

Abstract
Stem cell therapy has the potential to regenerate injured tissue. For stem cells to achieve their full therapeutic potential, stem cells must differentiate into the target cell, reach the site of injury, survive, and engraft. To fully characterize these cells, evaluation of cell morphology, lineage specific markers, cell specific function, and gene expression must be performed. To monitor survival and engraftment, cell fate imaging is vital. Only then can organ specific function be evaluated to determine the effectiveness of therapy. In this review, we will discuss methods for evaluating the function of transplanted cells for restoring the heart, nervous system, and pancreas. We will also highlight the specific challenges facing these potential therapeutic areas.
1. Introduction
Stem cell therapy is a novel method to replace injured tissue with healthy cells capable of restoring the function of damaged organs. Recent studies have shown the potential of this technique to regenerate the heart, nervous system, and pancreas following injury. Although preliminary results have been promising, clinical trials are either not yet underway or have had inconsistent or unconvincing results (1, 2).
Stem cells need to reach the site of injury, survive, and engraft in order to restore function. A previous study has shown that cells delivered systemically can become entrapped in the lung or microvasculature (3). In addition, poor circulation can limit effective delivery. If cells are injected directly into tissue, the ideal site would be near the area of tissue damage, which can often be difficult to identify. Despite successful delivery, as many as 90%of transplanted cells typically die after implantation, as a result of physical stress, inflammation, hypoxia, or immunogenic rejection (4, 5). Tissue specific differentiation has also been challenging. For example, after delivery, adult progenitor cells often fail to transdifferentiate into target tissues (6).
The use of human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) as alternative sources for cell therapy can bypass the problems associated with transdifferentiation. Because these cells can be differentiated into appropriate cell types in vitro, they can be administered directly to the patient. However, hESCs and iPSCs have a low efficiency of directed in vitro differentiation into therapeutic cell types, presenting an additional obstacle for clinical implementation (7, 8).
Thus, in order to fully evaluate whether transplanted stem cells actually improve function, it will be important to demonstrate that these cells can (1) differentiate into tissue specific cells, (2) survive and engraft in the target tissue, as well as (3) restore function and alleviate disease. A schematic of the key steps in evaluating the lineage, fate and function of transplanted hESCs and/or iPSCs is shown in Figure 1. In this review, we will discuss the advantages and limitations of current techniques used to evaluate the functional effects of transplanted stem cells.
Figure 1.

Schematic of the key steps in evaluating the lineage, fate, and function of transplanted stem cells for the treatment of cardiovascular disease. In this schematic, human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) undergo differentiation into cardiomyocytes in vitro prior to injection into the mouse heart. Abbreviations: MRI, magnetic resonance imaging
2. Confirmation of Cell Specific Differentiation and Lineage Commitment
Differentiation dramatically changes a cell’s size, shape, membrane potential, metabolic activity, and responsiveness to signals. These changes are largely due to highly controlled modifications in gene expression and rarely involve changes to the actual genome. Thus, different cell types can have very different characteristics despite having the same DNA sequence. Whether stem cells differentiate in vivo after transplantation (e.g., adult progenitor cells) or in vitro prior to transplantation (e.g., ESCs and iPSCs), it is important to confirm cell specific differentiation and lineage commitment. If not performed adequately, cells incapable of providing the appropriate function will be given to patients. For example, although early attempts to derive β-islet cells directly from ESCs appeared successful in generating insulin-containing cells (9, 10) in mice with streptozotocin-induced diabetes, further studies revealed that these cells did not produce insulin or respond to glucagon or glucose (11, 12) in vitro. The confirmation of stem cell differentiation into the target cell type can be performed by demonstrating appropriate 1) cell morphology, 2) lineage specific markers, 3) gene expression, and 4) cell function.
2.1. Cell morphology
Specific cell types have distinct morphological or functional forms, which can be examined under a fluorescent or electron microscope. For example, murine ESC-derived cardiomyocytes have an appearance similar to neonatal cardiomyocytes when cultured in vitro (13). Specifically, fully mature cardiomyocytes are elongated, mononucleated, and rod shaped with well-organized myofibrils and sarcomeres as well as nascent intercalated disks, facia adherens, desmosomes, and gap junctions (14). In contrast, murine iPSC-derived neurons have a cell body with extending dendrites (15), whereas hESC-derived pancreatic β-islet cells appear as cells with an eccentric nuclei and cytoplasmic insulin granules (16). Relying on cell morphology alone, however, is not sufficient because cells must be functional (e.g., be responsive to appropriate signaling and be able to perform cell specific function) in order to be therapeutic.
