

Abstract
Complement activation, a key component of neuroinflammation, has been reported in both Parkinson's disease (PD) and Alzheimer's disease (AD). However, it is unclear whether complement activation and neuroinflammation in general are distinctly different from each another in major neurodegenerative disorders. In the present study, cerebrospinal fluid complement 3 (C3) and factor H (FH) were measured and evaluated together with amyloid-β42 (Aβ42), which in recent investigations was decreased in patients with PD, in particular those with cognitive impairment. The study included 345 participants: 126 patients with PD at various stages with or without cognitive impairment, 50 with AD, and 32 with multiple-system atrophy, and 137 healthy control individuals. In addition to changes in Aβ42 concentrations, there were clear differences in the patterns of complement profiles among neurodegenerative disorders. The C3/FH ratio demonstrated high sensitivity and specificity in differentiating patients with multiple-system atrophy from those with AD or PD and control individuals. In addition, the C3/Aβ42 and FH/Aβ42 ratios not only correlated with PD severity approximated using the Unified Parkinson's Disease Rating Scale but also with the presence of cognitive impairment or dementia in PD. Both C3 and FH correlated with the severity of impairment in AD as indicated using Mini-Mental State Examination scores.
The primary synthesis site for complement proteins is the liver, and for many years, the brain was considered an immune-privileged organ. However, recent studies have demonstrated that a complete and functional set of complement system components are synthesized in the central nervous system (CNS)1–3 and in human neuronal cells in vitro.4 Accumulating evidence suggests that activation of the complement system, a pivotal process of neuroinflammation, occurs in almost all major neurodegenerative disorders including both Parkinson's disease (PD) and Alzheimer's disease (AD). Complement 3 (C3) has a central role in the complement cascade, and its proteolytic fragments aid in stimulation of proinflammatory responses. In conjunction with other complement proteins, C3 acts as a C5 convertase, whereas the primary action of factor H (FH) is regulation of C3 activity.5
Alterations in several components of the complement system in human cerebrospinal fluid (CSF) have been reported in both PD and AD; however, few studies have demonstrated consistent results. Most investigations have been performed in small cohorts without controlling for major variables such as age, sex, and potential contamination of CSF by blood, a major source of complement components.6–8 None of these previous studies have determined whether complement activation is associated with disease severity or progression of PD and AD or whether complement activation is the same or different among various neurodegenerative diseases.
A decrease in CSF amyloid-β (Aβ) species, including Aβ42, which is thought to be especially toxic, along with an increase in total tau and/or phosphorylated tau, is traditionally considered a defining feature of amyloidopathy and tauopathy associated with AD.9,10 The significance of abnormalities in Aβ has not been recognized in PD until recently, when several groups reported independently that CSF Aβ42 tended to decrease in the absence of increased tau or phosphorylated tau in patients with PD, especially those with cognitive impairment.11–14 Aβ42 deposition in the CNS, with a resulting decrease in CSF Aβ42, is clearly associated with increased neuroinflammation in AD.15,16 Thus, it was hypothesized that complement profile and its activation in the CNS may differ in synucleinopathy such as PD and multiple-system atrophy (MSA) versus amyloidopathy and tauopathy in AD. To test this hypothesis, in the present investigation, both C3 and FH, as indices of complement regulation at key control points in this system, were measured in human CSF using established robust assays, with three major goals: (1) to address some of the problems associated with complement profile and its activation in PD and AD, (2) to further understand specific mechanisms of inflammation in various neurodegenerative disorders, and (3) to evaluate the usefulness of CSF C3 and FH, with and without considering CSF Aβ42 concentration, as biomarkers for differentiation of these disorders or for determination of disease severity or progression.
Materials and Methods
Study Participants
The study was approved by the institutional review boards of Baylor College of Medicine, Oregon Health and Science University, the University of California at San Diego, Veterans Affairs Puget Sound Health Care System at Seattle and the University of Washington, and the National Institutes of Health. The 345 participants included 126 patients with PD, 50 with AD, and 32 with MSA, and 137 healthy control individuals. All CSF samples were obtained with informed patient consent, and all patients underwent evaluations consisting of medical history, physical and neurologic examinations, laboratory tests, and neuropsychologic assessment. Inclusion and exclusion criteria for patients with AD or PD and for healthy controls have been described previously for this cohort.17
MSA was diagnosed according to generally accepted clinical criteria.18,19 The parkinsonian form of MSA was identified by the presence of symptoms or signs of autonomic failure coupled with rigidity and bradykinesia with or without clinical evidence of cerebellar failure, and the cerebellar form by the presence of autonomic and cerebellar failure without parkinsonism. All patients underwent testing while receiving their usual medications. To ensure that treatment with levodopa or carbidopa did not influence neurochemical findings, CSF data were included only for patients in whom the plasma dopamine concentration was less than 2500 pg/mL (<12.7 nmol/L) during supine rest. Orthostatic hypotension was defined as a decrease in systolic blood pressure of at least 20 mm Hg and in diastolic pressure of at least 10 mm Hg between supine rest for at least 15 minutes and upright posture for 5 minutes, unless symptomatic or rapid hypotension necessitated return to the supine position before 5 minutes of upright posture. Orthostatic hypotension was determined to be neurogenic by abnormal beat-to-beat blood pressure associated with performance of the Valsalva maneuver. The finding of normal 6-[18F]fluorodopamine–derived radioactivity in the left ventricular myocardium was also used to differentiate MSA from PD with orthostatic hypotension because all PD patients with orthostatic hypotension demonstrate neuroimaging evidence of cardiac sympathetic denervation. All patients with MSA included in the study underwent formal evaluation of orthostatic hypotension at tilt table testing. All patients also underwent 6-[18F]fluorodopamine PET scanning to evaluate cardiac sympathetic innervation, and all had evidence of intact innervation.
