

Abstract
Maturation of dendritic cells (DC) to competent APC is essential for the generation of acquired immunity and is a major function of adjuvants. dsRNA, a molecular signature of viral infection, drives DC maturation by activating TLR3, but the size of dsRNA required to activated DC and the expression patterns of TLR3 protein in DC subsets have not been established. Here we show that cross-priming CD8α+ and CD103+ DC subsets express much higher levels of TLR3 than other DC. In resting DC, TLR3 is located in early endosomes and other intracellular compartments but migrates to LAMP1+ endosomes upon stimulation with a TLR3 ligand. Using homogeneous dsRNA oligonucleotides (ONs) ranging in length from 25 to 540 bp, we observed that a minimum length of about 90 bp was sufficient to induce CD86, IL12p40, IFN-β, TNF-α, and IL-6 expression and to mature DC into APC that cross-presented exogenous antigens to CD8+ T cells. TLR3 was essential for activation of DC by dsRNA ONs, and the potency of activation increased with dsRNA length and varied between DC subsets. In vivo, dsRNA ONs, in a size-dependent manner, served as adjuvants for the generation of antigen-specific CTL and for inducing protection against lethal challenge with influenza virus when given with influenza nucleoprotein as an immunogen. These results provide the basis for the development of TLR3-specific adjuvants capable of inducing immune responses tailored for viral pathogens.
Introduction
Successful immunization requires both an antigen and a signal that induces dendritic cells (DC) to mature into competent APC (1,2). DC take up and process antigens from peripheral microenvironments, then migrate to the draining lymph nodes (LNs) where they present the processed antigens on MHC-I and MHC-II molecules to T cells (3–7). Under homeostatic conditions, DC tolerize T cells to self antigens. However, at sites of infection or immunization, pathogen-derived substances or adjuvants activate DC, inducing expression of co-stimulatory molecules and cytokines, which together with MHC-antigen complexes induce cognate T-cells to differentiate into antigen-specific CTL and helper T cells (8–11). DC recognize pathogen-associated molecular patterns (PAMPs) and many adjuvants using receptors of the innate immune system including members of the TLR family (12–17). Subsets of DC vary in TLR expression and responses to different PAMPs (18). Therefore, by activating specific subsets of DC, TLR ligands can promote immune responses appropriate for particular types of pathogens (19,20).
TLR3 is an endosomal receptor that recognizes dsRNA, a common viral replication intermediate and a potent indicator of viral infection (16,21–26). The recognition of dsRNA by TLR3 is independent of its base sequence (27,28) which prevents viruses from avoiding detection through genomic mutations. dsRNA from virally infected cells enters TLR3-containing endosomes of neighboring uninfected cells by direct uptake from the medium or by phagocytosis of infected cells (29,30). There, it activates TLR3 and triggers defensive responses, including cytokine and chemokine production and DC maturation. Because dsRNA is a viral PAMP, it is reasonable to hypothesize that recognition of dsRNA by TLR3 will induce antiviral responses and that a TLR3 agonist, given as an adjuvant with viral immunogens, would generate immune responses that are tailored to preventing viral infection and dissemination. The most commonly used experimental TLR3 agonist is polyI:polyC (pIC), which is a large dsRNA-like synthetic polymeric complex. pIC preparations vary in distribution of strand lengths, solubility, and biological properties including toxicity (25,31,32). Recently we characterized the interaction of homogeneous dsRNA oligonucleotides (ONs) of defined lengths with TLR3 extracellular domain (ECD) protein (27) and here we examine their capacity to serve as adjuvants in generating immune responses. We previously showed that the shortest dsRNA ON capable of binding TLR3 is ~45 bp in length, and that TLR3 binds as dimers to 45 bp segments of dsRNA (27,28). In transfected cells that express high amounts of TLR3, a 45 bp ON was sufficient for activation, but in cells expressing less TLR3, longer ONs were required for signaling (27). However, the minimum length of dsRNA ON required to activate TLR3 in primary cells, including DC, is currently unknown. Here, we studied the capacity of dsRNA ONs of defined size to activate DC subsets from WT and TLR3−/− mice, to induce antigen specific CTL responses, and to serve as adjuvants for an experimental influenza vaccine. We found that TLR3 is most highly expressed in cross-presenting DC subsets, and that homogeneous dsRNA ONs can serve as effective adjuvants capable of activating DC and promoting strong adaptive immunity.
Materials and methods
Animals
C57BL/6 mice and B6-Ly5.2 mice were purchased from the Frederick Cancer Research Center (Frederick, MD, USA), TLR3−/− mice were provided by Dr. Daniela Verthelyi (CDER/FDA, Bethesda, MD, USA), melanoma differentiation-associated gene 5 deficient (MDA5−/−) mice were provided by Dr. Marco Colonna (Washington University, St. Louis, MO, USA), and OT-I mice were purchased from The Jackson Laboratory. Female mice were used at 6–10 weeks of age. All mice were cared for in accordance with NIH guidelines and with the approval of the National Cancer Institute animal care and use committee.