2.2. Lineage specific markers
Lineage specific markers on the cell membrane (“surface receptors”) and in the cytoplasm (“intracellular proteins”) can be used to identify differentiated cells. Cell surface receptors are specialized proteins that can selectively bind or adhere to signaling molecules. Intracellular proteins, on the other hand, are involved in cell specific function. Both these proteins differ in their structure and affinity for different molecules (i.e., signaling molecules and antibodies) and thus can be used to distinguish different cell types. Lineage specific markers commonly used to identify cells in the heart, nervous system and pancreas are listed in Table 1 (14–17).
Table 1.
Lineage Markers for the Heart, Nervous System, and Pancreas
| Cardiac Markers | Significance |
|---|---|
| Nkx2.5 | Transcription factor expressed by embroid bodies |
| GATA-4 | Transcription factor which regulates myocardial differentiation |
| α1CaCH | α1 subunit of the L-type calcium channel which indicates early differentiation |
| Atrial natiuretic factor (ANF) | A protein hormone which is expressed by cardiac embroid bodies |
| Calsequestrin (CSQ) | A calcium binding protein of the sarcoplasmic reticulum |
| Connexin | Gap junction proteins including Cx40, Cx 43, Cx45, expressed by cardiospheres |
| Phospholamban | Integral membrane protein that regulates the Ca2+ pump in cardiac muscle |
| Sarcomeric proteins | Protein components of the sarcomere, the basic unit of the myofibril, which indicate the presence of differentiated cardiomyocytes. These include myosin heavy chain (α,β), myosin light chain (MLC-2v), actin, titin, troponin T, tropomyosin, desmin, and M protein. |
| Markers of Nervous System | Significance |
| CD133 | Cell-surface protein that identifies neural stem cells |
| Glial fibrillary acide protein (GFAP) | Protein specifically produced by astrocyte |
| Microtubulue-associated protein-2 (MAP-2) | Dendrite-specific MAP; protein found specifically in dendritic branching of neuron |
| Myelin basic protein (MPB) | Protein produced by mature oligodendrocytes; located in the myelin sheath surrounding neuronal structures |
| Nestin | Intermediate filament structural protein expressed in primitive neural tissue |
| Neural tubulin | Important structural protein for neuron; identifies differentiated neuron |
| Neurofilament (NF) | Important structural protein for neuron; identifies differentiated neuron |
| Noggin | A neuron-specific gene expressed during development |
| O4 | Cell-surface marker on immature, developing oligodendrocytes |
| O1 | Cell-surface marker that characterizes mature oligodendrocytes |
| Synaptophysin | Neuronal protein located in synapses; indicates connections between neurons |
| Tau | Type of MAP; helps maintain structure of axon |
| Pancreatic Markers | Significance |
| Cytokeratin 19 (CK 19) | Expressed by specific pancreatic epithelial cells that are progenitors for islet and ductal cells |
| Glucagon | Expressed by alpha-islet cell |
| Insulin | Expressed by beta-islet cell |
| Insulin-promoting factor-1 (PDX-1) | Transcription factor expressed by beta-islet cell |
| Nestin | Structural filament protein indicative of progenitor cells |
| Pancreatic polypeptide | Expressed by gamma-islet cell |
| Somatostatin | Expressed by delta-islet cell |
Adapted from the National Institutes of Health, Regenerative Medicine, Appendix E: Stem cell Markers (17)
In order to identify cells, a fluorescent tag is attached to a signaling molecule or antibody with special affinity to a lineage specific marker, providing exquisite binding specificity. Many fluorescent tags are available that emit light with different colors and intensities. Cells labeled by fluorescent markers can be sorted in vitro and in vivo using fluorescent activated cell sorting (FACS) and identified ex vivo by immunofluorescence staining.
