

Abstract
Colorectal carcinomas (CRC) might be organized hierarchically and contain a subpopulation of tumorigenic, putative cancer stem cells that are CD133 positive. We studied the biological and genetic characteristics of such cells in CRC cell lines and primary tumors. Three CRC cell lines were sorted in CD133 positive and negative fractions. The respective genetic aberration profiles were studied using array comparative genomic hybridization (aCGH) and expression profiling. Tumorigenicity for each cellular population was tested by injection into nude mice. Additionally, we compared CD133+ and CD133− cells of 12 primary colorectal tumors using laser capture microdissection and aCGH. Three of five CRC cell lines displayed both CD133+ and CD133− cells, but tumorigenicity of these subfractions did not differ significantly and aCGH revealed essentially identical genomic imbalances. However, 96 genes were differentially expressed between the two populations. Array comparative genomic hybridization analysis after laser capture microdissection of CD133+ and CD133− areas in primary colorectal tumors revealed genetic differences in 7 of 12 cases. The use of cell lines for studying genomic alterations that define cancer stem cell characteristics, therefore, seems questionable. In contrast, CD133+ cells in primary cancer samples showed a unique genomic aberration profile. In conclusion, our data suggest that CD133 positivity defines a genetically distinct cellular compartment in primary CRC, which potentially includes tumor initiating cells.
Traditional models of carcinogenesis assert that cancer can originate in virtually any cell of a given tissue through a series of genetic events that promote cellular proliferation. Malignant transformation is the eventual result of increased cellular proliferation and inhibited apoptosis.1 Regarding colorectal cancer (CRC), this process begins in epithelial cells lining the gastrointestinal tract undergoing sequential mutations in specific key genes including APC, MYC, KRAS, P53, and SMAD2.2 These mutations, in concert with specific chromosomal aneuploidies drive the transition of functional colonic epithelia to dysplastic cells and finally to colorectal cancer. This process is called the “adenoma-carcinoma sequence.”3–5
Until a few years ago, all neoplastic cells in a malignant neoplasm were considered to have tumorigenic potential, but recent findings suggested a hierarchy, hypothesizing that only a more or less rare population of cancer stem cells (CSCs) can in fact replenish a tumor.6 CSCs were first described in human leukemia. Lapidot et al7 demonstrated that human acute myeloid leukemia harbored a subfraction of cells exclusively capable of tumor initiation in severe combined immunodeficiency mice. More recently, compelling evidence suggested that a wide variety of solid tumors,8–14 including colorectal cancers, also contain a tumor initiating fraction of CSCs.15,16 It has yet to be investigated whether cancer stem cells exhibit the same chromosomal changes as the other cells in the tumor or present with a specific genetic makeup.
Supposedly, CSCs can be identified and enriched by staining for specific cell surface markers. One of the most frequently used markers is CD133, which has been implied as a marker for CSC in different tumor entities.17,18 Originally described in the context of normal hematopoietic stem cells,19 CD133 gained recognition as a marker for CSCs in medulloblastoma and glioblastoma,13 and subsequently for tumors of epithelial origin, such as breast, lung, and pancreas.20–22 CD133, also known as PROM1 (prominin 1) or AC133, maps to chromosome 4p15 and codes for a 120 kDa transmembrane pentaspan protein. The precise function is still unclear. Studies of consanguineous pedigrees from India with retinal degeneration revealed a frame shift mutation in the PROM1 gene in these individuals.23 PROM1 is concentrated in the membrane evaginations at the base of the outer segment of rod photoreceptor cells. Therefore, it has been proposed that this protein has a role in establishing and/or maintaining certain plasma membrane protrusions, which is consistent with the apical membrane expression pattern in CRC cells.24
Recently, two groups identified two subsets of cells selected from colon cancer samples based on CD133 expression. In a series of studies, CD133-positive (CD133+) cells were shown to be capable of initiating tumor growth in murine xenograft models, while CD133-negative (CD133−) cells were not. Therefore, the authors concluded that the propagation of colorectal cancer depends on this small subset of CD133+ CSCs.25,26
However, this hypothesis was challenged by Shmelkov et al27 who observed that CD133− cells isolated from colon cancer metastases were also able to initiate tumors in nonobese diabetic/severe combined immunodeficiency mice. Furthermore, the same group found that CD133 is not only expressed in CSCs but also in differentiated tumor cells.27
In summary, the exact role of CD133 as a CSC marker for colorectal cancers still remains elusive.28,29 The goal of the present study was to investigate the biological role and in particular the genetic characteristics of CD133+ and CD133− cells in CRC cell lines and primary tumor samples. We analyzed isolated cell populations, both from CRC cell lines and primary tumors using array comparative genomic hybridization (aCGH) to determine whether CD133+ from CD133− cells exhibit distinct differences in their genomic aberration profiles.
Materials and Methods
Tissue Collection and Cell Lines
The five CRC cell lines (Caco-2, HCT 116, NCI-H508, LS174T, and HT-29) were purchased from the American Type Culture Collection (ATCC, Manassas, VA).
All cell lines were cultured in complete media (with fetal bovine serum) as recommended by ATCC. Paraffin–embedded tissues from diagnostic colon cancer samples were obtained from the archive of the Institute for Pathology, Paracelsus Medical University, Salzburg, Austria. The study was conducted in accordance with the regulation of the local ethics committee. All specimens were diagnosed according to the latest TNM classification by two board certified pathologists (see Supplemental Table S1 at http://ajp.amjpathol.org).
Immunohistochemistry and Immunofluorescence
Immunohistochemistry (IHC) and immunofluorescence were performed on 4-μm thick sections of formalin-fixed, paraffin-embedded tumor samples. Anti-CD133 rabbit monoclonal antibody (1:100 for IHC; 1:20 for IF; clone C24B9; Cell Signaling, Danvers, MA) and anti-CD133, mouse monoclonal antibody (1:40, clone AC133, Miltenyi Biotech, Bergisch Gladbach, Germany) were used as primary antibodies for CD133 detection. Slides were deparaffinized and endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 15 minutes. Slides were subjected to citrate-based antigen retrieval (0.01 mol/L of citric acid, pH 6.0, for 5 minutes) in a pressure cooker (Keystone Manufacturing, Buffalo, NY) followed by slow cooling for 20 minutes, and incubation at 4°C for 12 hours with a primary antibody diluted in PBS containing 0.2% bovine serum albumin and 5% goat serum. Subsequently, the slides were washed in 1 × PBS. For immunofluorescence, the slides were incubated for 1 hour at room temperature with a goat anti-rabbit IgG-fluorescein isothiocyanate as secondary antibody (dilution 1:200; clone 4030-02; SouthernBiotech, Birmingham, AL). For IHC, the EnVision-Plus Kit (DAKO, Carpinteria, CA) with diaminobenzidine as chromogen was used for detection.
