promoter regions. The two translation start sites (ATG) are 7 kbp apart, the “longer” form (PTPN6 1A) is expressed mostly in epithelial cells, whereas the slightly shorter transcript (PTPN6 1B) is expressed only in hematopoietic cells (Figure 1A).2,3
Figure 1.

Detailed analysis of the PTPN6 gene in a patient with familial SW (patient SW1) and PCR detection of splice variants in an additional seven patients with SW. A: Schematic of the PTPN6 transcript. Two transcripts of the epithelial (with exon 1A) and hematopoietic (with exon 1B) forms are indicated; whereas the other 16 exons are the same in both forms. The symbol // indicates the site of a 1.7-kbp intronic deletion between exons 1A and 1B in patient SW1 and his father, which may affect the transcription of the hematopoietic form in combination with a SNP (E441G, shown in the red box) involving the catalytic domain of PTPN6. Arrows (above the schematic structure) show the locations of forward and reverse primers in the flanking exons used to amplify PCR fragments of exon 5 (421 bp encoding part of the SH2C domain of PTPN6 protein) and exon 11 (497 bp within the PTP domain). The protein domains SH2N (blue line), SH2C (green line), and PTP domain (red line) are indicated under the corresponding coding exons. Primer sequences are listed in Table 1. B: PCR products showing the most abundant alterations, including deletions of exons 2, 5, 11, and 15 in patient SW1 (P) and in a healthy control (H). In each panel, the top arrow shows the PCR product without any exon deletion, and the bottom arrow shows the smaller PCR fragment with an exon deletion. The sequence of the exon deletion site is shown on the right of each panel. C: Schematic of the different splicing variants of PTPN6 in patient SW1, his mother, his father, and a healthy control. Transcripts were semiquantitatively detected with overhanging primers (listed in Table 1). The horizontal green arrows show the locations of the primers and red triangles indicate the deleted exons. The vertical red arrows point to the location of the native stop codon in exon 15, whereas the small red crosses represent premature stop codons. All agarose electrophoreses are in black background, and the ethidium bromide-stained PCR bands are white. D: Western blot comparing the “full-length” PTPN6 in cell lysates of a healthy individual, patient SW1, and the mother of patient SW1. E: PCR amplification of cDNA from patients with SW (patients SW2 through SW8) using a forward primer in exon 4 and a reverse primer in exon 14. The 1089-bp PCR product and three shorter splice variants are indicated by arrows.
PTPN6 plays an important role in cell proliferation and signaling that involves cells of the innate and adaptive immune systems.4–6 The absence or impaired function of Ptpn6 in the homozygous state causes the development of the motheaten phenotype in mice, an autosomal recessive condition with focal skin inflammation and the patchy absence of hair. Failure of neutrophils to undergo apoptosis results in the accumulation of these cells in the peripheral blood, skin, lung, and spleen of affected mice (See Nesterovitch et al7 in this issue of The American Journal of Pathology). A pathologically similar extensive skin infiltration by neutrophils is present in pyoderma gangrenosum (PG) and Sweet's syndrome (SW), two relatively uncommon neutrophilic dermatoses of unknown origin. SW (acute febrile neutrophilic dermatosis) appears in several clinical forms: idiopathic, tumor associated, postinfectious, and drug induced (eg, after administration of granulocyte-macrophage colony-stimulating factor).8 Both SW and PG have strong associations with hematological tumors.8 Recent studies have shown that patients with lymphoma and leukemia had heavily methylated promoter P2 (upstream of exon 1B) in the PTPN6 gene, causing the absence of PTPN6 protein.9,10 Also, a point mutation in the PTPN6 gene leading to amino acid changes has been described in acute lymphoid leukemia.11 Splicing variants of the human PTPN6 transcript with retention of introns 1, 2, or 3 cause frameshift and premature stop codons, resulting in truncated proteins in patients with acute myeloid leukemia and lymphoma.11,12 Overall, the lack or diminished function of PTPN6 appears to be involved in different forms of lymphoma and leukemia, and changes in PTPN6 function warrant consideration as a potential etiological factor in different forms of neutrophilic dermatoses. In the current study, we investigated the PTPN6 gene for potential abnormalities in patients with idiopathic PG and SW.
Materials and Methods
Patients and Peripheral Blood Samples
During the past 4 years, we collected peripheral blood samples from 14 consenting patients (patients PG1 through PG14) diagnosed as having PG at Rush University Dermatology Clinic (Chicago, IL), 1 patient (patient SW1) with a familial inflammatory disorder with a skin biopsy specimen resembling SW13 from Children's Hospital (Detroit, MI), and 8 patients (patients SW2 through SW9) with SW from the Dermatology Clinic at the University of Michigan (Ann Arbor, MI). Available clinical information about the patients is summarized in Supplemental Table S1 at http://ajp.amjpathol.org. In addition, peripheral blood samples from nine healthy individuals (age range, 32 to 60 years) were used as controls. Blood (15 mL) was collected into Vacutainer tubes with Lithium Heparin (BD, Franklin Lakes, NJ) and processed within 1 to 2 hours (PG patients) or shipped overnight in a ThermoSafe Diagnostic Shipper (Tegrant Co., Arlington Heights, IL) and processed the next morning. Collection of blood was approved by the institutional review board of Rush University Medical Center (Chicago, IL), University of Michigan Medical School (Ann Arbor, MI), and Wayne State University, Children's Hospital (Detroit, MI). Plasma was separated, snap frozen, and stored at −80°C. Erythrocytes were lysed in cold water, and the white blood cells were washed with calcium- and magnesium-free PBS and used for flow cytometric analysis and isolation of RNA, genomic DNA (gDNA), and protein.
