

Abstract
Fibrosis can occur in any human tissue when the normal wound healing response is amplified. Such amplification results in fibroblast proliferation, myofibroblast differentiation, and excessive extracellular matrix deposition. Occurrence of these sequelae in organs such as the eye or lung can result in severe consequences to health. Unfortunately, medical treatment of fibrosis is limited by a lack of safe and effective therapies. These therapies may be developed by identifying agents that inhibit critical steps in fibrotic progression; one such step is myofibroblast differentiation triggered by transforming growth factor-β1 (TGFβ1). In this study, we demonstrate that TGFβ1-induced myofibroblast differentiation is blocked in human fibroblasts by a candidate endogenous aryl hydrocarbon receptor (AhR) ligand 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE). Our data show that ITE disrupts TGFβ1 signaling by inhibiting the nuclear translocation of Smad2/3/4. Although ITE functions as an AhR agonist, and biologically persistent AhR agonists, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin, cause severe toxic effects, ITE exhibits no toxicity. Interestingly, ITE effectively inhibits TGFβ1-driven myofibroblast differentiation in AhR−/− fibroblasts: Its ability to inhibit TGFβ1 signaling is AhR independent. As supported by the results of this study, the small molecule ITE inhibits myofibroblast differentiation and may be useful clinically as an antiscarring agent.
Scarring, or fibrosis, can occur in any human tissue as a result of injury, surgery, certain drugs, or infection.1 Fibrosis results from an amplification of the normal wound healing response, in which fibroblasts proliferate, differentiate to myofibroblasts, and deposit collagen to rebuild damaged tissue.2–5 In fibrosis, an imbalance exists between the production of extracellular matrix components (eg, fibronectin and collagen I) and their degradation.1–3,5–7 In certain organs, such as the eye and lung, fibrosis can result in complete failure of tissue function and significant morbidity. Scarring in the orbit can result as a complication of thyroid eye disease (TED). TED is a condition in which intense inflammation leads to remodeling and expansion of the connective tissues of the orbit.8 The increased volume of orbital connective tissue leads to forward protrusion of the eye (exophthalmos), accompanied by nerve and muscle damage.9–11 In patients with severe TED, the initial inflammation subsides, but infiltration of muscle fibers by fibroblasts leads to fibrosis of the extraocular muscles, which limits their motility.8,10,11 The lung is another organ in which the consequences of scarring are particularly severe. Idiopathic pulmonary fibrosis (IPF) is characterized by fibrotic remodeling of lung tissue, including excessive deposition of extracellular matrix components.5,12 This process thickens and stiffens the alveolar walls, interfering with gas exchange and ventilation.3–5,7,13 IPF is a progressive disease, with poor prognosis and a median survival of only 2 to 5 years from the time of diagnosis.12 The consequences of both ocular and pulmonary fibrosis are severe, and effective clinical treatments are urgently needed, but not currently available.1,2,4,14,15 To develop such therapies, we need to establish a firm understanding of fibrotic mechanisms that might be exploited in fibrosis prevention or treatment.
Fibroblasts and myofibroblasts play major regulatory roles in both the normal wound-healing process and the development of fibrosis.4,5,12,16,17 Myofibroblasts are differentiated fibroblasts with contractile properties similar to smooth muscle cells.12 Myofibroblast contractility is conferred by their expression of smooth muscle proteins, such as α-smooth muscle actin (α-SMA) and calponin.2,5,12 Myofibroblasts are also a major source of extracellular matrix proteins, including collagen and fibronectin.3,6,12,13 Because of their prominent role in the fibrotic response, myofibroblast differentiation is a promising target for the development of novel antifibrotic therapies.12 We have used transforming growth factor-β1 (TGFβ1) to stimulate myofibroblast differentiation in primary cultures of human fibroblasts.3–5,12,14,15 This serves as a useful model system for the identification of potential small molecule inhibitors of myofibroblast differentiation.12
The results of many studies support the idea that TGFβ1, produced during the normal wound healing response, is a master regulator of fibrosis.2,5,7,12,14,18 Because of the prominent role of TGFβ1 signaling in myofibroblast differentiation and fibrosis, this pathway is an obvious target for potential antifibrotic therapies.1,18 TGFβ1 signaling is mediated primarily by members of the Smad family of signal transduction molecules.19 TGFβ1 binding to its receptors results in the phosphorylation of receptor-regulated Smads, including Smad2/3.18–21 Phosphorylated receptor-regulated Smads dissociate from the receptor and recruit the common-mediator, Smad4.21 This association allows for translocation of the Smad2/3/4 complex to the nucleus and initiation of TGFβ1-induced transcriptional responses.20,21 Although the overall mechanism of TGFβ1 signaling has been well studied, unrecognized regulatory factors that modulate the pathway may remain. This study is the first to suggest that the aryl hydrocarbon receptor (AhR) ligand 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) may play such a role.
The AhR is a ligand-dependent transcription factor that has been well-conserved throughout vertebrate evolution and is constitutively expressed in most cell types.19,22 Studies using AhR knock-out (AhR−/−) mice have revealed that the AhR plays a role in processes as diverse as cell proliferation, differentiation, migration, development, tissue homeostasis, vasculogenesis, and cancer.23–26 From the perspective of TGFβ1 signaling, it is interesting to note that AhR−/− mice are prone to develop bile duct fibrosis.23,25 Candidate endogenous ligands of the AhR include ketocholesterol, bilirubin, indigo, lipoxin A4, and tryptophan derivatives such as ITE.22,27–32 However, the AhR is better-known for its ligation by a diverse group of xenobiotics, the most potent of which is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).22 Agonist binding induces nuclear translocation of the AhR, which allows it to regulate expression of a variety of genes, including those of the cytochrome P450 family of drug-metabolizing enzymes.33 Until now, there has been no report of the effect that AhR ligands have on TGFβ1 signaling in primary human fibroblasts.
Here, we present our exciting findings that the AhR agonist ITE can block TGFβ1-induced myofibroblast differentiation and extracellular matrix production. These new findings suggest that ITE acts to modulate TGFβ1 signaling and to inhibit fibrosis. Furthermore, this small molecule may find potential therapeutic use as an antiscarring agent.
Materials and Methods
Cells and Reagents
Primary orbital and Tenon's capsule fibroblasts were isolated from individual patients undergoing eye surgery. Primary corneal fibroblasts were derived from anatomically normal cornea tissue from corneas donated to the Rochester/Finger Lakes Eye and Tissue Bank. Primary lung fibroblasts were isolated from anatomically normal lung tissue from patients undergoing surgical resection for benign hamartoma or for small peripheral nodules. In each case, patient samples were obtained with informed consent under the approval of the Institutional Review Board of the University of Rochester. Primary mouse fibroblasts (corneal and lung) were generated from wild-type (AhR+/+) and AhR−/− C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). All primary fibroblasts were established by standard explant techniques, as previously described.34 Strains were cultured in Minimum Essential Medium (MEM; Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hy Clone Labs, Logan, UT), 2 mmol/L l-glutamine, penicillin (100 units/mL), streptomycin (100 μg/mL), and amphotericin (0.25 μg/mL) (Invitrogen) at 37°C in 7% CO2. In all cases, the resulting cells were morphologically consistent with fibroblasts: they were adherent, did not express CD45, factor VIII, or cytokeratin, but did express vimentin and types I and III collagen.35 Strains were stored in liquid N2 until needed and were used between passages 4 and 10. ITE (Sigma Aldrich, St. Louis, MO) was prepared as a 20 mmol/L stock in DMSO. It was added to cell cultures to the final concentrations indicated. Recombinant human TGFβ1 was purchased from R&D Systems (Minneapolis, MN).