Flow Cytometry and Cell Sorting
Cells from CRC cell lines were detached using 0.25% Trypsin-EDTA (Invitrogen, Carlsbad, CA) in PBS, counted with a hemocytometer and washed in 0.1% bovine serum albumin in PBS. Cells from the mouse xenografts were prepared using a protocol for preparation of single-cell suspensions.30
At least 500,000 cells (in 100 μL PBS/0.5% bovine serum albumin) were incubated with phosphatidylethanolamine-labeled mouse anti-human CD133 monoclonal antibody (1:10; clone AC133; Miltenyi Biotech) at 4°C for 30 minutes in the dark. Unstained cells and cells stained with an isotype control were used as reference. After the washing steps, labeled cells were analyzed by flow cytometry using a FACS-Calibur (BD Biosciences, San Jose, CA). A minimum of 20,000 membrane intact cells was recorded and analyzed with CellQuest Pro (BD Biosciences) or FloJo software (Tree Star Inc, Ashland, OR). CD133+ and CD133− cells were sorted by fluorescence-activated cell sorting for further experiments using a FACSAria II system and FACSDiva software. The staining protocol was chosen according to earlier studies with primary tumor material to allow direct comparison with the original CD133-related CRC literature.25–27 An improved staining procedure has been published by us recently.31
aCGH from Flow Cytometry-Sorted Cells
After fluorescence-activated cell sorting (FACS), DNA was isolated from CD133+ and CD133− HT-29, Caco-2, and HCT 116 cells, and hybridized to aCGH arrays with genomically normal DNA as a reference following published protocols.32 Briefly, 3 μg test DNA and 3 μg normal genetic reference DNA (Genomic DNA, Promega, Madison, WI) were differentially labeled with dCTP-Cy5 and dCTP-Cy3, respectively (Perkin Elmer, Waltham, MA). Genome wide analysis of DNA copy number changes was performed using Human Genome CGH Microarray Kit 105A (Agilent, Santa Clara, CA) with 21.7 Kb overall median probe spacing resolution according to the manufacturer's protocol version 6.0 (Agilent). Slides were scanned with microarray scanner G2505B (Agilent) and analyzed using CGH Analytics software 4.0.76.
Laser Microdissection or FFPE Block Punch Biopsy and aCGH
Five consecutive sections each (first and last section on glass slides, section two to four on membrane slides) were prepared from each of the 12 primary CRC cases exhibiting a CD133+ subfraction. The first and the fifth slide were stained for CD133 by IHC and used to guide the laser capture microdissection (LCM) performed on the sections two to four mounted on membrane slides. This approach ensured that IHC-positive cells were selectively dissected without compromising their DNA quality due to staining artifacts. CD133+ and CD133− tumor cells were selected and the glands were dissected using Arcturus XT (Arcturus Engineering Inc., Mountain View, CA) with a UV and IR laser. To rule out random genomic heterogeneity in the tumor samples we also performed punch biopsies (0.6 mm in diameter) from eight tissue blocks. We choose two areas (0.6 mm in diameter and approximately 5 mm between the two areas) in either the CD133+ or CD133− section and dissected the tissue with a needle (see Supplemental Figure S1 at http://ajp.amjpathol.org). The tissue punches were placed in a tube containing 1 mL xylene. Proteinase K digestions were performed at 56°C. After DNA preparation using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) the samples were labeled with a Bioprime Array CGH Genomic Labeling Kit according to the manufacturer's instructions (Invitrogen). Briefly 200 ng test DNA and 200 ng normal genomic reference DNA were differentially labeled with dCTP-Cy5 and dCTP-Cy3, respectively (GE Health care, Piscataway, NJ). Genome wide analysis of DNA copy number changes was performed using SurePrint G3 Human CGH Microarray Kit 8 × 60K (Agilent) with 41.5 Kb overall median probe spacing resolution according to the manufacturer's protocol version 6.0 (Agilent).
Expression Array
One μg of total RNA of each FACS sorted CD133+ and CD133− cells (HT-29, Caco-2, and HCT 116) were labeled with Cy3, using a T7 RNA polymerase according to the manufacturer's protocol version 6.0 (Agilent), and hybridized to the 44 K oligonucleotide-based Whole Human Genome Microarray (Agilent). Microarrays were washed and processed using a G2565BA scanner. Data were quality controlled and extracted using Technologies' Feature Extraction (Agilent, version 9.1).
Transplantation of Cancer Cells and Tumorigenicity Assay
Each 2000 and 20,000 CD133± sorted cells (HT-29, Caco-2, and HCT 116) were resuspended in 50 μL of PBS after sorting, and cell aliquots were diluted in 1:1 with Growth Factor Reduced Matrigel Matrix (BD Biosciences) before subcutaneous injection into the flanks of athymic NCr-nu/nu (nude) mice (five mice per cell line and per CD133 fraction).
All mice were bred and housed in a pathogen-free environment and used in experiments in accordance with institutional guidelines at the Center for Cancer Research, National Cancer Institute, National Institutes of Health. All experimental procedures conducted in this study were approved by the Animal Care and Use Committee (National Institutes of Health). Tumor sizes were measured in two dimensions two times per week, and volumes were calculated using the formula for a rotational ellipsoid v = π/6 × a × b2.33 Mice were sacrificed once the tumor diameter had reached 2 cm in either length or width.
Tumors were removed and prepared for flow cytometric analyses.30 A fraction of each specimen was also fixed in 10% neutral buffered formalin for histopathological examination.
Statistical Analyses
Differences between groups were estimated by Student's t-test and repeated measures analysis of variance analysis. Normality tests were done on the gene expressions for samples from each CD133+ or CD133− cell line using the Shapiro-Wilk test. After adjusting the P values for multiple comparisons using the false discovery rate method of Benjamini and Hochberg34, no genes passed significance for non-normality. For multivariate analysis, possible factors correlating with CD133 IHC were identified by multivariate linear regression analysis. All differences were deemed significant when reaching the level of P < 0.05. For each cell line, we selected genes differentially expressed between CD133+ versus CD133− groups using Student's t-test with a threshold P value < 0.05 (using R version 2.10).
Results
CD133 Expression Pattern in Primary Colorectal Cancers and CRC Cell Lines
We first examined the expression and the topology of CD133 in tissue sections of primary human tumors using immunohistochemistry. Consistent with previous reports, CD133 expressing cells were located in the apical luminal surface, and/or in the intraglandular lumen (Figure 1).35 Both antibodies (C24B9 and AC133) used for IHC showed comparable staining patterns supporting previous reports.35 Twelve of 15 stained tumor samples (80%) showed CD133+ tumor cells with percentages ranging from 5 to 95% positive cells. The remaining three tumors showed no CD133 expression (see Supplemental Table S1 at http://ajp.amjpathol.org). CD133 positivity was not correlated to clinical parameters, including nodal status or TNM classification (data not shown). As a next step, we investigated whether the frequency of CD133+ cells in the primary tumors was maintained in CRC cell lines. Using flow cytometry under standardized serum supplemented conditions we found that three of five cell lines showed clear expression of the marker with both positive and negative CD133 populations. The proportion of cells above isotype with a clear CD133+ staining ranged from 9% ± 3 (HT-29), 62 ± 8 (HCT 116) to 80% ± 15 (Caco-2). LS174T and NCI-H508 did not contain a CD133+ fraction (see Supplemental Figure S2 at http://ajp.amjpathol.org). With the exception of HT-29, these percentages are consistent with previous reports.31,36,37 The differences observed for HT-29 are likely due to different culture conditions, frozen subclones, or the staining procedure.
