

Abstract
Fast Photochemical Oxidation of Proteins (FPOP) is a mass-spectrometry-based protein footprinting method that modifies proteins on the microsecond time scale. Highly reactive •OH, produced by laser photolysis of hydrogen peroxide, oxidatively modifies the side chains of approximately one half the common amino acids on this time scale. Owing to the short labeling exposure, only solvent-accessible residues are sampled. Quantification of the modification extent for the apo and holo states of a protein-ligand complex should provide structurally sensitive information at the amino-acid level to compare the structures of unknown protein complexes with known ones. We report here the use of FPOP to monitor the structural changes of calmodulin in its established binding to M13 of the skeletal muscle myosin light chain kinase. We use the outcome to establish the unknown structures resulting from binding with melittin and mastoparan. The structural comparison follows from a comprehensive examination of the extent of FPOP modifications as measured by proteolysis and LC-MS/MS for each protein-ligand equilibrium. The results not only show that the three calmodulin-peptide complexes have similar structures but also reveal those regions of the protein that became more or less solvent-accessible upon binding. This approach has the potential for relatively high throughput, information-dense characterization of a series of protein-ligand complexes in biochemistry and drug discovery when the structure of one reference complex is known, as is the case for calmodulin and M13 of the skeletal muscle myosin light chain kinase, and the structures of related complexes are not,.
INTRODUCTION
Mass spectrometry (MS) has become an important tool for studying protein structure, dynamics, interactions, and function.1 In particular, detailed characterization of protein-ligand interactions is now possible,2 a critical step toward understanding biological function. One MS-based approach is protein footprinting,3 When a protein binds with a ligand, the solvent-accessible surface areas of its residues are affected by the resulting conformational changes and direct ligand interactions. Residues with reduced solvent accessibility, such as those at the binding site in the holo state, show attenuated labeling or cleavage relative to the comparison state, and this signal change is readily resolved by MS. Thus, protein footprinting is well suited to the study of protein-ligand interaction, although its outcome is of lower resolution than that provided by X-ray crystallography and NMR spectroscopy. An advantage of MS-based footprinting over these methods is that protein-ligand structure can be interrogated in a near physiological setting (i.e., at dilute concentration in solution with appropriate buffer, ionic strength, and bimolecular milieu).
Footprinting was first reported as a cleavage-based strategy to detect protein-DNA binding specificity.4 This demonstration inspired similar methods to be developed for the analysis of proteins5; Hanai6 and co-workers first demonstrated that modification reagents could footprint proteins. Two classes of modification strategies have emerged: reversible and irreversible modification.7 The most popular reversible modification is hydrogen deuterium (H/D) exchange,8 which detects solvent accessibility of exchangeable amide hydrogens on the backbone of proteins. PLIMSTEX9 and SUPREX,10 two HD exchange protocols, were developed to investigate protein-ligand interactions and affinities. The major disadvantage of reversible modification is that this labeling cannot survive if extensive purification and handling is needed to give residue-resolved information.
Besides reversible modification, several reagents can label irreversibly certain amino acids.11 One method is hydroxyl-radical (•OH) based protein footprinting, which was first introduced by Chance and coworkers12. Hydroxyl radicals can be generated by synchrotron radiolysis of water,13 laser photolysis of hydrogen peroxide14 and other methods.15, 16 Fast Photochemical Oxidation of Proteins (FPOP) employs laser photolysis of hydrogen peroxide to label proteins on the microsecond timescale.17 The advantages of •OH compared to other irreversible labeling reagents are its high reactivity, with approximately one half the common amino acids, and its size, which is similar to that of water. The growth in application of •OH based protein footprinting was recently reviewed.18 Both reversible and irreversible strategies elicit a real incentive for further development given the powerful analytical capabilities of modern MS-based proteomics.
In this paper, we use FPOP to show the effect of peptide ligands on calmodulin conformation. Our intention is to set forth a method for comparing structures of protein/ligand complexes when one or more reference structures are available. We chose as a model calmodulin (CaM), a small acidic calcium binding protein.19 Increased Ca2+ concentration promotes specific CaM-Ca2+ binding; with the uptake of four Ca2+ ions, CaM undergoes a conformational changes to a wholly different state capable of ligand binding.20 As a Ca2+ sensor and signal transducer, Ca2+-bound CaM binds to target proteins to alter their functions, acting as part of a calcium signal transduction pathway.21,22 CaM mediates processes such as inflammation, metabolism, muscle contraction, intracellular movement, short-term and long-term memory, nerve growth and the immune response.23 One family of CaM targets is Ca2+/Calmodulin–dependent protein kinases; an example is skeletal muscle myosin light chain kinase (SK-MLCK).24 Some peptides also bind CaM to inhibit the calcium signal transduction pathway. Two such peptides are melittin (Mel) and mastoparan (Mas). Mel, the principal component of honeybee venom, is a 26-residue peptide,25 whereas Mas is a 14-residue peptide toxin from wasp.26
To date, no high resolution 3D structures of CaM-Mel or CaM-Mas have been published, although studies by NMR,27 H/D exchange,28 fluorescence,29 enzyme cleavage,30 cross-linking31 and chemical modification were reported.32, 33 Previous studies demonstrated that both Mel and Mas share a target recognition and activation mechanism with the SK-MLCK binding domain (M13 peptide) (Figure 1), suggesting their structures are similar.34,35, 36
Figure 1.