Western Blots for α-SMA, Calponin, Fibronectin, Collagen I, Smad2/3, Smad4, AhR, CYP1B1, pErk1/2, and pAkt
Primary human fibroblasts were cultured to confluence, serum starved for 24 hours, and treated with TGFβ1 and/or ITE as described. Cell lysates were prepared using either sample buffer (50 mmol/L dithiothreitol, 0.01% bromophenol blue, 62.5 mmol/L Tris-HCl, pH 6.8, 10% glycerol, 2% SDS) or the commercially available Nuclear Extract kit (Active Motif, Carlsbad, CA). Lysates containing 2 to 15 μg total protein were separated using precast 4% to 15% SDS-PAGE gradient gels (Bio-Rad, Hercules, CA), transferred to PVDF membranes (Millipore, Billerica, MA), and probed for the proteins of interest by Western blotting (Immobilon Western, Millipore) and chemiluminescence detection via HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Lysates were examined for expression of α-SMA (1:2000 dilution; Sigma Aldrich #A2547 for human samples and Sigma Aldrich #A5228 for mouse samples), calponin (1:5000 dilution; Dako, Carpinteria, CA; #M3556), fibronectin (1:5000 dilution; Sigma Aldrich #F3648), collagen I (1:1000 dilution; Santa Cruz, Carlsbad, CA; #sc8783), Smad2/3 (1:1000 dilution; Cell Signaling, Beverly, MA; #3102), pSmad2/3 (1:1000 dilution; Cell Signaling #3101), Smad4 (1:1000 dilution; Cell Signaling #9515), AhR (1:5000 dilution; Biomol, Plymouth Meeting, PA; #SA-210), CYP1B1 (1:1000 dilution; Santa Cruz #sc32882), pErk1/2 (1:1000 dilution; Cell Signaling #9101), or pAkt (1:1000 dilution; Cell signaling #4060). GAPDH (1:10,000 dilution; Calbiochem, Gibbstown, NJ; #CB1001), β-tubulin (1:2000 dilution; Cell Signaling #2146) and/or Lamin A (1:5000 dilution; Sigma Aldrich #L1293) were used as loading controls. Densitometry of the resulting bands was performed using Quantity One Imaging Software (Bio-Rad) and normalized to the loading control.
Slot-Blot for Collagen I
A goat anti-human collagen type I antibody (1:2000 dilution; SouthernBiotech, Birmingham, AL, #1310-01) was used for immunodetection of collagen I in the conditioned supernatants of human orbital fibroblasts. Supernatants (5 μl per sample in triplicate) were applied to a nitrocellulose membrane using a manifold apparatus (Schleicher and Schuell, Keene, NH). The antibody-bound collagen I was visualized by use of an anti-goat IgG secondary antibody and enhanced chemiluminescence kit (Western Lightning, Perkin Elmer, Wellesley, MA), according to the manufacturers' guidelines.
Real-Time RT-PCR
RNA was isolated from homogenized primary human orbital fibroblast cultures using RNeasy according to the manufacturer's protocol (Qiagen, Valencia, CA). RNA (0.5 μg) was incubated with PCR buffer, 0.5 μg oligo (dT)12-18 primer (Invitrogen) and 10 mmol/L each dNTP for 10 minutes at 70°C and 5 minutes in ice water followed by addition of 40 U recombinant RNasin ribonuclease inhibitor (Promega, Madison, WI), 0.1 mmol/L dithiothreitol, and 200 U Superscript III reverse transcriptase (RT; Invitrogen). The mixture was further incubated for 5 minutes at room temperature, 60 minutes at 50°C, and 15 minutes at 70°C. The reaction contents were diluted to 80 μl volume and stored at −20°C. Negative controls contained no RT enzyme. Quantitative real-time RT-PCR reactions were performed using a Bio-Rad iCycler with SYBR Green Supermix (Bio-Rad) according to the supplier's recommended protocol with the following modifications. For amplification of the collagen I (COL1A1) mRNA, the reaction contained 4 mmol/L MgCl2 and 0.1 μmol/L of each primer, whereas for collagen III (COL3A1), α-SMA (ACTA2), and GAPDH, the reactions contained 3 mmol/L MgCl2 and 0.2 μmol/L of each primer. Oligomer primers were ordered from Real Time Primers (COL1A1, Elkins Park, PA) and Integrated DNA Technologies (COL3A1, ACTA2 and GAPDH, Coralville, IA). The primer sequences were as follows. COL1A1 sense: 5′-GGACACAGAGGTTTCAGTGG-3′; antisense: 5′-CCAGTAGCACCATCATTTCC-3′. GAPDH sense: 5′-AAGGTCGGAGTCAACGGATTTGGT-3′; antisense: 5′-AGCCTTGACGGTGCCATGGAATTT-3′. ACTA2 sense: 5′-TCTGGAGATGGTGTCACCCACAAT-3′; antisense: 5′-AATAGCCACGCTCAGTCAGG-3′. COL3A1 sense: 5′-GGAGAATGTTGTGCAGTTTGCCCA-3′; antisense 5′-TCTGAGGACCAGTAGGGCATGATT-3′.
Cell Viability Assay
Primary human orbital fibroblasts were cultured and treated with TGFβ1 and/or ITE as described. Culture medium was removed from cells, AlamarBlue solution (10%; Biosource, Carlsbad, CA; #DAL1100) was added according to the manufacturer's instructions, and cultures were incubated at 37°C for 3 hours. Fluorescence was measured at excitation 530 nm and emission 590 nm using a Thermol electron corporation plate reader with Varioskan Flash 2.4.3 software. Cell viability was calculated as percent control.
Imaging Flow Cytometry
Primary human orbital fibroblasts were cultured and treated as described above. At harvest, cells were trypsinized, washed twice with PBS, and fixed with 100% methanol for 30 minutes at 4°C. The cells were then washed twice with PBS/0.1% Tween-20 (PBS-T) and blocked with 5% normal mouse serum for 20 minutes at room temperature. Blocked cells were washed once with PBS-T and incubated with FITC-labeled mouse anti-α-SMA antibody (1:400 dilution; Sigma Aldrich #F3777) in PBS-T + 1% BSA, overnight at 4°C. The next day, cells were washed twice with PBS-T and once with PBS, and stained with Draq5 (Axxora, San Diego, CA) at a 1:200 dilution in PBS. Samples were analyzed directly on an ImageStream System 100 (Amnis Corporation, Seattle, WA). Signals from brightfield, α-SMA-FITC and Draq5 were collected in channels 5, 3, and 6, respectively. The area and aspect ratio of the brightfield image were used in a gating strategy to isolate single cells from clumps and debris. The aspect ratio was calculated based on the ratio of cellular width to height. A single, round cell has an aspect ratio close to 1.0, whereas clumps of cells have a lower aspect ratio. A region was drawn on a dot-plot of all collected events (aspect ratio versus area) to enclose mostly single, round objects of average size. The accuracy of this strategy was verified by examining the brightfield images from events inside and outside of the resulting single cell region. Subsequently, objects from this region were subjected to α-SMA expression analysis. α-SMA expression was defined by the total intensity of the FITC (α-SMA) signal and the area of the cell covered by the α-SMA signal.
Immunofluorescence Microscopy for α-SMA and Collagen I or Smad3
Primary human fibroblasts were cultured and treated with TGFβ1 and/or ITE as described above and in figure legends. Cells were then fixed in 4% paraformaldehyde for 15 minutes (α-SMA and collagen I) or in −20°C methanol for 10 minutes (Smad3) and labeled for α-SMA (1:800 dilution; Sigma Aldrich #A2547) and collagen I (1:50 dilution; Santa Cruz #sc8783) or Smad3 (1:100 dilution; Cell Signaling #9523) using an immunofluorescent procedure in which biotinylated secondary antibodies were visualized with Avidin D fluorochrome conjugates (FITC-Avidin D for collagen I, Texas Red-Avidin D for α-SMA and Fluorescein Avidin DCS for Smad3). TO-PRO−3 iodide (642/661; Invitrogen #T3605) or DAPI (Vector Laboratories #H-1200) was used for nuclear counterstain. Samples were observed with an Olympus BX51 fluorescence microscope (Olympus Imaging America, Center Valley, PA) or a Zeiss Axio Imager MI microscope (Carl Zeiss, Oberkochen, Germany), and images were captured using AxioVision Rel 4.6 software. All images were acquired using the same intensity and photodetector gain to allow quantitative comparisons of relative levels of immunoreactivity between samples.