Figure 1.
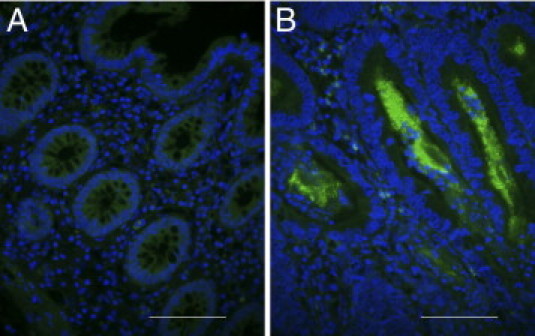
Immunohistochemistry of primary tissue sections from normal mucosa (A) and colorectal cancer (B) with an antibody against CD133 (fluorescein isothiocanate) and nuclear counterstaining (DAPI). While normal mucosa did not stain for CD133, a strongly CD133+ tumor (herein 85% CD133+ glands) shows apical luminal staining of the tumor cells and staining of intraglandular debris. Scale bars = 100 μm.
After renewed, short-term culture of the different fractions isolated from HCT 116, the percentages of CD133+ versus CD133− cells changed only slightly and confirmed earlier observations.31 However, in long-term cultures, we observed changes in the CD133 distribution throughout culturing (see Supplemental Figure S3 at http://ajp.amjpathol.org). Taken together, these results confirm the presence of CD133+ cell fractions in primary human colon cancers and CRC cell lines. The heterogeneity among the investigated samples indicated that CD133+ may potentially mark genetically or epigenetically distinct tumor populations and served as a basis for our subsequent analyses. The three CRC cell lines that contained positive fractions were used for subsequent experiments.
Tumorigenic Capacity of CD133+ and CD133− Cells after Xenotransplantation
The most important functional property ascribed to CSC is the capacity to initiate tumor growth in vivo. To examine whether the status of CD133 results in differences in tumor initiation in vivo, we transplanted flow sorted CD133+ and CD133− cells from three cell lines (HCT 116, Caco-2, and HT-29) subcutaneously into the flank of nude mice. For Caco-2, which is considered to be weakly tumorigenic,38 only 2 of 20 injections resulted in a tumor after 8 weeks. All other injections failed to initiate tumor growth (observation time >5 months) (see Supplemental Table S2 at http://ajp.amjpathol.org). For HCT 116, all 20 injections resulted in a tumor after 3 to 4 weeks. There was no difference in tumor initiating capacity (P = 0.997) or growth rate (P = 0.238; P = 0.994) between the two fractions in the athymic NCr-nu/nu indicating that for these two cell lines CD133 is indeed unable to discriminate between tumorigenic and nontumorigenic cells (Figure 2A and B, and Supplemental Table S2 at http://ajp.amjpathol.org).
Figure 2.
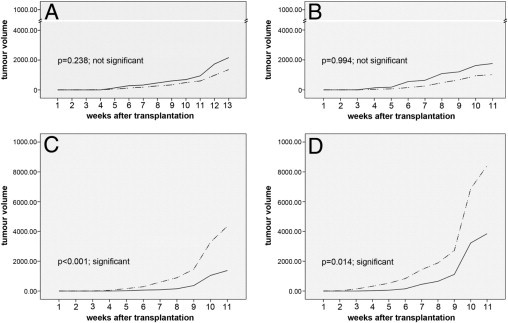
Evaluation of the tumor initiating potential of CD133+ (dotted line) and CD133− (solid line) cells of colon cancer cell lines on subcutaneous injection of cells into nude mice. Y-axis, tumor volume in mm3. X-axis, time after injection in weeks. A: HCT 116, 2000 cells. B: HCT 116, 20,000 cells. C: HT-29, 2000 cells. D: HT-29, 20,000 cells.
For HT-29, 6 of 10 injections with CD133− and 9 of 10 injections with CD133+ cells resulted in tumor growth, again with no significant difference between the fractions (P = 0.135) (see Supplemental Table S2 at http://ajp.amjpathol.org). However, the CD133+ injected cells showed a faster tumor growth rate than the CD133− cells (Figure 2, C and D) (P ≤ 0.001; P = 0.014), independent of the injected tumor cell numbers (2000 or 20,000).
Next, we examined whether the CD133 status of the injected cells was maintained in the xenografts by dissociating the tumors into single cell suspensions followed by staining for CD133 using flow cytometry. After injection and tumor formation the percentages changed: primary CD133+ injected cells were able to generate the corresponding negative cell fraction. The CD133+ cell fraction of HCT 116 showed only 29% ± 5 CD133+ cells after xenotransplantation, originally the cell line contained 62% ± 8 CD133+. The CD133+ cell fraction of HT-29 showed 49% ± 7 CD133+ cells after xenotransplantation, originally this cell line contained only 9% ± 3% CD133+ cells. These data indicate a high variability for CD133 expression in nude mice assays, probably caused by the changes in environmental conditions. Conversely, the CD133− cell population also showed an enrichment of CD133+ tumor cells (see Supplemental Figure S4 at http://ajp.amjpathol.org) after injection and tumor formation.
Gene Expression Profiles of CD133+ and CD133− Cells in HCT 116, Caco-2, and HT-29
Then, we went on to examine whether the CD133 status in our CRC cell lines would be reflected in specific gene expression profiles. To select those genes that are consistently differentially expressed between CD133+ and CD133− fractions, we built two intersection gene lists, with each intersection gene list retaining only those genes that are i) up-regulated in CD133+ fraction for the three cell lines or ii) down-regulated in CD133+ fraction for the three cell lines.
We used a χ2 goodness of fit test to evaluate if the intersection gene lists are larger than expected by random chance and found that the P value is highly significant (P value < 2.2 × 10−16). We identified 96 genes (86 up-regulated and 10 down-regulated) that were consistently differentially expressed between CD133+ and CD133− fractions for all three cell lines.
Reassuringly, CD133 ranked among the most significant up-regulated genes (see Supplemental Table S3 at http://ajp.amjpathol.org). While the gene expression differences between the fractions were not as pronounced as the one from one cell line to another (Figure 3), a principal component analyses based on the 96 genes could differentiate the CD133+ and CD133− fractions (Figure 4).
Figure 3.
