

Abstract
AMP-activated protein kinase (AMPK) regulates cellular energy homeostasis and multiple biological processes in cell growth and survival, hence an attractive drug target. AMPK is a heterotrimeric protein consisting of α catalytic, β and γ regulatory subunits; two isoforms of each subunit are present in the heart. Studies using both genetic and pharmacological approaches have demonstrated important roles of AMPK in protecting the heart during ischemia/reperfusion injury as well as in pathological hypertrophy and failure. There is also emerging evidence suggesting isoform-specific function of AMPK, e.g. mutations of the γ2 subunit cause human cardiomyopathy. Thus, strategies avoiding the undesirable effects of altering γ2-AMPK activity, such as isoform selective activation of AMPK may lead to cardioprotective therapies with greater efficacy and safety.
Keywords: AMPK, energy homeostasis, drug target, isoform, cardiomyopathy
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase and is phylogenetically conserved from yeast to mammals. AMPK is sensitive to a broad spectrum of stresses, especially those that cause changes in cellular energy status. Activation of AMPK stimulates ATP-generating pathways, such as glucose uptake, glycolysis and fatty acid oxidation, and inhibits ATP-consuming pathways, such as fatty acid and cholesterol synthesis [1, 2]. Therefore, the kinase is originally considered a master metabolic switcher that controls the cellular and whole body energy homeostasis. More recent work has also revealed an important role of AMPK in cell growth and survival through its interactions with mitochondrial biogenesis, protein synthesis and degradation pathways [3-5]. Thus, there has been intense interest in targeting AMPK for the treatment of multiple prevalent diseases, such as diabetes and obesity, cancer and cardiovascular diseases.
AMPK structure and activity
AMPK is a heterotrimeric complex composed of a catalytic α- subunit and two regulatory β- and γ- subunits. Each subunit exists in multiple isoforms encoded by separate genes (α1, α2, β1, β2, γ1, γ2, and γ3), and their combination give rise to a variety of AMPK holoenzymes (Figure 1). Phosphorylation of Threonine 172 in the catalytic domain of the α subunit is required for AMPK activation. The C-terminal domain is required for binding with the β and γ subunits. The β subunit has scaffold/docking properties and contains two domains, glycogen binding domain and the C-terminal domain for binding with the α and γ subunits [6]. The β subunit also contains several sites for post-translational modifications such as myristoylation and phosphorylation which may regulate the subcellular localization of the heterotrimer [7]. The γ subunit contains four cystathionine-β-synthase (CBS) motifs in the C-terminus. Each pairs of the CBS motifs tightly associate in a pseudo dimeric arrangement through their β-sheets forming a so-called Bateman domain. Nucleotide binding assays suggested that each Bateman domain bound one molecule of AMP or ATP in an exclusive manner [8]. This observation was supported by a recent crystal structure study which in addition revealed a third site containing a non-exchangeable AMP [9] . The binding of AMP activates while the binding of ATP inactivates the kinase, thus, the two exchangeable sites on the γ subunit are responsible for the sensitivity of AMPK to the AMP/ATP ratio in the environment.
Figure 1.
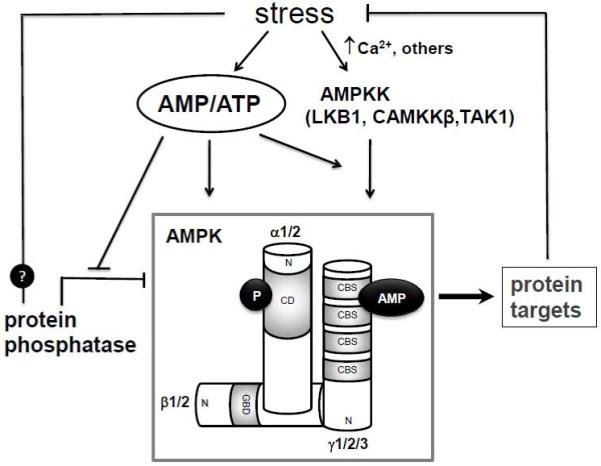
Regulation of the AMPK signaling cascade. The intracellular AMP/ATP ratio is a primary regulator of the system. Increased AMP/ATP activates AMPK via three mechanisms 1) allosteric activation; 2) promotes the Thr 172 phosphorylation of the α- AMPK by its upstream kinase (AMPKK) such as LKB1, and 3) prevents protein phosphatase from dephosphorylating AMPK. Stress can also directly activate the CAMKKβ via increased intracellular calcium levels. It is proposed that certain stresses may directly affect the dephosphorylation process by yet undetermined mechanisms. AMPK, AMP-activated protein kinase; AMPKK, AMPK-kinase; CaMKKβ, Ca2+/calmodulin-dependent protein kinase kinase β; TAK1, transforming growth factor-β activated kinase 1; CD, catalytic domain; GBD, glycogen binding domain; N, NH2-terminus of subunits; CBS, cystathionine-β-synthase motif.