FACS, however, has some limitations, which may compromise its accuracy. A potential problem that limits qualitative accuracy is that stem cells may fuse with host endogenous cells, making them appear as if they have differentiated (18, 19). Problems for correct quantitative interpretation include contamination from other fluorescent tags and non-specific binding. Ideally, each fluorescent tag should produce an image that is not contaminated by the emission of other tags. However, in practice, this rarely occurs because spectral overlap between dyes is common. A narrow-band of excitation or emission filters can be used and positioned to have minimal spectral overlap, but this can compromise the amount of signal allowed to pass through. Another common issue is non-specific binding (“background staining”), which can reduce contrast, making it difficult to find weakly expressing signals. Although indirect labeling can boost signal intensity, it has been associated with extensive non-specific binding. Direct instead of indirect labeling can thus be used to minimize these effects. These potential limitations require analysis of gene expression to confirm the presence of cell specific proteins.
2.3. Gene expression analysis
During cell specific differentiation, certain genes are upregulated and others downregulated. For example, hESCs initially express pluripotent genes, including Oct4, Sox2, NANOG, Crypto and LCK (13, 20) during in vitro differentiation. Once hESCs differentiate into the beating embroid body stage, they express high levels of mesodermal regulators (i.e., Twist1, Tbx5, and Meox), as well as enriched levels of cardiogenic specific regulators (i.e., IsL1, Hand, GATA4-5-6, MEF2C), and cardiac specific myosins (13). Fully differentiated cardiomyocytes, however, downregulate early mesodermal genes and express more cardiac specific structural genes (13).
Conversely, differentiation of murine neural progenitor cells in vitro involves five steps: 1) cessation of proliferation, 2) attachment of the neurosphere to extracellular matrix, 3) detachment of cells from the neurosphere cluster, 4) migration of cells away from sphere, and 5) differentiation into different cell types (21). For neuron specific differentiation, there is upregulation of Nsg2, chomogranin B, synataphilin, reticulon, and RIKEN cDNA 4930565N16 gene homologous to rat Tomosyn. For astrocyte specific differentiation, there is upregulation of S100B but downregulation of deiodinase iodothyronine type II. For oligodendrocyte-specific differentiation, there is upregulation of myelin components.
For differentiation into pancreatic islet cells in vitro, hESCs pass through the following stages: 1) mesendoderm (i.e., upregulation of BRA, FGF4, WNT3, and NCAD), 2) definitive endoderm (i.e., upregulation of SOX17, CER, FOXA2, and CXCR4), 3) primitive gut tube (i.e., upregulation of HNF1B and HNF-4A), 4) posterior foregut (i.e., upregulation of PDX1, HNF6, PROX1, and SOX 9), and 5) pancreatic endoderm (i.e., upregulation of NKX6-1, PF1A, NGN3 and NKX2-2) (16).
These unique patterns of “gene expression” can be detected by reporter genes, quantitative real time polymerase chain reaction (RT-PCR), and microarray analysis. Reporter genes can be used to follow stem cell differentiation in animal models (22). In this technique, specific reporter gene constructs are transferred to cells via viral or non-viral transfection. For example, a reporter gene that expresses a fluorescent protein can be inserted into the stem cell (22–24). The gene is only activated or “reports” when cells are undifferentiated and is turned off once they become specialized. Once activated, the gene directs the stem cells to produce a fluorescent color, which can be sorted by FACs. The feasibility of this approach has been shown in a previous study (24), which demonstrated that ESCs transfected with a reporter gene construct differentiated into endothelial cells that were detectable by FACS analysis.
This technique may address the question of whether stem cells truly differentiate or merely fuse with host endogenous cells. If stem cells fuse with the host cells, it is possible they could mimic features of endogenous cells. Tissue reporter gene expression would not change unless nuclear fusion occurs which has been reported in up to 1% of cells (25).
Both quantitative RT-PCR and DNA microarray analysis can measure levels of gene expression. Although quantitative RT-PCR can detect whether a single gene is being expressed, it rapidly becomes impractical if many genes within the sample are being studied. Using DNA microarray analysis, the expression of many genes can be measured at once, enabling efficient evaluation of cell specific differentiation. Since an array can contain thousands of probes, a microarray experiment can perform many genetic tests in parallel, dramatically accelerating gene expression profiling.