Immunocytochemistry for α-SMA
Primary fibroblasts cultured as described above at a density of 2000 cells per well on four-well chamber slides were treated for 96 hours with recombinant human TGFβ1 (1 ng/mL) and/or with ITE (1 μmol/L). Cells were then fixed with methanol and sequentially treated with mouse monoclonal α-SMA antibody (1:400 dilution; Sigma Aldrich #A2547) or IgG2A isotype control (1:90 dilution; R&D Systems #MAB-003), biotinylated goat anti-mouse (1:200 dilution; Vector Labs, Burlingame, CA; #BA-9200), and HRP-streptavidin conjugate (1:1000 dilution; Jackson Immunoresearch; #016-030-084). Cells were then stained with aminoethyl carbazole (Invitrogen) and counterstained with Gill's hematoxylin no. 1 (Sigma Aldrich) before mounting with Immunomount (Shandon, Thermo Fisher Scientific, Waltham, MA).
Collagen Gel Contraction Assay
Collagen gels were prepared from lyophilized rat-tail collagen (Roche, Switzerland). Collagen was dissolved in 0.2% acetic acid (v/v) and mixed with complete medium and neutralized with NaOH for a final collagen concentration of 0.75 mg/mL. A total of 5000 primary human orbital fibroblasts were added to each collagen gel, and gels were allowed to polymerize at 37°C and 5% CO2 in 24-well plates preblocked with complete medium. After 1 hour, gels were floated in medium containing ITE (0 to 1 μmol/L) and TGFβ1 (0 to 1 ng/mL). After 4 days, the gels were weighed and percent contraction, as compared with gels without cells, was calculated.
Luciferase Assay
Primary human orbital fibroblasts were plated in a 24-well plate at 40 × 103 cells/well 1 day before transfection. Fugene HD (Roche, Switzerland) was used as per the manufacturer's instructions to transfect 0.8 μg Smad Binding Elements-luciferase plasmid (SBE-luc). SBE-luc was a kind gift from Dr. Jian-Dong Li (University of Rochester, School of Medicine and Dentistry, Rochester NY).36 After transfection, the cells were allowed to grow for 24 hours; they were then pretreated with 0 to 1 μmol/L ITE for 1 hour and treated with 0 to 1 ng/mL TGFβ for 48 hours. Subsequently, luciferase activity was measured in cell lysates using the Luciferase Assay System (Promega, Madison, WI) and the DTX 880 multimode detector (Beckman Coulter, La Brea, CA) according to the manufacturer's instructions.
Results
ITE Inhibits TGFβ1-Induced Myofibroblast Differentiation and Extracellular Matrix Production in Primary Human Orbital Fibroblasts
Primary cultures of human orbital fibroblasts were treated with TGFβ1 (1 ng/mL) to stimulate myofibroblast differentiation. ITE was included in some cultures to assess its ability to inhibit this process. Western blotting confirmed that AhR protein is expressed in this cell type (Figure 1A). Furthermore, the expressed AhR is responsive to ligand-induced activation, as shown by both the induction of the cytochrome P450 protein CYP1B1 and the degradation of AhR in response to ITE treatment. Figure 1A also shows that TGFβ1 treatment does not influence AhR expression in human orbital fibroblasts. Once we knew that AhR protein was expressed and active in this cell type, we conducted dose-response experiments of AhR activation based on induction of CYP1B1 to choose the concentrations of ITE to use in subsequent experiments (data not shown).
Figure 1.
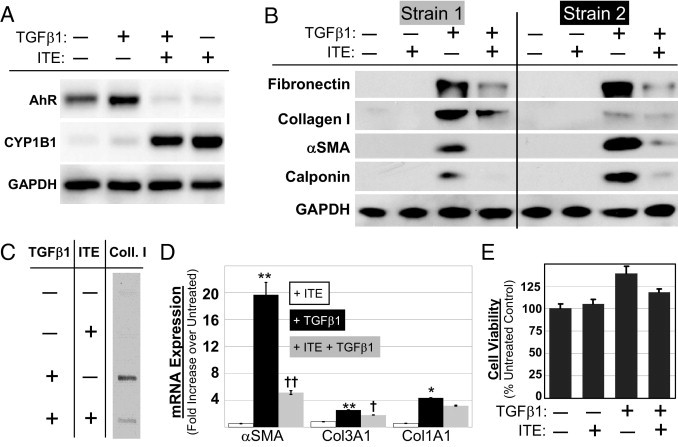
ITE potently suppresses TGFβ1-induced expression of myofibroblast marker proteins but is not toxic in primary human orbital fibroblasts. Primary cultures of human orbital fibroblasts were treated for 96 hours with TGFβ1 (1 ng/mL) and ITE (1 μmol/L). A: Whole cell lysates were Western blotted using antibodies specific for AhR, CYP1B1, and GAPDH, as indicated. B: Whole cell lysates from two separate strains of fibroblasts (ie, fibroblasts derived from two separate individuals) were Western blotted using antibodies specific for fibronectin, collagen I, α-SMA, calponin, and GAPDH, as indicated. C: Cell culture supernatants were slot-blotted and probed using a collagen I antibody. D: Primary human orbital fibroblasts were treated with TGFβ1 alone (1 ng/mL) or with TGFβ1 and ITE (1 μmol/L) and mRNA was harvested after 48 hours. α-SMA, collagen III (COL3A1), and collagen I (COL1A1) mRNAs were quantified using real time RT-PCR. *P < 0.01 compared with untreated; **P < 0.001 compared with untreated; †P < 0.01 compared with TGFβ1 alone; ††P < 0.001 compared with TGFβ1 alone (analysis of variance). E: Percent control cell viability was measured using the AlamarBlue cell viability assay.
Myofibroblast differentiation was measured using Western blot analysis to monitor the expression of the canonical myofibroblast marker α-SMA as well as the smooth muscle protein calponin and extracellular matrix components, collagen I and fibronectin. Collagen I was also detected in cell culture supernatants via slot blot analysis. Real time RT-PCR was used to measure steady state mRNA levels of α-SMA, collagen III (COL3A1) and collagen I (COL1A1). ITE (1 μmol/L) inhibited TGFβ1-induced fibronectin, collagen I, α-SMA, and calponin expression, as shown by Western blot (Figure 1B). Slot blot analysis revealed that levels of collagen I in cell culture supernatants were increased in TGFβ1-treated cultures and that this increase was inhibited in the presence of ITE (Figure 1C). ITE was also shown by real time RT-PCR to reduce the expression of TGFβ1-induced α-SMA and collagen III (Figure 1D). Collagen I mRNA levels were also reduced, but the magnitude of the reduction failed to reach statistical significance (P > 0.05).
ITE Treatment Is Not Toxic to Primary Human Orbital Fibroblasts
After treatment for 96 hours with 1 ng/mL TGFβ1 and/or 1 μmol/L ITE, the AlamarBlue assay was used to measure cell viability in primary cultures of human orbital fibroblasts (Figure 1E). AlamarBlue is a cell-permeable compound that fluoresces only when it is reduced after entry into a viable cell. Compared with untreated cultures, treatment with ITE alone had no impact on AlamarBlue fluorescence while treatment with TGFβ1 increased fluorescence by about 40%. However, it is more likely that the increased fluorescence observed in the TGFβ1-treated samples resulted from increased fibroblast proliferation rather than enhanced viability for the following reasons: (1) the cells were cultured for 4 days before the assay was run, (2) AlamarBlue essentially provides a measure of viable cell number, and (3) TGFβ1 is known to be an important mitogenic signal for fibroblasts.37 The addition of ITE to TGFβ1-treated cultures did reduce AlamarBlue fluorescence relative to samples treated with only TGFβ1. However, AlamarBlue fluorescence was still greater than control in these co-treated cultures, confirming that ITE treatment of fibroblasts produces no toxicity.
Cellular Imaging Confirms that ITE Inhibits TGFβ1-Induced α-SMA Expression in Primary Human Fibroblasts
In addition to the standard techniques for measuring myofibroblast differentiation described above, we have also developed and used a protocol to monitor α-SMA expression via imaging flow cytometry. This technique has proved to be invaluable for the quantification of cells expressing the myofibroblast phenotype, a parameter that has, until now, been inadequately addressed by available techniques. Imaging flow cytometry uses the Amnis ImageStream instrument to collect fluorescent images from thousands of cells per sample. These images are used to make quantitative measurements of protein expression based on fluorescence pattern and intensity. Imaging flow cytometry effectively combines the advantages of both microscopic and flow cytometric analyses, allowing for quantitative measurement of α-SMA expression on a per-cell basis with an event count sufficient for statistical analysis. Myofibroblasts were distinguished from fibroblasts based on the area and intensity of α-SMA staining, as described in Materials and Methods. TGFβ1 treatment for 96 hours resulted in myofibroblast differentiation in 79% of cultured orbital fibroblasts (Figure 2A). Addition of 1 μmol/L ITE to the cultures resulted in a decrease in the percentage of differentiated cells to only 40%. Figure 2, B and C show examples of the images gathered by the ImageStream, which form the basis for the quantitative data in Figure 2A.