Dendritic cells
Dendritic cells were differentiated either from wild-type (WT), TLR3−/− or MDA5−/− bone marrow cells as described (33,34). For fms-like tyrosine kinase-3 ligand (FLT-3L)-generated DC (FL-DC), bone marrow cells were cultured for 10 days in 6-well plates at 1×106 cells/ml, 5ml/well in complete medium (RPMI containing 10% FCS, 55 μM 2-ME, glutamine, antibiotics, nonessential amino acids, and sodium pyruvate) containing 50 ng/ml FLT3-L (Pepro Tech, Rocky Hill, NJ). For GM-CSF-generated DC (GM-DC), cells were cultured for 7 days in 10 cm dishes at 0.5 × 106 cells/ml 20 ml/dish in complete medium containing 10 ng/ml GM-CSF (GeneScript, Piscataway, NJ). Splenic and LN DC were positively selected using CD11c microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s instructions (purity >80%).
Flow Cytometry
Antibodies for flow cytometry were purchased from either from BD Biosciences (San Jose, CA), Miltenyi Biotec, or eBioscience (San Diego, CA), with the exception of the anti-mTLR3 mAb (clone 11F8) which was generated in rats with the aid of Green Mountain Antibodies (Burlington, VT) using the mTLR3 extracellular domain as the immunogen. The 11F8 antibody is a rat IgG2a, and was initially selected by ELISA for binding to purified TLR3 extracellular domain protein, and then for the ability to label HEK293 cells transfected with mTLR3. Purified anti-TLR3 antibody was labeled using an Alexa 647 Monoclonal Antibody Labeling Kit (Invitrogen/Molecular Probes., Carlsbad, CA). For intracellular staining, cells were fixed and permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences) according to the manufacturer’s instructions. To characterize OVA-specific CD8 T cells, staining with SIINFEKL H-2Kb-pentamer (ProImmune, Bradenton, FL) was performed according to the manufacturer’s instructions. All antibody staining was performed in the presence of purified 2.4G2 antibody (Fc block; BD Biosciences). Flow cytomety was performed using a FACSCalibur or LSRII instrument (BD Biosciences) and data were analyzed using FlowJo (Tree Star, Inc., Ashland, OR). FL-DC subsets were sorted based on CD24 and CD11b expression using a BD FACSAria (BD Biosciences).
Confocal microscopy
FL-DC were seeded on CC2-treated Lab-TekRII Chamber Slides (Nalge Nunc/Thermo Fisher Scientific, Rochester, NY) for 24 h and treated with 10 μg/ml of dsRNA ONs or pIC (InvivoGen, San Diego, CA) or left untreated. Then cells were fixed for 1 h with 2% PFA and permeabilized for 30 min with block/perm buffer (PBS containing 1% BSA, 0.05% saponin, 10mM glycine and 10% FBS). Cells were stained either with anti-calnexin, anti-EEA1, and anti-LAMP-1 primary antibodies (Abcam, Cambridge, MA) followed by goat anti-rabbit IgG-Alexa 546 (Invitrogen) together with 11F8 (anti-TLR3) followed by goat anti-rat IgG-Alexa 488 (Invitrogen). Staining was done in the presence of block/perm buffer. Slides were mounted using ProLong Gold antifade reagent (Invitrogen). Images were acquired with a Zeiss LSM510 META confocal microscope (Carl Zeiss MicroImaging Inc, Thornwood, NY) equipped with a 63x Plan-apochromat (N.A. 1.4) oil immersion objective lens.
DC activation
DC were activated for 24 hours with pIC or homogeneous dsRNA ONs, 25, 60, 62, 90, 139, and 540 bp in length (10 μg/ml for unseparated DC or 25 μg/ml when sorted subsets were treated), synthesized as described (27). Preliminary experiments indicated that these concentrations of ON or pIC were on the or near the plateau of the dose response curve. CD86 up-regulation was determined by FACS, and cytokine production was analyzed using a CBA inflammation kit (BD Biosciences) except for IFN-β and IL-12p40 which were measured by standard ELISA (R&D Systems, Minneapolis, MN).
In vitro cross-presentation
DC (1 × 106/ml) were incubated overnight with 0.1 μg/ml OVA (Acros Organics, Geel, Belgium) in the presence of either 10 μg/ml pIC, 10 μg/ml dsRNA ONs, or 2 μg/ml CpG 1d (35). The following day CD8+ T cells were isolated from OT-I spleens and LN using a MACS CD8+ T-cell isolation kit (Miltenyi Biotec), and then labeled with 1 μM CFSE (Invitrogen). DC were washed extensively, and then co-cultured with an equal number (1 × 105) of CFSE-labeled OT-I CD8+ T cells in complete medium in 96-well round bottom plates. After 60 hours 3 × 104 polystyrene beads (15μm, Fluka/Sigma-Aldrich, St. Louis, MO) were added to each well. Cells and beads were recovered and CFSE dilution was measured by FACS. The number of beads measured in each sample, relative to the total number added to the well was used to calculate the total number of dividing OT-I cells per well for each sample.