The AMP/ATP ratio regulates AMPK activity by several mechanisms (Figure 1). First, the binding of AMP in the γ subunit allosterically activates the kinase. Increased AMP binding also facilitates the phosphorylation of and protects against the dephosphorylation of Thr 172, thus promotes and sustains the kinase activity [10]. The combined effects of the allosteric activation and Thr 172 phosphorylation increase AMPK activity by 1000-fold [11]. Since the [ATP] in the cell is several orders of magnitude higher than [AMP], AMPK activity is very low in unstressed conditions. However, because of the large difference in free concentrations of the nucleotides, a small decrease in [ATP] can lead to a large increase of [AMP] (ATP → ADP → AMP) resulting in a great change of AMP/ATP. In this way, AMPK functions as an ultra-sensitive gauge of cellular energetic status [12].
Multiple upstream kinases of AMPK (AMPKK) have been reported, including LKB1, Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ), and transforming growth factor-β activated kinase 1 (TAK1) (Figure 1). LKB1, encoded by the Peutz-Jeghers syndrome tumor suppressor gene, exists in a heterotrimetric complex associated with two accessory proteins, mouse protein 25 (MO25α/β) and Ste20-related adaptor protein (STRADα/β). LKB1 complex functions as a constitutively active kinase that phosphorylates AMPK in the presence of AMP [13]. LKB1-AMPK pathway exists in majority of cell and tissue types except in Hela cells where LKB1 is absent [14]. CaMKKβ has considerable sequence and structural homology with LKB1 but its activity is dependent on intracellular Ca2+ levels. Recent studies suggest that CaMKKβ regulates AMPK in a Ca2+/calmodulin-dependent and AMP-independent manner [11]. The expression of CaMKKβ in the heart is very low but the CaMKKβ-AMPK cascade likely has an important role in the neurological tissues [15, 16]. TAK1 was first shown to activate the yeast AMPK homologue Snif1 and purified mammalian AMPK and more recently in human cell culture [17, 18]. Deletion of TAK1 results in decreased phosphorylation of AMPK in mice [19].
Recent studies have also proposed that the AMPKKs may be constitutively active and the regulation of AMPK phoshporylation and activity is, instead, achieved by the dephosphorylation process. Increased AMP has been shown to prevent the dephosphorylation and inactivation of AMPK by protein phosphatase [10] but the specific phosphatase for the AMPK in vivo as well as its regulation remains to be defined.
Several endocrine factors/hormones have been reported to regulate AMPK activity, such as adiponectin, leptin, ciliary neurotrophic factor and ghrelin [20-23]. The mechanisms of action for these factors/hormones are complex involving both central nervous system and direct effects on the peripheral tissue. It is though very unlikely these factors/hormones interact directly with the AMPK complex.
Tissue Distribution of AMPK
Although AMPK is widely distributed, tissue-specific expressions of selective isoforms have been reported [24-26]. Among the seven isoforms, the α1, β1 and γ1 isoforms are ubiquitously expressed. The α2 and β2 isoforms are highly expressed in heart and skeletal muscle. The γ2 isoform is expressed in several tissues including the heart, whereas the γ3 isoform is exclusively expressed in skeletal muscle. The differential expression pattern of the isoforms enables a tighter regulation of the AMPK activity in a tissue dependent manner. It also provides a unique opportunity to modulate AMPK activity in selective tissues or to target isoform-specific AMPK function(s) using pharmacological approaches.