Isolation of RNA and DNA and Methods for RT-PCR
For RNA and gDNA isolation, we used the QIAamp RNA Blood Mini-kit and the QIAamp DNA Blood Midi-kit, respectively, following the manufacturer's guidelines (Qiagen, Valencia, CA). The quality of RNA was tested using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Total RNA samples (1 μg) were pretreated with DNase I (Invitrogen, Carlsbad, CA) and reverse transcribed with oligo(dT) primer using the SuperScript First-Strand Synthesis System (Invitrogen) following the manufacturer's instructions. PCR fragments were amplified from cDNA or gDNA using the Core PCR Kit (Qiagen). The PCR primers (listed in Table 1) were designed to be inside of flanking exons or overhanging two distant exons; for an example, the forward primer overhanging exons 1 and 3 (skipping exon 2) were paired with a reverse primer in exon 5 (Figure 1C). PCR products were separated in a 1.5% agarose gel, purified with the GeneClean Spin kit (MP, Solon, OH) and sequenced on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA) with the BigDye Terminator Cycle Sequencing Ready Reaction kit v.2.0 (Applied Biosystems). Sequence chromatograms were analyzed using the Vector NTI Advance 11 software (Invitrogen).
Table 1.
PTPN6 PCR Primers Used in the Study
| PTPN6 | Primers | Sequence | cDNA, bp WT | cDNA, bp minus exon | gDNA, bp retention |
|---|---|---|---|---|---|
| Exon-specific primers used in Figure 1B | |||||
| ex2del | ex1fw1 | 5′-CCCAGGCCAGTGGAGTGGCAG-3′ | 461 | 338 | 800 |
| ex3rv8 | 5′-GGGTACTTGAGGTGGATGATGG-3′ | ||||
| ex3del | ex2fw1 | 5′-GTGGTTTCACCGAGACCTCAGT-3′ | 498 | 303 | 3367 |
| ex4rv5 | 5′-ACCTTGATGTGGGTGACC-3′ | ||||
| ex4del | ex3fw2 | 5′-GGTGGAGTACTACACTCAGCA-3′ | 409 | 219 | 3192 |
| ex5rv5 | 5′-CTGCCGCAGGTAGACAAAGG-3′ | ||||
| ex5del | ex4fw2 | 5′-GTGGTACCATGGCCACATGTC-3′ | 421 | 304 | 681 |
| ex6rv1 | 5′-CTCAAACTCCTCCCAGAAGCCA-3′ | ||||
| ex6del | ex5fw1 | 5′-GGTGGACGCTACACAGTGGGT-3′ | 320 | 206 | 598 |
| ex7rv4 | 5′-ATGTTCTTGTAGCGGTTCTTGC-3′ | ||||
| ex7del | ex6fw1 | 5′-AGGGTGAATGCGGCTGACATTG-3′ | 240 | 143 | 799 |
| ex8rv1 | 5′-GATGTTACTGTCCCGTCC-3′ | ||||
| ex8del | ex7fw2 | 5′-CAGGAGGTGAAGAACTTGC-3′ | 312 | 232 | 894 |
| ex9rv1 | 5′-GCCTTTCTCCACCTCTCGGGTG-3′ | ||||
| ex9del | ex8fw9 | 5′-TGACCACAGCCGAGTGATCC-3′ | 361 | 211 | 1634 |
| ex10rv1 | 5′-ATTGTCCAGCGGGGAGACCTG-3′ | ||||
| ex10del | ex9fw1 | 5′-GAACAGCCGTGTCATCGTCATG-3′ | 300 | 168 | 1518 |
| ex11rv1 | 5′-CTTTCCTGCCGCTGGTTGATCT-3′ | ||||
| ex11del | ex10fw1 | 5′-AACAAATGCGTCCCATACTGG-3′ | 341 | 186 | 2327 |
| ex12rv1 | 5′-TTCTCCATGAGCATGTCG-3′ | ||||
| ex12del | ex11fw1 | 5′-GGAGACCTGATTCGGGAGATCT-3′ | 365 | 297 | 2311 |
| ex13rv7 | 5′-TCCAGCTTCTTCTTAGTGG-3′ | ||||
| ex13del | ex12fw1 | 5′-CCATCATTGTCATCGACATGC-3′ | 260 | 108 | 457 |
| ex14rv2 | 5′-GGCATTCTTCATGGCTGG-3′ | ||||
| ex14del | ex13fw7 | 5′-GACTGTGACATTGACATCC-3′ | 331 | 239 | 805 |
| ex15rv3 | 5′-TGCTCTTCTCCTTGTCTGC-3′ | ||||
| ex15del | ex14fw1 | 5′-TCGCAGAAGGGCCAGGAGT-3′ | 410 | 270 | 960 |
| ex16rv6 | 5′-GGGCTTTATTTACAAGAGG-3′ | ||||
| Overhanging primers used in Figure 1C | |||||
| Exon 2 | ex1-3fw | 5′-CCAGGATGGTGAG–GGTGGG-3′⁎ | |||
| Exon 3 | ex2-4fw | 5′-CTCTCCGTCAG–GTGGTACCATG-3′ | |||
| Exon 4 | ex3-5fw | 5′-TCCGATCCCACTAGTGAGAG–GGTGGAC-3′ | |||
| Exon 4 + 5 | ex3-6fw | 5′-TCCGATCCCACTAGTGAGAG–CCGTAC-3′ | |||
| Exon 5 | ex4-6fw | 5′-ACATCAAGGTCATGTGCGAG–CCGTAC-3′ | |||
| Exon 6 | ex5-7fw | 5′-ACCTGCGGCAG–AGTTTGCAGAAG-3′ | |||
| Exon 7 | ex6-8fw | 5′-GAGGAGTTTGAG–TTGACCACAGC-3′ | |||
| Exon 8 | ex9-7rv | 5′-GCAGCTGGTT–AGGGGAGAATGTTC-3′ | |||
| Exon 9 | ex10-8rv | 5′-GGGACGCATTTGTT–CTTGATGTAG-3′ | |||
| Exon 10 | ex11-9rv | 5′-TCCCGAATCAGGTCTCC–CCGGCCTTT-3′ | |||
| Exon 11 | ex12-10rv | 5′-GGCCGATGCCGGCG–ATTGT-3′ | |||
| Exon 10 + 11 | ex12-9rv | 5′-GATGCCGGCG–CCGGCCTTTCT-3′ | |||
| Exon 12 | ex13-11rv | 5′-AGTCCAGGC–CTGCAGTGCACG-3′ | |||
| Exon 13 | ex14-12rv | 5′-CCTTCTGCGA–CCTTGGTGGAG-3′ | |||
| Exon 14 | ex15-13rv | 5′-CCTCCTTGTGT–CTGCAGGACC-3′ | |||
| Exon 15 | ex16-14rv | 5′-ACAGGGTCAGGGCTGAGG–TTGGA-3′ | |||
| A | SB715Fw | 5′-GGTGTTGATGTTTGTATTTGGAATTA-3′ | |||
| SB715Rv | 5′-CATCTCTCCCTCCTCCACC-3′ | ||||
| B | ex1fw | 5′-ATCTGAGGCTTAGTCCCTGAGCT-3′ | |||
| ex15rv | 5′-TGAGGACAGCACCGCTCACTT-3′ | ||||
ex, exon; fw, forward primer; rv, reverse primer; intron (n-1)/ intron n/ both introns retention PCR product size; WT, wild type.