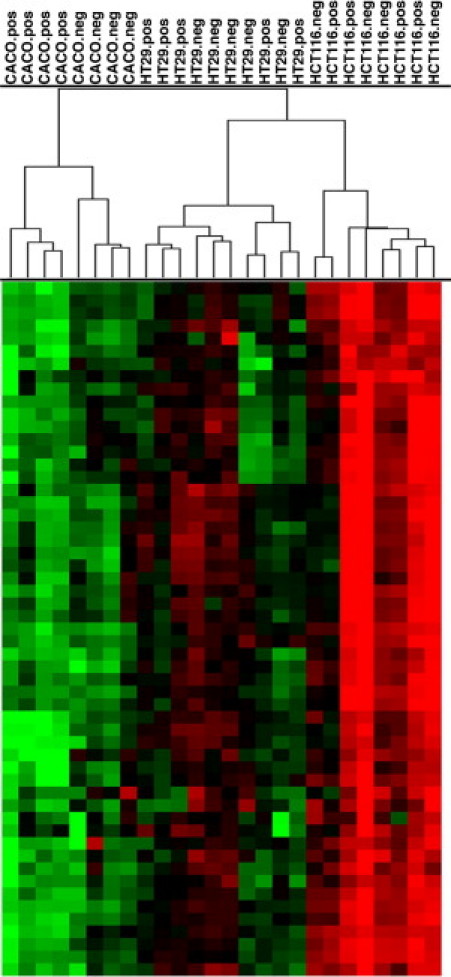
Unsupervised hierarchical clustering based on gene expression profiles of CD133+ and CD133− fractions from Caco-2, HCT 116, and HT-29 cell lines, data expressed using Euclidean metrics and the complete linkage algorithm. Samples cluster by cell line first. Only in the cell line Caco-2 were the different CD133 fractions clearly separated.
Figure 4.
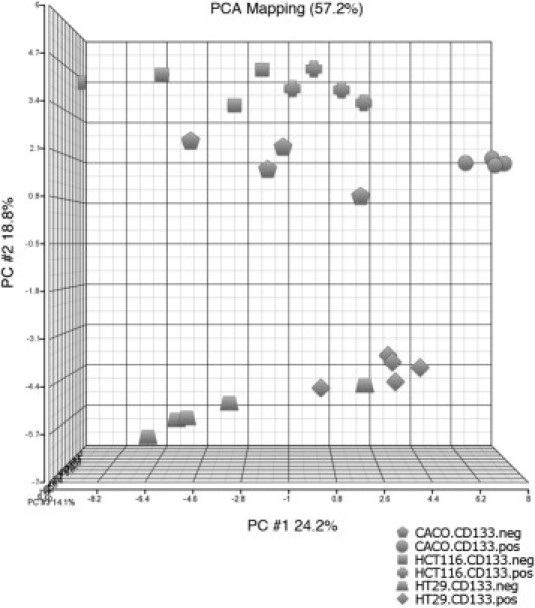
Supervised principal component analysis (PCA) of gene expression arrays using 96 genes up or down-regulated in CD133+ versus CD133− fractions for HCT 116 (n = 4), Caco-2 (n = 4), and HT-29 (n = 5). The supervised PCA shows a clear separation of CD133+ and CD133− along the first principal component for all samples, except one outlier (one HT-29 CD133− sample).
The corresponding functional annotation of the 96 differentially expressed genes and their affiliation with specific genetic pathways was interrogated using the Ingenuity Pathway Analysis Software (Ingenuity Systems, Redwood City, CA) and revealed the genes up-regulated in CD133+ cells mapped to the pathways lipid metabolism, small molecule biochemistry, and cancer (P < 0.05).
Because of the growth rate differences observed for CD133+ versus CD133− in HT-29, we also performed an Ingenuity Pathway Analysis Software (Ingenuity Systems) analyses for these subsets, demonstrating that the stem cell proliferation pathway is enhanced in the CD133+ cells. Key players in this pathway are APC, CTNNB1, and WNT3A (Figure 5). This is in line with findings that CTNNB1 was upregulated in CD133+ SW620 cells, thereby also explaining growth rate differences.39
Figure 5.
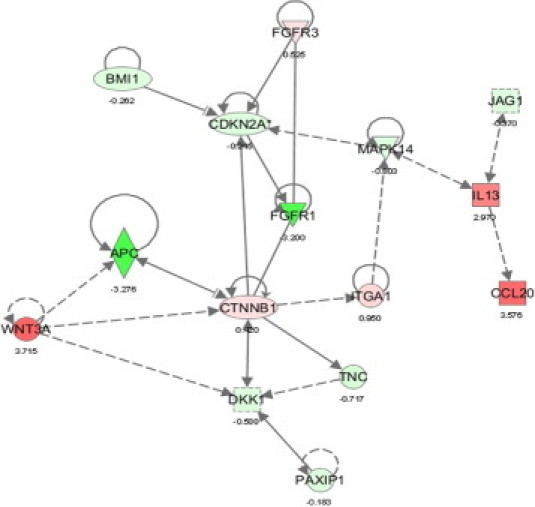
Network annotation of genes differentially expressed according to the CD133 status in HT-29 using ingenuity pathway analyses. Red, genes up-regulated in CD133+; green, genes down-regulated in CD133+. Dark red or green shade, genes with > threefold differential expression; light red or green shade, genes with lower difference in expression. All genes spotted were deregulated significantly (P < 0.0001).
aCGH from Flow Cytometry-Sorted Cells from Colon Cancer Cell Lines
To determine whether the observed changes in gene expression could be explained by distinct genomic aberration profiles, we performed aCGH from unsorted and sorted CD133+ and CD133− cells from HCT 116, HT-29, and Caco-2. We observed no differences among the fractions (Figure 6). These results imply that the specific transcriptional differences we observed were not attributable to differences in the underlying genomic aberrations.
Figure 6.

Ideogram of array comparative genomic hybridization chromosomal gains and losses for HT-29. (A) CD133+ and (B) CD133− cells. The aberration profile looked identical.
Genetic Profiling of CD133+ and CD133− Populations in Primary Human Colorectal Cancers by Array CGH after LCM
Since our aCGH analysis of CRC cell lines did not reveal genomic differences between CD133+ and CD133− cells, we extended our experiments to tissue sections of primary tumor specimens. CD133+ crypts, identified using IHC, from 12 samples were microdissected with LCM and analyzed by aCGH.
All 12 cancer samples had chromosomal imbalances that were characteristic for colon cancer.4 Copy number increases were most prominent on chromosomes 13 (67%), 7 (58%), 8q (50%), 1q (33%), and 20 (33%), and chromosomal losses on chromosomes 8p (50%), 18 (42%), 1p (42%), 15 (42%), and 4 (33%) (Table 1). We detected differences between the CD133+ and CD133− cell fraction in 7 of 12 (58%) cases. The remaining cases showed identical aberration profiles (Table 1). The significance of these findings was further substantiated by our control experiments, in which we extracted DNA from two randomly selected punch biopsies of tissue sections from eight patients (in four cases from CD133+ areas, and in another set of four from CD133− areas on the slides). In all cases, aCGH showed identical aberration profiles. The differences between the different CD133 fractions can therefore not be attributed to general sample heterogeneity.