In vivo CTL generation
WT and TLR3−/− mice were immunized in the footpads with OVA alone (0.25 μg), OVA plus 2 μg dsRNA ONs (90bp, 139bp or 540bp) or OVA plus 2 μg pIC. After seven days CD8+ T cells from draining popliteal LNs were labeled with SIINFEKL H-2Kb-pentamer to measure OVA-specific T cells. On day 6 after immunization, spleen cells from B6-Ly5.2 (CD45.1+) mice were loaded with 1μM OVA peptide (Genscript, Piscataway, NJ) for one hour or left untreated, then cells were labeled with 5 or 0.5 μM CFSE for 5 minutes at 37°C, respectively. Labeled splenocytes were mixed in a 1 to 1 ratio and injected i.v. into immunized mice (107 cells/mouse). The following day popliteal LNs were harvested and the ratios of the CFSE bright to dim peaks in the CD45.1+ population were calculated. Percent-specific lysis was calculated using the formula: [1 (CFSE ratio in mice immunized with adjuvant/CFSE ratio of mice immunized without adjuvant)] × 100 as described by Ingulli (36).
Immunization against influenza infection
Mice were primed with 20 μg recombinant influenza A nucleoprotein (rNP; Imgenex, San Diego, CA) alone, with 540 bp dsRNA ON alone (10 μg) or with rNP (20 μg) in combination with 90, 139, 540 bp dsRNA ONs or pIC (10 μg each) i.m into the rear quadriceps, 7–10 mice/group. Four weeks later animal groups were boosted using the same protocol. Three weeks after boosting, mice were challenged with 10 LD50 of influenza strain A/PR/8/34 (H1N1) intranasally under ketamine anesthesia and monitored for survival.
Statistical analysis
Statistical significance between different experimental groups was assessed using either simple or unbalanced analysis of variance for independent data and repeated measures analysis of variance for multiple responses in the same mice. Data distributions were positively skewed, and appropriate transformations (primarily logarithmic and square root) were applied before analysis to maintain consistency with model assumptions. A binomial-gaussian mixed form likelihood method was used for data with a high proportion of zero values. Significance of differences in survival was tested by the exact two-tailed logrank test. The P values were corrected for multiple simultaneous comparisons by the method of Hochberg and by Dunnett’s test against a common control. P values less than 0.05 were considered statistically significant. Results are represented as means +SEM.
Results
High TLR3 expression in cross-presenting DC
Although TLR3 mRNA expression has been measured in isolated DC subsets (33,37), the relative levels of TLR3 protein have not been analyzed at the single cell level. Therefore, we examined TLR3 protein expression in lymphocytes, macrophages, and DC subsets by intracellular FACS staining using a novel anti-mTLR3 mAb. We found that splenic T and NK cells express no TLR3, B cells express low levels, and peritoneal macrophages expressed moderate amounts of TLR3 (Fig. 1A). By contrast, TLR3 expression in DC was heterogeneous, with a minor fraction expressing high amounts of TLR3. All DC in the spleen are “resident” DC, and are derived from blood precursors. They contain three major subsets (all CD11c+); B220+ plasmacytoid DC (pDC), and CD11b+ CD8α − and CD11b− CD8α+ conventional DC (cDC) (18,38,39). As shown in Fig. 1B, the CD8α+ cDC express high amounts of TLR3, while the CD8α − subset expresses low levels of TLR3, and the pDC express none. FL-DC, similar to splenic DC, contain pDC and two subsets of cDC with the CD24high (CD8α+ equivalent (33,40)) subset expressing high amounts of TLR3 (Fig. 1C). GM-DC, which represent monocyte-derived inflammatory DC (39,41), express uniformly low levels of TLR3 (Fig. 1D).
Figure 1. High TLR3 expression in cross-presenting DC.

Cells were fixed, permeabilized, and stained for surface markers and TLR3. The same populations of cells from TLR3−/− mice were used as negative controls. (A) TLR3 expression in splenic T cells (CD3ε+), NK cells (DX5+), B cells (B220+), peritoneal macrophages (MΦ, CD11b+) and DC (CD11c+). (B-D) TLR3 expression in (B) CD11c-enriched splenic DC subsets, (C) FL-DC subsets, (D) GM-DC, and (E) CD11c-enriched LN DC subsets. All cells were electronically gated for CD11c expression, and further gated as indicated in the figure. Histograms are representative of at least 3 experiments.
In addition to resident DC subsets, LNs contain DC that migrate from peripheral tissues to the LNs via the lymphatics. These migratory DC express DEC205 (CD205), and can be subdivided on the basis of CD103 expression. The CD103− subset includes classic dermal DC and Langerhans cells, while the CD103+ subset derives from langerin+ dermal DC (38). As seen in Fig. 1E, the CD103+ subset of migratory DC expresses high levels of TLR3, whereas the CD103− migratory DC express much lower amounts. We conclude that most cDC express low levels of TLR3, but two subsets of DC, the CD8+ resident cDC, and the CD103+ DEC205+ CD8−migratory cDC, express much higher levels of TLR3 than other DC. These two DC subsets and the CD24high FL-DC uniquely cross-present antigens to CD8+ T cells (42–45). Thus, we conclude that high TLR3 expression is a phenotypic characteristic of cross-presenting DC within LN and splenic CD11c+ DC populations.