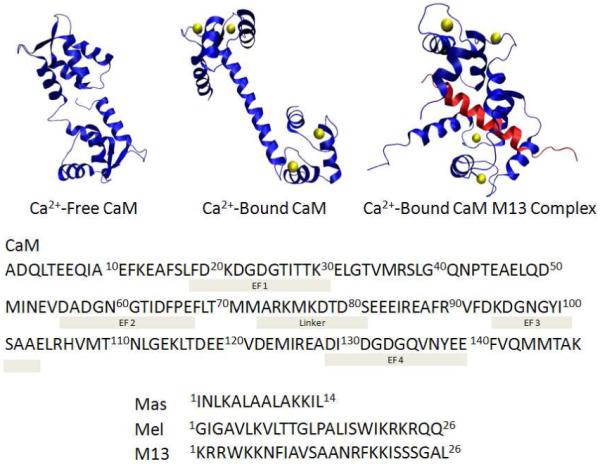
3D structure of CaM and CaM-peptide complex: Ca2+-free CaM structure from the average NMR structure (PDB ID: 1CFD). Ca2+-bound CaM structure from the x-ray crystal structure (PDB ID: 1CLL). Ca2+-bound CaM-M13 complex structure from the average NMR structure (PDB ID: 2BBM). M13 peptide is in pink. Ca2+ is displayed as yellow balls.
We tested the hypothesis of structural homology among the three CaM-ligand complexes by comparing their quantitative FPOP outcomes using spectra-contrast-angle evaluation.37 This method utilizes the CaM-M13 complex as a well-understood template structure for the interpretation of FPOP signals of the CaM-Mel and CaM-Mas complexes. By this example, we demonstrate a method for the detailed characterization of protein-ligand interactions. Such an approach should be applicable to protein-ligand systems where sub libraries of ligands that are known to bind can be screened for similarity of interaction relative to a standard protein-ligand complex; the approach has higher throughput than NMR and X-ray crystallography. Application of this method for the rapid analysis of protein-ligand complexes may prove useful for studies of disease-related protein complexes, especially in drug discovery.
MATERIALS AND METHODS
Materials
Bovine CaM (calcium free) was obtained from Ocean Biologics Co. (Edmonds, WA). Hydrogen peroxide, water, acetonitrile, formic acid, calcium chloride, phosphate buffered saline powder, EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetra-acetic acid), L-methionine, L-glutamine, melittin from honeybee venom (MW 2846), mastoparan from Vespula lewisii (MW 1478), catalase from bovine liver, and trypsin from porcine pancreas at the highest purity available were purchased from Sigma-Aldrich (St. Louis, MO). Skeletal muscle myosin light chain kinase peptide (M13), 1KRRWKKNFIAVSAANRFKKISSSGAL26, was purchased from AnaSpec, Inc. (Fremont, CA).
Protein and peptide stock solution
All proteins and peptides were received as lyophilized powder. A stock solution of CaM was prepared in water, and its concentration determined by UV-absorbance at 280 nm (ε = 2980 M−1 cm−1). All FPOP samples contained 10 μM CaM in 10 mM phosphate buffered saline (PBS buffer, 138 mM NaCl, 2.7 mM KCl, pH = 7.4) and 20 μM L-glutamine. Peptides, Mel, Mas, M13, were used without further purification.
Formation of Complexes
Eight types of samples were prepared from the CaM stock solution and PBS buffer: Ca2+-free-CaM (100 μM EGTA), Ca2+-free-CaM-Mel (100 μM EGTA, 20 μM Mel), Ca2+-free-CaM-Mas (100 μM EGTA, 20 μM Mas), Ca2+-free-CaM-M13 (100 μM EGTA, 20 μM M13), Ca2+-bound-CaM (100 μM calcium chloride), Ca2+-bound-CaM-Mel complex (100 μM calcium chloride, 20 μM Mel), Ca2+-bound-CaM-Mas complex (100 μM calcium chloride, 20 μM Mas), and Ca2+-bound-CaM-M13 complex (100 μM calcium chloride, 20 μM M13). The protein-peptide ratio in the sample solutions was 1:2. All samples were incubated overnight at 22 °C to allow for equilibration prior FPOP labeling.