Table 1.
List of Genetic Alterations in Primary Colorectal Tumor Samples Depending on the CD133 Immunophenotype
| Pt. number | Genetic alterations in all tumor cells | Δ CD133+ | Δ CD133− |
|---|---|---|---|
| H12017/09 | 2+, 7+, 8+, 17p−, 17q+, X+ | 13q+ | − |
| H14515/09 | 1p−, 3p−, 4−, 5q−, 6q−, 7q+, 8−, 9q−, 10−, 15q−, 17−, 18−, 21q−, 22q− | 14q− | 1q− |
| H21254/09 | 1p−, 4q+, 7+, 9p+, 13q+, 15q−, 16p−, 17−, 18−, 19−, 20+, 21q−, 22q− | 8+ | 8p− |
| H35810/07 | 1p−, 4−, 5q−, 6−, 7+, 8p−, 8q+, 9−, 13q+, 14q− | 10q− | 5p+ |
| H1676/08 | 7+, 8+, Y+ | − | − |
| H12291/09 | 1q+, 18q+, Y+ | − | − |
| H13103/09 | 5q−, 7q+, 8p−, 10p−, 11q−, 13q+, 14q−, 15q−, 18p−, 20q+, 21q−, 22q− | − | − |
| H25110/08 | 1p−, 4−, 8p−, 8q+, 15q−, 17p−, 18− | 13q+ | − |
| H25542/08 | 1p−, 1q+, 2q+, 4p−, 6q−, 7+, 9p−, 13q+, 15q−, 18−, 20q+ | − | − |
| H32373/08 | 1q+, 2+, 3q+, 5q+, 7+, 9q+, 10+, 11+, 12+, 15q+, 16+, 20+ | 13q+ | 6q+ |
| H33961/07 | 1+, 4q−, 7+, 8p−, 8q+, 13q+, 17−, 18q−, X− | − | − |
| H35810/07 | 8q+ | − | 9+ |
Δ CD133+, genetic alterations only observed in CD133+ tumor cells; Δ CD133−, alterations only observed in CD133− tumor cells; Pt. number, individual patient tumor histology number.
The gain of 13q occurred in three of the seven cases with genetic differences (Table 1). We evaluated whether the presence of this unique difference between CD133+ and CD133− was higher than what would be expected by chance. For this analysis, we used a re-sampling method to compute a P value. For each patient we pooled the chromosome arms that are changed in either CD133+ or CD133− samples. Then we selected zero or one arm that is randomly assigned as being changed in CD133+ but not in CD133− cells. We also selected zero or one arm that is randomly assigned as changed in CD133− but not in CD133+. This was done in a way that ensures there are exactly 11 arms changed in one region but not in the other when summed across all seven patients (R script is available on request). This re-sampling approach resulted in a P value of 0.024 for the probability that the same gain or loss emerges as different between the two regions in three or more of the seven patients. These data indicate that CD133 in the majority of colorectal cancer cases studied herein (58%) marked genetically distinct cell populations.
Discussion
Colorectal cancer is the third most common cancer in the United States resulting in approximately 50,000 deaths every year.40 While curative surgical treatment is possible at early stages, the presence of synchronous metastases at the time of diagnosis dramatically worsens prognosis.41,42 Adjuvant chemotherapy often leads to temporary remission but is frequently followed by disease recurrence. This could possibly be due to the fact that the majority of anti-cancer therapies are targeting rapidly dividing cells. In other words, conventional chemotherapies only target the transit amplifying and differentiated cells that form more than 99% of the tumor, yet spare the resting tumor initiating cells. CSCs, which are supposed to initiate new tumors, are slow cycling and are therefore less affected by anti-proliferative therapies.43–45 In addition to chemotherapy resistance,46 CSCs are often refractory to standard radiotherapy regimes.47
In colon cancer, CSCs were described as being contained in a fraction of cells positive for the surface marker CD133.25,26 Furthermore, it has been reported that CD133+ CSCs increase in proportion after therapy.43,45,48–50 It is tempting to speculate that residual colon cancer stem cells are responsible for loco regional recurrence, a hypothesis supported by the finding that indeed high levels of CD44 and CD133 expression were associated with poor prognosis in colorectal cancer.51,52
There is a pressing need to develop new therapies that can target this unique subpopulation of cancer cells. One of the first steps to achieve this would be a molecular characterization of these stem cells.
The maintenance of a recurrent pattern of chromosomal aneuploidy in the bulk of the tumor suggests that cells with this specific pattern of aberrations have a growth advantage. Therefore, one could also argue that this very pattern of aneuploidies originates in the stem cell compartment, and when present, converts a stem cell into a tumor stem cell. This hypothesis could be tested by analyzing the genomic aberration profile of suspected stem cell populations.
We found, in concordance with the literature, that not all CRC lines contained CD133+ cells. Of the five examined cell lines, three showed both populations.31 The high percentage of CD133+ cells in HCT 116 and Caco-2, however, is somewhat surprising because stem cells were thought to be rare.53 An explanation is that cell culture conditions provide a suitable milieu for amplifying cancer stem cells, and therefore artificially increase the stem cell compartment.
Our aCGH analyses that followed FACS for CD133± revealed an identical genomic aberration profile in HT-29, Caco-2, and HCT 116, arguing against a hierarchical organization based on different subclones. This is consistent with our observation that there were no differences in the tumorigenic potential of CD133+ versus CD133− subpopulations in the three cell lines.31,54 We only noticed a subtly faster tumor growth rate for CD133+ cells versus CD133− cells in HT-29, which was also observed by Ieta et al.54
However, an increased tumor growth rate for the CD133+ HT-29 fraction is not an argument for stemness, because tumor initiation, the most important defining feature of stem cells, is not necessarily linked to proliferative activity. In addition, the morphology of tumors after xenotransplantation was identical regardless of the CD133 status.
Analyses of the xenografted tumors with flow cytometry revealed that CD133+ tumors had now gained a significant percentage of CD133− cells, while CD133− tumors acquired a certain number of CD133+ tumor cells, therefore reconstituting the biphasic distribution in the cell lines before sorting, possibly reflecting differences in the growth environment.
To investigate the molecular consequences of CD133 positivity, we performed global gene expression analyses of FACS fractions. This revealed an overexpression of genes involved in lipid metabolism in CD133+ cells. This finding is intuitive because PROM1 is associated with a cholesterol-based membrane microdomain in which PROM1 interacts directly and specifically with plasma membrane cholesterol.55–57
In conclusion, the in vitro results demonstrate a high variability of CD133 expression and do not support an association with stemness.