Summarized in Table 1 are reported distributions of each subunit isoform in mammalian tissues. In cardiac and skeletal muscle, α2 AMPK complexes accounted for 70-80% of total AMPK activity while α1 complexes accounted for the remaining 20-30% [26]. In contrast, α1 complexes accounted for 60-90% of total AMPK activity with α2 complexes for the remaining 10-40% in lung, kidney, testis and brain. Liver had the same contribution of α1 and α2 AMPK to the total activity. Yang et al. have demonstrated that α1 subunit isoform was more abundantly expressed in adipose tissue, peritoneal macrophages, and spleen compared to muscle and might account for AMPK activation in those tissues [27]. The distribution of γ-subunit isoform was different. The γ1 complexes accounted for the major part (80-90%) of total AMPK activity in liver, lung, kidney, pancreas, cardiac and skeletal muscles in rodents. The γ2 complexes accounted for 10-20% of the total activity in these tissues [26]. Even though γ3 mRNA level was readily detected in the skeletal muscle, the activity of γ3 complexes was elusive [26]. A recent study showed that ~20% of α2 complexes and none of the α1 complexes in human skeletal muscle contained γ3-subunit [28-30].
Table 1.
Tissue distribution of AMPK subunit isoforms
| Tissue | α-subunit | β-subunit | γ-subunit |
|---|---|---|---|
| Adipose tissue | α1: 90% a α2: 10% a |
ND | ND |
| Brain | α1: 75% b α2: 25% b |
ND | γ1: 36% b γ2: 36% b |
| Bone Osteoblast Osteoclast |
α1: 100%c α1: 100%d |
ND β1:100%d |
γ1:100%d γ1:100%d |
| Heart Endothelial cell Smooth muscle |
α1: 30% b α2: 70%b α1>α2f ND |
β1<β2e β1>β2f ND |
γ1: 85% b γ2: 15%b γ1<γ2f ND |
| Kidney | α1: 80% b α2: 20%b |
β1<β2e | γ1: 80% b γ2: 20%b |
| Liver | α1: 45% b α2: 55%b |
β1>β2e | γ1: 90% b γ2: 10%b |
| Lung | α1: 90% b α2: 10%b |
β1>β2e | γ1: 80% b γ2: 20%b |
| Skeletal muscle | α1: 20% b α2: 80%b |
β1<β2e | γ1: 90% b γ2: 10%b γ3: ND |
determined by activity assay in rodent [25]
determined by RT-PCR in rodent [90]
determined by Western blot in rodent [71]
determined by Northern and Western blots in human and rodent [24]
determined by RT-PCR and Western blot in rodent [91]
ND: the isoforms are present but the relative abundance is not determined.
The functional significance of the isoform composition of AMPK was not recognized until recently when studies of human skeletal muscle have shown that only 3 of the 12 possible AMPK complexes were present (α1β2γ1, α2β2γ1, and α2β2γ3), and their expressions were differentially regulated during exercise depending on strength and duration [28-30]. Furthermore, exercise caused nuclear translocation of α2-AMPK, which likely mediated the transcriptional regulation of glucose metabolism in human skeletal muscle [31, 32]. In mouse C2C12 myoblasts, nuclear translocation of α2-AMPK by leptin occurred only in complexes containing β2 subunit while α2-AMPK containing the β1 subunit was retained in the cytoplasm [33]. The subcellular redistribution of AMPK complexes in the heart has not been explored. In addition, mechanisms responsible for the isoform-specific responses to various physiological or pathological stimuli are largely unclear at present. A thorough understanding of these questions holds a great promise to achieve isoform-selective targeting of AMPK for therapeutic purposes, and is thus highly warranted.
AMPK function in the heart
AMPK activity in the heart is very low under normal conditions; its function in the unstressed heart is not clearly defined. However, activation of AMPK is rapid and robust in response to a variety of stresses such as ischemia, exercise and chronic pressure overload [34-36]. A number of studies using pharmacological activators of AMPK or transgenic mice expressing inactive catalytic subunit of AMPK have been carried out to understand the functional role of AMP during stresses.
Gain of Function Studies
Most of the studies in the literature used AICAR or metformin to activate AMPK. AICAR is a nucleoside 5-aminoimidazole-4-carboxamide riboside that is taken up by the cells and then converted into the AMP analog, ZMP. The ZMP activates AMPK by binding to the γ subunit as an AMP mimetic. Metformin is a biguanide derivative that has been used for the treatment of hyperglycemia in diabetes with unknown mechanisms. Recent studies have shown that metformin indirectly activates AMPK by increasing AMP/ATP in hepatocytes through inhibition of complex I in the mitochondrial respiratory chain [37].