Intracellular localization of TLR3 in FL-DC
To examine where TLR3 localizes in resting DC and how activation affects its intracellular trafficking, unstimulated and pIC-stimulated FL-DC were stained for the ER marker calnexin, the early endosome marker EEA-1, and the late endosome/lysosome marker LAMP-1 and analyzed for TLR3 co-expression. In resting DC, TLR3 appeared to co-localize with the early endosome marker, and to a lesser extent, with the ER and late endosomes markers; TLR3 was not expressed on the plasma membrane or in the nucleus, as expected. (Fig. 2A). By contrast, in DC activated by pIC, almost all TLR3 co-localized with LAMP-1. We next asked whether dsRNA ONs could effectively induce TLR3 redistribution. As seen in Fig 2B, the capacity of dsRNA ONs to induce TLR3 redistribution increased with the length of the ON. Thus the 25 and 62 bp ONs failed to induce TLR3 redistribution, while the 90 and 139 bp ONs caused partial redistribution, and the 540 bp ON was as effective as pIC. These results suggest that TLR3 resides in the ER, early and late endosomes in resting DC, but following ligation with dsRNA ONs greater than 100 bp in length and pIC, TLR3 traffics to late endosomes and lysosomes.
Figure 2. Intracellular localization of TLR3 in FL-DC.
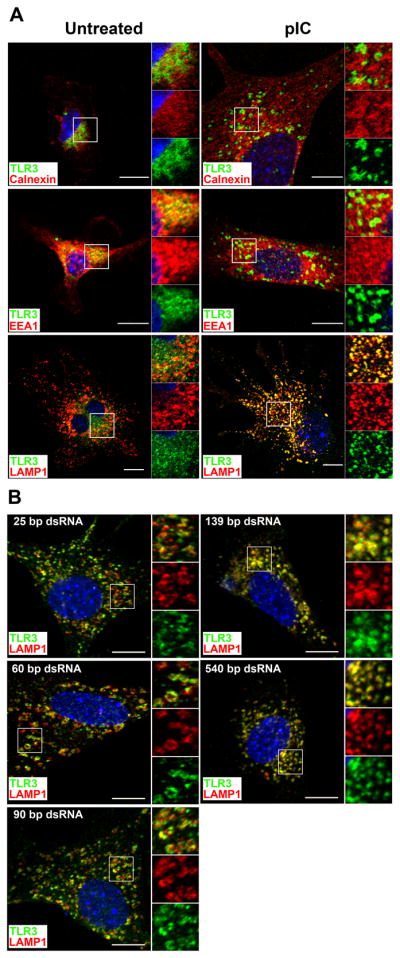
A. Unstimulated or pIC-stimulated FL-DC were fixed, permeabilized, and stained intracellularly for the ER-resident protein calnexin, early endosome marker EEA-1 or late endosome/lysosome marker LAMP-1 (red) together with anti-TLR3 (green) and examined by confocal microscopy. B. FL-DC were stimulated with 25, 60, 90, 139, or 540 bp dsRNA ONs, and co-stained for TLR3 and LAMP-1. Scale bars represent 10μm. Indicated squares are shown magnified on the right of each panel. Bottom magnification, TLR3 alone; middle magnification, vesicle marker alone; top magnification, TLR3 and vesicle markers merged.
TLR3-dependence of activation of DC by dsRNA
We next asked whether DC from spleen and bone marrow cultures would respond to dsRNA in a TLR3-dependent manner. dsRNA can trigger the release of type I interferons and inflammatory cytokines and upregulation of co-stimulatory molecules using either TLR3 or two related cytoplasmic receptors, retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5), collectively known as RIG-I like receptors (RLRs) (46,47). Because TLR3 and RLRs trigger the same types of responses, the loss of responsiveness to dsRNA in TLR3 or RLR deficient mice would indicate that the deleted receptor alone mediates the response to dsRNA. We expected that the DC response to exogenous ONs would be predominantly TLR3 dependent, since exogenously added dsRNA ONs are taken into endosomes, the intracellular location of TLR3, rather than into the cytoplasm where RLRs reside. To test this, spleen cells, FL-DC and GM-DC were treated with medium, dsRNA (540 bp), or pIC, and after 24 hr, CD11c+ cells were examined for up-regulation of the costimulatory molecule, CD86. As shown in Fig. 3, dsRNA and pIC induced CD86 expression in splenic DC and in FL-DC from WT mice but not from TLR3−/− mice, as expected. By contrast, the dsRNA ON failed to activate GM-DC from either WT or TLR3−/− mice, but surprisingly, pIC activated GM-DC from both WT and TLR3−/− mice, suggesting that RLRs might be responsible for the GM-DC response to pIC. It has been shown that MDA-5, but not RIG-I responds to pIC (46,47). We therefore asked whether the induction of CD86 by pIC required MDA-5. As seen in Fig 3, deletion of MDA-5 abrogated the responsiveness of GM-DC, but not splenic DC or FL-DC to pIC, suggesting that pIC enters the cytoplasm in GM-DC, where it activates MDA-5. Thus FL-DC and splenic DC respond differently from GM-DC to dsRNA and pIC stimulation, and pIC can activate at least one type of DC independently of TLR3.
Figure 3. TLR3 dependence of activation of splenic and bone marrow derived DC.

CD86 expression of WT, MDA5−/− and TLR3−/− splenic DC, FL-DC and GM-DC after 24 h treatment with dsRNA (540 bp), pIC or medium alone (Ctrl). Data shown are mean +SEM of triplicate DC cultures and are representative of four independent experiments.