PD was staged using the Unified Parkinson's Disease Rating Scale (UPDRS).20 In addition, patients with PD were classified on the basis of extent of cognitive impairment, that is, PD with no cognitive impairment (PD-NCI; n = 49), PD with cognitive impairment but no dementia (PD-CIND; n = 65), and PD with dementia (PD-D; n = 11). The diagnosis of PD-D was determined using published criteria.21 Core features of these criteria include a diagnosis of PD,22 dementia syndrome in the context of established PD, impairment in more than one cognitive domain, a decline from premorbid level of cognitive function, and deficit severe enough to impair daily life. These criteria also include the “1-year” rule for differentiation from dementia with Lewy bodies; that is, dementia must occur 1 year after the onset of motor parkinsonism. The diagnosis of PD-CIND was made in patients with a diagnosis of PD,22 a clinical dementia rating of 0.5,23 and in the absence of dementia as determined using PD-D criteria.
Collection of CSF and Quality Control
All CSF samples were obtained via lumbar puncture in the morning. As much as 25 mL of CSF was taken from each individual, and every 5 mL was pooled into one fraction. Samples were aliquoted and stored at −80°C. Before analysis, all CSF samples were only thawed once when 10% protease inhibitor cocktail (Sigma, St Louis, MO) was added and samples were further aliquoted; details have been described previously.17 The hemoglobin (Hb) concentration in CSF samples was chosen as an index of the degree of red blood cell contamination, and was measured as previously described.17
Bead-Based Luminex Assays
CSF C3 and FH concentrations were measured using a kit (Millipore Corp, Billerica, MA), following the manufacturer's instructions with minor modifications. In brief, CSF samples were treated with an equal volume of 2× radioimmunoprecipitation assay buffer for 1 hour, diluted with 0.1% bovine serum albumin and PBS (pH 7.2) to a final dilution of 1:400, and incubated with capturing antibody–coupled beads overnight (18 hours) at 600 rpm on a plate shaker. The total initial volume in each well was 75 μL. CSF samples were analyzed using a LiquiChip Luminex 200 Workstation (Qiagen Corp., Valencia, CA). All CSF incubation was performed in 96-well MultiScreen Filter plates (Millipore Corp.). CSF Aβ42 also was measured using a Luminex assay, and the data have been published recently.13
Statistical Analysis
All analyses were performed with commercially available software (Prism 4.0, GraphPad Software, Inc., San Diego, CA, and SPSS version 18.0, SPSS, Inc., Chicago, IL). Linear regression analysis was used to determine the relationships between age, Hb, and C3 or FH. One-way analysis of variance followed by the Tukey test was used to compare differences between groups to determine age cutoff. One-way analysis of covariance (ANCOVA) also was performed on disease groups using Hb, age, and sex as covariates, with the exception of comparisons between sexes, in which Hb and age were used as covariates, followed by simple contrasts between disease groups to compare differences between groups. In addition, relationships between the analytes (C3 and FH) and Hb, age, sex, UPDRS (a clinical measure of PD motor severity), and Mini-Mental State Examination (MMSE; a clinical measure of dementia severity) were analyzed at bivariate correlation using the Pearson correlation coefficients. A receiver operating characteristic (ROC) curve was used to calculate the relationship between sensitivity and specificity for the disease groups versus healthy controls, and hence to evaluate the diagnostic performance of the analytes. The C3 and FH values were log-transformed (log 10) to induce a gaussian distribution, and Hb values were root-transformed to reduce heteroscedasticity. P < 0.05 was considered statistically significant.
Results
Effects of Age, Sex, and Blood Contamination
Linear regression analysis revealed an age-dependent effect for both C3 and FH (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org) in the control (C3, r = 0.43, P < 0.0001; and FH, r = 0.51, P < 0.0001) and PD (C3, r = 0.18, P < 0.05; and FH, r = 0.27, P < 0.01) groups. This effect seems to be primarily due to a larger age range in these two groups (21 to 90 years in the control group, and 37 to 84 years in the PD groups). There was no significant difference in age among any groups when individuals younger than 50 years were excluded (analysis of variance, data not shown).
A previous study demonstrated that blood contamination of CSF, which occasionally occurs during collection via lumbar puncture, could significantly affect CSF concentrations of certain proteins, namely, α-synuclein and DJ-1.17 To control for this variable, the Hb concentration was evaluated in all CSF samples to determine the extent of contamination. Using linear regression analysis of CSF concentrations, there was an appreciable increase in C3 and FH concentrations at higher Hb concentrations (Figure S1, C and D). This increase was significant for C3 when all individuals were included (r = 0.17, P < 0.01), and in the control (r = 0.18, P < 0.05), PD (r = 0.27, P < 0.01), and AD (r = 0.34, P < 0.05) groups. Similarly, a statistically significant increase was observed for FH in all individuals (r = 0.19, P < 0.001) and in each group (control, r = 0.24, P < 0.01; PD, r = 0.26, P < 0.01; AD, r = 0.29, P < 0.05; and MSA, r = 0.36, P < 0.05). No significant association was observed between Hb and C3 or FH in any of the groups when samples with Hb concentrations of 200 ng/mL or greater were excluded.