The results also indicate that established cell lines are most likely not appropriate to study stemness defining molecular features, and that such analyses require the use of primary human colorectal cancers, including recurring and metastatic tumors. This considerable amount of variability of CD133 expression could be possibly due to an adaptation of cells to tissue culture conditions. In three-dimensional cultures (spheroids) of primary tumors, such differences might not be as pronounced.
Then we approached the question as to whether CD133+ cells are different from negative fractions in primary colorectal carcinomas by combining IHC, LCM, and aCGH on tissue sections. The percentage of CD133 IHC-positive tumor cells in our FFPE-embedded CRC cases ranged from 0 to nearly 95% and revealed earlier findings.35 Usually, several CD133+ tumor glands were grouped together with intervening CD133− glands.
In tumor glands with CD133+ tumor cells, the intraglandular cellular debris was always also CD133+. The amount of positive cells was considerably higher than the approximately 2 to 5% positive glands published by Ricci-Vitiani et al.25 The cause of that discrepancy could be a different IHC staining procedure and embedding strategy. Ricci-Vitiani et al25 used fresh frozen cryostat-sectioned material and not FFPE, which might affect IHC results.
After LCM of the CD133+ and CD133− tumor cells and aCGH of each fraction, we observed a genomic aberration profile in all 24 samples that was typical for colorectal carcinoma.58 Specifically, each of the 12 analyzed cancer samples showed recurrent losses (≥ 35%) of chromosomes 5q, 8p, 17p, 18p, 18q, and 20p, and recurrent gains (≥ 35%) of chromosomes 7p, 7q, 8q, 11q, and 20q.
While 42% (5 of 12) of the cases showed an identical aberration profile in the subfractions, we found different patterns in 58% (7 of 12) of the cases. To exclude that genetic differences in the subfractions were caused by polygenomic tumors without any association to a surface marker, we also performed independent aCGHs in the same tumor area. None of the performed control cases showed genetic differences within the chosen areas, thereby ruling out that the observed differences were attributed to random heterogeneity.
Our observations could be explained by the existence of different cell clones in primary tumors. The distribution of these clones does not appear to be coincidental, but defined by CD133 status. By showing a distinct aberration profile in CD133+ cells, we provide an explanation for the different properties of CD133+ cancer cells observed by Ricci-Vitiani et al.25 and O'Brien et al.26
However, in 42% of the tumor samples, the genomic aberrations profile was identical in the two fractions, possibly suggesting that CD133 status does not identify a putative CSC fraction in all colorectal tumors. This has also been shown in breast cancer.59 Therefore, it might be necessary to group colorectal tumors in two general classes of tumors, namely the monogenomic or polygenomic ones. It would be interesting to correlate this observation to therapy response and disease-free and overall survival.
Among the genetic alterations found, gain of 13q was detected in 3 of 7 cases (43%) that showed genetic differences between the CD133 positive and negative subfractions, thereby reaching a significant level. The gain of 13q is one of the major factors associated with the progression from colorectal adenoma to adenocarcinoma in chromosomal instable tumors.60 It was also demonstrated that 13q gain correlates particularly with metastasis, hence underlining that this is an important genetic region.61 Furthermore, it has been demonstrated that gain of 13q is associated with increased microRNA-17 to 92 cluster expression.62
Our results could now, for the first time, link this chromosomal aberration to cancer stem cells, which might have important implications for future therapeutic studies. Further studies in our laboratory will identify the role and function of candidate genes located on the chromosome of interest and potentially demonstrate an association with cancer stem cell pathways.
In summary, to the best of our knowledge, we were able to demonstrate for the first time that cancer stem cells, defined by the surface marker CD133, show a different genetic profile than the rest of the tumor cells in primary tumor samples.
While cell lines do not seem to be an appropriate model system for the characterization of cancer stem cells, CD133 expression in primary cancer samples was able to define a cell type that carries a specific aberration pattern in the majority of tumors investigated in this study. Given that not all cancer samples showed genetic differences in CD133± cells, it will be crucial to characterize the samples with gene expression and epigenetic assays. These data support the interpretation that CD133 positivity defines a tumor hierarchy, based on a distinct aberration profile, which could denote the tumor initiating cellular compartment.
Acknowledgments
The authors thank Buddy Chen for preparing figures and information technology-related support, Jens Marquardt for critical comments on the manuscript, and Hulia Waurig for technical assistance.
Footnotes
Supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. T.G. is supported by a fellowship within the post doctorate program of the Mildred Scheel Foundation. The work performed by the group of LAKS (Tumor Pathophysiology, OncoRay) was supported by the German Research Foundation (DFG) through a grant (KU 971/7-1). OncoRay is funded by the Bundesministerium fuer Bildung und Forschung in the program “Center for Innovation Competence.”
Supplemental material for this article can be found http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.12.036.
Supplementary Data
A, B: Control group. From eight FFPE blocks, two needle biopsies of approximately 0.5-cm distance in either the CD133+ or CD133− tumor area were cut and analyzed with aCGH for genetic differences (E). After immunohistochemistry for CD133, (C) the CD133+ and CD133− cells were laser microdissected (D) and analyzed with array comparative genomic hybridization for genetic differences (E).
Staining was performed according to a conventional set-up used for primary CRC cells in the literature.25,26
The proportion of CD133+/CD133− in HCT-116 cultures was not significantly different to the set-up in Supplemental Figure 2. The staining procedure allowed identifying small changes in the CD133 distribution throughout culturing. In short-term cultures after separation, the populations preserved their CD133 expression profile. At later stages, we observed a redistribution of the CD133+ and CD133− cell populations in the cultured CD133+ fraction. Given are the number of passages (P) and of cumulative population doublings (CPD) after sorting. The culture doubling time for the two fractions was identical.
CD133+/CD133− distributions after tumor growth in xenografts derived from injection of either CD133+ or CD133− subpopulation estimated by FACS (A) after HT−29 cell injection and (B) after HCT 116 cell injection.