Activation of AMPK in the heart by ischemia or AICAR stimulates glucose uptake and glycolysis by translocating the glucose transporters to the cell membrane and activating phosphofrutokinase-2 via insulin-independent mechanism(s) [38, 39]. These effects of AMPK have been shown to maintain energy homeostasis and protect against ischemic injury in the heart [40, 41]. AMPK also stimulates fatty acid oxidation (FAO) via phosphorylation and inactivation of acetyl-CoA carboxylase 2 (ACC2) [42]. Although increased FAO promotes ATP production, previous studies have shown that a high level of FAO contributes to reperfusion injury, thus, suggesting that increased AMPK activity during reperfusion is not desirable [43, 44]. However, failure to activate AMPK in vivo results in more cell death and larger infarct size, suggesting that the biological effects of AMPK during the ischemia/reperfusion insult may have extended beyond the acute regulation of cell metabolism [40]. Recent evidence suggest that AMPK modulates mTOR signaling and autophagy, both are closely linked to cell survival during stress [16, 45].
Studies in the last decade have also suggested a cardioprotective role of AMPK during chronic stresses that lead to pathological cardiac hypertrophy. Increased AMPK activity was first found in hypertrophied hearts with impaired energetics leading to the hypothesis that activation of AMPK is a compensatory response to restore energy balance [36]. Activation of AMPK by pharmacological compounds such as, AICAR or metformin, has been shown to inhibit cardiac hypertrophy, blunt cardiac remodeling and delay the development of heart failure [46, 47]. Several signaling mechanisms downstream of the AMPK cascade have been implicated in cardiac hypertrophy, including metabolic pathways, protein synthesis and degradation mechanisms, mitochondrial biogenesis, and NO signaling [47-49]. It has been shown that activation of AMPK in neonatal rat cardiac myocytes by AICAR or metformin leads to phosphorylation and inactivation of eEF-2 kinase thus blunting the hypertrophic response to phenylepherine [47]. Metformin treatment in mouse models of myocardial infarction improved mitochondrial function and reduced LV remodeling in an AMPK and eNOS dependent mode [50]. Even though AMPK is activated during cardiac stress, phamarcological activation that induces an early and sustained increase of AMPK activity likely provides additional beneficial effects.
It should be noted that although effective and widely used as AMPK activators, pharmacological compounds such as metformin or AICAR have significant off-target effects [51-54]. For example, recent studies have shown that metformin affects hepatic gluconeogensis and mTOR signaling via AMPK-independent mechanisms [55, 56]. Thus, results from these studies need to be interpreted with caution and best to be confirmed with genetic approaches.
Activation of AMPK by genetic approach appeared to be more complicated than expected. There has been no report of mouse models of increased AMPK activity by simply overexpressing the catalytic subunits likely due to the fact that the kinase is tightly regulated and a heterotrimer is required for the activity [57, 58]. Recently, mutations of the γ regulatory subunit have been found to abolish the sensor function of the Bateman domains thus altering the kinase activity [8, 59]. Mutations of the γ3 subunit (encoded by PRKAG3) caused gain of function changes of AMPK resulting in glycogen storage in the skeletal muscle of pigs and mice [60-62]. Similar mutations of the γ2 subunit (encoded by PRKAG2) cause human cardiomyopathy with glycogen storage, severe cardiac hypertrophy and arrhythmias [63-65]. The PRKAG2 cardiomyopathy has been attributed to the aberrant activation of AMPK that causes metabolic derangement and glycogen storage [63, 66, 67]. However, decreased AMPK activity has also been observed in mouse hearts expressing some PRKAG2 mutants, possibly due to feedback inhibition of the kinase activity [68, 69]. Interestingly, the PRKAG2 mutations present primarily a cardiac phenotype suggesting a cardiac-specific function of this particular isoform.
The heart is made up of cardiomyocytes and non-myocyte cell types. AMPK is expressed in the cardiomyocytes as well as fibroblasts, endothelial cells and smooth muscle cells in the heart. The functional role of AMPK in non-cardiomyocytes is just being understood. AMPK in endothelial cells has been shown to be involved in maintaining endothelial function through the anti-inflammatory and anti-atherogenic properties [70-72]. Previous studies showed that AMPK activated eNOS (endothelial NO synthase) in endothelial cells in response to shear stress and myocardial ischemia contributing to the regulation of vascular tone under these condtions [73, 74]. AMPK activation inhibited the vascular smooth muscle cell hypertrophy induced by angiotensin II [75], and activation of AMPK in cardiac fibroblasts was also reported to be cardioprotective [76]
Loss of Function Studies
There are a number of genetic models for reduced AMPK activity including deletions of α1, α2, β1, β2 or γ3 subunit of AMPK in mice [60, 77-79]. Mice expressing dominant negative or kinase inactive catalytic subunit of AMPK in the heart or in the heart and skeletal muscles are also available [40, 41]. There is, however, no report of genetic models with complete loss of AMPK activity in the heart at the present time. Many studies with genetic null mice for AMPK have shown that the reduced AMPK activity did not cause any cardiac dysfunction in normal conditions but did affect on cardiac function during stress. Mice deficient of α2 catalytic subunit of AMPK exhibited exacerbated left ventricular hypertrophy and dysfunction during chronic pressure-overload [80]. In isolated perfused mouse hearts with the kinase inactive mutant of α2 subunit or complete deletion of α2 subunit, the reduced AMPK activation developed a more rapid and severe contracture of the ischemic hearts and was detrimental for the heart during ischemia [40, 41, 81]. But, the response to ischemia/reperfusion was somewhat dependent on the perfusion condition [40, 82].