A dash sign in primer sequences represents a border between two flanking exon sequences. PCR were run in 25 μL of total reaction volume with an initial denaturing conditions of 95°C for 5 minutes, followed by 35 cycles of 96°C for 30 seconds, 55°C for 20 seconds, 72°C for 30 seconds, finishing with a final 72°C incubation period for 4 minutes in C1000 Thermo Cycler (Bio-Rad, Hercules, CA).
Protein Isolation, Western Blot Analysis, and Phosphatase Activity Assay
White blood cells (5 × 106) were lysed in 0.5 mL of cold RIPA buffer containing the Halt Protease Inhibitors Cocktail (both from Thermo Scientific, Rockford, IL) and then sonicated on ice for 15 seconds using a Virsonic Digital 550 (VirTis, Gardiner, NY). The protein content was measured using the Bicinchoninic Acid Protein Assay kit (Pierce/Thermo Scientific).
Proteins (10 μg per lane) were separated on SDS polyacrylamide gel electrophoresis (10%) gels and electrophoretically transferred onto nitrocellulose membranes (BioRad Laboratories, Hercules, CA). The membranes were incubated with peroxidase-conjugated rabbit anti-PTPN6 antibody (C-19) (Santa Cruz Biotechnology, Santa Cruz, CA) or with rabbit anti-PTPN6 antibody (ab2020) (Abcam, Cambridge, MA) followed by peroxidase-conjugated anti-rabbit secondary antibody. The bands were visualized by enhanced chemiluminescence (Pierce/Thermo Scientific).
Proteins (10 μg) of white blood cell lysates from patients PG2 through PG9 were analyzed with the RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (Invitrogen) preloaded with 6,8-difluoro-4-methyllumbelliferyl phosphate according to the manufacturer's instructions. The results of the phosphatase activity assay were statistically analyzed using the Student's t-test and the one-way analysis of variance to compare means. All statistical analyses were performed using the SPSS (version 16.0) statistical software package (SPSS, Chicago, IL). P < 0.05 was considered statistically significant.
Cytokine Measurements
Duplicate plasma samples were tested for the following factors: IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, interferon-γ, TNF-α, TNF receptor-1, TNF receptor 2, monocyte chemoattractant protein-1, lymphocyte inhibitory factor, regulated on activation normal T-cell expressed and presumably secreted, granulocyte/macrophage colony-stimulating factor, granulocyte colony-stimulating factor, and Fas-L (TNF superfamily member 6). Samples were measured with Cytometric Bead Arrays (BD Biosciences, San Diego, CA) in a FACS Canto-II flow cytometer and analyzed using FCAParray software (BD Biosciences).
Results
Combination of Heterozygous Mutations of the PTPN6 Gene in the Familial Case with Skin Lesions Resembling SW
The most abundant abnormalities in cDNA and gDNA were found in patient SW1.13 We sequenced cDNA isolated from the peripheral blood of this patient and found a nonsynonymous heterozygous mutation (resulting in an E441G replacement) in exon 11 of PTPN6 (Figure 1A). The amino acid glutamate E441 is located distal to the α3 loop and proximal to the β12 sheet of the phosphatase catalytic center of the PTPN6 protein.14 The E441G mutation changes the hydrophilic, negatively charged amino acid glutamate to the hydrophobic nonpolar, aliphatic amino acid glycine, thereby potentially affecting the tertiary structure of PTPN6.14 The mother of patient SW1, who had no history of skin disease, had the same heterozygous mutation (E441G), whereas the father of the patient did not have this mutation. However, the gDNAs from both patient SW1 and his father had 1.7-kbp deletions (Figure 1A) 1418 bp upstream of the PTPN6 P2 transcription initiation site.15,16 This deletion also occurs in healthy individuals with a reported heterozygous frequency for this polymorphism of 0.18 in the white population.15 In summary, patient SW1 had both a heterozygous nonsynonymous E441G mutation and a heterozygous 1.7-kbp deletion (Figure 1A). In addition, we found polymorphisms in the PTPN6 gene sequence of SW1 and his father's DNA, but none of them altered the amino acid sequence (data not shown).
Splicing Variants of the PTPN6 Transcript in SW1
The full-length native PTPN6 transcript (1894 bp) amplified with PCR primers in exons 1B and 15 (Table 1) was present in the parents of patient SW1 and in healthy controls but was almost undetectable in an agarose gel using cDNA from SW1 (Figure 1B). Therefore, the PCR product mixture was cloned into the TOPO TA-vector and sequenced. We found 24 different PTPN6 splice variants in the cDNA sample of patient SW1. Only two of 43 sequenced clones of SW1 had no exon splicing. Ten of 43 clones had frameshifts with premature stop codons that might be translated into a truncated protein lacking a phosphatase domain or no protein at all.