Despite the advantage of efficiently screening a wide array of genes and its relatively low cost, DNA microarray analysis has its limitations. First, only genes on the microarray chip will be tested. Second, DNA microarrays reveal only relative gene expression; thus, quantitative RT-PCR must be performed to provide quantitative information. In addition, the amount and quality of RNA in the sample remains a challenge. DNA microarray technology requires a relatively large amount of RNA to produce adequate signal-to-noise ratios, especially when levels of gene expression are low. Fluorescent detection requires 10 μg of total RNA (equivalent to a million cells), whereas radiation detection requires 0.1 μg of total RNA (equivalent to ten thousand cells). RT-PCR thus may be required prior to DNA microarray analysis to amplify the amount of RNA available. This creates additional opportunities for compromising RNA integrity, which is critical for maintaining the accuracy of DNA microarray data. mRNA also degrades very quickly, which can affect data quality if the sample is not processed correctly. Because of the many factors that can disrupt data integrity, DNA microarray analysis is often performed in triplicate, followed by RT-PCR to confirm results.
2.4. Cell specific function
Although genetic expression analysis is a valuable tool to evaluate stem cell specific differentiation, it is still not sufficient to fully inform us about gene function. Functional testing is required for a complete analysis of the effectiveness of stem cell therapy. The function of differentiated stem cells can be characterized by their electrophysiological properties, which regulate cell specific activities. The “patch clamp” technique is used to study excitable cells such as cardiomyocytes, neurons, and pancreatic β-islet cells. Patch clamp recording uses an electrode that is sealed to the cell membrane surface, enabling the electronic isolation of currents to be measured across the membrane patch with little competing noise. The patch clamp technique can be used to study ion channels and action potentials.
Ion channels are pore-forming proteins that help establish and control the small voltage currents across the plasma membrane of all living cells by regulating the flow of ions across the cell membrane. Ion channels are specific to different cell types and their activity changes as cells mature. Ion channels, thus, generate cell and age specific action potentials (14, 26–28). For example, as murine iPSCs differentiate into fully mature cardiomyocytes in vitro, the density of voltage gated Ca2+ and K+ channels increases, whereas the number of Na+ channels remains the same (26). As cardiomyocytes mature, most cells display ventricular action potentials with fewer atrial action potentials. In contrast, as murine ESC-derived neurocytes mature in vitro, the density of Na+ channels increases significantly, causing an alteration in the steady-state activation and inactivation. These changes result in enhanced excitability and overshooting action potentials in response to depolarization in fully differentiated and functional neurons (27). Interestingly, as murine ESC-derived β-islet cells mature in vitro, they display a burst of activity when exposed to glucose, a response which is regulated by ATP-modulated K+ channels (28). Thus, through modulation of the cell membrane potential, ion channels enable rapid changes in cells that result in a variety of biological processes including spontaneous contraction in cardiomyocytes (14), “neurotransmitter-activated” conduction across neural synapses (15), and pancreatic β-islet cell insulin release in response to glucose (28).
3. Imaging Survival and Engraftment
In addition to appropriate cell differentiation, transplanted stem cells must reach the site of injury, survive, and engraft in target tissue. Several techniques have been used to identify, localize, and monitor stem cells after delivery (29–31). Before the advent of molecular imaging, determination of cell fate relied on post-mortem histological analysis, performed at pre-determined time points following cell transplantation. Molecular imaging now enables the in vivo tracking of stem cell fate longitudinally by labeling cells with specific markers, including iron particles, radionuclides, and reporter genes. Labeled stem cells can be visualized non-invasively using multiple imaging modalities, such as magnetic resonance imaging (MRI) (32, 33), single photon emission tomography (SPECT) (34), positron emission tomography (PET) (35, 36), and bioluminescent imaging (BLI) (5). A schematic of these approaches can be found in Figure 2. Each imaging technique has its advantages and disadvantages as shown in Table 2 (37). An alternative strategy to monitor stem cell fate is imaging programmed cell death by labeling apoptosis markers. which will be discussed later in Section 3.4.
Figure 2.