Several AMPK inhibitors have been used in a number of in vitro studies. The most widely used AMPK inhibitor, Compound C, is a cell permeable pyrazolopyrimidine compound and acts as ATP-competitive inhibitor of AMPK [37]. Like many other kinase inhibitors, this inhibitor is not strictly specific to AMPK. Several studies have reported that Compound C can inhibit a number of other protein kinases and hence inhibits various biological events independently of AMPK activity [83, 84]. Furthermore, the very short half-life of the Compound C significantly limits its use for studies in vivo. Thus, specific AMPK inhibitors and/or additional loss of function models are necessary for our further understanding of the kinase function in vivo.
AMPK as a potential therapeutic target
AMPK is a key enzyme that regulates cardiac energy homeostasis and multiple biological processes in cell growth and survival, hence an attractive drug target. A number of pharmacological compounds currently on the market have been shown to activate AMPK, including the anti-diabetic drugs metformin, thiazolidinediones (TDZ) and the cholesterol lowering drug statins [70]. It is likely that these compounds activate AMPK through an indirect effect of increasing AMP/ATP, and in the case of metformin via inhibition of mitochondrial respiration [85]. To achieve potent and specific activation of AMPK, “direct” activators would be most desirable. By definition, a direct activator should directly interact with the kinase to increase its activity. Several compounds in this category have been reported in cell culture or animal studies. A-769662, a compound developed by Abbot Laboratory, is a nonnucleoside thienopyridone derivative that directly and exclusively activates AMPK complexes containing the β1 subunit [86]. Although it may not achieve maximal activation of AMPK in tissues expressing both isoforms of β subunit, it presents the exciting possibility of implementing AMPK-isoform selective therapeutics. A-769622 was well tolerated in small animals but a recent study showed that it caused inhibition of 26S proteasome function and arrest of cell cycle progression via an AMPK independent mechanism [87]. Another small molecule compound, PT1, is shown to activate both α1 and α2 isoforms through the relief of autoinhibition in the catalytic α subunits [88]. It is thus possible that PT1 can activate all 12 AMPK complexes although the in vivo studies of PT1 are still pending.
There are several concerns of activating AMPK systemically as a therapeutic strategy. AMPK activity in hypothalamus is positively correlated with food intake [23, 89]. The potential effectiveness of perturbing energy homeostasis via AMPK activation was also challenged by recent studies demonstrating that direct targeting of ACC2, the primary downstream target of AMPK, minimally affects whole body energy balance [90, 91]. This, however, does not exclude the possibility of achieving metabolic modulation via other AMPK downstream targets. Furthermore, these concerns do not detract from the potential benefit of AMPK activation for cardioprotection. The conceptual obstacle in developing cardiovascular selective AMPK activators is the observation that the gain of function mutation in the γ subunit of AMPK causes glycogen storage in the skeletal muscle (γ3 subunit) and the heart (γ2 subunit) in both animals and human [61, 62, 64-66]. The glycogen storage cardiomyopathy is associated with severe cardiac hypertrophy and arrhythmias suggesting that activation of γ2-containing AMPK in the heart may not be desirable. In the regard, it becomes critical to understand the isoform-specific function of the γ-subunit. If the PRKAG2 cardiomyoapthy represents a unique phenotype of altered γ2-AMPK function, selective activation of γ1-AMPK will be a viable therapeutic strategy. These considerations further emphasize the significance of developing tissue selective and isoform specific activators of AMPK. Therefore, understanding of the isoform specific functions of the AMPK complexes is an important focus of future research in the field.