FPOP Protocol
The protocol used here was based on a previous report38 with minor changes. A 248 nm KrF excimer laser (GAM Laser Inc, Orlando, FL) tuned to 40 mJ/pulse, was used to irradiate the sample solution. The laser was focused through a 250 mm convex lens (Edmunds Optics, Barrington, NJ) onto 150 μm i.d. fused silica tubing (Polymicro Technologies, Pheonix, AZ) located 125 mm from the lens, giving a 2.5 mm irradiation window; the fused silica polyimide coating in this region was first removed by using a propane torch. The flow rate and pulse frequency were adjusted to guarantee that1.2 x the reaction volume vacated the window between laser shots. The laser pulse frequency was controlled by an external pulse generator (B&K Precision, Yorba Linda, CA).
Hydrogen peroxide was added to each sample to a final concentration of 15 mM just prior to FPOP. Samples of 50 μL were infused through the apparatus and collected in tubes containing 10 μL of 50 nM catalase and 20 μM methionine. Residual hydrogen peroxide was decomposed by catalase treatment at room temperature for 10 min before storage at 4 °C. FPOP labeling was done in triplicate for each sample type.
ESI MS Analysis with a Q-TOF Mass Spectrometer
All ESI mass spectra were acquired in the positive-ion mode on a Waters (MicroMass) Q-TOF Ultima (Manchester, U.K.) equipped with a Z-spray ESI source. The instrument setup for protein analysis was similar to that reported previously.39
Trypsin Proteolysis Protocol
Digestion of CaM FPOP sample was conducted according to the previously reported protocol without modification.40
LC-ESI-MS/MS Analysis of Protein Digest
Digested samples (1 μL) were diluted in 100 μL water with 0.1% formic acid. An aliquot of 5 μL diluted solution was loaded onto a silica capillary column with a PicoFrit™ tip (New Objective, Inc., Woburn, MA) that was custom-packed with C18 reverse phase material (Magic, 0.075 mm × 150 mm, 5 μm, 120 Å, Michrom Bioresources, Inc., Auburn, CA). The gradient was pumped with an Eksigent NanoLC-1D ultra (Eksigent Technologies, Inc. Livermore, CA) at 260 nL/min, from 2% to 60% solvent B (acetonitrile, 0.1% formic acid) over 60 min, then to 80% solvent B for 10 min, followed by a 12 min re-equilibration step by 100% solvent A (water, 0.1% formic acid). The flow was directed by a PicoView Nanospray Source (PV550, New Objective, Inc., Woburn, MA) to an LTQ Orbitrap (Thermo-Scientific, San Jose, CA) with a spray voltage of 1.8-2.0 kV, and a capillary voltage of 27 V. The LTQ Orbitrap was operated in the standard, data-dependent acquisition mode controlled by Xcalibur 2.0.7 software. Peptide mass spectra (m/z range: 350-2000) were acquired at high mass resolving power (60,000 for ions of m/z 400) in FT mode. The top six most abundant multiply charged ions with minimal signal intensity at 1000 counts were subjected to CID (collision-induced dissociation) in the linear ion trap. Precursor activation was performed with an isolation width of 2 Da and an activation time of 30 ms. The normalized collision energy was set at 35%. The automatic gain control target value was regulated at 1 × 106 for the FT analyzer and 3 × 104 for ion-trap analyzer with a maximum injection time of 1000 ms for FT and 200 ms for ion trap.
LC-MS Feature Annotation
The LC-MS/MS data were searched for modified and unmodified CaM tryptic peptides by using Mascot 2.2.06 (Matrix Science, London, UK)41 and the NCBI database. All known •OH-side chain reaction products were added into modification database for searching as variable modifications. Modification site assignments were validated by manual inspection of production spectra.
LC-MS/MS .raw files were imported into the Rosetta Elucidator system (v3.3.0.0.220, Rosetta Biosoftware, Seattle, WA) for retention time alignment of shared LC-MS features.42 The aligned retention time and peak volume of all high resolution extracted ion chromatogram features across a 5-70 min window were output to a spreadsheet for each sample. Each product-ion spectrum was paired with its LC-MS feature by using its retention time and precursor m/z; a table of this pairing was used by the Excel macro written in our lab to annotate all Elucidator-determined LC-MS features with the Mascot assignments linked to their product-ion spectra.
Calculation of Modification Extent
The fraction modified (Fox1 in Eq. 1) was calculated for any specific residue as the ratio of signal intensities of each peptide (Iox1) modified at this residue to the total intensities of modified and unmodified peptide signals spanning this residue (I + Iox1 + Iox2 + …+ Ioxn). The changes in modification (R) at a site between Ca2+-free and Ca2+-bound states (Eq. 2) were calculated at the peptide level by using the modification fraction (FCa free and FCa bound) of each peptide, wherein the signal from all modifications on the peptide contribute to the numerator in equation 1.
| Eq. 1 |
| Eq. 2 |
To identify changes induced by complexation, results from CaM (no complex) were used as controls in both comparisons.