When amplified with primers located in the flanking exons (Table 1), we were able to detect and sequence all 17 exons of the PTPN6 transcript in patient SW1. Although all exons were present in intact form as well, we also found PCR fragments with deletions of exons 2, 3, 4, 5, 11, 12, and 15. Exons 6, 7, 8, 9, 10, 13, and 14 were always intact and were never spliced out. The variants with a deletion of exons 2, 5, 11, or 15 (Figure 1B) and retention of intron 1 and intron 5 (data not shown) were the most abundant splicing variants. Intron 1 retention appeared to be a relatively frequent mutation in PTPN6 and has been described in the T-lymphoma cell line HUT78 originating from a patient with Sézary syndrome,12,17 in a T-cell line (KIT225) from a patient with chronic T-lymphoid leukemia,12 in the human T-lymphoblastic lymphoma cell line SUPT-1,12 and in the early germ cell line JKT.12 Interestingly, cell lines HUT78 and KIT225 had no native PTPN6 transcripts, and the only detected form was the abnormal splicing variant with intron 1 retention.12
Next, we used overhanging PCR primers in different combinations (Table 1) to identify splicing variants in patient SW1 (Figure 1C). We detected a single deletion of exons 2, 5, 11, or 15, a double deletion of exons 4 and 5 or 10 and 11, and a triple deletion of exon 5 plus exons 10 and 11 (Figure 1C). The maternal sample had an exon-skipping PCR profile similar to that of the patient, but the paternal sample carried fewer variants. The splicing variants with deletions of exons 11, 15, or 10 and 11 were also detected in the cDNA samples of some healthy controls (Figure 1C). Corresponding to these results, PTPN6 protein was hardly detectable in patient SW1, with a smaller amount in his mother, when compared with leukocyte cell lysates of healthy individuals (Figure 1D).
The Coding Sequence of PTPN6 in Patients with SW and PG
Significantly fewer splice variants occurred in other patients with SW (patients SW2 through SW8) (Figure 1E) and PG (patients PG1 through PG13) (Figure 2), and the full-length coding sequence of PTPN6 was present in all 14 PG and 8 nonfamilial SW patients. The full-length transcripts were present in a comparable amount in all cases. We also sequenced the entire PTPN6 gene from gDNA isolated from peripheral blood leukocytes of patients with PG or SW. We found no alterations in the coding nucleotide sequences that could change the amino acid sequences, except in patient SW1 (E441G). However, a number of single nucleotide polymorphisms (SNPs) were detected, which may not necessarily be specific to either PG or SW (data not shown). In patient PG2, two SNPs in introns 1B and intron 8 were located in close proximity (within 6 bp) to exon-intron boundaries, but these most likely had no effect on intron splicing.
Figure 2.
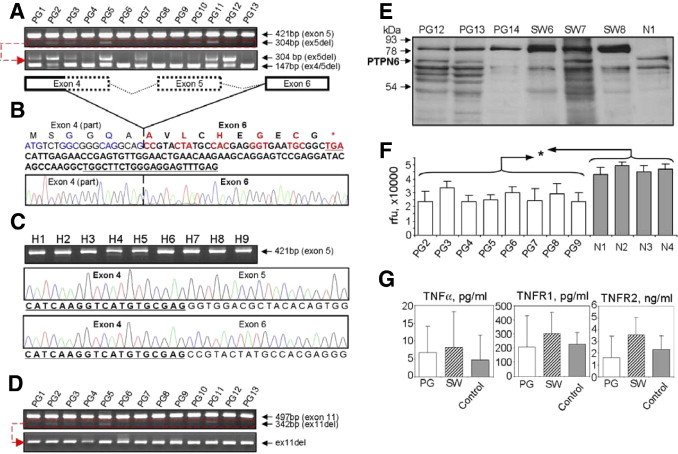
Analysis of the PTPN6 gene in patients with PG. A: PCR products in a 1.5% agarose gel show the presence of two fragments [one with native exon 5 present (421 bp) and one with exon 5 deleted (ex5del, 304 bp)] in PG samples (top panel).The bottom gel image shows nested PCRs with amplification of the splicing variants with either an exon 5 deletion (ex5del, 304 bp) or with a partial exon 4 and full exon 5 deletion (ex4part/5del, 147 bp). B: Amino acid and nucleotide sequences of the splicing variant with a deletion of part of exon 4 (regular font) and the entire exon 5 (dotted lines on the schematic) with a frameshift and a premature stop codon (TGA, asterisk). Amino acids with their coding nucleotides (triplets) are shown in alternating blue and black fonts (part of exon 4) and in red and black boldface fonts (exon 6). Five nucleotides (3′ end) of the forward and reverse primers used to amplify this 147-bp PCR fragment are underlined (all primers are listed in Table 1). C: PCR panel of nine healthy individuals and corresponding sequences showing the normal exons 4 and 5 sequences or the rare splice variant having no exon 5 but complete sequence of exon 4 followed by exon 6. D: The bottom two images of PCR products of 13 PG patients show the presence of two fragments with native exon 11 present (497 bp) and with exon 11 deleted (ex11del, 342 bp, top panel). The small amounts of PCR products were extracted from agarose gels and reamplified to demonstrate an exon 11 deletion in all samples (bottom panel) with a forward primer in exon 10 and a reverse primer in exon 12. E–F: Analysis of leukocyte PTPN6 in PG and SW patients. E: Western blotting of leukocyte cell lysates of three PG and three SW patients and one control sample stained with rabbit anti-PTPN6 antibody (Abcam, ab2020, see Supplemental Table S2 at http://ajp.amjpathol.org). The full-length PTPN6 (68 kDa) is indicated by an arrow. F: Relative fluorescence levels (phosphatase PTPN6 activities) in leukocyte cell lysates of patients PG2 through PG9 compared with the four normal samples. All PG samples expressed significantly reduced (*P < 0.05) phosphatase activity (relative fluorescence units) compared with any of the four healthy individuals. G: Serum/plasma levels of 3 TNF family members in patients with PG (n = 14) or SW (n = 9) and healthy individuals (n = 9) measured by flow cytometric bead array. Because of the low number of samples and large variations in patients, the differences did not reach significance. Although individual plasma samples occasionally showed large differences, when the data were pooled, significant differences disappeared between patients and healthy controls. This result may be due to the heterogeneous sample collection and was characteristic of some other cytokines as well (eg, IL-1β, IL-4, IL-10, etc).