Conclusions
AMPK is a master protein kinase that mediates energy homeostasis and has been considered as a drug target for obesity and metabolic syndrome. The accumulating evidence indicates that AMPK is also critically involved in cardiovascular diseases such as cardiac hypertrophy and myocardial ischemia. Future research should be directed to further understanding of cell type-specific and isoform-specific AMPK activation in the pathogenesis as well as the treatment of cardiovascular diseases. Advances in the development of isoform-selective and direct activators of AMPK will likely deliver the therapy with greater efficacy and safety.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, et al. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003 Feb;31(Pt 1):162–8. doi: 10.1042/bst0310162. [DOI] [PubMed] [Google Scholar]
- [2].Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004 Jan;29(1):18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- [3].Hardie DG. AMPK and Raptor: matching cell growth to energy supply. Mol Cell. 2008 May 9;30(3):263–5. doi: 10.1016/j.molcel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- [4].Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. Mar;6(3):457–70. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007 Feb 16;100(3):328–41. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- [7].Warden SM, Richardson C, O’Donnell J, Jr., Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001 Mar 1;354(Pt 2):275–83. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004 Jan;113(2):274–84. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007 Sep 27;449(7161):496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- [10].Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995 Dec 27;377(3):421–5. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- [11].Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006 Oct 27;281(43):32207–16. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- [12].Hardie DG, Salt IP, Hawley SA, Davies SP. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J. 1999 Mar 15;338(Pt 3):717–22. Pt 3. [PMC free article] [PubMed] [Google Scholar]
- [13].Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab. 2004 Aug;287(2):E310–7. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- [14].Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2(4):28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008 May;7(5):377–88. doi: 10.1016/j.cmet.2008.02.011. [DOI] [PubMed] [Google Scholar]
- [16].Lee JY, Jeon BT, Shin HJ, Lee DH, Han JY, Kim HJ, et al. Temporal expression of AMP-activated protein kinase activation during the kainic acid-induced hippocampal cell death. J Neural Transm. 2009 Jan;116(1):33–40. doi: 10.1007/s00702-008-0158-9. [DOI] [PubMed] [Google Scholar]
- [17].Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006 Sep 1;281(35):25336–43. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- [18].Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. Embo J. 2009 Mar 18;28(6):677–85. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, et al. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006 Nov 14;103(46):17378–83. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005 Oct;11(10):1096–103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002 Jan 17;415(6869):339–43. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- [22].Steinberg GR, Watt MJ, Fam BC, Proietto J, Andrikopoulos S, Allen AM, et al. Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice. Endocrinology. 2006 Aug;147(8):3906–14. doi: 10.1210/en.2005-1587. [DOI] [PubMed] [Google Scholar]
- [23].Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004 Mar 26;279(13):12005–8. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- [24].Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996 Jan 12;271(2):611–4. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- [25].Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. 1998 May 15;273(20):12443–50. doi: 10.1074/jbc.273.20.12443. [DOI] [PubMed] [Google Scholar]
- [26].Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000 Mar 15;346(Pt 3):659–69. [PMC free article] [PubMed] [Google Scholar]
- [27].Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. Jun 18;285(25):19051–9. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wojtaszewski JF, Birk JB, Frosig C, Holten M, Pilegaard H, Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J Physiol. 2005 Apr 15;564(Pt 2):563–73. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Treebak JT, Birk JB, Rose AJ, Kiens B, Richter EA, Wojtaszewski JF. AS160 phosphorylation is associated with activation of alpha2beta2gamma1- but not alpha2beta2gamma3-AMPK trimeric complex in skeletal muscle during exercise in humans. Am J Physiol Endocrinol Metab. 2007 Mar;292(3):E715–22. doi: 10.1152/ajpendo.00380.2006. [DOI] [PubMed] [Google Scholar]
- [30].Birk JB, Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006 Dec 15;577(Pt 3):1021–32. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003 Apr;52(4):926–8. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- [32].McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009 Dec 15;587(Pt 24):5951–8. doi: 10.1113/jphysiol.2009.181065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007 Jun;27(12):4317–27. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li J, Coven DL, Miller EJ, Hu X, Young ME, Carling D, et al. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006 Oct;291(4):H1927–34. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- [35].Musi N, Hirshman MF, Arad M, Xing Y, Fujii N, Pomerleau J, et al. Functional role of AMP-activated protein kinase in the heart during exercise. FEBS Lett. 2005 Apr 11;579(10):2045–50. doi: 10.1016/j.febslet.2005.02.052. [DOI] [PubMed] [Google Scholar]