Analysis also was conducted to determine whether there was sex dependence for either of the CSF analytes (Figure 1). Significant correlations were observed between sex and C3 concentration when all groups were included (P < 0.001) or when PD (P < 0.0001), AD (P < 0.05), and MSA (P < 0.05) groups were analyzed separately. This sex dependence was also found when comparing FH and sex, whether all groups were included (P < 0.0001) or when the control (P < 0.01), PD (P < 0.0001), AD (P < 0.01), and MSA (P < 0.05) groups were analyzed independently. When the C3/FH ratio was analyzed, no significant sex dependence was observed for any of the groups studied. After eliminating individuals younger than 50 years and/or in whom the Hb concentration was greater than 200 ng/mL, an association between C3 concentration and sex remained, whether all groups were included (P < 0.0001) or when the control (P < 0.05), PD (P < 0.001), AD (P < 0.05), and MSA (P < 0.01) groups were analyzed separately. Similarly, FH sex dependence persisted when all groups were analyzed together (P < 0.0001) or when the control (P < 0.0001), PD (P < 0.001), AD (P < 0.05), and MSA (P < 0.05) groups were analyzed individually. Significant sex dependence was also observed for the C3/FH ratio, but only in the control group (P < 0.01).
Figure 1.
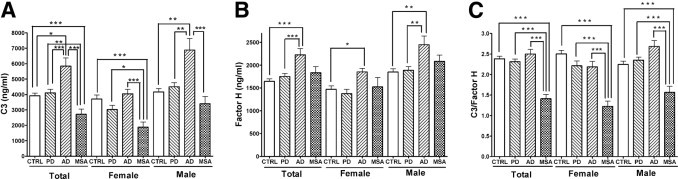
Cross-sectional examination of CSF C3 and FH using a Luminex assay. Concentrations of C3 and FH in CSF were measured using a Luminex assay, and were log-transformed (log 10) before further analysis. Comparisons of the mean concentrations of C3 (A), FH (B), and C3/FH (C) in patients with AD, PD, or MSA and in age-matched healthy control individuals were performed using ANCOVA, with Hb and age as covariates in male and female participants. Sex was also considered a covariate when data for male and female (Total) participants were analyzed together. Samples with high blood contamination (cutoff value ≥200 ng/mL) were eliminated from these analyses. Case numbers included in each group are given in Table 1. Data are given as mean (SD). *P < 0.01. **P < 0.001. ***P < 0.0001.
Diagnostic Usefulness of C3 and FH
Samples from individuals younger than 50 years or with an Hb concentration of 200 ng/mL or greater were eliminated when determining diagnostic usefulness, to minimize the effects of age and blood contamination. ANCOVA, with age, sex, and CSF Hb concentration as covariables, revealed no difference between the PD and control groups, but a significant decrease in C3 concentration in the MSA group compared with the control and PD groups (MSA versus control, P < 0.0001; MSA versus PD, P < 0.001), and a significant increase in AD when comparing it with all other groups studied (AD versus control, P < 0.01; AD versus MSA, P < 0.0001; AD versus PD, P < 0.0001) (Figure 1A and Table 1). In contrast, FH concentration, although not altered significantly in patients with PD, tended to increase in both the MSA and AD groups compared with the control and PD groups, and statistical significance was achieved when differentiating AD from control (P < 0.0001) and from PD (P < 0.0001) after adjustment for age, sex, and Hb. Significant differences also were observed when data for male and female participants were analyzed separately (Figure 1B and Table 1). However, none of the ROC calculations yielded acceptable (<60%) sensitivity and/or specificity for classifying any diseases when C3 or FH was used alone (data not shown).
Table 1.
Summary of Demographic Data and CSF Marker Values of Donors⁎
| Control | PD | AD | MSA | |
|---|---|---|---|---|
| No. of participants | 137 | 126 | 50 | 32 |
| Age ≥ 50 and Hgb < 200 ng/mL only | 91 | 86 | 38 | 20 |
| Sex, F/M | 0.8 | 0.35 | 0.61 | 0.52 |
| Age ≥ 50 and Hgb < 200 ng/mL only | 1.17 | 0.37 | 0.58 | 0.82 |
| Age, years | 58.61 (18.59) | 63.84 (10.42) | 68.12 (9.512) | 60.34 (9.008) |
| Age ≥ 50 and Hgb < 200 ng/mL only | 67.34 (10.62) | 64.33 (8.80) | 67.47 (9.27) | 61.95 (6.93) |
| C3, ng/mL† | ||||
| All‡§¶∥** | 3917 (1638) | 4111 (2029) | 5842 (3269) | 2718 (1510) |
| Femaleঠ| 3704 (1781) | 3026 (1293) | 4046 (1071) | 1884 (967.4) |
| Male§∥** | 4166 (1433) | 4508 (2111) | 6890 (3665) | 3400 (1563) |
| FH, ng/mL† | ||||
| All§∥ | 1645 (524.7) | 1751 (634.9) | 2228 (818.1) | 1833 (585.3) |
| Female§∥ | 1471 (510.9) | 1376 (444.0) | 1851 (291.5) | 1527 (606.8) |
| Male§∥ | 1847 (469.7) | 1887 (641.6) | 2448 (944.1) | 2085 (449.8) |
| C3/FH† | ||||
| All‡¶** | 2.384 (0.5790) | 2.314 (0.5703) | 2.501 (0.6841) | 1.411 (0.4681) |
| Female‡¶** | 2.502 (0.6128) | 2.213 (0.5524) | 2.187 (0.5053) | 1.224 (0.3871) |
| Male‡¶** | 2.246 (0.5100) | 2.351 (0.5766) | 2.685 (0.7164) | 1.564 (0.4890) |
| C3/Aβ42† | ||||
| All†‡§¶∥** | 11.48 (7.35) | 14.54 (9.71) | 32.61 (27.24) | 78.40 (50.04) |
| Female†‡§¶∥** | 10.39 (6.82) | 10.43 (4.30) | 22.61 (13.78) | 43.15 (18.44) |
| Male†‡§¶∥** | 12.76 (7.82) | 16.04 (10.70) | 38.45 (31.46) | 107.24 (49.53) |
| FH/Aβ42† | ||||
| All‡§¶∥** | 0.0030 (0.0015) | 0.0035 (0.0015) | 0.0054 (0.0021) | 0.0297 (0.011) |
| Female‡§¶∥** | 0.0028 (0.0011) | 0.0036 (0.0015) | 0.0055 (0.0023) | 0.0246 (0.0072) |
| Male‡§¶∥** | 0.0032 (0.0018) | 0.0035 (0.0015) | 0.0054 (0.0021) | 0.0339 (0.0120) |
AD, Alzheimer's disease; C3, complement 3; CSF, cerebrospinal fluid; FH, factor H; Hgb, hemoglobin; MSA, multiple-system atrophy; PD, Parkinson's disease.