Additional Exon Splicing Variants of PTPN6: Partial Deletion of Exon 4 with Complete Deletion of Exon 5 Is a Characteristic Alteration in Patients with PG or SW
We detected PCR fragments with deletions of exon 5 (SH2C domain) and exon 11 (PTP domain), similar to those found in patient SW1 (Figures 1, B and C), in cDNA samples from other patients with SW (patients SW3, SW4, SW7, and SW8) (Figure 1E) or PG (patients PG2, PG3, PG5, PG10, PG11, PG12, and PG13) (Figure 2, A and B). The exon 5 deletion does not cause a frameshift, but it results in the translation of a shorter protein with a 41-amino acid deletion in the SH2C domain. In contrast, the exon 11 deletion causes a frameshift and a premature stop codon in exon 13, thus translating the sequence into a truncated protein having only the SH2N and SH2C domains. gDNA sequencing identified no mutations that could interfere with normal splicing in either intron 4 or 5 or in boundaries between exon 5 and the adjacent introns. We also sequenced the exon-intron boundaries of exon 11 and completely sequenced introns 10 and 11 in all of the samples and found two SNPs in intron 11: rs2071079 (319 bp upstream of exon 11) and rs11608598 (482 bp downstream of exon 12), which seem too far from exon-intron boundaries to interfere with intron splicing.
We isolated and purified cDNA with an exon 11 deletion (342-bp band) from all PG and SW samples (Figure 2D). Even if the lower band was hardly visible on the agarose gel after the first PCR, we repeated the PCR with the same primers and found exon 11 in all of the samples (including healthy controls) (Figure 2D). Using visualized or technically invisible PCR products as templates with repeated amplification with the same primers, we also found that all PG (Figure 2A) and SW samples (data not shown) had a partial deletion of exon 4 and complete deletion of exon 5 (Figure 2B). The deletion of the 3′-end half of exon 4 plus exon 5 caused a frameshift that led to a premature stop codon in exon 6 (Figure 2B). These changes were not detected in any of the nine healthy samples, although the exon 5 was also spliced out in less than 5% of the total PTPN6 transcript, but this deletion did not generate frameshift and premature stop codon in the translated protein (Figure 2C).
PTPN6 Proteins and Phosphatase Activities in Patients with PG and SW
PTPN6 is a cytoplasmic 68-kDa protein. Western blotting of patients PG12, PG13, and SW7 with anti–PTPN6-specific antibodies (C-19) detected the presence of the 68-kDa band along with numerous smaller bands that could represent alternative splicing variants (Figure 2E). The samples from patients PG14, SW6, and SW8 did not have detectable amounts of the 68-kDa protein (Figure 2E) (normalized to β-actin). Interestingly, the samples from patients PG12, PG13, PG14, SW6, SW7, and SW8 had another major band at approximately 80 kDa that could be detected with two different anti-PTPN6 antibodies (C-19 and ab2020). When tyrosine phosphatase activity in the PG samples was assessed, we found that all of the patient samples (patients PG2 thorough PG9) had significantly lower (1.3- to 1.9-fold) levels of phosphatase activity (P < 0.05) than did healthy controls (Figure 2F). Cytokine analysis did not show any significant specificity for any disease group (Figure 2G), except for a few newly diagnosed cases in patients with PG (data not shown).
Discussion
Neutrophilic dermatoses constitute a group of rare skin diseases that potentially represent autoinflammatory conditions. Autoinflammatory diseases are arising from mutations of genes that regulate innate immunity and are characterized by recurrent, unprovoked inflammatory events without evidence of autoimmunity or infection.18–21 PG occurs in approximately one to five per 100,000 people per year. In a period of 3 years, 14 new cases of PG (of 37,000 dermatology patient visits per year) were diagnosed at Rush University Dermatology Clinic (Chicago, IL). SW is even less frequent, estimated to involve one to two patients per 10,000 new dermatology cases,22,23 and only a few hundred cases have been reported thus far.24 Approximately 10% to 20% of all SW cases have been associated with hematopoietic tumors.8 Increased tyrosine phosphorylation of intracellular proteins was detected in neutrophils from patients with PG25 or other autoinflammatory skin diseases,26 which suggests a possible defect(s) in tyrosine phosphatase function. Aberrant splicing forms and point mutations in PTPs, including PTPN6, have been reported in a number of hematopoietic tumors (leukemia and lymphoma) associated with neutrophilic dermatoses.8 One possible explanation for the development of neutrophilic dermatoses could be the prolonged survival of otherwise short-lived neutrophils, due to the aberrant phosphorylation/dephosphorylation of the apoptosis-related enzyme caspase 8.27 Although the ratios of early and late apoptotic/dead (annexin V+/−/7-AAD+) neutrophils in one patient with PG and another with SW were comparable to age-matched healthy controls, we found fewer apoptotic and dead neutrophils after 24 hours of cultures of these patients' cells compared with controls (data not shown).
Homozygous motheaten mice with mutations in the Ptpn6 gene exhibit neutrophil infiltration in the skin, similar to the phenotype seen in PG and SW patients.7 Different mutations in the Ptpn6 gene in different phenotypes of motheaten mice are inherited in an autosomal recessive pattern, where heterozygous animals do not have skin lesions.7,28–30 The motheaten phenotype also appears in double-mutant immunodeficient (scid,31 xid,32 nude,33 or Rag-1–deficient34) mice that are also homozygous for a Ptpn6 gene deficiency. These observations suggest that a Ptpn6 deficiency could alter innate immunity, leading to an autoinflammatory phenotype, similar to that seen in neutrophilic dermatoses.35–37 However, although motheaten mice carry inherited mutations in the Ptpn6 gene in all cell types (monogenic disease), patients with neutrophilic dermatoses may be more likely to acquire somatic mutations in a limited number of cell types, generally in cells of hematopoietic origin (monocytes, lymphocytes, and granulocytes). This is supported by the “clonal restriction” effect described for the monoclonal/oligoclonal accumulation of neutrophil populations in patients with SW or PG,22,38 as well as the therapeutic effect of allogeneic bone marrow transplantations in PG patients39,40 and successful stem cell transplantation in SW patients.41
Animals carrying either a single homozygous mutation (Ptpn6spin/spin mice30) or a heterozygous combination of two different mutations of Ptpn6 (Ptpn6meb2/me, Ptpn6meb2/mev, or Ptpn6mev/me mice) develop extensive tissue neutrophil infiltration.7 Thus, it is conceivable that patient SW1's combination of a heterozygous mutation (E441G) in the catalytic domain of PTPN6 and a 1.7-kbp deletion resulting in a change of PTPN6 expression15 could be responsible for the development of his disease. In contrast to the case of patient SW1, we were unable to find similar mutations in PTPN6 cDNA and gDNA sequences from peripheral blood leukocytes in 14 PG and 8 SW patients. Rather, we found a large number of splice variants of PTPN6 in almost all of these patients. Most of these splice variants could not be translated into functionally active PTPN6 (Figures 1C and Figure 3B). PTPN6 phosphatase activity was reduced in leukocytes of patients with PG and SW, most likely due to the large number of isoforms competing with the native form of PTPN6. Because the gDNA sequences had no mutation that could explain the aberrant splicing, we hypothesize that the splicing machinery controlling PTPN6 transcription might be affected by other gene(s).