References
- 1.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Leslie A., Carey F.A., Pratt N.R., Steele R.J. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89:845–860. doi: 10.1046/j.1365-2168.2002.02120.x. [DOI] [PubMed] [Google Scholar]
- 3.Habermann J.K., Paulsen U., Roblick U.J., Upender M.B., McShane L.M., Korn E.L., Wangsa D., Kruger S., Duchrow M., Bruch H.P., Auer G., Ried T. Stage-specific alterations of the genome, transcriptome, and proteome during colorectal carcinogenesis. Genes Chromosomes Cancer. 2007;46:10–26. doi: 10.1002/gcc.20382. [DOI] [PubMed] [Google Scholar]
- 4.Ried T., Knutzen R., Steinbeck R., Blegen H., Schrock E., Heselmeyer K., du Manoir S., Auer G. Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer. 1996;15:234–245. doi: 10.1002/(SICI)1098-2264(199604)15:4<234::AID-GCC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B., Fearon E.R., Hamilton S.R., Kern S.E., Preisinger A.C., Leppert M., Nakamura Y., White R., Smits A.M., Bos J.L. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 6.Wicha M.S., Liu S., Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. discussion 1895–1886. [DOI] [PubMed] [Google Scholar]
- 7.Lapidot T., Sirard C., Vormoor J., Murdoch B., Hoang T., Caceres-Cortes J., Minden M., Paterson B., Caligiuri M.A., Dick J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 8.Wang S., Garcia A.J., Wu M., Lawson D.A., Witte O.N., Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci USA. 2006;103:1480–1485. doi: 10.1073/pnas.0510652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;117:3155–3163. doi: 10.1172/JCI33295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stingl J., Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7:791–799. doi: 10.1038/nrc2212. [DOI] [PubMed] [Google Scholar]
- 12.Singh S.K., Clarke I.D., Terasaki M., Bonn V.E., Hawkins C., Squire J., Dirks P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 13.Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 14.Jordan C.T. Cancer stem cell biology: from leukemia to solid tumors. Curr Opin Cell Biol. 2004;16:708–712. doi: 10.1016/j.ceb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 15.O'Brien C.A., Kreso A., Dick J.E. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19:71–77. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Visvader J.E., Lindeman G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 17.Mizrak D., Brittan M., Alison M.R. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 18.Ferrandina G., Petrillo M., Bonanno G., Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets. 2009;13:823–837. doi: 10.1517/14728220903005616. [DOI] [PubMed] [Google Scholar]
- 19.Yin A.H., Miraglia S., Zanjani E.D., Almeida-Porada G., Ogawa M., Leary A.G., Olweus J., Kearney J., Buck D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 20.Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S., Roz E., Caserini R., Lo Vullo S., Camerini T., Mariani L., Delia D., Calabro E., Pastorino U., Sozzi G. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Immervoll H., Hoem D., Sakariassen P.O., Steffensen O.J., Molven A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008;8:48. doi: 10.1186/1471-2407-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright M.H., Calcagno A.M., Salcido C.D., Carlson M.D., Ambudkar S.V., Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maw M.A., Corbeil D., Koch J., Hellwig A., Wilson-Wheeler J.C., Bridges R.J., Kumaramanickavel G., John S., Nancarrow D., Roper K., Weigmann A., Huttner W.B., Denton M.J. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet. 2000;9:27–34. doi: 10.1093/hmg/9.1.27. [DOI] [PubMed] [Google Scholar]
- 24.Corbeil D., Roper K., Fargeas C.A., Joester A., Huttner W.B. Prominin: a story of cholesterol, plasma membrane protrusions and human pathology. Traffic. 2001;2:82–91. doi: 10.1034/j.1600-0854.2001.020202.x. [DOI] [PubMed] [Google Scholar]
- 25.Ricci-Vitiani L., Lombardi D.G., Pilozzi E., Biffoni M., Todaro M., Peschle C., De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien C.A., Pollett A., Gallinger S., Dick J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 27.Shmelkov S.V., Butler J.M., Hooper A.T., Hormigo A., Kushner J., Milde T., St Clair R., Baljevic M., White I., Jin D.K., Chadburn A., Murphy A.J., Valenzuela D.M., Gale N.W., Thurston G., Yancopoulos G.D., D'Angelica M., Kemeny N., Lyden D., Rafii S. CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111–2120. doi: 10.1172/JCI34401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y., Wu P.Y. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev. 2009;18:1127–1134. doi: 10.1089/scd.2008.0338. [DOI] [PubMed] [Google Scholar]
- 29.LaBarge M.A., Bissell M.J. Is CD133 a marker of metastatic colon cancer stem cells. J Clin Invest. 2008;118:2021–2024. doi: 10.1172/JCI36046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jungblut M., Oeltze K., Zehnter I., Hasselmann D., Bosio A. Standardized Preparation of Single-Cell Suspensions from Mouse Lung Tissue using the gentleMACS Dissociator. J Vis Exp. 2009:e1266. doi: 10.3791/1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dittfeld C., Dietrich A., Peickert S., Hering S., Baumann M., Grade M., Ried T., Kunz-Schughart L.A. CD133 expression is not selective for tumor-initiating or radioresistant cell populations in the CRC cell lines HCT-116. Radiother Oncol. 2009;92:353–361. doi: 10.1016/j.radonc.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 32.du Manoir S., Speicher M.R., Joos S., Schrock E., Popp S., Dohner H., Kovacs G., Robert-Nicoud M., Lichter P., Cremer T. Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum Genet. 1993;90:590–610. doi: 10.1007/BF00202476. [DOI] [PubMed] [Google Scholar]
- 33.Euhus D.M., Hudd C., LaRegina M.C., Johnson F.E. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31:229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 34.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 35.Horst D., Kriegl L., Engel J., Kirchner T., Jung A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br J Cancer. 2008;99:1285–1289. doi: 10.1038/sj.bjc.6604664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corbeil D., Roper K., Hellwig A., Tavian M., Miraglia S., Watt S.M., Simmons P.J., Peault B., Buck D.W., Huttner W.B. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. 2000;275:5512–5520. doi: 10.1074/jbc.275.8.5512. [DOI] [PubMed] [Google Scholar]
- 37.Florek M., Haase M., Marzesco A.M., Freund D., Ehninger G., Huttner W.B., Corbeil D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005;319:15–26. doi: 10.1007/s00441-004-1018-z. [DOI] [PubMed] [Google Scholar]
- 38.Rousset M., Dussaulx E., Chevalier G., Zweibaum A. Growth-related glycogen levels of human intestine carcinoma cell lines grown in vitro and in vivo in nude mice. J Natl Cancer Inst. 1980;65:885–889. [PubMed] [Google Scholar]
- 39.Kawamoto H., Yuasa T., Kubota Y., Seita M., Sasamoto H., Shahid J.M., Hayashi T., Nakahara H., Hassan R., Iwamuro M., Kondo E., Nakaji S., Tanaka N., Kobayashi N. Characteristics of CD133(+) human colon cancer SW620 cells. Cell Transplant. 2010;19:857–864. doi: 10.3727/096368910X508988. [DOI] [PubMed] [Google Scholar]