Figure 2.
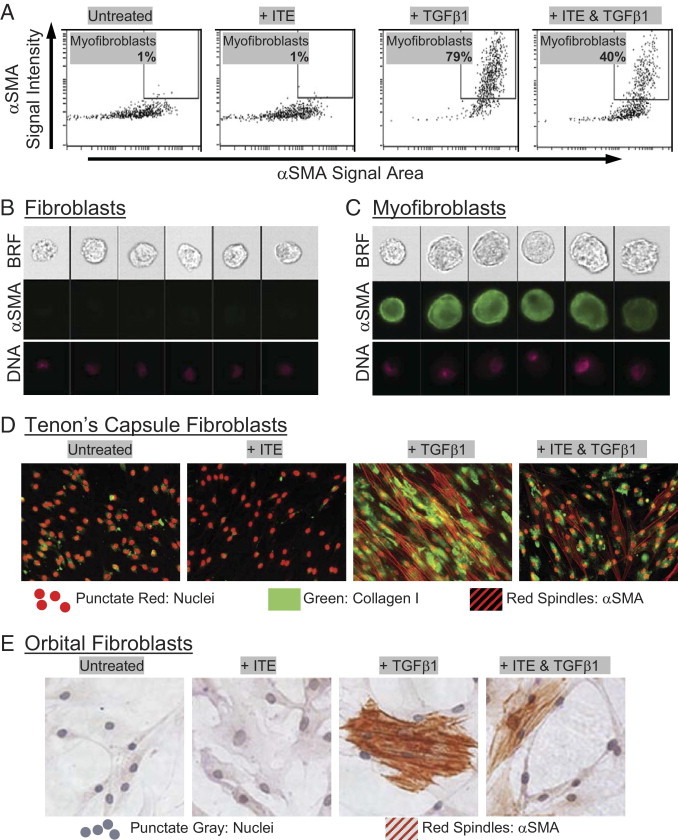
Cellular imaging confirms that ITE attenuates TGFβ1-induced myofibroblast differentiation. (A–C) Human orbital fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 96 hours. They were then fixed with methanol and stained for α-SMA using a FITC-conjugated antibody and analyzed on an Amnis ImageStream imaging flow cytometer. Live single cells were identified as described in Materials and Methods. A: Myofibroblasts were distinguished from fibroblasts based on the intensity (y axis) and area (x axis) of α-SMA staining. Numerical percentage value on each dot-plot refers to the percentage of cells contained within the myofibroblast region. Representative images of cells from both (B) outside and (C) inside the myofibroblast region are shown. Image panels show brightfield (BRF), α-SMA (green), and nuclei (DNA) stained with Draq5 (pink). D: Tenon's capsule fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 96 hours. Cells were stained with antibodies specific for collagen I and α-SMA then counterstained with TO-PRO−3 iodide to detect nuclei. Green staining indicates the presence of collagen I. Red spindles represent positive staining for α-SMA. Nuclei are also stained red, but they can easily be distinguished morphologically from α-SMA. E: Primary cultures of human orbital fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 96 hours. Cells were stained with α-SMA antibody and counterstained with hematoxylin. Red spindles represent positive staining for α-SMA. Magnification, ×200.
More traditional cellular imaging techniques were also used to assess myofibroblast marker expression in fibroblasts treated with TGFβ1 and/or ITE. Here, primary cultures of human fibroblasts from Tenon's capsule (Figure 2D) or orbit (Figure 2E) were treated with 1 ng/mL TGFβ1 to stimulate myofibroblast differentiation in the presence of ITE. Myofibroblast differentiation was measured by immunofluorescence microscopy for α-SMA and collagen I (Figure 2D) or by immunocytochemistry for α-SMA (Figure 2E). The results of these techniques confirm that ITE treatment attenuates TGFβ1-induced myofibroblast marker expression in human fibroblast cultures.
ITE Inhibits TGFβ1-Induced Myofibroblast Differentiation in Primary Human Fibroblasts Derived from a Wide Variety of Tissue Types
Thus far in this study, we have established that ITE inhibits TGFβ1-induced myofibroblast differentiation in primary cultures of human orbital fibroblasts. However, our laboratory and others have shown that fibroblasts from different human tissues are truly heterogeneous, and have different biosynthetic repertoires and responses to provocation with activating agents.38 Therefore, we wondered whether the inhibitory effects of ITE on TGFβ1 signaling would prove to be specific to orbital fibroblasts or would generalize to fibroblasts derived from a variety of human tissues. As shown above, we were able to demonstrate that ITE exerts an effect on Tenon's capsule fibroblast cultures that is similar to that observed in cultures of orbital fibroblasts (Figure 2D). However, to further confirm the generality of this effect, primary cultures of human fibroblasts from cornea (Figure 3A) and lung (Figure 3B) were treated with 1 ng/mL TGFβ1 to stimulate myofibroblast differentiation in the presence of 0 to 20 μmol/L ITE. AhR activation by ITE was confirmed in lung fibroblasts via CYP1B1 Western blot (Figure 3B). Myofibroblast differentiation was measured by Western blotting for α-SMA, calponin and fibronectin in corneal (Figure 3A) and lung (Figure 3B) fibroblasts. The cumulative results of these studies show that ITE inhibited myofibroblast differentiation in a variety of primary human fibroblast cultures, including those derived from orbit, Tenon's capsule, cornea, and lung.
Figure 3.
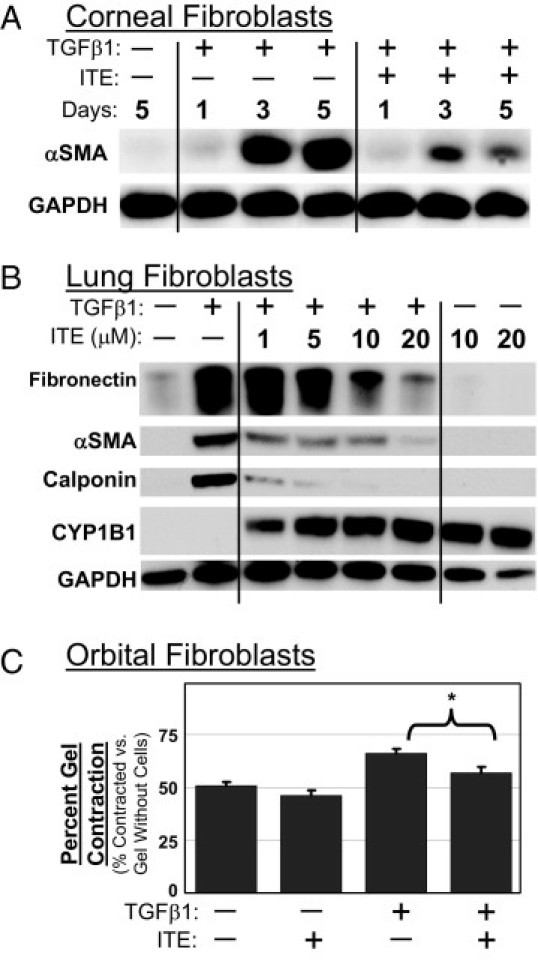
Inhibition of TGFβ1-induced myofibroblast differentiation by ITE is not specific to orbital fibroblasts, and it results in decreased myofibroblast activity, as demonstrated by collagen gel contraction assay. A: Human corneal fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 1, 3, or 5 days. Cell lysates were Western blotted using antibodies specific for α-SMA and GAPDH. B: Human lung fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (0 to 20 μmol/L) for 96 hours. Cell lysates were Western blotted using antibodies specific for fibronectin, α-SMA, calponin, CYP1B1, and GAPDH. C: Collagen gels were prepared with primary human orbital fibroblasts and treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 4 days. Collagen gels were weighed and percent contraction, as compared with gels without cells, is shown. *P < 0.001 compared with TGFβ1 alone (Student's t-test).