Schematic for non-invasive imaging of stem cell fate in the myocardium using iron particle labeling, radionuclide labeling and reporter gene labeling. Abbreviations: SPIO, superparamagnetic iron oxide; 18F-FDG, 18F-fluorodeoxyglucose; 99mTc, 99mTechnetium; 111ln-Oxine, 111 Indium-Oxine.
Table 2.
Comparison of Techniques for Imaging Cell Survival and Engraftment
| Characteristic | MRI with Iron Particles | PET/SPECT with Radionuclide Labeling | BLI with Reporter Gene Labeling |
|---|---|---|---|
| Sensitivity | 10−3–10−5 mol/L | 10−10–10−12 mol/L | 10−15–10−17 mol/L |
| Spatial Resolution | 25–100 μm | 1–2 mm | 3–5 mm |
| Label Loss | Yes | Yes | No |
| Cell Division Dilution | Yes | Yes | No |
| Signal Interference | Yes | No | No |
| Causing False Positives | |||
| Signal Corresponds to Cell Viability | Maybe | Maybe | Yes |
| Signal Corresponds to Cell Proliferation | Maybe | Maybe | Yes |
| Duration of Tracking | Cell Lifetime (Diluted Over Time) | Dependent on Isotope Half Life | Cell Lifetime |
| Ability to be used in Human Imaging | Yes | Yes | No |
| Toxicity | Relatively Safe | Radiation Effects | Genetic Modification |
3.1. Iron particle labeling
Iron particle labeling has been used to visualize transplanted stem cells in vivo. Because most stem cells are non-phagocytotic, they must be induced to take up these MRI contrast agents; therefore, labeling occurs ex vivo prior to transplantation. Labeled cells must generate sufficient contrast to be distinguishable from background. The most commonly used agents are super paramagnetic iron oxide (SPIO) particles (38), which generate a hypointense signal that allows visualization of cells.
Two methods have emerged for cell surface labeling with SPIO particles: 1) magnetofection and 2) magnetoelectroporation (MEP). Magnetofection combines the SPIO particles with a transfection agent, which is then incubated with cells. One disadvantage of this technique is its relatively long incubation time (~24 hours). The use of a transfection agent is also controversial as it may encounter challenges for regulatory approval. Magnetoelectroporation, on the other hand, is a more rapid technique (~15 minutes) to label stem cells and does not use a transfection agent. In this technique, low voltage pulsations are used to induce cells to take up contrast agents, which can occur in a matter of minutes.
The feasibility of tracking SPIO labeled stem cells has been demonstrated previously. In animal models of myocardial infarction, changes in signal intensity and volume by MRI were indicative of ESC migration within the infarct (39). Similarly, in animal models of stroke (40, 41), animals given injections of SPIO-labeled stem cells have shown migration of cells to the ischemic area, which correlated with histologic and functional findings (Figure 3). An initial study in animal models of diabetes has shown that transplanted islet cells can be detected using iron particles (42).
Figure 3.