Subjects younger than 50 years or with CSF Hgb ≥200 ng/mL were excluded. Group comparisons were made using ANCOVA, with Hgb and age as covariates in male and female subjects, respectively. Sex was also considered a covariate when male and female (All) subjects were analyzed together. P < 0.05 for PD versus Control†, MSA versus Control‡, AD versus Control§, PD versus MSA¶, PD versus AD∥, and MSA versus AD**.
Unless otherwise indicated, data are given as mean (SD).
Because C3 and FH represent two different mechanisms for complement activation, potential application of the C3/FH ratio was evaluated as a diagnostic marker. This ratio enabled differentiation of MSA from control, AD, and PD (P < 0.0001 for all individuals, or when data for male and female participants were analyzed separately (Figure 1C and Table 1). ROC analysis revealed high sensitivity and specificity (MSA versus control, sensitivity 85% and specificity 81%; MSA versus PD, sensitivity 80% and specificity 87%; and MSA versus AD, sensitivity 70% and specificity 95%) after eliminating individuals younger than 50 years or in whom the CSF Hb concentration was 200 ng/mL or higher (Figure 2). The cut-off values used to calculate sensitivity and specificity were 1.87, 1.78, and 1.62, respectively, for the control, PD, and AD groups.
Figure 2.
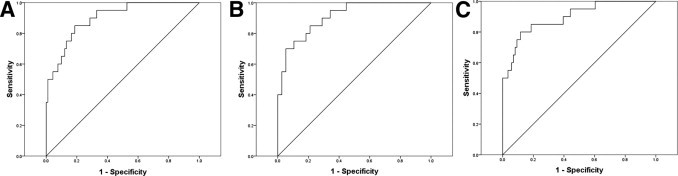
Receiver operating characteristic evaluation of C3/FH as a biomarker of MSA. A: MSA versus control. B: MSA versus AD. C: MSA versus PD.
Given that CSF Aβ42 is significantly reduced in patients with AD and tends to decrease in patients with PD, in particular those with cognitive impairment,11–14 the value of C3/Aβ42 and FH/Aβ42 was also evaluated as a function of disease diagnosis. The results demonstrated that C3/Aβ42 and FH/Aβ42 were altered significantly between all pairs of diagnostic groups except for FH/Aβ42 in the PD group versus the control group (Table 1). However, the diagnostic usefulness of the ratios as determined at ROC analysis did not improve significantly over those of C3 or FH alone. The only exception was AD versus control, where high sensitivity and specificity were achieved (C3/Aβ42, sensitivity 92.1% and specificity 76.9%; FH/Aβ42, sensitivity 92.1% and specificity 80.2%).
Disease Severity or Progression Correlations
Because diagnostic and prognostic markers can be different,17 the usefulness of C3 and FH was examined, as well as their ratios, as markers of PD severity approximated using the UPDRS score. When age, Hb, and sex were controlled for, an increase was observed in both C3 and FH levels in PD samples, with an increasing UPDRS score; however, it was not statistically significant for either analyte (data not shown). Furthermore, no statistical significance was achieved when correlating Aβ42 alone with UPDRS score (data not shown). When either C3/Aβ42 or FH/Aβ42 was analyzed, a statistically significant correlation with UPDRS scores was observed (C3/Aβ42, r = 0.26, P < 0.01; FH/Aβ42, r = 0.27, P < 0.01) (Figure 3,A and B).
Figure 3.

Correlation of C3/Aβ42 and/or FH/Aβ42 with PD severity as approximated using UPDRS motor scores and cognitive states. Log-transformed C3/Aβ42 (A) or FH/Aβ42 (B) concentrations were plotted against UPDRS motor scores for PD severity. A statistically significant correlation with UPDRS scores was observed for C3/Aβ42 (r = 0.26; P < 0.01) and FH/Aβ42 (r = 0.27; P < 0.01). Mean concentrations of C3/Aβ42 (C) and FH/Aβ42 (D) were also compared in 49 patients with PD-NCI [sex (F/M) ratio, 0.36; mean (SD) age, 61.10 (11.26) years], 65 with PD-CIND [sex (F/M) ratio, 0.38; age, 64.32 (9.23) years], and 11 with PD-D [sex (F/M) ratio, 0.22; age, 71.45 (8.12) years].