Additional genes may be involved in the pathogenesis of different forms of neutrophilic dermatoses, and PTPN6 is probably only one of these candidate genes. A polygenic combination leading to altered PTPN6 function may result in different forms of neutrophilic dermatoses. Familial PG and SW have been reported,13,42–46 and SW has been associated with familial Mediterranean fever.47–49 PG and SW are also linked to many autoimmune and hematopoietic diseases,8,21,49–53 suggesting the polygenic character of these disorders, in contrast to the monogenic inheritance of mice with the motheaten phenotype. This may explain the extremely rare familial association of PG and SW. The heterogeneous clinical phenotypes, even within one group of neutrophilic dermatoses, the variability of response to therapy, and the diversity and subtypes of the neutrophilic dermatoses suggest that more than one gene is involved in the pathogenesis. Nonetheless, the findings in motheaten mice and patients with SW or PG suggest that PTPN6 function is important in certain subtypes of neutrophilic dermatoses, but alterations in other genes is also necessary for the full clinical expression of these disorders.
Acknowledgments
We thank Anita C. Gilliam, M.D., Ph.D. (Dermatology, Case Western Reserve University), and Andrea Gonda (Dermatology, University of Debrecen, Health Science Center, Hungary) for the paraffin-embedded sections from patients with SW.
Footnotes
Supported by The Grainger Foundation (Forest Park, IL) (T.T.G.) and in part by The Clark W. Finnerud, M.D., endowed chair (Department of Dermatology) (M.D.T.), and J.O. Galante, M.D., D.Sc., endowed chair (Department of Orthopedic Surgery, Rush University Medical Center, Chicago, IL) (T.T.G.).
The authors did not disclose any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2010.12.035.
Supplementary data
References
- 1.Banville D., Stocco R., Shen S.H. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics. 1995;27:165–173. doi: 10.1006/geno.1995.1020. [DOI] [PubMed] [Google Scholar]
- 2.Yi T.L., Cleveland J.L., Ihle J.N. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol Cell Biol. 1992;12:836–846. doi: 10.1128/mcb.12.2.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wlodarski P., Zhang Q., Liu X., Kasprzycka M., Marzec M., Wasik M.A. PU. 1 activates transcription of SHP-1 gene in hematopoietic cells. J Biol Chem. 2007;282:6316–6323. doi: 10.1074/jbc.M607526200. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–359. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasler P., Zouali M. B cell receptor signaling and autoimmunity. FASEB J. 2001;15:2085–2098. doi: 10.1096/fj.00-0860rev. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J., Somani A.-K., Siminovitch K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signaling. Semin Immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 7.Nesterovitch A.B., Szanto S., Gonda A., Bardos T., Kis-Toth K., Adarichev V.A., Olasz K., Ghassemi-Najad S., Hoffman M.D., Tharp M.D., Mikecz K., Glant T.T. Spontaneous insertion of a B2 element in the Ptpn6/Shp1 gene drives a systemic autoinflammatory disease in mice resembling neutrophilic dermatosis in humans. Am J Pathol. 2011;178:1701–1714. doi: 10.1016/j.ajpath.2010.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen P.R. Sweet's syndrome–a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34–62. doi: 10.1186/1750-1172-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oka T., Ouchida M., Koyama M., Ogama Y., Takada S., Nakatani Y., Tanaka T., Yoshino T., Hayashi K., Ohara N., Kondo E., Takahashi K., Tsuchiyama J., Tanimoto M., Shimizu K., Akagi T. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–6394. [PubMed] [Google Scholar]
- 10.Oka T., Yoshino T., Hayashi K., Ohara N., Nakanishi T., Yamaai Y., Hiraki A., Sogawa C.A., Kondo E., Teramoto N., Takahashi K., Tsuchiyama J., Akagi T. Reduction of hematopoietic cell-specific tyrosine phosphatase SHP-1 gene expression in natural killer cell lymphoma and various types of lymphomas/leukemias: Combination analysis with cDNA expression array and tissue microarray. Am J Pathol. 2001;159:1495–1505. doi: 10.1016/S0002-9440(10)62535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beghini A., Ripamonti C.B., Peterlongo P., Roversi G., Cairoli R., Morra E., Larizza L. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet. 2000;9:2297–2304. doi: 10.1093/oxfordjournals.hmg.a018921. [DOI] [PubMed] [Google Scholar]
- 12.Ma X.Z., Jin T., Sakac D., Fahim S., Zhang X., Katsman Y., Bali M., Branch D.R. Abnormal splicing of SHP-1 protein tyrosine phosphatase in human T cells: implications for lymphomagenesis. Exp Hematol. 2003;31:131–142. doi: 10.1016/s0301-472x(02)01025-1. [DOI] [PubMed] [Google Scholar]
- 13.Parsapour K., Reep M.D., Gohar K., Shah V., Church A., Shwayder T.A. Familial Sweet's syndrome in 2 brothers, both seen in the first 2 weeks of life. J Am Acad Dermatol. 2003;49:132–138. doi: 10.1067/mjd.2003.328. [DOI] [PubMed] [Google Scholar]