RESULTS AND DISCUSSION
ESI MS of calmodulin submitted to FPOP
Beginning with CaM in the presence of Mel or Mas, we detected by ESI MS multiply charged CaM molecules (Figure S1) that have a Gaussian charge-state distribution centered at the most abundant 15+ ion. We also saw ions for Mel and Mas at lower m/z, consistent with dissociation of the complex under ESI conditions. Trace signals for oxidized Mel/Mas were observed as well. The deconvoluted mass spectrum showed that the molecular weight (MW) of CaM is 16790 Da, consistent with CaM having the expected post-translational modifications of N-terminal acetylation (+42 Da) and trimethylation of K115 (+42 Da).40
After submitting CaM to FPOP, we found evidence in the mass spectra for significant oxidative modification (Figure S1b) as mass shifts +16, 32, 48… Da. The distribution of oxidation products is Poisson-like, serving as an indication that FPOP is fast and probed only a single state of the target protein. This conclusion is consistent with recent results43 showing that the distribution of oxidation products of CaM and two other proteins sensitive to oxidative-induced unfolding are well modeled by a Poisson distribution. The conformation sampled by FPOP for these experiments is singular and invariant during labeling (i.e., the protein did not unfold on the labeling timescale).
The abundances of the +16, +32, and +48 Da modification products, as estimated from their +15 charge state intensities (Figure S1b-d), show that slightly more protein is modified in the Ca2+-bound, ligand-free state than the Ca2+-free and Ca2+-bound CaM-ligand states. The Ca2+-free and Ca2+-bound structures are markedly different. Given that the global FPOP product distributions do not substantially distinguish these two, we turned to peptide and residue-level analysis for their and CaM-peptide complexes’ structural footprints.
Information Content of LC-ESI-MS/MS proteolyzed FPOPsamples
Mascot processing of the LC-MS/MS data of the trypsin-proteolyzed FPOP samples showed 93% sequence coverage for the three CaM complexes. The aforementioned PTMs were also identified by using Mascot. Of the 12 peptides identified, nine had product-ion spectra for both the oxidatively modified and unmodified forms for all samples, spanning over 70% of protein sequence. Calmodulin 38-74, a large tryptic peptide, was not detected probably owing to high retention upon sample handling and chromatography. This peptide was not included among the peptides used for quantitative analysis. Small tryptic peptides from ligand peptides (Mel/Mas/M13) were also observed. Owing to the excess ligand present at equilibrium prior to FPOP, labeling occurs for both bound and free ligands; their differential analysis is consequently not possible.
Minimization of post-FPOP oxidative-modification bias
Several factors give rise to post-FPOP oxidation on peptides, including methionine oxidation during proteolytic digestion, sample handling,44 and electrospray ionization (ESI).45 Of these modification-biasing signals, ESI-induced oxidation is the easiest to distinguish and thereby to be excluded in our analysis. The reason that we are not misled by ESI-induced oxidation is the hydrophilicity of a peptide is nearly always increased with oxidative-modification so that its reverse-phase LC retention time is earlier than that of its unmodified peptide (Figure S2). We routinely observe this in all FPOP labeling experiments (data not shown). As coelution of an oxidized peptide with its unmodified counterpart is unlikely, we attribute any oxidatively modified peptides co-eluting with the unmodified counterpart to ESI-induced oxidation.
A recent study of post-FPOP sample handling showed that proteins stored in millimolar levels of hydrogen peroxide can oxidize while frozen and at lower temperatures.46 As a precaution, we stored FPOP-treated samples at 4 °C after removing hydrogen peroxide with catalase immediately following FPOP labeling. To this mixture was added free methionine to curtail further any post-FPOP oxidation.47
Changes of modification at peptide and residue level
The extents of modification can be compared for CaM in the presence of various peptides with and without bound calcium (Figure 2). Comparisons between CaM-Mel/Mas/M13 complexes (Figure 2b-d) and CaM (Figure 2a) reveal that CaM peptides fall into three groups as discussed in following paragraphs.
Figure 2.

Changes of modification extent from Ca2+-free to Ca2+-bound states for various CaM tryptic peptides from different CaM complexes. (a) CaM itself, (b) CaM-Mel Complex, (c) CaM-Mas Complex, (d) CaM-M13 complex.
Peptides 76-86, 78-86 and 31-37 belong to group I; they have similar trends of modification change for both CaM and CaM-Mel/Mas/M13 complexes. Peptides 76-86 and 78-86 peptides originate from the linker region of CaM and display increased labeling in their Ca2+-bound vs. the Ca2+-free states, whereas peptide 31-37 shows decreased labeling. More importantly, their modification extents are invariant with addition of the peptide ligands (Mel/Mas/M13), indicating that the regions represented by these peptides are not directly involved in any CaM-Mel/Mas/M13 interaction.