The same ratios were analyzed based on the cognitive state of patients with PD stratified as having PD-NCI, PD-CIND, or PD-D. Both analysis of variance and ANCOVA (adjustment for age, sex, and CSF Hb) revealed a significant increase in C3/Aβ42 in the PD-D group compared with the PD-NCI or PD-CIND groups (PD-D versus PD-NCI, P < 0.001; PD-D versus PD-CIND, P < 0.001) (Figure 3, C and D). Similarly, a statistically significant increase was observed for FH/Aβ42 in the PD-D group compared with the PD-NCI or PD-CIND groups (PD-D versus PD-NCI, P < 0.001; PD-D versus PD-CIND, P < 0.001). Although the ratios (C3/Aβ42 or FH/Aβ42) provided more robust results, a significant decrease in Aβ42 alone was also observed in the PD-D group compared with the PD-NCI or PD-CIND groups (PD-D versus PD-NCI, P < 0.05; PD-D versus PD-CIND, P < 0.05; analysis of variance and ANCOVA), an observation recently reported in several studies.11–14 Furthermore, neither tau, phosphorylated tau, or either species/Aβ42 provided better results than C3/Aβ42 or FH/Aβ42 in terms of correlating with the presence of cognitive impairment or dementia in PD. C3/β42 and FH/Aβ42 did not seem to enhance the performance of each other (data not shown).
Although there is no established scale to stage MSA clinically, CSF complement concentrations were analyzed against the concentrations of CSF DOPAC (3,4-dihydroxyphenylacetic acid) and DHPG ([S]-3,5-dihydroxyphenylglycine), the major metabolites of dopamine and norepinephrine, respectively. No significant correlation was appreciated between C3, FH, or C3/FH with DOPAC or DHPG. Furthermore, factoring in Aβ42 did not improve the performance of C3, FH, or C3/FH (data not shown).
The usefulness of C3 and FH was analyzed, as well as C3/FH, against the MMSE score only in patients with AD with CSF Hb concentration less than 200 ng/mL (Figure 4,A and B). There was a statistically significant correlation between lower MMSE score and increased concentrations of both CSF C3 (r = 0.27; P < 0.001) and FH (r = 0.31; P < 0.0001). However, like the MSA data, factoring in Aβ42 did not improve the performance of C3, FH, or C3/FH (data not shown).
Figure 4.
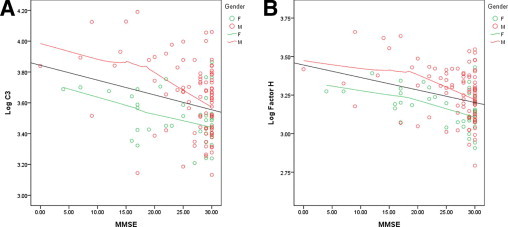
Correlation of C3 and/or FH with MMSE scores in patients with AD. CSF C3 and FH concentrations were log-transformed (log 10) because of their skewed distribution. Samples with high blood contamination (cutoff value, >200 ng/mL Hb) were excluded from the analysis. Significant correlation was observed for both C3 (A) (r = 0.27; P < 0.001) and FH (B) (r = 0.31; P < 0.0001).
Discussion
In this relatively large cohort of patients, along with healthy control individuals, use of total C3 and FH was explored, with and without considering CSF Aβ42, as potential biomarkers for PD, AD, and MSA. It was observed that although C3 or FH alone and their ratios cannot be used as effective diagnostic markers for PD or AD, C3/FH could be used to differentiate patients with MSA from those with PD or AD and healthy controls, with greater than 80% to 90% sensitivity and specificity. In addition, C3/Aβ42 or FH/Aβ42 correlated well with PD severity and the presence of cognitive impairment. A significant correlation was also identified between the MMSE score and C3 and FH concentrations in patients with AD.
Recent reports have described a role for neuroinflammation, including complement activation, in the development and progression of neurodegenerative disorders.24–26 In the present investigation, the pattern of changes in complement concentration was apparently different between the groups, with C3/FH as a marker that can be used to differentiate patients with MSA from those with PD or AD and healthy controls, with high sensitivity and specificity. Although further validation is needed, this finding could aid clinical practice substantially because, aside from imperfect clinical evaluations even with experienced physicians, the best diagnostic test for differentiating major forms of parkinsonism remains the structural and functional neuroimaging techniques27,28 including magnetic resonance imaging and positron emission tomography. However, most of these methods have not proved reliable, cost-effective, or readily accessible even in major medical centers in developed countries. In addition, although MSA and PD are both considered synucleinopathies, in MSA, α-synuclein aggregates (glial cytoplasmic inclusions) are found largely in oligodendrocytes, whereas in PD, they are predominantly localized in neurons.7 Thus, the differential pattern of alterations in complement concentration could potentially provide clues leading to the discovery of novel mechanisms involved in the pathogenesis of the two distinct diseases. C3 is not only critically involved in the classical and lectin pathways, but is also associated with the alternative pathway, along with various cofactors including FH, which serves as a negative regulator in the alternative pathway. Thus, C3/FH may indicate the overall activation of both classical and alternative pathways.
In a previous study in which complement components including FH and the proteolytic cleavage fragment of C3 (C3b) were examined in CSF from a relatively small cohort of patients with PD, AD, multiple sclerosis, and neurosyphilis using 2-dimensional gel electrophoresis, compared with healthy controls, patients with PD exhibited a significant decrease in concentrations of total C3b or its 2-dimensional isoform 2 and concentrations of FH isoform 1.7 A decrease in C3, C4a, and three 2-dimensional isoforms of haptoglobin in CSF from patients with PD compared with healthy controls has also been reported recently.29 In the present study, no decrease in C3 was observed. Other than a difference in cohort size, an important variable is that only total C3 and FH concentrations were measured. In addition, in the two previous investigations,7,29 it seemed that blood contamination, a factor that could contribute significantly to CSF complement concentration (Figure S1, C and D), was not controlled for.