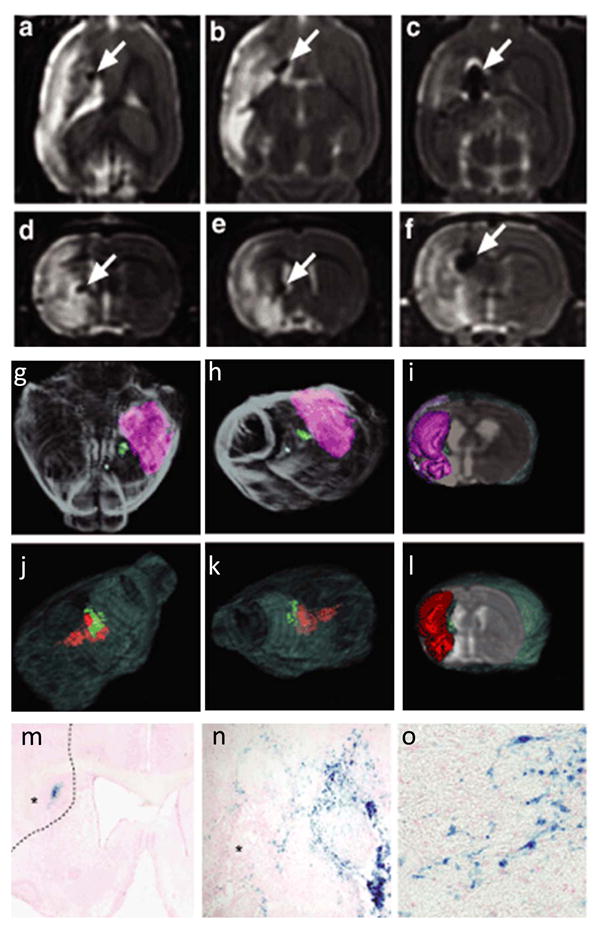
Tracking of transplanted stem cells by iron particle labeling with histological confirmation in an experimental stroke model. (a-c) Horizontal and (d-f) frontal MRI scans show dose-dependent size of iron oxide labeled grafts as hypointense arrows in the striatum (arrow) medially in the penumbral zone of the stroke, which appears as strongly hyperintense areas on T2-weighted images. The cell doses are as follows: (a,d) 50,000 cells (low dose), (b,e) 200,000 cells (intermediate dose), and (c,f) 400,000 cells (high dose). (g-i) Three dimensional surface rendering reconstruction of high resolution T2-weighted MRI of brain shows grafts in green and stroke in pink and red from the low dose (g-i) and intermediate dose group (j-l). (m-o) Histological analysis using Prussian blue staining for iron particles shows cytosolic deposition of blue crystals in the grafted stem cells and migration toward the infarcted brain (asterisk in m, n). The interrupted line in (o) shows the boundary of infarcted brain. Bars= (n) 50 μm; (o) 20 μm. Reprinted with permission from M. M. Daadi, Molecular and magnetic resonance imaging of human embryonic stem cell-derived neural stem cell grafts in ischemic rat brain, Mol Ther 17 (2009) 1282–91 (41).
Conventional magnetic cell labeling techniques which rely on surface attachment of iron particles, however, result in rapid reticuloendothelial recognition and clearance of cells, limiting their utility in vivo. Alternatively, SPIOs can be conjugated to cell-membrane particles, such as the Tat protein-derived peptide sequence that can deliver these particles into the cytoplasm or the nucleus. This approach has been described in a previous study (43) that demonstrated efficient internalization of SPIO particles conjugated to short HIV-Tat peptides into hematopoietic and neural progenitor cells. Iron incorporation did not affect stem cell viability, differentiation, or proliferation. In vivo migration of cells was tracked at the single cell level in immunodeficient mice.
Direct labeling using SPIOs coupled with MRI provides high spatial resolution and appears to be safe. Labeling with SPIO appears relatively nontoxic; however, one previous study has suggested that it may affect chondrogenesis (44). MRI also does not expose subjects to harmful radiation. The detection threshold of labeled cells depends on a number of factors, including the magnetic field strength, labeling efficiency (amount of iron that accumulates inside each cell), number of cells implanted, relaxivity of the particle, and spatial resolution of the image (29). A relatively low sensitivity of 105 cells, however, has been reported using conventional clinical scanners (30). This sensitivity may be enhanced to the single cell level using higher magnetic fields >11 Tesla, but exposure to such high magnetic fields is not safe in humans. Another potential issue is that labeling of transplanted cells is diluted by cell division (30). Specifically, if cells engraft and divide, the label may no longer be detectable. In addition, labels may be detected even after cell death, which can lower signal specificity (45). A final limitation with MRI is that it is contraindicated in patients with implantable devices (e.g., pacemakers and defibrillators), who often have refractory symptoms and will be in greater need of novel stem cell therapy.
3.2. Radionuclide labeling
Recent improvements in spatial resolution (1–2 mm) of small animal PET and SPECT cameras have enabled the implementation of specialized systems for in vivo tracking of radionuclide labeled cells (46). Compared to MRI, PET and SPECT have significantly higher sensitivity (10−10-10−12 mol/L probe) despite a lower spatial resolution (37, 47). PET and SPECT can track cells for several hours to days (47), depending on the half-life of radioisotopes used.