- [36].Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001 Oct 2;104(14):1664–9. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- [37].Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001 Oct;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000 Oct 19;10(20):1247–55. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- [39].Russell RR, 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999 Aug;277(2 Pt 2):H643–9. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- [40].Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004 Aug;114(4):495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003 Aug 1;278(31):28372–7. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- [42].Dyck JR, Kudo N, Barr AJ, Davies SP, Hardie DG, Lopaschuk GD. Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5′-AMP activated protein kinase. Eur J Biochem. 1999 May;262(1):184–90. doi: 10.1046/j.1432-1327.1999.00371.x. [DOI] [PubMed] [Google Scholar]
- [43].Lopaschuk GD, Spafford MA. Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol. 1990 May;258(5 Pt 2):H1274–80. doi: 10.1152/ajpheart.1990.258.5.H1274. [DOI] [PubMed] [Google Scholar]
- [44].Hendrickson SC, St Louis JD, Lowe JE, Abdel-aleem S. Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem. 1997 Jan;166(1-2):85–94. doi: 10.1023/a:1006886601825. [DOI] [PubMed] [Google Scholar]
- [45].Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006 Sep 8;126(5):955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- [46].Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, et al. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007 Apr 1;100(5):1086–99. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- [47].Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004 Jul 30;279(31):32771–9. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- [48].Chen BL, Ma YD, Meng RS, Xiong ZJ, Wang HN, Zeng JY, et al. Activation of AMPK inhibits cardiomyocyte hypertrophy by modulating of the FOXO1/MuRF1 signaling pathway in vitro. Acta Pharmacol Sin. Jul;31(7):798–804. doi: 10.1038/aps.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dolinsky VW, Morton JS, Oka T, Robillard-Frayne I, Bagdan M, Lopaschuk GD, et al. Calorie Restriction Prevents Hypertension and Cardiac Hypertrophy in the Spontaneously Hypertensive Rat. Hypertension. 2010 Aug 9;:9. doi: 10.1161/HYPERTENSIONAHA.110.154732. [DOI] [PubMed] [Google Scholar]
- [50].Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009 Feb 13;104(3):403–11. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Longnus SL, Wambolt RB, Parsons HL, Brownsey RW, Allard MF. 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am J Physiol Regul Integr Comp Physiol. 2003 Apr;284(4):R936–44. doi: 10.1152/ajpregu.00319.2002. [DOI] [PubMed] [Google Scholar]
- [52].Vincent MF, Marangos PJ, Gruber HE, Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes. 1991 Oct;40(10):1259–66. doi: 10.2337/diab.40.10.1259. [DOI] [PubMed] [Google Scholar]
- [53].Musi N, Goodyear LJ. Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes. Curr Drug Targets Immune Endocr Metabol Disord. 2002 Jul;2(2):119–27. [PubMed] [Google Scholar]
- [54].Dargie HJ, Hildebrandt PR, Riegger GA, McMurray JJ, McMorn SO, Roberts JN, et al. A randomized, placebo-controlled trial assessing the effects of rosiglitazone on echocardiographic function and cardiac status in type 2 diabetic patients with New York Heart Association Functional Class I or II Heart Failure. J Am Coll Cardiol. 2007 Apr 24;49(16):1696–704. doi: 10.1016/j.jacc.2006.10.077. [DOI] [PubMed] [Google Scholar]
- [55].Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. Jul 1;120(7):2355–69. doi: 10.1172/JCI40671. 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. May 5;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dyck JR, Gao G, Widmer J, Stapleton D, Fernandez CS, Kemp BE, et al. Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. J Biol Chem. 1996 Jul 26;271(30):17798–803. doi: 10.1074/jbc.271.30.17798. [DOI] [PubMed] [Google Scholar]
- [58].Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998 Dec 25;273(52):35347–54. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- [59].Zou L, Shen M, Arad M, He H, Lofgren B, Ingwall JS, et al. N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ Res. 2005 Aug 19;97(4):323–8. doi: 10.1161/01.RES.0000179035.20319.c2. [DOI] [PubMed] [Google Scholar]
- [60].Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, et al. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004 Sep 10;279(37):38441–7. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- [61].Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, et al. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science. 2000 May 19;288(5469):1248–51. doi: 10.1126/science.288.5469.1248. [DOI] [PubMed] [Google Scholar]