Collagen Gel Contraction Assay Provides Functional Confirmation that ITE Inhibits TGFβ1-Induced Myofibroblast Differentiation in Primary Human Orbital Fibroblasts
The collagen gel contraction assay is used to study the interaction between cells and the extracellular matrix protein collagen. On TGFβ1 treatment, collagen gels containing primary human orbital fibroblasts contracted due to the increased contractile properties of myofibroblasts relative to undifferentiated fibroblasts (Figure 3C). ITE treatment significantly attenuated the collagen gel contraction induced by TGFβ1.
Inhibition of Myofibroblast Differentiation by ITE Is AhR Independent
To assess the importance of AhR expression to the antifibrotic activity of ITE, we repeated our experiments using AhR−/− mouse fibroblast strains from corneal tissue and from lung. In an initial experiment, AhR−/− mouse corneal fibroblasts were treated with 1 ng/mL TGFβ1 in the presence of 1 μmol/L ITE. Western blotting revealed an ITE-dependent attenuation of TGFβ1-induced α-SMA up-regulation even in the complete absence of AhR expression (Figure 4A). To build on this finding, a similar protocol was followed using AhR+/+ and AhR−/− mouse lung fibroblasts. These cells were also treated with TGFβ1 and ITE, but in this expanded experiment, we blotted lysates for CYP1B1, collagen I, fibronectin, and calponin. Here, we found that a lack of AhR expression and activity (as indicated by a lack of CYP1B1 induction in response to ligand) did not significantly reduce the ability of ITE to inhibit TGFβ1-induced myofibroblast differentiation, as measured by α-SMA expression (Figure 4, B and C). Because we eliminated AhR signaling as a factor in the antifibrotic mechanism of this compound, we sought to identify the point in the TGFβ1 signaling pathway targeted by ITE.
Figure 4.
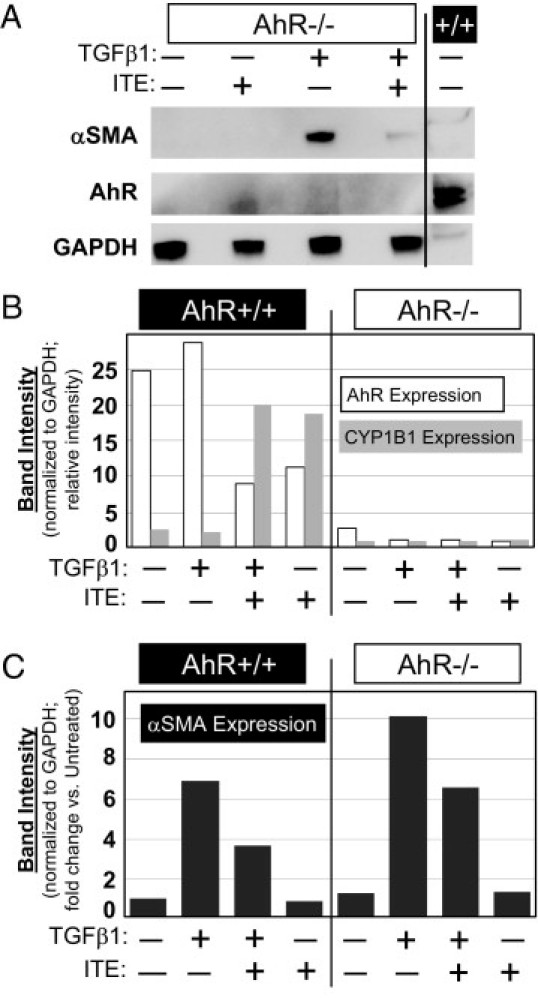
ITE functions through an AhR-independent pathway to inhibit TGFβ1-induced myofibroblast marker expression. A: AhR−/− mouse corneal fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 96 hours. Cell lysates were Western blotted using antibodies specific for α-SMA, AhR, and GAPDH. Far right lane contains lysate from untreated AhR+/+ mouse fibroblasts to demonstrate the ability to detect AhR under these blotting conditions. B and C: AhR+/+ and AhR−/− mouse lung fibroblasts were treated with TGFβ1 (1 ng/mL) and ITE (1 μmol/L) for 96 hours. Cell lysates were Western blotted using antibodies specific for AhR, CYP1B1, α-SMA, and GAPDH. Band intensities of AhR, CYP1B1, and α-SMA blots were quantified by densitometry and normalized to GAPDH. C: Band intensities are expressed as fold change versus untreated fibroblasts.
ITE Inhibits TGFβ1-Induced Nuclear Translocation of Smad2, Smad3, and Smad4 in Primary Human Orbital Fibroblasts
One mechanism by which ITE may inhibit TGFβ1 signaling is by interference with the Smad pathway. Primary human orbital fibroblasts were pretreated with ITE for 2 hours, then treated with TGFβ1 and harvested after 1 hour. Western blot analysis demonstrated that TGFβ1 treatment significantly increased pSmad2/3 levels in primary human orbital fibroblasts and that pretreatment with ITE did not inhibit TGFβ1-induced phosphorylation of Smad2/3 in these cells (Figure 5A). Neither total Smad 2/3 nor Smad4 levels were altered by either TGFβ1 or ITE treatment (Figure 5A). These results indicate that activation of AhR signaling does not block TGFβ1-induced phosphorylation of Smad2/3, and that disruption of Smad2/3 phosphorylation does not account for the inhibitory effects of ITE on TGFβ1-induced myofibroblast differentiation.
Figure 5.
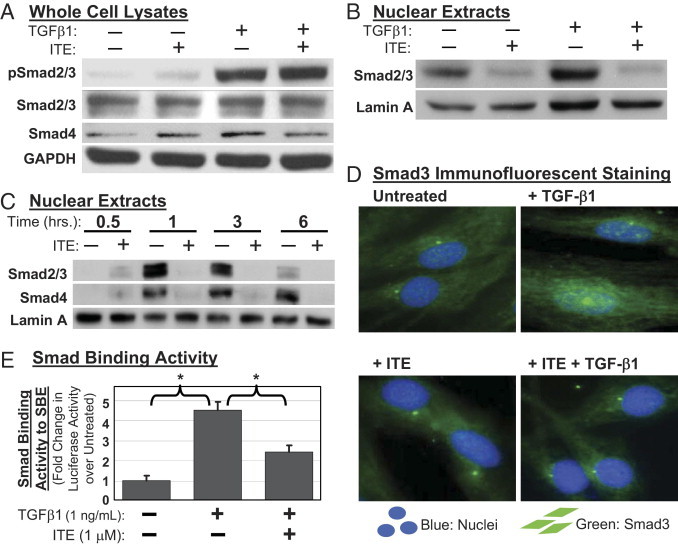
TGFβ1-induced nuclear translocation of Smad2, Smad3, and Smad4 is inhibited by ITE. Primary cultures of human orbital fibroblasts were pretreated with ITE (1 μmol/L) followed by treatment with TGFβ1 (1 ng/mL). A: Whole cell lysates were Western blotted after 1 hour of TGFβ1 treatment using antibodies specific for phosphorylated-Smad2/3 (pSmad2/3), Smad2/3, Smad4, and GAPDH. B: Nuclear extracts were Western blotted after 1 hour of TGFβ1 treatment using antibodies specific for Smad2/3 and Lamin A. C: Nuclear extracts were harvested after 0.5, 1, 3, or 6 hours of TGFβ1 treatment and Western blotted using antibodies specific for Smad2/3, Smad4, and Lamin A. D: After 20 minutes of TGFβ1 treatment, cells were stained with an antibody specific for Smad3 and counterstained with DAPI to detect nuclei. Green staining indicates the presence of Smad3. Nuclei are shown in blue. E: After 48 hours of TGFβ1 treatment, cells transfected with SBE-luc were lysed and assayed for luciferase activity. Shown here is the fold change in luciferase activity normalized to total protein in treated versus untreated fibroblasts. *P < 0.001 compared with TGFβ1 alone (analysis of variance).