Peptides 14-21, 22-30, 107-126, and 127-148 belong to group II; they exhibit different labeling trends for the CaM-Mel/Mas/M13 complexes than for CaM itself. The extents of oxidative modification decrease for peptides 14-21, 107-126 and 127-148 when complexed with peptide ligands (Figure 2b,c,d) compared to Ca2+-bound CaM in the absence of peptide ligands, where increases occur (Figure 2a). Reverse trends pertain to peptide 22-30. The most significant decrease in oxidative modification occurs for peptide 14-21. The differential labeling between CaM and the CaM-Mel/Mas/M13 complexes indicates that, upon forming the complexes, these regions of CaM are either buried allosterically or directly protected by the ligand.
Group III peptides 1-13 and 95-106 do not share a single labeling trend when CaM binds to peptides. The N-terminus peptide, 1-13, from CaM and the CaM-Mel bound complex (Figure 2a, b) show a decrease in oxidative modification whereas that peptide from CaM-Mas and CaM-M13 complexes (Figure 2c,d) undergo increased labeling. For peptide 95-106, the trend for CaM, CaM-Mel, and CaM-Mas (Figure 2a,b,c) showed decreased labeling whereas the CaM-M13 (Figure 2d) showed increased oxidative labeling.
Taken together, we propose that the patterns for all nine peptides compose a unique “fingerprint” of the protein-ligand complex. Although these data are not residue-resolved, they are readily accessible for comparing structures of the complexes because the modification extent of a peptide is a function of the aggregate of its modifiable residues’ solvent accessibilities (Figure 3a). Examining these fingerprints should establish whether the CaM-Mel/Mas structures are similar to the known structure of CaM/M13 complex.
Figure 3.

FPOP footprinting results on CaM structures. (a) Changes of oxidation are labeled on peptides with different color. M13 is black. (b) Bottom view of CaM-M13 complex. Residues in pink are several modified residues detected by LC-MS experiment. M13 is in the center of structure with ball shape.
Turning to product-ion spectra (Figure S3), we identified 14 modified residues. Seven are at the N-terminal domain, two from the linker region, and five from the C-terminal domain. Nine have side chains that contain sulfur (M36, M76, M109, M124, M144, and M145) or aromatic rings (F16, F19, Y99), all of which are highly reactive with •OH. The extent of modification is highest for Met (with modification fractions > 0.2). Five aliphatic amino acids (L4, I9, L18, I27, and I85) undergo detectable amounts of modification. The relative modification yields between these residues agree well with those reported in previous studies of the •OH reactivity of amino acids.48 Although we can detect other low-abundance modifications, we did not include them because their assignments based on product-ion spectra were problematic. Instead, we focused on the 14 oxidized residues as a source of comparative structural information of CaM complexes (Figure 4). Peptides containing more than one highly reactive site and undergoing double modifications are considered separately.
Figure 4.

Extent of modification for Ca2+-free (white) and Ca2+-bound (black) states of each modified residue for various CaM complexes. (a) CaM itself (b) CaM-Mel Complex (c) CaM-Mas Complex (d) CaM-M13 complex.
Higher resolution provided by residue level FPOP data taken by MS/MS should afford more detailed views of CaM-peptide complexes. Those “zoom-in” views help to elucidate residue-level interactions between the protein and the peptide ligand, a view that is missing in the peptide mass spectra. In the case of peptide 14-21, its modified residues F16, L18, and F19, however, do not share the same trends shown for the full peptide. L18 and F19 show higher modification levels for CaM than for the complexes, which is the same trend seen at the peptide level. F16, however, is modified to nearly the same extent for CaM as for the complexes. Thus, one model of complexation puts L18 and F19 in direct interaction with the peptide ligand and F16 away from this interface. These data for peptide 14-21 show that analysis at the amino-acid level, made efficient because the modifications are irreversible, provides a more resolved picture of protein-ligand interaction than methodologies that only use peptide-level data. Integrating high-resolution views with a reference structure of CaM-M13 can allow the structures of unknown CaM-Mel/Mas complexes to be compared (Figure 3b).
Spectral-contrast-angle comparison of the modification patterns of peptides
At this point, one may conclude from analysis at both the peptide level and the amino-acid residue level that the modification extents in the peptide-bound and free states for the three complexes are similar. To test more rigorously the similarity, we employed a spectral-contrast-angle (θ) analysis of the nine peptide modification trends. This spectra-contrast-angle approach can validate the comparison process by providing a confident value (θ) related to similarity. It was used previously to compare the product-ion spectra of oligodeoxynucleotide isomers.37 The changes of oxidative modification of the various peptides from CaM can be treated in similar way. Although the sequence coverage of the tryptic peptides is ~70%, this statistical approach still is suitable for making comparisons and does not require full coverage. For comparing the CaM-peptide complex with the peptide-free sample, the set of peptide signals can be represented as a basis vector in an N-dimensional space (ai). In this analysis, each signal is the change in modification level for the calcium-bound vs. calcium-free state. A comparison vector (bi) is comprised of the same signal data from a different sample replicate or CaM-peptide mixture. The spectral-contrast-angle (Eq. 3) of these vectors provides a single parameter reflecting their similarity; as the similarity increases, θ ➔0.