Radionuclide labeling of autologous white blood cells with 111In-oxyquinoline and 99mTc-hexamethylpropylene amine oxime is already utilized in clinical practice to localize inflammatory sites (48). The process requires an initial incubation period to allow the isotope to be carried into the cell by a lipophilic chelator. Once inside the cell, the lipophilicity of the molecule is lost, and the isotope is retained. The cells are then washed to remove any unbound activity and injected into the host. The labeling efficiency can approach 100%, although this varies with the cellular isotope concentration, which is dependent on the cell type, incubation time, and initial administered concentration.
This technique has also been applied to track human endothelial progenitor cells (49), rat hematopoietic progenitor cells (50), and human mesenchymal stem cells (51) after delivery to damaged organs. For tracking stem cells in the heart, a previous study using 111In-radiotracer observed that only 4.7% of injected human endothelial progenitor cells were retained in the infarcted myocardium of athymic nude rats (49). Similar findings were seen in a study using 99mTc-labeled bone-marrow derived rat MSCs (52). In the first human study, the [18F]-FDG PET tracer was used to follow the intracoronary delivery of bone marrow cells in patients after myocardial infarction (53). Only 14–39% of CD34+ bone marrow cells were found in the infarcted myocardium 1 hour after intracoronary delivery.
A major concern of using radioactive labeling agents is the potential radiation exposure (49), which can lead to decreased cell viability, function and differentiation. A recent study in nude rats demonstrated a significant impairment of proliferation and function of labeled human hematopoetic progenitor cells despite accurate homing to the infarcted myocardium (50). Another major problem is the short half-life of the radiotracer, which limits the duration for long-term cell tracking. Finally, a potential limitation is that labels can efflux out of cells over time, resulting in label loss from viable cells and errors in the detection of surviving cells.
3.3. Reporter gene labeling
Another imaging approach for cell trafficking is reporter gene imaging. In this technique, specific reporter gene constructs are transfected into stem cells. After transplantation, cells can be detected by their gene expression (e.g., fluorescent proteins) or by using reporter probes that target the reporter gene products (e.g., intracellular proteins or surface receptors). Specifically, if stem cells are viable and functional, transcription and translation lead to the production of reporter proteins, which can produce an imaging signal or require interaction with reporter probes to generate a detectable imaging signal (54). If the stem cells proliferate, the reporter gene will be passed onto daughter cells if stably integrated, and the corresponding imaging signals will increase in intensity. However, if cells are apoptotic or dead, they will cease to emit signals.
There are several advantages of this approach. First, because reporter protein expression requires intact DNA transcription and RNA translation, the imaging signal will indicate the presence of viable cells. Unlike iron particle or radionuclide labeling, there is no risk of probe dilution or leakage that can lead to erroneous results (55). Second, the reporter gene can be integrated into the cellular chromosome via integrating vectors and can be passed from the mother to daughter cell, which allows monitoring of cellular proliferation and repeat imaging over long periods of time. In contrast, radionuclide labeling is limited by the half-life of the radioisotopes used. Finally, reporter gene constructs driven by various promoters (e.g., constitutive, tissue specific and inducible) can also be made.
Report gene constructs have been developed for multiple imaging modalities. For example, fluorescent reporter proteins (e.g., monomeric red fluorescent protein, enhanced green fluorescent protein, and enhanced cyan fluorescent protein) allow imaging at the single-cell level by fluorescence microscopy as well as isolation of stable cell populations by FACS. Bioluminescence reporter proteins (e.g., firefly luciferase, renilla luciferase, and click beetle luciferase), on the other hand, can be used for high-throughput BLI. Fluorescence and BLI, however, rely on low energy photons that become attenuated within deeper tissues. Imaging of large animals and humans requires PET imaging, which uses the herpes simplex virus thymidine kinase (HSV-tk) reporter gene (53).
Reporter gene imaging can be used to monitor stem cell survival, migration, and proliferation. For example, in a previous study, bone marrow mononuclear cells were harvested from transgenic mice constitutively expressing both firefly luciferase (FLuc) and enhanced green fluorescent protein (eGFP) reporter gene and were injected into wild type mice randomized to sham surgery or ischemia reperfusion injury (56). FLuc and eGFP correlated with cell number as measured by BLI (Figure 4a) as well as fluorescent microscopy (Figure 4b) and FACS analysis (Figure 4c), respectively. BLI also showed preferential homing of cells to damaged tissue, as shown in Figure 4d (56). Studies in larger animal models of myocardial infarction using PET have also shown that reporter gene expression can quantitatively and serially track porcine bone marrow derived mesenchymal stem cells with high detection sensitivity (57).