The second major observation of the present study is that C3/Aβ42 or FH/Aβ42 correlated well with PD motor severity and with the presence of cognitive impairment in patients with PD. To fully understand the meaning of these ratios, first consider patients with AD, in which both C3 and FH increased with advancing AD severity (MMSE score), with or without the adjustment for CSF Aβ42 concentration (Figure 4). Inasmuch as complement activation is a key component of neuroinflammation, it is not surprising to observe increased C3 and FH levels in AD and their association with MMSE score. The observed increase in CSF C3 and FH in patients with AD is also consistent with a previous report of up-regulation of C3 mRNA and protein concentrations in the brain of patients with AD compared with healthy controls.7 In addition, with 2-dimensional gel electrophoresis, two isoforms of the C4b protein (isoforms 2 and 3) were increased in AD.29 Thus, it is possible that an increase in CSF C3 and FH might reflect the extent of complement activation as disease progresses. In contrast, it is well-recognized that Aβ deposition, with enhanced neuroinflammation in the CNS, in patients with AD is correlated with a significant decrease in CSF Aβ42.30 However, Aβ42 alone does not correlate with AD severity, probably because of “floor effects.”31–33 This phenomenon likely explains the observation that C3/Aβ42 or FH/Aβ42 did not improve the performance of C3 or FH alone related to AD severity. It should be stressed that there was a clear difference in CSF complement proteins in AD compared with PD or MSA, which suggests that it is likely that a different pathway (e.g., classical versus alternate complement activation) might be involved in AD.
A decrease in CSF Aβ42 in patients with PD, in particular those with cognitive impairment, in the absence of increased CSF tau or phosphorylated tau, is a relatively new observation made by several independent groups.11–14, 34–36 It has also been demonstrated that α-synuclein is decreased in patients with PD compared with healthy controls; however, CSF α-synuclein did not correlate with PD motor severity,17 again potentially owing to floor effects, as observed in Aβ in AD. Although it was suggested that α-synuclein and Aβ might interact with each other,37 it is unclear whether Aβ pathology interacts directly with α-synuclein pathology in PD, which is an area for future investigation. It is also uncertain whether a decrease in CSF Aβ42 in PD represents deposition of this peptide in the CNS, a process mimicking early stages of AD. Nonetheless, the fact that a combination of increased CSF C3 or FH with decreased Aβ42 correlated with PD motor severity and cognitive impairment seems to suggest that PD progression is intimately associated with enhanced neuroinflammation and altered metabolism of Aβ42, but in a way that may be different from AD. This also warrants further investigation, not only in terms of biomarker validation but also for the pathogenesis of PD and AD initiation and progression.
In summary, in this primarily discovery investigation, it was demonstrated that CSF C3 and FH can enable differentiation of patients with MSA from those with PD or AD and healthy controls, with high sensitivity and specificity, and, thus, has potential as a useful and cost-effective marker of MSA. In addition, C3/Aβ42 and FH/Aβ42 correlated well with PD severity and with the presence of cognitive impairment. C3 and FH concentrations, when used independently, correlated with AD severity, and could be useful as potential AD progression markers, which are urgently needed. In addition to validating these observations in a large and independent cohort, future areas of investigation should include examining other complement components and complement proteolytic fragments directly involved in complement activation. In addition, further characterization of the complement system might aid in elucidating mechanisms underlying PD, AD, and MSA; developing therapies targeted at modulating inflammation; and determining response to treatment.
Acknowledgments
We thank those who donated their CSF for the study and Mr. Thomas Quinn for assistance in preparation of the manuscript.
Footnotes
Supported by grants ES004696, NS057567, AG025327, AG033398, and NS060252 (J.Z.), NS062684 (T.J.M., C.P.Z., J.B.L., and J.Z.), AG005136 (E.R.P. and T.J.M.), and AG008017 (K.A.C. and J.F.Q.) from the National Institutes of Health, and grants from the Michael J. Fox Foundation and the Cheng-Mei Shaw Endowment (J.Z.) and the Nancy and Buster Alvord Endowment (T.J.M.).
Y.W. and A.M.H. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.006.
Supplementary data
Effects of age and blood contamination on C3 and FH concentrations in CSF. CSF C3 and FH were measured in individual samples from healthy control subjects and patients with AD, PD, or MSA using a Luminex assay, and Hb concentration (as an index of red blood cell contamination in CSF) was measured using an enzyme-linked immunosorbent assay kit. C3 and FH concentrations were log-transformed (log 10), and Hb data were root-transformed because of their positively skewed distributions. The following significant correlations between age and C3 (A) or FH (B) concentrations were observed: C3 in healthy controls (r = 0.43; P < 0.0001) and patients with PD (r = 0.18; P < 0.05); and FH in controls (r = 0.51; P < 0.0001) and patients with PD (r = 0.27; P < 0.01). The Hb concentration was significantly correlated with C3 (C) in patients with AD (r = 0.34; P < 0.05), PD (r = 0.27; P < 0.01), and controls (r = 0.18; P < 0.05), and with FH (D) in all groups: control (r = 0.24; P < 0.01), PD (r = 0.26; P < 0.01); AD (r = 0.29; P < 0.05), and MSA (r = 0.36; P < 0.05).