The first four entries in Table 1 are the average θ for the three pair-wise comparisons of replicates from the independent triplicates of a CaM-peptide or absent-peptide FPOP experiments. These entries provide an expectation level for near identity of θ < 35. The θ determined from the average of each CaM vs CaM-peptide comparison is significantly larger (Table 1, 5th entry). This implies a significant change in structure with peptide binding, as is expected, and the small standard deviation conveys the overall similarity of this change among the three peptide complexes. The pair-wise comparisons between each CaM-peptide complex give an average θ (Table 1 last entry), which is similar to the smallest angle from replicate experiments. This similarity further supports the conclusion that the two unknown CaM-peptide complexes have similar structures and that they resemble the known CaM-M13 structure. We propose that this statistical approach is a useful means of comparing protein/ligand structures for a series of ligands when one structure is known and can be used as a reference.
| Eq. 3 |
Table 1.
Spectral Contrast Angle Similarity Evaluation on Changes of Oxidation
| Items of Comparison | Contrast angle (°) |
|---|---|
| CaM | 34±5 |
| CaM-Mel | 32±2 |
| CaM-Mas | 31±3 |
| CaM-M13 | 18±6 |
| CaM vs. CaM-Peptide* | 75±1 |
| CaM-Peptide vs. CaM-Peptide | 42±5 |
Peptide = Mel, Mas, M13
Comparative structure assignment from FPOP data
The approach of utilizing the “fingerprint” of changes in extent of modification at the peptide and residue levels is more empirical than but complements that of Chance and coworkers49, 50 who showed that the combination of •OH surface mapping data with computational modeling provides important protein structural data. Our approach not only allows conclusions about the similarities of the protein-ligand structures but also permits establishment of some structural details. Proteins or peptides (including Mel, Mas) that bind to CaM are likely to bind to both N-term and C-term hydrophobic clefts by forming a long alpha helix in a manner similar to that of SK-MLCK M13 peptide.34 In addition to the NMR structure (PDB ID: 2BBM) of CaM-M13 complex, the structures of Ca2+-free CaM from NMR (PDB ID: 1CFD) and Ca2+-bound CaM from x-ray crystal structure (PDB ID: 1CLL) are also known (Figure 1). The FPOP results for the established structure of the CaM-M13 complex provide a basis for a more detailed structural analysis. For example, we see that L18, F19, M109, M124, M144 and M145 from both N and C terminal domains of the three complexes are protected against •OH reaction upon complex formation with the various ligand peptides. All six are hydrophobic and are located at the interface between CaM and M13, as seen in the NMR structure (Figure 3b). M109, M124, M144 and M145 are located inside the hydrophobic cleft of the C-terminus and make contact with the M13 hydrophobic side chains. For the CaM-M13 structure, L18 and F19, which are at the hydrophobic-cleft center of the N-terminus, become less solvent-accessible when the Ca2+-bound CaM binds with M13. On the other hand, F16 is not part of the hydrophobic cleft in the CaM-M13 complex, but it is hydrophobic and close in proximity to L18 and F19 (Figure 3b).
The L4 and I9 side chains have relatively similar solvent accessibilities in CaM and the CaM-M13 complex, suggesting that these residues share similar conformations in CaM and in the peptide complexes. Two other interesting sites are I27 and Y99, which are located in the EF hands. Y99 is in a short anti parallel beta sheet between adjacent calcium binding loops. The aromatic side chain of Y99 points outside the molecule. Owing to its high solvent accessibility and its inherent high •OH reactivity, this tyrosine residue is particularly sensitive to slight changes in structure. This suggests the peptide-binding-induced deprotection when M13 binds and induced protection when Mel and Mas bind are small changes although significantly different. I27, however, is on the small loop connecting two alpha helices of the first EF hand motif. Its side chain is inside the hydrophobic cleft in all three of the high resolution CaM structures (Ca2+-free CaM, Ca2+-bound CaM and CaM-M13 complex), consistent with the low reactivity and lack of difference between Ca2+-loaded CaM and the peptide complexes.