Dependence of DC activation on dsRNA ON size
In view of the large differences in TLR3 expression between DC subsets, we asked how different subsets would respond to dsRNA. For these experiments CD24high and CD11bhigh FL-DC were isolated by cell sorting and treated with 25, 60, 90, 139, and 540 bp dsRNA ONs or pIC at concentrations predetermined to lie at or near the plateau on the dose response curve. The CD24high/TLR3high FL-DC subset responded strongly to the dsRNA stimuli, and the responses increased with dsRNA length and required TLR3 (Fig 4 left panels). The 60 bp ON was sufficient to up-regulate CD86 expression but larger dsRNA ONs were required to induce IL-12p40, TNF-α, IL-6, and IFN-β. The CD11bhigh/TLR3low subset of FL-DC (Fig 4 right panels) constitutively expressed high levels of CD86 (normalized in Fig 4) and TNF-α which were marginally increased by dsRNA. These cells gave a very weak IL-12p40 response to dsRNA but did secrete IFN-β and IL-6 in a TLR3-dependent manner, indicating that in spite of their low TLR3 expression, the CD11bhigh DC are able to respond to dsRNA. Taken together, these data show that qualitatively different responses are generated by treating DC with different sized dsRNA ONs, and that the responses vary between DC subsets.
Figure 4. DC activation increases with dsRNA length.

FACS-sorted CD24high and CD11bhigh FL-DC subsets from WT and TLR3−/− mice were stimulated with 25 bp, 60 bp, 90 bp, 139 bp and 540 bp dsRNA, with pIC or were left untreated (Ctrl). After 24 h, CD86 upregulation and cytokine production were determined. pIC and dsRNA ON concentrations were at or near the plateau on dose response curves. Values depicted are means +SEM from 3 independent experiments. * designates significant differences compared to untreated control (*P<0.05; **P<0.01; ***P<0.001), whereas † indicates significant differences between WT and TLR3−/− DC (†P<0.05; ††P<0.01; †††P<0.001).
Because dsRNA ONs stimulate CD86 and cytokine expression in CD24high/TLR3high FL-DC, we asked whether ONs could induce FL-DC to efficiently cross-present exogenous antigens to CD8+ T cells. FL-DC were incubated with OVA and dsRNA ONs, then cultured with naïve CD8+ T cells from OT-I mice that express a TCR transgene specific for OVA peptide bound to H-2kb. Fig. 5 shows that DC treated with OVA alone or OVA plus 25 or 62 bp dsRNA were unable to induce proliferation of the OT-I CD8+ T cells. However ONs ≥ 90 bp and pIC were able to mature WT, but not TLR3−/− DC into competent cross-presenting APC capable of activating T-cells in vitro and the antigen presenting capacity of DC increased with dsRNA length. As a control, the TLR9 ligand CpG ODN was able to induce maturation of both WT and TLR3−/− DC, indicating that the TLR3−/− DC had not lost the capacity to mature in response to other stimuli. We conclude that dsRNA ONs induce DC maturation by processes that require TLR3 and depend upon dsRNA length.
Figure 5. dsRNA size-dependently induces cross-presentation.

WT and TLR3−/− FL-DC were incubated overnight with OVA alone (Ctrl) or OVA plus dsRNA ONs with specified length, pIC, or CpG, then co-cultured with CFSE-labeled OT-I CD8+ T cells. Proliferation was detected by CFSE dilution. Panel (A) shows representative histograms of proliferating T cells induced by DC that had been treated with OVA and the indicated stimulants. (B) Shows number of proliferating T cells; n=6 from two independent experiments, means + SEM * designates significant differences compared to OVA alone control (***P<0.001), whereas † indicates significant differences between T cells stimulated with WT and TLR3−/− DC (††P<0.01; †††P<0.001).
dsRNA ONs induce antigen-specific CTL in vivo
Because dsRNA ONs drive DC maturation in vitro, we asked whether the same ONs could serve as adjuvants for the generation of antigen-specific CD8+ T cells in vivo. Mice were immunized with OVA and dsRNA ONs or pIC, and seven days later CD8+ cells from draining LNs were examined for OVA specificity by pentamer staining. As seen in Fig. 6A, dsRNA ONs served as effective adjuvants for the generation of OVA-specific T cells. No expansion of antigen-specific T cells was seen in the absence of dsRNA, and even the smallest ON tested (90 bp) showed significant adjuvant activity. The 540 bp ON was as effective as pIC, and both exhibited some TLR3-independent activity. OVA-immunized mice were also tested for the generation of CTL activity, using an in vivo cytotoxicity assay (Fig. 6B, C). We observed that all ONs tested and pIC were effective at inducing OVA-specific cytotoxicity. The 139 bp ON produced a near optimal CTL response that was completely TLR3-dependent, but pIC and 540 bp dsRNA were able to induce CTL responses by both TLR3-dependent and TLR3-independent pathways.
Figure 6. In vivo CTL induction by dsRNA.

WT and TLR3−/− mice were immunized with OVA alone (Ctrl) or OVA plus the indicated dsRNA ON or pIC. (A) Percentages of OVA-specific (pentamer positive) CD8+ T cells seven days after immunization. (B) Representative histograms showing in vivo cytotoxicity assay. Six days after immunization mice were injected with OVA peptide- coated target cells labeled with high amounts of CFSE and control cells labeled with low amounts of CFSE. One day later cytotoxicity was detected as a loss in CFSE bright cells relative to CFSE dull cells. (C) Percent specific lysis. Data are from at least 3 mice per group (mean + SEM). * designates significant differences compared to control (**P<0.01; ***P<0.001), whereas † indicates significant differences between WT and TLR3−/− mice (†P<0.05; †††P<0.001).