- 14.Yang J., Liang X., Niu T., Meng W., Zhao Z., Zhou G.W. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:28199–28207. doi: 10.1074/jbc.273.43.28199. [DOI] [PubMed] [Google Scholar]
- 15.Nehls M., Schorpp M., Boehm T. An intragenic deletion in the human PTPN6 gene affects transcriptional activity. Hum Genet. 1995;95:713–715. doi: 10.1007/BF00209495. [DOI] [PubMed] [Google Scholar]
- 16.Nakase K., Cheng J., Zhu Q., Marasco W.A. Mechanisms of SHP-1 P2 promoter regulation in hematopoietic cells and its silencing in HTLV-1-transformed T cells. J Leukoc Biol. 2009;85:165–174. doi: 10.1189/jlb.0608383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leon F., Cespon C., Franco A., Lombardia M., Roldan E., Escribano L., Harto A., Gonzalez-Porque P., Roy G. SHP-1 expression in peripheral T cells from patients with Sezary syndrome and in the T cell line HUT-78: implications in JAK3-mediated signaling. Leukemia. 2002;16:1470–1477. doi: 10.1038/sj.leu.2402546. [DOI] [PubMed] [Google Scholar]
- 18.Shin K., Gurish M.F., Friend D.S., Pemberton A.D., Thornton E.M., Miller H.R., Lee D.M. Lymphocyte-independent connective tissue mast cells populate murine synovium. Arthritis Rheum. 2006;54:2863–2871. doi: 10.1002/art.22058. [DOI] [PubMed] [Google Scholar]
- 19.Galeazzi M., Gasbarrini G., Ghirardello A., Grandemange S., Hoffman H.M., Manna R., Podswiadek M., Punzi L., Sebastiani G.D., Touitou I., Doria A. Autoinflammatory syndromes. Clin Exp Rheumatol. 2006;24:S79–S85. [PubMed] [Google Scholar]
- 20.Kanazawa N., Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601–618. doi: 10.1111/j.1346-8138.2007.00342.x. [DOI] [PubMed] [Google Scholar]
- 21.Farasat S., Aksentijevich I., Toro J.R. Autoinflammatory diseases: clinical and genetic advances. Arch Dermatol. 2008;144:392–402. doi: 10.1001/archderm.144.3.392. [DOI] [PubMed] [Google Scholar]
- 22.Magro C.M., Kiani B., Li J., Crowson A.N. Clonality in the setting of Sweet's syndrome and pyoderma gangrenosum is not limited to underlying myeloproliferative disease. J Cutan Pathol. 2007;34:526–534. doi: 10.1111/j.1600-0560.2006.00654.x. [DOI] [PubMed] [Google Scholar]
- 23.Zamanian A., Ameri A. Acute febrile neutrophilic dermatosis (Sweet's syndrome): a study of 15 cases in Iran. Int J Dermatol. 2007;46:571–574. doi: 10.1111/j.1365-4632.2005.02688.x. [DOI] [PubMed] [Google Scholar]
- 24.Mizoguchi M., Matsuki K., Mochizuki M., Watanabe R., Ogawa K., Harada S., Hino H., Amagai M., Juji T. Human leukocyte antigen in Sweet's syndrome and its relationship to Behcet's disease. Arch Dermatol. 1988;124:1069–1073. [PubMed] [Google Scholar]
- 25.Adachi Y., Kindzelskii A.L., Cookingham G., Shaya S., Moore E.C., Todd R.F., III, Petty H.R. Aberrant neutrophil trafficking and metabolic oscillations in severe pyoderma gangrenosum. J Invest Dermatol. 1998;111:259–268. doi: 10.1046/j.1523-1747.1998.00311.x. [DOI] [PubMed] [Google Scholar]
- 26.Mahoney D.J., Mulloy B., Forster M.J., Blundell C.D., Fries E., Milner C.M., Day A.J. Characterization of the interaction between tumor necrosis factor-stimulated gene-6 and heparin: implications for the inhibition of plasmin in extracellular matrix microenvironments. J Biol Chem. 2005;280:27044–27055. doi: 10.1074/jbc.M502068200. [DOI] [PubMed] [Google Scholar]
- 27.Jia S.H., Parodo J., Kapus A., Rotstein O.D., Marshall J.C. Dynamic regulation of neutrophil survival through tyrosine phosphorylation or dephosphorylation of caspase-8. J Biol Chem. 2008;283:5402–5413. doi: 10.1074/jbc.M706462200. [DOI] [PubMed] [Google Scholar]
- 28.Tsui H.W., Siminovitch K.A., de Souza L., Tsui F.W. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 29.Shultz L.D., Coman D.R., Bailey C.L., Beamer W.G., Sidman C.L. “Viable motheaten,” a new allele at the motheaten locus. Am J Pathol. 1984;116:179–192. [PMC free article] [PubMed] [Google Scholar]
- 30.Croker B.A., Lawson B.R., Rutschmann S., Berger M., Eidenschenk C., Blasius A.L., Moresco E.M., Sovath S., Cengia L., Shultz L.D., Theofilopoulos A.N., Pettersson S., Beutler B.A. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL-1: MyD88, and a microbial trigger. Proc Natl Acad Sci U S A. 2008;105:15028–15033. doi: 10.1073/pnas.0806619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominique V., Francis L. Interactions of the scid or beige mutations with the viable motheaten mutation. Autoimmunity. 1995;22:199–207. doi: 10.3109/08916939508995318. [DOI] [PubMed] [Google Scholar]
- 32.Scribner C.L., Hansen C.T., Klinman D.M., Steinberg A.D. The interaction of the xid and me genes. J Immunol. 1987;138:3611–3617. [PubMed] [Google Scholar]
- 33.Shultz L.D., Coman D.R., Lyons B.L., Sidman C.L., Taylor S. Development of plasmacytoid cells with Russell bodies in autoimmune “viable motheaten” mice. Am J Pathol. 1987;127:38–50. [PMC free article] [PubMed] [Google Scholar]