CONCLUSION
Comparison of FPOP data taken for a protein complex of known structure to the FPOP-induced modifications of unknowns by using spectral-contrast-angle provides a way to test whether a protein has similar or different structures when it binds to various ligands. Specifically, a statistical analysis of the modification extents peptide regions of CaM extricated by tryptic digestion shows that two peptide ligands (i.e., Mel and Mas) bind similarly in the presence of Ca2+ as does a third ligand (M13) that forms a known structure with CaM. We chose not to increase the certainty of the comparison by using more proteases in the digestion of FPOP samples to obtain more data for comparison, a suggestion made by one of the reviewers. We wished to show that this comparative approach is effective for distinguishing the structural differences even when the sequence coverage isn’t 100%, as is the case here. An advantage of FPOP coupled with MS is that one can examine the modification extents at the amino-acid level to identify those residues that are involved in the conformational changes induced by ligand binding. Those residues that show differential modification levels in the structure of CaM-M13 complex are those that are expected on the basis of the high resolution 3D structure from NMR.
The MS-based FPOP method has the ability to probe structures on a comparative basis for small amounts of protein (pmoles) in relatively short times (fractions of a week) compared to NMR or X-ray crystallography. These simple comparisons may have application in screening many protein/ligand interactions including those in drug development. The FPOP data serve as a “fingerprint” for the ligand interactions. Combining the “fingerprints” with those from H/D exchange, specific chemical modification (e.g., acetylation of Lys), and cross linking can give detailed information about protein-ligand interactions with reasonable throughput.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Justin Sperry for helpful suggestions and Dr. Henry Rohrs for assistance with the LTQ-Orbitrap studies. This research was supported by the National Centers for Research Resource of the NIH (Grant NO. P41RR000954). Additional support was provided by Merck; MLG is a consultant for Merck.
REFERENCES
- (1).Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- (2).Schermann SM, Simmons DA, Konermann L. Expert Rev Proteomics. 2005;2:475–485. doi: 10.1586/14789450.2.4.475. [DOI] [PubMed] [Google Scholar]
- (3).Jay DG. J Biol Chem. 1984;259:15572–15578. [PubMed] [Google Scholar]
- (4).Mirzabekov AD, Melnikova AF. Mol Biol Rep. 1974;1:379–384. doi: 10.1007/BF00385669. [DOI] [PubMed] [Google Scholar]
- (5).Sheshberadaran H, Payne LG. Proc Natl Acad Sci U S A. 1988;85:1–5. doi: 10.1073/pnas.85.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hanai R, Wang JC. Proc Natl Acad Sci U S A. 1994;91:11904–11908. doi: 10.1073/pnas.91.25.11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fitzgerald MC, West GM. J Am Soc Mass Spectrom. 2009;20:1193–1206. doi: 10.1016/j.jasms.2009.02.006. [DOI] [PubMed] [Google Scholar]
- (8).Wales TE, Engen JR. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- (9).Zhu MM, Rempel DL, Du Z, Gross ML. J Am Chem Soc. 2003;125:5252–5253. doi: 10.1021/ja029460d. [DOI] [PubMed] [Google Scholar]
- (10).Roulhac PL, Powell KD, Dhungana S, Weaver KD, Mietzner TA, Crumbliss AL, Fitzgerald MC. Biochemistry. 2004;43:15767–15774. doi: 10.1021/bi0481848. [DOI] [PubMed] [Google Scholar]
- (11).Mendoza VL, Vachet RW. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Maleknia SD, Brenowitz M, Chance MR. Anal Chem. 1999;71:3965–3973. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- (13).Xu G, Chance MR. Anal Chem. 2004;76:1213–1221. doi: 10.1021/ac035422g. [DOI] [PubMed] [Google Scholar]
- (14).Sharp JS, Becker JM, Hettich RL. Anal Chem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- (15).Zhu Y, Guo T, Park JE, Li X, Meng W, Datta A, Bern M, Lim SK, Sze SK. Mol Cell Proteomics. 2009;8:1999–2010. doi: 10.1074/mcp.M900081-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tong X, Wren JC, Konermann L. Anal Chem. 2007;79:6376–6382. doi: 10.1021/ac070724u. [DOI] [PubMed] [Google Scholar]