Protection against influenza virus infection
Finally we investigated whether dsRNA ONs could improve the effectiveness of an experimental influenza virus subunit vaccine as a first step in their development as immunization adjuvants. As the immunogen, we used (rNP), which is weakly immunogenic but highly conserved between virus strains (48). Mice were immunized and boosted four weeks later with rNP either alone or in the presence of pIC or dsRNA ONs, 90, 139, or 540 bp in length. Two weeks after the boost, mice were challenged with 10 LD50 of H1N1 influenza virus, strain A/PR/8/34, and survival was monitored thereafter. As seen in Fig. 7, no mice survived past nine days in the groups that were either not immunized, or that were given 540 bp dsRNA alone. Immunization with rNP alone prolonged survival slightly. By contrast, long-term survival was seen when mice were immunized with rNP using either pIC or dsRNA ONs as adjuvants. Adjuvant effectiveness increased with dsRNA length (Fig. 7B) and the 540 bp ON was about as effective as pIC (Fig 7A). Thus, dsRNA ONs can provide protection against infection when administered with a viral antigen.
Figure 7. dsRNA ONs serve as effective adjuvants for an influenza vaccine.

WT mice were primed and boosted with rNP plus pIC or 540 bp dsRNA (A) or rNP plus 90, 139 or 540 bp dsRNA ONs (B). Controls (dashed lines) include untreated naïve mice, mice treated with rNP alone and mice treated with 540 bp dsRNA alone. Three weeks after the boost mice were challenged with 10 LD50 of influenza virus and survival was monitored thereafter. Asterisks indicate a significant difference in the percentage of survival between naïve and immunized mice (*P<0.05; **P<0.01; ***P<0.001).
Discussion
Recent studies have demonstrated that TLR3 forms dimeric complexes with 45 bp segments of dsRNA and that single TLR3 dimers formed with a 48 bp dsRNA ON are capable of activating transfected cells expressing high amount of TLR3 (27). A goal of the current study is to determine how TLR3:dsRNA complexes, formed with dsRNA ONs of varying lengths, function in primary cells and to translate these findings into the development of well-defined TLR3-dependent adjuvants. Here we examined the activation of DC by dsRNA ONs because this response is key for inducing acquired anti-viral immunity. We have previously shown that the interaction of TLR3-ECD protein with dsRNA is highly dependent upon dsRNA length and TLR3-ECD concentration (27) which implies that the ability of a specifically sized dsRNA ON to bind TLR3 and activate a cell is governed in part by the membrane density of TLR3 in the endosomes. Thus, the large differences in TLR3 expression levels that we observed between DC subsets are predicted to play an important role in determining their responsiveness to dsRNA ONs. Two subsets of DC that expressed the highest levels of TLR3 are the resident CD8+ cDC and the migratory CD103+ dermal DC. These two subsets are known to cross-present exogenous antigens to CD8+ T cells (38,43–45,49,50) and are related by requiring Batf3 and IRF8 for their development (50). The human DC subset equivalent to murine CD8+ cDC has recently been identified and shown to express TLR3 mRNA (51–54). However, it is not known whether this human DC subset expresses TLR3 protein at the distinctively high levels that we observed in mice. It has been shown that CD8+ DC phagocytose virally infected cells and undergo maturation in a TLR3/dsRNA-dependent process. These DC then cross-present viral antigens to CD8+ T cells, inducing them to proliferate and differentiate into virus-specific CTL (30). Thus, the high expression of TLR3 in cross-presenting DC represents one way in which a viral molecular pattern can induce a virus-specific response.
As expected from their high TLR3 expression, cross-presenting DC were highly responsive to dsRNA ONs in vitro. ONs as short as 60 bp induced phenotypic maturation of the CD24high cross-priming subset of FL-DC in a TLR3-dependent manner, and 90 bp dsRNA matured FL-DC into potent APC that activated CD8+ T cells. By contrast, GM-DC, which express low levels of TLR3, were not activated by a 540 bp dsRNA ON but were activated by pIC. However, activation of GM-DC by pIC was mediated by MDA-5, a cytosolic receptor for dsRNA, and not by TLR3. This suggests that pIC was taken into the cytoplasm of GM-DC, whereas the 540 bp ON was not, or alternatively, both pIC and the ON entered the cytoplasm, but only pIC activated MDA-5 (55,56). In vivo, the 90 bp dsRNA ON was capable of inducing antigen-specific CD8+ T cell proliferation and CTL generation. This was most likely mediated by the cross-presenting DC subsets because previous studies using mice deficient in CD8α+ and CD103+ DC showed that these subsets were essential for cross presentation of exogenous antigens to CD8+ T cells (49,50). While the 90 and 139 bp ONs induced antigen-specific CD8+ T cell expansion and CTL generation in a TLR3-dependent manner, pIC and to a lesser extent 540 bp dsRNA operated in part through a TLR3-independent pathway. This suggests that long pieces of dsRNA and pIC may exert a qualitatively different effect on the immune system than shorter dsRNA ONs.