References
- 1.Morgan B.P., Gasque P. Expression of complement in the brain: role in health and disease. Immunol Today. 1996;17:461–466. doi: 10.1016/0167-5699(96)20028-f. [DOI] [PubMed] [Google Scholar]
- 2.Rus H., Cudrici C., David S., Niculescu F. The complement system in central nervous system diseases. Autoimmunity. 2006;39:395–402. doi: 10.1080/08916930600739605. [DOI] [PubMed] [Google Scholar]
- 3.Morgan B.P., Gasque P., Singhrao S., Piddlesden S.J. The role of complement in disorders of the nervous system. Immunopharmacology. 1997;38:43–50. doi: 10.1016/s0162-3109(97)00059-3. [DOI] [PubMed] [Google Scholar]
- 4.Thomas A., Gasque P., Vaudry D., Gonzalez B., Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. 2000;12:1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- 5.Ingram G., Hakobyan S., Hirst C.L., Harris C.L., Pickersgill T.P., Cossburn M.D., Loveless S., Robertson N.P., Morgan B.P. Complement regulator factor H as a serum biomarker of multiple sclerosis disease state. Brain. 2010;133:1602–1611. doi: 10.1093/brain/awq085. [DOI] [PubMed] [Google Scholar]
- 6.Einstein L.P., Hansen P.J., Ballow M., Davis A.E., III, Davis J.S., IV, Alper C.A., Rosen F.S., Colten H.R. Biosynthesis of the third component of complement (C3) in vitro by monocytes from both normal and homozygous C3-deficient humans. J Clin Invest. 1977;60:963–969. doi: 10.1172/JCI108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finehout E.J., Franck Z., Lee K.H. Complement protein isoforms in CSF as possible biomarkers for neurodegenerative disease. Dis Markers. 2005;21:93–101. doi: 10.1155/2005/806573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasch M.C., Van Den Bosch N.H., Daha M.R., Bos J.D., Asghar S.S. Synthesis of complement components C3 and factor B in human keratinocytes is differentially regulated by cytokines. J Invest Dermatol. 2000;114:78–82. doi: 10.1046/j.1523-1747.2000.00841.x. [DOI] [PubMed] [Google Scholar]
- 9.Hampel H., Burger K., Teipel S.J., Bokde A.L., Zetterberg H., Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer's disease. Alzheimers Dement. 2008;4:38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Dubois B., Feldman H.H., Jacova C., Dekosky S.T., Barberger-Gateau P., Cummings J., Delacourte A., Galasko D., Gauthier S., Jicha G., Meguro K., O'Brien J., Pasquier F., Robert P., Rossor M., Salloway S., Stern Y., Visser P.J., Scheltens P. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 11.Alves G., Bronnick K., Aarsland D., Blennow K., Zetterberg H., Ballard C., Kurz M.W., Andreasson U., Tysnes O.B., Larsen J.P., Mulugeta E. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson's disease: the Norwegian Park West study. J Neurol Neurosurg Psychiatry. 2010;81:1080–1086. doi: 10.1136/jnnp.2009.199950. [DOI] [PubMed] [Google Scholar]
- 12.Compta Y., Marti M.J., Ibarretxe-Bilbao N., Junque C., Valldeoriola F., Munoz E., Ezquerra M., Rios J., Tolosa E. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson's disease. Mov Disord. 2009;24:2203–2210. doi: 10.1002/mds.22594. [DOI] [PubMed] [Google Scholar]
- 13.Montine T.J., Shi M., Quinn J.F., Peskind E.R., Craft S., Ginghina C., Chung K.A., Kim H., Galasko D.R., Jankovic J., Zabetian C.P., Leverenz J.B., Zhang J. CSF Abeta(42) and tau in Parkinson's disease with cognitive impairment. Mov Disord. 2010;25:2682–2685. doi: 10.1002/mds.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siderowf A., Xie S.X., Hurtig H., Weintraub D., Duda J., Chen-Plotkin A., Shaw L.M., Van Deerlin V., Trojanowski J.Q., Clark C. CSF amyloid (beta) 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fioravanzo L., Venturini M., Di Liddo R., Marchi F., Grandi C., Parnigotto P.P., Folin M. Involvement of rat hippocampal astrocytes in beta-amyloid–induced angiogenesis and neuroinflammation. Curr Alzheimer Res. 2010;7:591–601. doi: 10.2174/156720510793499020. [DOI] [PubMed] [Google Scholar]
- 16.Ager R.R., Fonseca M.I., Chu S.H., Sanderson S.D., Taylor S.M., Woodruff T.M., Tenner A.J. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer's disease. J Neurochem. 2010;113:389–401. doi: 10.1111/j.1471-4159.2010.06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong Z., Shi M., Chung K.A., Quinn J.F., Peskind E.R., Galasko D., Jankovic J., Zabetian C.P., Leverenz J.B., Baird G., Montine T.J., Hancock A.M., Hwang H., Pan C., Bradner J., Kang U.J., Jensen P.H., Zhang J. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain. 2009;133:713–726. doi: 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilman S., Low P.A., Quinn N., Albanese A., Ben-Shlomo Y., Fowler C.J., Kaufmann H., Klockgether T., Lang A.E., Lantos P.L., Litvan I., Mathias C.J., Oliver E., Robertson D., Schatz I., Wenning G.K. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 19.Gilman S., Wenning G.K., Low P.A., Brooks D.J., Mathias C.J., Trojanowski J.Q., Wood N.W., Colosimo C., Durr A., Fowler C.J., Kaufmann H., Klockgether T., Lees A., Poewe W., Quinn N., Revesz T., Robertson D., Sandroni P., Seppi K., Vidailhet M. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahn S., Elton R., UPDRS Committee . Unified Parkinson's Disease Rating Scale. In: Fahn S., Marsden C.D., Calne D.B., Goldstein M., editors. Recent Developments in Parkinson's Disease, vol 2. Macmillan Healthcare Information; Florham Park, NJ: 1987. pp. 153–163. 293–304. [Google Scholar]