- (17).Hambly DM, Gross ML. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- (18).Xu G, Chance MR. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- (19).Weinstein H, Mehler EL. Annu Rev Physiol. 1994;56:213–236. doi: 10.1146/annurev.ph.56.030194.001241. [DOI] [PubMed] [Google Scholar]
- (20).Wriggers W, Mehler E, Pitici F, Weinstein H, Schulten K. Biophys J. 1998;74:1622–1639. doi: 10.1016/S0006-3495(98)77876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tanaka T, Hidaka H. J Biol Chem. 1980;255:11078–11080. [PubMed] [Google Scholar]
- (22).Degrado WF. Adv Protein Chem. 1988;39:51–124. doi: 10.1016/s0065-3233(08)60375-7. [DOI] [PubMed] [Google Scholar]
- (23).Bachs O, Agell N, Carafoli E. Biochim Biophys Acta. 1992;1113:259–270. doi: 10.1016/0304-4157(92)90041-8. [DOI] [PubMed] [Google Scholar]
- (24).Blumenthal DK, Takio K, Edelman AM, Charbonneau H, Titani K, Walsh KA, Krebs EG. Proc Natl Acad Sci U S A. 1985;82:3187–3191. doi: 10.1073/pnas.82.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kataoka M, Head JF, Seaton BA, Engelman DM. Proc Natl Acad Sci U S A. 1989;86:6944–6948. doi: 10.1073/pnas.86.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Matsushima N, Izumi Y, Matsuo T, Yoshino H, Ueki T, Miyake Y. J Biochem. 1989;105:883–887. doi: 10.1093/oxfordjournals.jbchem.a122773. [DOI] [PubMed] [Google Scholar]
- (27).Seeholzer SH, Cohn M, Putkey JA, Means AR, Crespi HL. Proc Natl Acad Sci U S A. 1986;83:3634–3638. doi: 10.1073/pnas.83.11.3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhu MM, Rempel DL, Zhao J, Giblin DE, Gross ML. Biochemistry. 2003;42:15388–15397. doi: 10.1021/bi035188o. [DOI] [PubMed] [Google Scholar]
- (29).Chapman ER, Alexander K, Vorherr T, Carafoli E, Storm DR. Biochemistry. 1992;31:12819–12825. doi: 10.1021/bi00166a016. [DOI] [PubMed] [Google Scholar]
- (30).Scaloni A, Miraglia N, Orru S, Amodeo P, Motta A, Marino G, Pucci P. J Mol Biol. 1998;277:945–958. doi: 10.1006/jmbi.1998.1629. [DOI] [PubMed] [Google Scholar]
- (31).Schulz DM, Ihling C, Clore GM, Sinz A. Biochemistry. 2004;43:4703–4715. doi: 10.1021/bi036149f. [DOI] [PubMed] [Google Scholar]
- (32).Steiner RF, Albaugh S, Fenselau C, Murphy C, Vestling M. Anal Biochem. 1991;196:120–125. doi: 10.1016/0003-2697(91)90127-f. [DOI] [PubMed] [Google Scholar]
- (33).Wong JW, Maleknia SD, Downard KM. J Am Soc Mass Spectrom. 2005;16:225–233. doi: 10.1016/j.jasms.2004.11.009. [DOI] [PubMed] [Google Scholar]
- (34).Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- (35).Heidorn DB, Seeger PA, Rokop SE, Blumenthal DK, Means AR, Crespi H, Trewhella J. Biochemistry. 1989;28:6757–6764. doi: 10.1021/bi00442a032. [DOI] [PubMed] [Google Scholar]
- (36).Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- (37).Wan KX, Vidavsky I, Gross ML. J Am Soc Mass Spectrom. 2002;13:85–88. doi: 10.1016/S1044-0305(01)00327-0. [DOI] [PubMed] [Google Scholar]
- (38).Hambly D, Gross ML. International Journal of Mass Spectrometry. 2007;259:124–129. [Google Scholar]
- (39).Sperry JB, Shi X, Rempel DL, Nishimura Y, Akashi S, Gross ML. Biochemistry. 2008;47:1797–1807. doi: 10.1021/bi702037p. [DOI] [PubMed] [Google Scholar]
- (40).Sharp JS, Tomer KB. Biophys J. 2007;92:1682–1692. doi: 10.1529/biophysj.106.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- (42).Neubert H, Bonnert TP, Rumpel K, Hunt BT, Henle ES, James IT. J Proteome Res. 2008;7:2270–2279. doi: 10.1021/pr700705u. [DOI] [PubMed] [Google Scholar]
- (43).Gau BC, Sharp JS, Rempel DL, Gross ML. Analytical Chemistry. 2009;81 doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xu G, Kiselar J, He Q, Chance MR. Anal Chem. 2005;77:3029–3037. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- (45).Boys BL, Kuprowski MC, Noel JJ, Konermann L. Anal Chem. 2009;81:4027–4034. doi: 10.1021/ac900243p. [DOI] [PubMed] [Google Scholar]
- (46).Hambly DM, Gross ML. Anal Chem. 2009;81:7235–7242. doi: 10.1021/ac900855f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Saladino J, Liu M, Live D, Sharp JS. J Am Soc Mass Spectrom. 2009;20:1123–1126. doi: 10.1016/j.jasms.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Takamoto K, Chance MR. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- (49).Zheng X, Wintrode PL, Chance MR. Structure. 2008;16:38–51. doi: 10.1016/j.str.2007.10.019. [DOI] [PubMed] [Google Scholar]
- (50).Kamal JK, Chance MR. Protein Sci. 2008;17:79–94. doi: 10.1110/ps.073071808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.