dsRNA ONs have several properties that render them advantageous as adjuvants. First, dsRNA is stable, because it is resistant to digestion by the ubiquitous RNAses that rapidly degrade single-stranded RNA, a ligand for TLRs 7 and 8 (57,58). dsRNA ONs are soluble, homogeneous molecules that can be manufactured in a consistent fashion. By contrast, other TLR3-stimulating adjuvants including pIC, polyI:C12U (59), poly I:CLC (60), and PIKA (61) are heterogeneous and may vary between batches in both chemical structure and biological function, particularly with regard to their capacity to promiscuously activate non-TLR3 dependent pathways with unpredictable consequences. The efficacy of a dsRNA ON as an activator of TLR3 depends upon its length and not its nucleotide sequence, in contrast to CpG ODNs that are being tested as TLR9-based adjuvants (62). Thus, it may be possible to fine-tune the adjuvant properties of dsRNA ONs simply by varying their lengths.
Adjuvant design is especially important for developing effective subunit protein-based vaccines. Virus subunit proteins are easy to produce, stable during storage, and safe relative to live attenuated or killed virus vaccines, but these proteins are poorly immunogenic and therefore require adjuvants to generate protective immunity (19,20). To test whether dsRNA ONs could serve as effective adjuvants in an experimental subunit vaccine, we immunized mice with rNP with or without dsRNA ONs or pIC and then challenged the mice with influenza virus. NP varies much less between influenza subtypes than do the major antibody epitopes on the hemaglutanin and neuraminidase subunits. For this reason, a strong anti-NP response could be cross-protective against multiple influenza A subtypes (63,64). However, NP is a poor immunogen when it is administered as a recombinant protein rather than expressed in situ from plasmid or viral vectors (48). We found that rNP alone did not confer protection against lethal influenza infection but that co-administration of rNP with dsRNA ONs enhanced survival and that protection increased with the length of the dsRNA. The level of protection induced by the 540bp ON was similar to pIC. Thus dsRNA ONs can serve as effective adjuvants in an experimental vaccination system.
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute and by a NIH/FDA Intramural Biodefense award from the National Institute of Allergy and Infectious Diseases.
We thank Dr. Daniela Verthelyi (FDA Center for Drug Evaluation and Research, Rockville, MD, USA) for TLR3−/− mice and Dr. Marco Colonna (Washington University School of Medicine, St Louis, MO, USA) for MDA5−/− mice. We also thank Dr. Chia Yun Lo, Dr. Julia Ann Misplon (Center for Biologics Evaluation and Research, FDA, Rockville, Maryland, USA) and Ms. Lana Stepanian (Bioqual Inc, Rockville, MD, USA) for their expertise in immunizing and handling mice. We thank Dr. Michael Kruhlak (Experimental Immunology Branch, Center for Cancer Research, NCI, NIH, Bethesda, Maryland, USA) for his help with confocal imaging, image processing and figure preparation.
Abbreviations in this paper
- DC
dendritic cell
- cDC
conventional DC
- ECD
extracellular domain
- LN
lymph node
- MDA5
melanoma differentiation-associated gene 5
- ODN
oligodeoxnucleotide
- ON
oligonucleotide
- PAMP
pathogen-associated molecular patterns
- pDC
plasmacytoid DC
- pIC
polyI:polyC
- rNP
recombinant influenza A nucleoprotein
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
Reference List
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007;37(Suppl 1):S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 3.Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2009;9:15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- 4.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 5.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 6.Segura E, Villadangos JA. Antigen presentation by dendritic cells in vivo. Curr Opin Immunol. 2009;21:105–110. doi: 10.1016/j.coi.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinman RM. The control of immunity and tolerance by dendritic cell. Pathol Biol (Paris) 2003;51:59–60. doi: 10.1016/s0369-8114(03)00096-8. [DOI] [PubMed] [Google Scholar]
- 9.Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, Iyoda T, Ravetch J, Dhodapkar M, Inaba K, Nussenzweig M. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. 2003;987:15–25. doi: 10.1111/j.1749-6632.2003.tb06029.x. [DOI] [PubMed] [Google Scholar]
- 10.Granucci F, Zanoni I, Ricciardi-Castagnoli P. Central role of dendritic cells in the regulation and deregulation of immune responses. Cell Mol Life Sci. 2008;65:1683–1697. doi: 10.1007/s00018-008-8009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lutz MB, Kurts C. Induction of peripheral CD4+ T-cell tolerance and CD8+ T-cell cross-tolerance by dendritic cells. Eur J Immunol. 2009;39:2325–2330. doi: 10.1002/eji.200939548. [DOI] [PubMed] [Google Scholar]
- 12.Hemmi H, Akira S. TLR signalling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. doi: 10.1159/000086657. [DOI] [PubMed] [Google Scholar]
- 13.Benko S, Magyarics Z, Szabo A, Rajnavolgyi E. Dendritic cell subtypes as primary targets of vaccines: the emerging role and cross-talk of pattern recognition receptors. Biol Chem. 2008;389:469–485. doi: 10.1515/bc.2008.054. [DOI] [PubMed] [Google Scholar]
- 14.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reis e Sousa Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin Immunol. 2004;16:21–25. doi: 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 17.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 18.Naik SH. Demystifying the development of dendritic cell subtypes, a little. Immunol Cell Biol. 2008;86:439–452. doi: 10.1038/icb.2008.28. [DOI] [PubMed] [Google Scholar]