- 21.Emre M., Aarsland D., Brown R., Burn D.J., Duyckaerts C., Mizuno Y., Broe G.A., Cummings J., Dickson D.W., Gauthier S., Goldman J., Goetz C., Korczyn A., Lees A., Levy R., Litvan I., McKeith I., Olanow W., Poewe W., Quinn N., Sampaio C., Tolosa E., Dubois B. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22:1689–1707. doi: 10.1002/mds.21507. quiz 1837. [DOI] [PubMed] [Google Scholar]
- 22.Gibb W.R., Lees A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris J.C. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 24.Bonifati D.M., Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 25.McGeer E.G., Klegeris A., McGeer P.L. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(Suppl 1):94–97. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Yasojima K., Schwab C., McGeer E.G., McGeer P.L. Up-regulated production and activation of the complement system in Alzheimer's disease brain. Am J Pathol. 1999;154:927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruck A., Aalto S., Nurmi E., Vahlberg T., Bergman J., Rinne J.O. Striatal subregional 6-[18F]fluoro-l-dopa uptake in early Parkinson's disease: a two-year follow-up study. Mov Disord. 2006;21:958–963. doi: 10.1002/mds.20855. [DOI] [PubMed] [Google Scholar]
- 28.Van Laere K., Clerinx K., D'Hondt E., de Groot T., Vandenberghe W. Combined striatal binding and cerebral influx analysis of dynamic 11C-raclopride PET improves early differentiation between multiple-system atrophy and Parkinson disease. J Nucl Med. 2010;51:588–595. doi: 10.2967/jnumed.109.070144. [DOI] [PubMed] [Google Scholar]
- 29.Guo J., Sun Z., Xiao S., Liu D., Jin G., Wang E., Zhou J., Zhou J. Proteomic analysis of the cerebrospinal fluid of Parkinson's disease patients. Cell Res. 2009;19:1401–1403. doi: 10.1038/cr.2009.131. [DOI] [PubMed] [Google Scholar]
- 30.Glass C.K., Saijo K., Winner B., Marchetto M.C., Gage F.H. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersson C., Blennow K., Almkvist O., Andreasen N., Engfeldt P., Johansson S.E., Lindau M., Eriksdotter-Jonhagen M. Increasing CSF phospho-tau levels during cognitive decline and progression to dementia. Neurobiol Aging. 2008;29:1466–1473. doi: 10.1016/j.neurobiolaging.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Vemuri P., Wiste H.J., Weigand S.D., Knopman D.S., Trojanowski J.Q., Shaw L.M., Bernstein M.A., Aisen P.S., Weiner M., Petersen R.C., Jack C.R., Jr Serial MRI and CSF biomarkers in normal aging MCI, and AD. Neurology. 2010;75:143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahlund L.O., Blennow K. Cerebrospinal fluid biomarkers for disease stage and intensity in cognitively impaired patients. Neurosci Lett. 2003;339:99–102. doi: 10.1016/s0304-3940(02)01483-0. [DOI] [PubMed] [Google Scholar]
- 34.Bibl M., Esselmann H., Mollenhauer B., Weniger G., Welge V., Liess M., Lewczuk P., Otto M., Schulz J.B., Trenkwalder C., Kornhuber J., Wiltfang J. Blood-based neurochemical diagnosis of vascular dementia: a pilot study. J Neurochem. 2007;103:467–474. doi: 10.1111/j.1471-4159.2007.04763.x. [DOI] [PubMed] [Google Scholar]
- 35.Parnetti L., Tiraboschi P., Lanari A., Peducci M., Padiglioni C., D'Amore C., Pierguidi L., Tambasco N., Rossi A., Calabresi P. Cerebrospinal fluid biomarkers in Parkinson's disease with dementia and dementia with Lewy bodies. Biol Psychiatry. 2008;64:850–855. doi: 10.1016/j.biopsych.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Shi M., Bradner J., Hancock A.M., Chung K.A., Quinn J.F., Peskind E.R., Galasko D., Jankovic J., Zabetian C.P., Kim H.M., Leverenz J.B., Montine T.J., Ginghina C., Kang U.J., Cain K.C., Wang Y., Aasly J., Goldstein D.S., Zhang J. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2010 doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandal P.K., Pettegrew J.W., Masliah E., Hamilton R.L., Mandal R. Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease. Neurochem Res. 2006;31:1153–1162. doi: 10.1007/s11064-006-9140-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of age and blood contamination on C3 and FH concentrations in CSF. CSF C3 and FH were measured in individual samples from healthy control subjects and patients with AD, PD, or MSA using a Luminex assay, and Hb concentration (as an index of red blood cell contamination in CSF) was measured using an enzyme-linked immunosorbent assay kit. C3 and FH concentrations were log-transformed (log 10), and Hb data were root-transformed because of their positively skewed distributions. The following significant correlations between age and C3 (A) or FH (B) concentrations were observed: C3 in healthy controls (r = 0.43; P < 0.0001) and patients with PD (r = 0.18; P < 0.05); and FH in controls (r = 0.51; P < 0.0001) and patients with PD (r = 0.27; P < 0.01). The Hb concentration was significantly correlated with C3 (C) in patients with AD (r = 0.34; P < 0.05), PD (r = 0.27; P < 0.01), and controls (r = 0.18; P < 0.05), and with FH (D) in all groups: control (r = 0.24; P < 0.01), PD (r = 0.26; P < 0.01); AD (r = 0.29; P < 0.05), and MSA (r = 0.36; P < 0.05).