- 34.Yu C.C.K., Tsui H.W., Ngan B.Y., Shulman M.J., Wu G.E., Tsui F.W.L. B and T cells are not required for the viable motheaten phenotype. J Exp Med. 1996;183:371–380. doi: 10.1084/jem.183.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujimoto E., Fujimoto N., Kuroda K., Tajima S. Leukocytapheresis treatment for pyoderma gangrenosum. Br J Dermatol. 2004;151:1090–1092. doi: 10.1111/j.1365-2133.2004.06249.x. [DOI] [PubMed] [Google Scholar]
- 36.Kanekura T., Kawahara K., Maruyama I., Kanzaki T. Treatment of pyoderma gangrenosum with granulocyte and monocyte adsorption apheresis. Ther Apher Dial. 2005;9:292–296. doi: 10.1111/j.1744-9987.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 37.Okuma K., Mitsuishi K., Hasegawa T., Tsuchihashi H., Ogawa H., Ikeda S. A case report of steroid and immunosuppressant-resistant pyoderma gangrenosum successfully treated by granulocytapheresis. Ther Apher Dial. 2007;11:387–390. doi: 10.1111/j.1744-9987.2007.00498.x. [DOI] [PubMed] [Google Scholar]
- 38.Magro C.M., De Moraes E., Burns F. Sweet's syndrome in the setting of CD34-positive acute myelogenous leukemia treated with granulocyte colony stimulating factor: evidence for a clonal neutrophilic dermatosis. J Cutan Pathol. 2001;28:90–96. doi: 10.1034/j.1600-0560.2001.280205.x. [DOI] [PubMed] [Google Scholar]
- 39.Blanc D., Schreiber M., Racadot E., Kahn J.Y., Flesch M., Viennet G., Lab M., Zultak M. Pyoderma gangrenosum in an allogeneic bone marrow transplant recipient. Clin Exp Dermatol. 1989;14:376–379. doi: 10.1111/j.1365-2230.1989.tb02590.x. [DOI] [PubMed] [Google Scholar]
- 40.Yamanaka K., Kuniyuki S., Maekawa N., Yoshida Y., Teshima H. Pyoderma gangrenosum with myelodysplastic syndrome treated with analogous bone marrow transplantation 8. Acta Derm Venereol. 2009;89:105–106. doi: 10.2340/00015555-0577. [DOI] [PubMed] [Google Scholar]
- 41.Ashida T., Mayama T., Higashishiba M., Kawanishi K., Miyatake J., Tatsumi Y., Kanamaru A. Successful reduced-intensity stem cell transplantation in a patient with myelodysplastic syndrome combined with Sweet's syndrome. Hematology. 2006;11:179–181. doi: 10.1080/10245330600667492. [DOI] [PubMed] [Google Scholar]
- 42.Shands J.W., Jr., Flowers F.P., Hill H.M., Smith J.O. Pyoderma gangrenosum in a kindred: Precipitation by surgery or mild physical trauma. J Am Acad Dermatol. 1987;16:931–934. [PubMed] [Google Scholar]
- 43.Al Rimawi H.S., Abuekteish F.M., Daoud A.S., Oboosi M.M. Familial pyoderma gangrenosum presenting in infancy. Eur J Pediatr. 1996;155:759–762. doi: 10.1007/BF02002902. [DOI] [PubMed] [Google Scholar]
- 44.Khandpur S., Mehta S., Reddy B.S. Pyoderma gangrenosum in two siblings: a familial predisposition. Pediatr Dermatol. 2001;18:308–312. doi: 10.1046/j.1525-1470.2001.01936.x. [DOI] [PubMed] [Google Scholar]
- 45.Alberts J.H., Sams H.H., Miller J.L., King L.E., Jr Familial ulcerative pyoderma gangrenosum: a report of 2 kindred. Cutis. 2002;69:427–430. [PubMed] [Google Scholar]
- 46.Goncalves J., Capon D.N., Laurent F., Batard M.L., Pellerin P. [Familial pyoderma gangrenosum following a mammoplasty reduction: a case report]: French. Ann Chir Plast Esthet. 2002;47:308–310. doi: 10.1016/s0294-1260(02)00124-3. [DOI] [PubMed] [Google Scholar]
- 47.Oskay T., Anadolu R. Sweet's syndrome in familial Mediterranean fever: possible continuum of the neutrophilic reaction as a new cutaneous feature of FMF. J Cutan Pathol. 2009;36:901–905. doi: 10.1111/j.1600-0560.2008.01158.x. [DOI] [PubMed] [Google Scholar]
- 48.Miyoshi T., Yamashita K., Ohno T., Izumi T., Takaori-Kondo A., Sasada M., Uchiyama T. Familial Mediterranean fever gene as a possible modifier of Sweet syndrome with chronic myelogenous leukemia. Acta Haematol. 2008;120:57–62. doi: 10.1159/000158578. [DOI] [PubMed] [Google Scholar]
- 49.Ferguson P.J., Chen S., Tayeh M.K., Ochoa L., Leal S.M., Pelet A., Munnich A., Lyonnet S., Majeed H.A., El Shanti H. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome) J Med Genet. 2005;42:551–557. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sapienza M.S., Cohen S., Dimarino A.J. Treatment of pyoderma gangrenosum with infliximab in Chron's disease. Dig Dis Sci. 2004;49:1454–1457. doi: 10.1023/b:ddas.0000042245.20042.4f. [DOI] [PubMed] [Google Scholar]
- 51.Page G., Miossec P. Paired synovium and lymph nodes from rheumatoid arthritis patients differ in dendritic cell and chemokine expression. J Pathol. 2004;204:28–38. doi: 10.1002/path.1607. [DOI] [PubMed] [Google Scholar]
- 52.Christophi G.P., Panos M., Hudson C.A., Christophi R.L., Gruber R.C., Mersich A.T., Blystone S.D., Jubelt B., Massa P.T. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab Invest. 2009;89:742–759. doi: 10.1038/labinvest.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gollol-Raju N., Bravin M., Crittenden D. Sweet's syndrome and systemic lupus erythematosus. Lupus. 2009;18:377–378. doi: 10.1177/0961203308100046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.