TGFβ1 treatment significantly stimulated nuclear translocation of Smad2/3 as indicated by increased band intensities on a Western blot of isolated nuclear extracts (Figure 5B). Pretreatment with ITE significantly attenuated nuclear translocation of Smad2/3 (Figure 5B). Purity of nuclear extracts was confirmed via Western blotting for the cytosolic marker β-tubulin (data not shown) and the nuclear marker lamin A (Figure 5B). When primary human orbital fibroblasts were exposed to TGFβ1 for 0.5 to 6 hours, the nuclear accumulation of Smad2/3/4 peaked at 1 to 3 hours and decreased but did not reach pretreatment levels by 6 hours of TGFβ1 treatment (Figure 5C). Thirty-minute pretreatment with ITE (1 μmol/L) decreased TGFβ1-induced Smad2/3/4 nuclear translocation at every time point tested (Figure 5C). These results were confirmed using immunofluorescence microscopy, which clearly shows nuclear translocation of Smad3 with TGFβ1 treatment, but not in the presence of ITE (Figure 5D).
To demonstrate the effect of ITE on Smad binding to Smad binding elements (SBE) located in gene promoters, we transfected primary human orbital fibroblasts with a SBE-luciferase plasmid (SBE-luc). Transfected fibroblasts were pretreated with ITE for 1 hour before 48 hour treatment with TGFβ1. Smad binding to SBE-luc was measured as luciferase activity in cell lysates. As expected, TGFβ1 treatment increased Smad binding to SBE-luc (Figure 5E). However, the addition of ITE to TGFβ1-treated cultures decreased Smad binding as is consistent with a reduction in Smad nuclear translocation.
ITE Does Not Inhibit TGFβ1-Induced Phosphorylation of Erk1/2 or Akt in Primary Human Orbital Fibroblasts
In addition to the Smad pathway, TGFβ1 activates additional signaling pathways including ras/MEK/ERK and PI3K/Akt.39,40 To investigate the possible role of these alternate pathways in ITE-mediated suppression of TGFβ1-induced myofibroblast differentiation, we assessed the phosphorylation of Erk1/2 and Akt in TGFβ1- and ITE-treated primary human orbital fibroblasts by Western blotting (Figure 6). Specific inhibitors, U0126 and LY294004 were used as positive controls to demonstrate inhibition of TGFβ1-induced Erk1/2 and Akt phosphorylation, respectively. ITE treatment failed to inhibit TGFβ1-induced phosphorylation of either Erk1/2 or Akt, suggesting that these pathways are not involved in inhibition of TGFβ1-induced myofibroblast differentiation by ITE.
Figure 6.
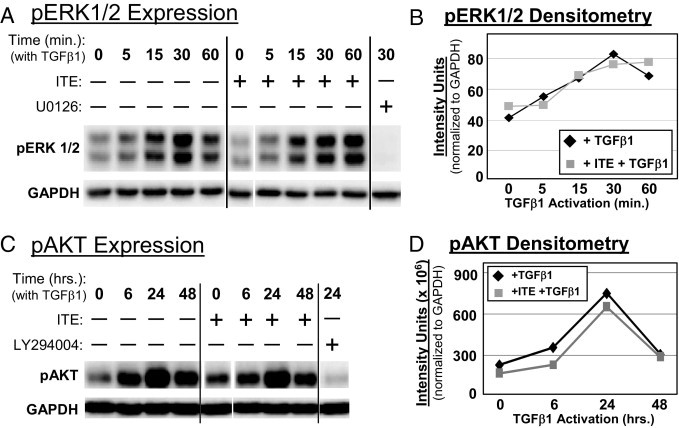
ITE does not inhibit TGFβ1-induced phosphorylation of Erk1/2 or Akt in primary human orbital fibroblasts. Primary cultures of human orbital fibroblasts were pretreated with ITE (1 μmol/L) for 2 hours. A: Whole cell lysates were harvested at 5, 15, 30, or 60 minutes after TGFβ1 addition and Western blotted using antibodies specific for pErk1/2 and GAPDH. Far-right lane contains positive control lysate from TGFβ1-treated fibroblasts pretreated with the MEK inhibitor U0126. B: Band intensities of pErk1/2 at each time point were quantified by densitometry and normalized to GAPDH. C: Whole cell lysates were harvested at 6, 24, or 48 hours after TGFβ1 addition and Western blotted using antibodies specific for pAkt and GAPDH. Far-right lane contains positive control lysate from TGFβ1-treated fibroblasts pretreated with the PI3K inhibitor LY294004. D: Band intensities of pAkt at each time point were quantified by densitometry and normalized to GAPDH.
Discussion
Fibrosis occurs when the normal wound healing response is amplified, resulting in aberrant myofibroblast persistence and excessive extracellular matrix deposition.1–3,5–7 The biological consequences of fibrosis can range from cosmetic to disabling to deadly, depending on the function of the affected tissue.1,3–5,7,12,13 Because they are urgently needed and not readily available, we seek to identify agents with the potential to act as safe, reliable, and efficacious therapies for the prevention or treatment of scarring.1,2,4,14,15 In this context, the myofibroblast is an important therapeutic target. We have found TGFβ1-challenged cultures of primary human fibroblasts to serve as a useful model system for the identification of small molecules that inhibit myofibroblast differentiation.12 Using this model, we have discovered that ITE possesses this ability and may form the basis for the development of a new antifibrotic drug.
The AhR ligand ITE was found to inhibit TGFβ1-induced myofibroblast differentiation in fibroblasts from several distinct tissue types as indicated by the following: (1) expression of smooth muscle proteins (α-SMA and calponin); (2) production of extracellular matrix components (collagen I, collagen III, and fibronectin); and (3) collagen gel contraction (Figures 1–3). ITE did not reduce cell viability, as measured by the AlamarBlue assay (Figure 1E). ITE did reduce the amount of AlamarBlue fluorescence generated in TGFβ1-treated cultures, but this result is not surprising in light of our overall findings of ITE-induced inhibition of TGFβ1-mediated effects. Although ITE is best known as an AhR agonist,22,27 we ultimately came to the conclusion that the antifibrotic activity of this compound is independent of AhR expression (Figure 4). We then sought to identify the point in the TGFβ1 signaling pathway targeted by ITE.
TGFβ1 is a key mediator of fibrotic tissue remodeling, and its increased expression correlates with the development of fibrotic disease.2,5,7,12,14,18 Activated TGFβ1 binds to a heteromeric receptor that transduces intracellular signals via Smad2/3 phosphorylation.18–21 Phosphorylation drives the activation of Smad2/3, which is necessary for recruitment of Smad4 and subsequent nuclear translocation of the Smad2/3/4 complex. If this complex does not enter the nucleus, it cannot bind to transcriptional co-activators and DNA to regulate the expression of downstream genes, including α-SMA and collagen I.20,21 Aside from phosphorylation by the activated TGFβ receptor complex, site-specific phosphorylation of Smad2/3 can also occur by a variety of other kinases and has been shown, depending on the context, to augment or interfere with downstream Smad signaling.41 This serves as a convergence point for different signaling pathways and is largely responsible for the diverse, context-specific effects of TGFβ1 signaling.42 It is possible that alterations in Smad2/3 phosphorylation outside of the sites targeted by the TGFβ receptor provide a mechanism by which ITE is able to interfere with TGFβ1 signaling.
In primary human orbital fibroblasts, we found that ITE inhibited TGFβ1-induced nuclear translocation of Smad2/3/4 and its subsequent binding to SBE (Figure 5 B–E). ITE did not inhibit TGFβ1-induced phosphorylation of Smad2/3, Erk1/2, or Akt (Figures 5A and 6). These findings suggest that future exploration should focus on mechanisms by which Smad2/3/4 nuclear translocation may be disrupted. For example, activation of the Ca2+/calmodulin-dependent CaM kinase II (CaMKII) has been shown to result in phosphorylation of Smad2 and inhibition of its TGFβ1-induced nuclear import.43 Perhaps ITE-induced CaMKII activity results in the phosphorylation of residues on Smad2/3 that disrupt their nuclear translocation, resulting in repression of transcriptional activation of TGFβ1-responsive promoters, such as those found on α-SMA and collagen I genes. Erk has also been shown to phosphorylate sites on Smad2/3 that are both independent of those targeted by the TGFβ receptor and able to inhibit TGFβ1-induced Smad 2/3 nuclear accumulation.44 The precise molecular basis for retention of Smad2/3/4 in the cytosol after ITE treatment remains to be identified.