- 19.Kwissa M, Kasturi SP, Pulendran B. The science of adjuvants. Expert Rev Vaccines. 2007;6:673–684. doi: 10.1586/14760584.6.5.673. [DOI] [PubMed] [Google Scholar]
- 20.Wilson-Welder JH, Torres MP, Kipper MJ, Mallapragada SK, Wannemuehler MJ, Narasimhan B. Vaccine adjuvants: current challenges and future approaches. J Pharm Sci. 2009;98:1278–1316. doi: 10.1002/jps.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichlmair A, Reis e Sousa Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Botos I, Liu L, Wang Y, Segal DM, Davies DR. The toll-like receptor 3:dsRNA signaling complex. Biochim Biophys Acta. 2009;1789:667–674. doi: 10.1016/j.bbagrm.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 27.Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci U S A. 2008;105:258–263. doi: 10.1073/pnas.0710779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diebold SS, Schulz O, Alexopoulou L, Leitner WW, Flavell RA, Reis e Sousa Role of TLR3 in the immunogenicity of replicon plasmid-based vaccines. Gene Ther. 2009;16:359–366. doi: 10.1038/gt.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 31.Homan ER, Zendzian RP, Schott LD, Levy HB, Adamson RH. Studies on poly I:C toxicity in experimental animals. Toxicol Appl Pharmacol. 1972;23:579–588. doi: 10.1016/0041-008x(72)90098-1. [DOI] [PubMed] [Google Scholar]
- 32.Robinson RA, V, DeVita T, Levy HB, Baron S, Hubbard SP, Levine AS. A phase I–II trial of multiple-dose polyriboinosic-polyribocytidylic acid in patieonts with leukemia or solid tumors. J Natl Cancer Inst. 1976;57:599–602. doi: 10.1093/jnci/57.3.599. [DOI] [PubMed] [Google Scholar]
- 33.Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O’Keeffe M, Shao QX, Chen WF, Villadangos JA, Shortman K, Wu L. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 34.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 36.Ingulli E. Tracing tolerance and immunity in vivo by CFSE-labeling of administered cells. Methods Mol Biol. 2007;380:365–376. doi: 10.1007/978-1-59745-395-0_23. [DOI] [PubMed] [Google Scholar]
- 37.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 38.Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol. 2009;10:1237–1244. doi: 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- 39.Steinman RM, Idoyaga J. Features of the dendritic cell lineage. Immunol Rev. 2010;234:5–17. doi: 10.1111/j.0105-2896.2009.00888.x. [DOI] [PubMed] [Google Scholar]
- 40.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234:45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol. 2007;179:7577–7584. doi: 10.4049/jimmunol.179.11.7577. [DOI] [PubMed] [Google Scholar]
- 42.Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR. Cutting edge: conventional CD8 alpha+ dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- 43.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, Brooks AG, Heath WR. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 45.Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev. 2010;234:18–31. doi: 10.1111/j.0105-2896.2009.00870.x. [DOI] [PubMed] [Google Scholar]
- 46.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Epstein SL. Control of influenza virus infection by immunity to conserved viral features. Expert Rev Anti Infect Ther. 2003;1:627–638. doi: 10.1586/14787210.1.4.627. [DOI] [PubMed] [Google Scholar]
- 49.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edelson BT, WKC, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, Bhattacharya D, Stappenbeck TS, Holtzman MJ, Sung SS, Murphy TL, Hildner K, Murphy KM. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, Vulink AJ, Hart DN, Radford KJ. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010 doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8{alpha}+ dendritic cells. J Exp Med. 2010 doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le MA, Faure F, Donckier V, Sancho D, Cerundolo V, Bonnet D, Reis e Sousa Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8{alpha}+ dendritic cells. J Exp Med. 2010 doi: 10.1084/jem.20092618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, Henn V, Kloetzel PM, Gurka S, Kroczek RA. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010 doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 56.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 58.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 59.Jasani B, Navabi H, Adams M. Ampligen: a potential toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine. 2009;27:3401–3404. doi: 10.1016/j.vaccine.2009.01.071. [DOI] [PubMed] [Google Scholar]
- 60.Poast J, Seidel HM, Hendricks MD, Haslam JA, Levy HB, Baron S. Poly I:CLC induction of the interferon system in mice: an initial study of four detection methods. J Interferon Cytokine Res. 2002;22:1035–1040. doi: 10.1089/107999002760624260. [DOI] [PubMed] [Google Scholar]
- 61.Lau YF, Tang LH, McCall AW, Ooi EE, Subbarao K. An adjuvant for the induction of potent, protective humoral responses to an H5N1 influenza vaccine with antigen-sparing effect in mice. J Virol. 2010 doi: 10.1128/JVI.00596-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCluskie MJ, Krieg AM. Enhancement of infectious disease vaccines through TLR9-dependent recognition of CpG DNA. Curr Top Microbiol Immunol. 2006;311:155–178. doi: 10.1007/3-540-32636-7_6. [DOI] [PubMed] [Google Scholar]
- 63.Epstein SL, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, Nabel GJ. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine. 2005;23:5404–5410. doi: 10.1016/j.vaccine.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 64.Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, Gromkowski SH, Deck RR, DeWitt CM, Friedman A. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–1749. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]