- [62].Costford SR, Kavaslar N, Ahituv N, Chaudhry SN, Schackwitz WS, Dent R, et al. Gain-of-function R225W mutation in human AMPKgamma(3) causing increased glycogen and decreased triglyceride in skeletal muscle. PLoS One. 2007;2(9):e903. doi: 10.1371/journal.pone.0000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002 Feb;109(3):357–62. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, et al. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 2001 Dec 18;104(25):3030–3. doi: 10.1161/hc5001.102111. [DOI] [PubMed] [Google Scholar]
- [65].Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001 May 15;10(11):1215–20. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- [66].Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005 Nov 15;112(20):3140–8. doi: 10.1161/CIRCULATIONAHA.105.550806. [DOI] [PubMed] [Google Scholar]
- [67].Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, et al. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest. 2007 May;117(5):1432–9. doi: 10.1172/JCI30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Folmes KD, Chan AY, Koonen DP, Pulinilkunnil TC, Baczko I, Hunter BE, et al. Distinct early signaling events resulting from the expression of the PRKAG2 R302Q mutant of AMPK contribute to increased myocardial glycogen. Circ Cardiovasc Genet. 2009 Oct;2(5):457–66. doi: 10.1161/CIRCGENETICS.108.834564. [DOI] [PubMed] [Google Scholar]
- [69].Davies JK, Wells DJ, Liu K, Whitrow HR, Daniel TD, Grignani R, et al. Characterization of the role of gamma2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am J Physiol Heart Circ Physiol. 2006 May;290(5):H1942–51. doi: 10.1152/ajpheart.01020.2005. [DOI] [PubMed] [Google Scholar]
- [70].Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006 Dec 12;114(24):2655–62. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- [71].Stahmann N, Woods A, Spengler K, Heslegrave A, Bauer R, Krause S, et al. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J Biol Chem. Apr 2;285(14):10638–52. doi: 10.1074/jbc.M110.108688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dixit M, Bess E, Fisslthaler B, Hartel FV, Noll T, Busse R, et al. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovasc Res. 2008 Jan;77(1):160–8. doi: 10.1093/cvr/cvm017. [DOI] [PubMed] [Google Scholar]
- [73].Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, et al. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006 Jun;26(6):1281–7. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- [74].Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999 Jan 29;443(3):285–9. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- [75].Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004 Jul 27;110(4):444–51. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- [76].Du JH, Xu N, Song Y, Xu M, Lu ZZ, Han C, et al. AICAR stimulates IL-6 production via p38 MAPK in cardiac fibroblasts in adult mice: a possible role for AMPK. Biochem Biophys Res Commun. 2005 Dec 2;337(4):1139–44. doi: 10.1016/j.bbrc.2005.09.174. [DOI] [PubMed] [Google Scholar]
- [77].Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004 Jan 9;279(2):1070–9. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- [78].Viollet B, Andreelli F, Jorgensen SB, Perrin C, Flamez D, Mu J, et al. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans. 2003 Feb;31(Pt 1):216–9. doi: 10.1042/bst0310216. [DOI] [PubMed] [Google Scholar]
- [79].Quinn JM, Tam S, Sims NA, Saleh H, McGregor NE, Poulton IJ, et al. Germline deletion of AMP-activated protein kinase beta subunits reduces bone mass without altering osteoclast differentiation or function. Faseb J. 2009 Jan;24(1):275–85. doi: 10.1096/fj.09-137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008 Nov;52(5):918–24. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, et al. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006 Dec;291(6):H2875–83. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- [82].Folmes CD, Wagg CS, Shen M, Clanachan AS, Tian R, Lopaschuk GD. Suppression of 5′-AMP-activated protein kinase activity does not impair recovery of contractile function during reperfusion of ischemic hearts. Am J Physiol Heart Circ Physiol. 2009 Jul;297(1):H313–21. doi: 10.1152/ajpheart.01298.2008. [DOI] [PubMed] [Google Scholar]
- [83].Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007 Dec 15;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Emerling BM, Viollet B, Tormos KV, Chandel NS. Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett. 2007 Dec 11;581(29):5727–31. doi: 10.1016/j.febslet.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. Jun 9;11(6):554–65. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008 Nov 24;15(11):1220–30. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- [87].Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008 Jul 23;582(17):2650–4. doi: 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- [88].Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008 Jun 6;283(23):16051–60. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004 Apr 1;428(6982):569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- [90].Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proc Natl Acad Sci U S A. Apr 20;107(16):7598–603. doi: 10.1073/pnas.0913492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hoehn KL, Turner N, Swarbrick MM, Wilks D, Preston E, Phua Y, et al. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. Jan;11(1):70–6. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]