In animal models of fibrotic disease, anti-TGFβ therapy has shown great promise. Anti-TGFβ antibodies prevented the progression of fibrosis in a murine model of scleroderma45 and in murine models of lung fibrosis induced by heat-killed Mycobacterium bovis (BCG)46 and bleomycin.47 In a rat model of pulmonary fibrosis triggered by adenovirus-mediated gene transfer of active TGFβ1, a TGFβ receptor I inhibitor blocked both fibrotic induction and progression.48 Transient gene transfer and expression of Smad7 or decorin, both negative regulators of TGFβ, prevented bleomycin-induced lung fibrosis in mice.49,50 There have been several studies of myofibroblast differentiation in response to photorefractive keratectomy (PRK) in which anti-TGFβ antibodies have been applied directly to the ocular surface of rabbits or cats. According to the results, blocking TGFβ signaling after PRK reduced corneal haze51,52 and enhanced optical quality.53 Although the efficacy of anti-TGFβ therapy has been demonstrated in animals and related pharmaceutical approaches to the treatment of IPF are close to clinical trial,54 an ideal antifibrotic therapeutic candidate has yet to be identified. In this study, ITE has proven to be a very effective inhibitor of TGFβ1 signaling in vitro. As a next step, preclinical animal models of fibrotic disease should be used to assess the efficacy of ITE for its potential as a novel antiscarring therapy.
This study clearly demonstrates for the first time that the AhR ligand ITE inhibits TGFβ1-stimulated myofibroblast differentiation in primary human fibroblasts from several different tissues, suggesting the potential use of this compound as an exciting new antifibrotic therapy. Of course, the AhR is infamous as the mediator of a number of toxic effects that result from exposure to its synthetic and highly stable ligands (eg, TCDD), including teratogenicity, immunotoxicity and tumor promotion.55–57 These toxicities may, at first, seem to contraindicate the use of AhR ligands as therapeutic agents. However, the discovery of a variety of potential, nontoxic, endogenous ligands for the AhR22,27–32 has opened up the possibility that these small molecules might be exploited in a safe and effective manner. ITE has been tested extensively by Henry et al both in vitro and in vivo to evaluate its potency as an AhR ligand and its potential to exert toxicity.22,58 These tests have shown ITE to be remarkably similar to TCDD in terms of its ability to bind and activate the AhR. The compounds show only two major differences. The first is that ITE is rapidly metabolized, whereas TCDD is exceptionally stable in biological systems. The second difference is that TCDD induces toxicities such as cleft palate and hydronephrosis in fetal mice, whereas ITE does not. These findings suggest that the toxic consequences of TCDD exposure may result not only from AhR activation per se but specifically from abnormal prolongation of AhR signaling that is not achievable with a less stable ligand such as ITE. Another clue to suggest that endogenous AhR ligands may exhibit a favorable safety profile is the fact that these ligands are produced endogenously. ITE, for example, was initially isolated from porcine lung.27 Thus, it is unlikely that it exerts toxicity, at least not in the lung at the concentrations at which it is found naturally. Furthermore, given its pulmonary origin and the results of this study, it is tempting to speculate that ITE may serve some natural function in the prevention of pulmonary fibrosis.
In this study, we have convincingly demonstrated the ability of the synthetic AhR agonist ITE to potently attenuate key TGFβ-mediated profibrotic effects, including myofibroblast differentiation and stimulation of extracellular matrix production. These data reveal the intriguing potential for this agent to play a role in antiscarring therapies.
Acknowledgments
We thank Dr. Timothy Bushnell for ImageStream instruction and assistance and Drs. Thomas A. Gasiewicz and Ellen C. Henry for reagents and guidance in using them.
Footnotes
Supported in part by the National Institutes of Health (grants EY017123, EY015836, HL095402, HL08832, HL075432, ES01247, and T32-HL66988), a Research to Prevent Blindness challenge grant, a Rochester/Finger Lakes Eye & Tissue Bank reserach grant, an American Lung Association research training fellowship, and a Parker B. Francis fellowship.
Current address of G.M.L., Hazardous Pollutant Assessment Group, National Center for Environmental Assessment, Office of Research and Development, United States Environmental Protection Agency, Research Triangle Park, North Carolina; of C.J.B., Meakins-Christie Laboratories, McGill University, Montreal, Quebec.
References
- 1.Ferguson M.W., O'Kane S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B Biol Sci. 2004;359:839–850. doi: 10.1098/rstb.2004.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hantash B.M., Zhao L., Knowles J.A., Lorenz H.P. Adult and fetal wound healing. Front Biosci. 2008;13:51–61. doi: 10.2741/2559. [DOI] [PubMed] [Google Scholar]
- 3.Phan S.H. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- 4.White E.S., Lazar M.H., Thannickal V.J. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol. 2003;201:343–354. doi: 10.1002/path.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferguson H.E., Kulkarni A., Lehmann G.M., Garcia-Bates T.M., Thatcher T.H., Huxlin K.R., Phipps R.P., Sime P.J. Electrophilic peroxisome proliferator activated receptor-{gamma} (PPAR{gamma}) ligands have potent anti-fibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 2009;41:722–730. doi: 10.1165/rcmb.2009-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selman M., Thannickal V.J., Pardo A., Zisman D.A., Martinez F.J., Lynch J.P., 3rd Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs. 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H.Y., Phan S.H. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1) Am J Respir Cell Mol Biol. 1999;21:658–665. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 8.Han R., Smith T.J. T helper type 1 and type 2 cytokines exert divergent influence on the induction of prostaglandin E2 and hyaluronan synthesis by interleukin-1beta in orbital fibroblasts: implications for the pathogenesis of thyroid-associated ophthalmopathy. Endocrinology. 2006;147:13–19. doi: 10.1210/en.2005-1018. [DOI] [PubMed] [Google Scholar]
- 9.Feldon S.E., O'Loughlin C.W., Ray D.M., Landskroner-Eiger S., Seweryniak K.E., Phipps R.P. Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am J Pathol. 2006;169:1183–1193. doi: 10.2353/ajpath.2006.060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldon S.E., Park D.J., O'Loughlin C.W., Nguyen V.T., Landskroner-Eiger S., Chang D., Thatcher T.H., Phipps R.P. Autologous T-lymphocytes stimulate proliferation of orbital fibroblasts derived from patients with Graves' ophthalmopathy. Invest Ophthalmol Vis Sci. 2005;46:3913–3921. doi: 10.1167/iovs.05-0605. [DOI] [PubMed] [Google Scholar]
- 11.Kahaly G., Forster G., Hansen C. Glycosaminoglycans in thyroid eye disease. Thyroid. 1998;8:429–432. doi: 10.1089/thy.1998.8.429. [DOI] [PubMed] [Google Scholar]
- 12.Burgess H.A., Daugherty L.E., Thatcher T.H., Lakatos H.F., Ray D.M., Redonnet M., Phipps R.P., Sime P.J. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 13.Sime P.J., O'Reilly K.M. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 14.Carrington L.M., Albon J., Anderson I., Kamma C., Boulton M. Differential regulation of key stages in early corneal wound healing by TGF-beta isoforms and their inhibitors. Invest Ophthalmol Vis Sci. 2006;47:1886–1894. doi: 10.1167/iovs.05-0635. [DOI] [PubMed] [Google Scholar]