- 40.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 41.Holzel D., Eckel R., Engel J. [Colorectal cancer metastasis. Frequency, prognosis, and consequences] Chirurg. 2009;80:331–340. doi: 10.1007/s00104-008-1603-x. [DOI] [PubMed] [Google Scholar]
- 42.O'Connell J.B., Maggard M.A., Ko C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer ed 4 staging. J Natl Cancer Inst. 2004;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 43.Saigusa S., Tanaka K., Toiyama Y., Yokoe T., Okugawa Y., Ioue Y., Miki C., Kusunoki M. Correlation of CD133: OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann Surg Oncol. 2009;16:3488–3498. doi: 10.1245/s10434-009-0617-z. [DOI] [PubMed] [Google Scholar]
- 44.Sussman R.T., Ricci M.S., Hart L.S., Sun S.Y., El-Deiry W.S. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol Ther. 2007;6:1490–1495. doi: 10.4161/cbt.6.9.4905. [DOI] [PubMed] [Google Scholar]
- 45.Todaro M., Alea M.P., Di Stefano A.B., Cammareri P., Vermeulen L., Iovino F., Tripodo C., Russo A., Gulotta G., Medema J.P., Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 47.Murat A., Migliavacca E., Gorlia T., Lambiv W.L., Shay T., Hamou M.F., de Tribolet N., Regli L., Wick W., Kouwenhoven M.C., Hainfellner J.A., Heppner F.L., Dietrich P.Y., Zimmer Y., Cairncross J.G., Janzer R.C., Domany E., Delorenzi M., Stupp R., Hegi M.E. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 48.Todaro M., Lombardo Y., Francipane M.G., Alea M.P., Cammareri P., Iovino F., Di Stefano A.B., Di Bernardo C., Agrusa A., Condorelli G., Walczak H., Stassi G. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008;15:762–772. doi: 10.1038/sj.cdd.4402305. [DOI] [PubMed] [Google Scholar]
- 49.Todaro M., Perez Alea M., Scopelliti A., Medema J.P., Stassi G. IL-4-mediated drug resistance in colon cancer stem cells. Cell Cycle. 2008;7:309–313. doi: 10.4161/cc.7.3.5389. [DOI] [PubMed] [Google Scholar]
- 50.Yasuda H., Tanaka K., Saigusa S., Toiyama Y., Koike Y., Okugawa Y., Yokoe T., Kawamoto A., Inoue Y., Miki C., Kusunoki M. Elevated CD133, but not VEGF or EGFR, as a predictive marker of distant recurrence after preoperative chemoradiotherapy in rectal cancer. Oncol Rep. 2009;22:709–717. doi: 10.3892/or_00000491. [DOI] [PubMed] [Google Scholar]
- 51.Peng J., Lu J.J., Zhu J., Xu Y., Lu H., Lian P., Cai G., Cai S. Prediction of treatment outcome by CD44v6 after total mesorectal excision in locally advanced rectal cancer. Cancer J. 2008;14:54–61. doi: 10.1097/PPO.0b013e3181629a67. [DOI] [PubMed] [Google Scholar]
- 52.Horst D., Scheel S.K., Liebmann S., Neumann J., Maatz S., Kirchner T., Jung A. The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J Pathol. 2009;219:427–434. doi: 10.1002/path.2597. [DOI] [PubMed] [Google Scholar]
- 53.Reya T., Morrison S.J., Clarke M.F., Weissman I.L. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 54.Ieta K., Tanaka F., Haraguchi N., Kita Y., Sakashita H., Mimori K., Matsumoto T., Inoue H., Kuwano H., Mori M. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann Surg Oncol. 2008;15:638–648. doi: 10.1245/s10434-007-9605-3. [DOI] [PubMed] [Google Scholar]
- 55.Corbeil D., Marzesco A.M., Fargeas C.A., Huttner W.B. Prominin-1: a distinct cholesterol-binding membrane protein and the organisation of the apical plasma membrane of epithelial cells. Subcell Biochem. 2010;51:399–423. doi: 10.1007/978-90-481-8622-8_14. [DOI] [PubMed] [Google Scholar]
- 56.Corbeil D., Marzesco A.M., Wilsch-Brauninger M., Huttner W.B. The intriguing links between prominin-1 (CD133), cholesterol-based membrane microdomains, remodeling of apical plasma membrane protrusions, extracellular membrane particles, and (neuro)epithelial cell differentiation. FEBS Lett. 2010;584:1659–1664. doi: 10.1016/j.febslet.2010.01.050. [DOI] [PubMed] [Google Scholar]
- 57.Florek M., Bauer N., Janich P., Wilsch-Braeuninger M., Fargeas C.A., Marzesco A.M., Ehninger G., Thiele C., Huttner W.B., Corbeil D. Prominin-2 is a cholesterol-binding protein associated with apical and basolateral plasmalemmal protrusions in polarized epithelial cells and released into urine. Cell Tissue Res. 2007;328:31–47. doi: 10.1007/s00441-006-0324-z. [DOI] [PubMed] [Google Scholar]
- 58.Grade M., Becker H., Liersch T., Ried T., Ghadimi B.M. Molecular cytogenetics: genomic imbalances in colorectal cancer and their clinical impact. Cell Oncol. 2006;28:71–84. doi: 10.1155/2006/173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navin N., Krasnitz A., Rodgers L., Cook K., Meth J., Kendall J., Riggs M., Eberling Y., Troge J., Grubor V., Levy D., Lundin P., Maner S., Zetterberg A., Hicks J., Wigler M. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hermsen M., Postma C., Baak J., Weiss M., Rapallo A., Sciutto A., Roemen G., Arends J.W., Williams R., Giaretti W., De Goeij A., Meijer G. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology. 2002;123:1109–1119. doi: 10.1053/gast.2002.36051. [DOI] [PubMed] [Google Scholar]
- 61.Neklason D.W., Tuohy T.M., Stevens J., Otterud B., Baird L., Kerber R.A., Samowitz W.S., Kuwada S.K., Leppert M.F., Burt R.W. Colorectal adenomas and cancer link to chromosome 13q22.1-13q31.3 in a large family with excess colorectal cancer. J Med Genet. 2010;47:692–699. doi: 10.1136/jmg.2009.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diosdado B., van de Wiel M.A., Terhaar Sive Droste J.S., Mongera S., Postma C., Meijerink W.J., Carvalho B., Meijer G.A. MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br J Cancer. 2009;101:707–714. doi: 10.1038/sj.bjc.6605037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, B: Control group. From eight FFPE blocks, two needle biopsies of approximately 0.5-cm distance in either the CD133+ or CD133− tumor area were cut and analyzed with aCGH for genetic differences (E). After immunohistochemistry for CD133, (C) the CD133+ and CD133− cells were laser microdissected (D) and analyzed with array comparative genomic hybridization for genetic differences (E).
Staining was performed according to a conventional set-up used for primary CRC cells in the literature.25,26
The proportion of CD133+/CD133− in HCT-116 cultures was not significantly different to the set-up in Supplemental Figure 2. The staining procedure allowed identifying small changes in the CD133 distribution throughout culturing. In short-term cultures after separation, the populations preserved their CD133 expression profile. At later stages, we observed a redistribution of the CD133+ and CD133− cell populations in the cultured CD133+ fraction. Given are the number of passages (P) and of cumulative population doublings (CPD) after sorting. The culture doubling time for the two fractions was identical.
CD133+/CD133− distributions after tumor growth in xenografts derived from injection of either CD133+ or CD133− subpopulation estimated by FACS (A) after HT−29 cell injection and (B) after HCT 116 cell injection.