- 15.Meltzer E.B., Noble P.W. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehmann G.M., Feldon S.E., Smith T.J., Phipps R.P. Immune mechanisms in thyroid eye disease. Thyroid. 2008;18:959–965. doi: 10.1089/thy.2007.0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehmann G.M., Garcia-Bates T.M., Smith T.J., Feldon S.E., Phipps R.P. Regulation of lymphocyte function by PPARgamma: relevance to thyroid eye disease-related inflammation. PPAR Res. 2008;2008:895–901. doi: 10.1155/2008/895901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Border W.A., Noble N.A. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Duran A., Carvajal-Gonzalez J.M., Mulero-Navarro S., Santiago-Josefat B., Puga A., Fernandez-Salguero P.M. Fitting a xenobiotic receptor into cell homeostasis: how the dioxin receptor interacts with TGFbeta signaling. Biochem Pharmacol. 2009;77:700–712. doi: 10.1016/j.bcp.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Massague J., Gomis R.R. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 21.Haarmann-Stemmann T., Bothe H., Abel J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol. 2009;77:508–520. doi: 10.1016/j.bcp.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Henry E.C., Bemis J.C., Henry O., Kende A.S., Gasiewicz T.A. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys. 2006;450:67–77. doi: 10.1016/j.abb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Salguero P., Pineau T., Hilbert D.M., McPhail T., Lee S.S., Kimura S., Nebert D.W., Rudikoff S., Ward J.M., Gonzalez F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 24.Corchero J., Martin-Partido G., Dallas S.L., Fernandez-Salguero P.M. Liver portal fibrosis in dioxin receptor-null mice that overexpress the latent transforming growth factor-beta-binding protein-1. Int J Exp Pathol. 2004;85:295–302. doi: 10.1111/j.0959-9673.2004.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson T.C., Hodgson P., Fernandez-Salguero P., Neumeister M., Gonzalez F.J. Hepatic fibrosis and cytochrome P450: experimental models of fibrosis compared with AHR knockout mice. Hepatol Res. 2000;17:112–125. doi: 10.1016/s1386-6346(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 26.Barouki R., Coumoul X., Fernandez-Salguero P.M. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007;581:3608–3615. doi: 10.1016/j.febslet.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 27.Song J., Clagett-Dame M., Peterson R.E., Hahn M.E., Westler W.M., Sicinski R.R., DeLuca H.F. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA. 2002;99:14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bittinger M.A., Nguyen L.P., Bradfield C.A. Aspartate aminotransferase generates proagonists of the aryl hydrocarbon receptor. Mol Pharmacol. 2003;64:550–556. doi: 10.1124/mol.64.3.550. [DOI] [PubMed] [Google Scholar]
- 29.Adachi J., Mori Y., Matsui S., Takigami H., Fujino J., Kitagawa H., Miller C.A., 3rd, Kato T., Saeki K., Matsuda T. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem. 2001;276:31475–31478. doi: 10.1074/jbc.C100238200. [DOI] [PubMed] [Google Scholar]
- 30.Savouret J.F., Antenos M., Quesne M., Xu J., Milgrom E., Casper R.F. 7-Ketocholesterol is an endogenous modulator for the arylhydrocarbon receptor. J Biol Chem. 2001;276:3054–3059. doi: 10.1074/jbc.M005988200. [DOI] [PubMed] [Google Scholar]
- 31.Schaldach C.M., Riby J., Bjeldanes L.F. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. 1999;38:7594–7600. doi: 10.1021/bi982861e. [DOI] [PubMed] [Google Scholar]
- 32.Sinal C.J., Bend J.R. Aryl hydrocarbon receptor-dependent induction of cyp1a1 by bilirubin in mouse hepatoma hepa 1c1c7 cells. Mol Pharmacol. 1997;52:590–599. doi: 10.1124/mol.52.4.590. [DOI] [PubMed] [Google Scholar]
- 33.Whitlock J.P., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 34.Fries K.M., Sempowski G.D., Gaspari A.A., Blieden T., Looney R.J., Phipps R.P. CD40 expression by human fibroblasts. Clin Immunol Immunopathol. 1995;77:42–51. doi: 10.1016/0090-1229(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 35.Baglole C.J., Reddy S.Y., Pollock S.J., Feldon S.E., Sime P.J., Smith T.J., Phipps R.P. Isolation and phenotypic characterization of lung fibroblasts. Methods Mol Med. 2005;117:115–127. doi: 10.1385/1-59259-940-0:115. [DOI] [PubMed] [Google Scholar]
- 36.Mikami F., Lim J.H., Ishinaga H., Ha U.H., Gu H., Koga T., Jono H., Kai H., Li J.D. The transforming growth factor-beta-Smad3/4 signaling pathway acts as a positive regulator for TLR2 induction by bacteria via a dual mechanism involving functional cooperation with NF-kappaB and MAPK phosphatase 1-dependent negative cross-talk with p38 MAPK. J Biol Chem. 2006;281:22397–22408. doi: 10.1074/jbc.M602124200. [DOI] [PubMed] [Google Scholar]
- 37.Powell D.W., Mifflin R.C., Valentich J.D., Crowe S.E., Saada J.I., West A.B. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol Cell Physiol. 1999;277:C1–C19. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 38.Fries K.M., Blieden T., Looney R.J., Sempowski G.D., Silvera M.R., Willis R.A., Phipps R.P. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin Immunol Immunopathol. 1994;72:283–292. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- 39.Leask A. Towards an anti-fibrotic therapy for scleroderma: targeting myofibroblast differentiation and recruitment. Fibrogenesis Tissue Repair. 2010;3:8. doi: 10.1186/1755-1536-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim G., Jun J.B., Elkon K.B. Necessary role of phosphatidylinositol 3-kinase in transforming growth factor β–mediated activation of Akt in normal and rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2002;46:1504–1511. doi: 10.1002/art.10314. [DOI] [PubMed] [Google Scholar]
- 41.Yakymovych I., Souchelnytskyi S. Regulation of Smad Function by Phosphorylation. In: Ten Dijke P., Heldin C.H., editors. Springer; Dordrecht: 2006. pp. 235–252. [Google Scholar]
- 42.Shi Y., Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 43.Wicks S.J., Lui S., Abdel-Wahab N., Mason R.M., Chantry A. Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20:8103–8111. doi: 10.1128/mcb.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kretzschmar M., Doody J., Timokhina I., Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormick L.L., Zhang Y., Tootell E., Gilliam A.C. Anti-TGF-{beta} treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999;163:5693–5699. [PubMed] [Google Scholar]
- 46.Denis M. Neutralization of transforming growth factor-beta 1 in a mouse model of immune-induced lung fibrosis. Immunology. 1994;82:584–590. [PMC free article] [PubMed] [Google Scholar]
- 47.Giri S.N., Hyde D.M., Hollinger M.A. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax. 1993;48:959–966. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonniaud P., Margetts P.J., Kolb M., Schroeder J.A., Kapoun A.M., Damm D., Murphy A., Chakravarty S., Dugar S., Higgins L., Protter A.A., Gauldie J. Progressive transforming growth factor {beta}1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 49.Nakao A., Fujii M., Matsumura R., Kumano K., Saito Y., Miyazono K., Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kolb M., Margetts P.J., Galt T., Sime P.J., Xing Z., Schmidt M., Gauldie J. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am J Respir Crit Care Med. 2001;163:770–777. doi: 10.1164/ajrccm.163.3.2006084. [DOI] [PubMed] [Google Scholar]
- 51.Jester J.V., Barry-Lane P.A., Petroll W.M., Olsen D.R., Cavanagh H.D. Inhibition of corneal fibrosis by topical application of blocking antibodies to TGFβ in the rabbit. Cornea. 1997;16:177–187. [PubMed] [Google Scholar]
- 52.Møller-Pedersen T., Cavanagh H.D., Petroll W.M., Jester J.V. Neutralizing antibody to TGFbeta modulates stromal fibrosis but not regression of photoablative effect following PRK. Curr Eye Res. 1998;17:736–747. [PubMed] [Google Scholar]
- 53.Buhren J., Nagy L., Swanton J.N., Kenner S., MacRae S., Phipps R.P., Huxlin K.R. Optical effects of anti-TGFbeta treatment after photorefractive keratectomy in a cat model. Invest Ophthalmol Vis Sci. 2009;50:634–643. doi: 10.1167/iovs.08-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ask K., Martin G.E., Kolb M., Gauldie J. Targeting genes for treatment in idiopathic pulmonary fibrosis: challenges and opportunities, promises and pitfalls. Proc Am Thorac Soc. 2006;3:389–393. doi: 10.1513/pats.200602-021TK. [DOI] [PubMed] [Google Scholar]
- 55.Fernandez-Salguero P.M., Hilbert D.M., Rudikoff S., Ward J.M., Gonzalez F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 56.Mimura J., Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–268. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu Y., Nakatsuru Y., Ichinose M., Takahashi Y., Kume H., Mimura J., Fujii-Kuriyama Y., Ishikawa T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2000;97:779–782. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henry E.C., Welle S.L., Gasiewicz T.A. TCDD and a putative endogenous AhR ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol Sci. 2010;114:90–100. doi: 10.1093/toxsci/kfp285. [DOI] [PMC free article] [PubMed] [Google Scholar]