Figure 4.

Bioluminescence and fluorescence imaging of transplanted stem cells. (a) Ex vivo analysis shows a linear correlation between cell number and bioluminescence imaging signal. The total count is noted above the corresponding well, with the color scale bar denoting the range of signal in photons per second per cm2 per steradian. (b) Fluorescent microscopy shows bright, cytosolic enhanced green fluorescent protein expression, with corresponding nuclei stained with blue 4, 6-diamidino-2-phenlindole. Scale bar: 5 μm. (c) FACS analysis demonstrates robust expression of enhanced green fluorescent protein by more than 87% of cells from FVB wild type mice and L2G transgenic mice. (d) Bioluminescence imaging shows that transplanted bone marrow mononuclear cells accurately home to areas of injury. Images of the sham (left) and ischemic reperfusion injury (right) are shown following stem cell transplantation (day 1 to 28). There is a persistent elevation of signal through day 14, which gradually decreases by day 28. (Please note the maximum values for scale bars in p/s/cm2/sr are different in the three rows). Abbreviations: p/s/cm2/sr, photons per second per cm2 per steradian. Reprinted with permission from Sheikh, A, et al. Molecular imaging of bone marrow mononuclear cell homing and engraftment in ischemic myocardium, Stem Cells 25 (2007) 2677–84 (56).
Similarly, reporter gene imaging has been used to monitor the fate of neural progenitor cells transfected with FLuc and eGFP in animal models of stroke. In a recent study using an experimental rat stroke model, human neural stem cells engineered to express FLuc and eGFP (41) were monitored successfully using BLI and fluorescence microscopy, respectively. For pancreatic β-islet cells in a murine diabetic model, initial studies using human and murine engineered islets have shown that cell survival and proliferation can be monitored using the luciferase and HSV-tk reporter gene constructs (Figure 5) (58).
Figure 5.

Tracking of survival and engraftment of transplanted islets by PET reporter labeling. (a) Immunohistological analysis of engineered HSV1-tk expressing islets, performed 15 days after transplantation, shows expression of thymidine kinase (shown in green) and insulin (red), observed in scattered islets. (b) Longitudinal micro PET imaging of a mouse with an intrahepatic islet graft shows that signals from the liver area of HSV1-tk expressing islets gradually decline over time. When imaged 40 days after transplantation, signals from the liver area were essentially at background levels. Signal loss was likely due to cellular death shortly after implantation and the transient nature of viral gene expression. Reprinted with permission from Y. Lu, et al., Noninvasive imaging of islet grafts using positron-emission tomography, Proc Natl Acad Sci U S A 103 (2006) 11294–9 (58).
Despite its advantages, several hurdles prevent reporter imaging from being applied in routine clinical practice. For example, appropriate reporter proteins that are not immunogenic need to be developed. In addition, appropriate vectors that allow site specific integration (rather than random insertion) are needed to minimize potential interference from stem cell function and differentiation. Finally, the effects of the reporter genes on the specific stem cell types will need to be carefully evaluated using genomic and proteonomic analyses, as previously described (59, 60).
3.4. Labeling markers of apoptosis
Similar to other developmental processes, stem cell renewal and proliferation is partially controlled by programmed cell death (apoptosis). Inhibition of apoptosis may increase survival of stem cells after transplantation; whereas, controlling apoptosis may prevent the development of teratomas. Thus, noninvasive imaging of apoptosis may be important for the clinical translation of stem cell therapy.
Noninvasive imaging of programmed cell death can be performed by labeling markers of apoptosis, followed by visualization and quantification using multiple imaging modalities. In addition to radiotracers and magnetic labeling, optical probes can be used to label apoptosis markers. With the advent of near infrared (NIR) imaging, fluorescence imaging can now be performed at the macroscopic level in vivo (61). Compared to visible light, NIR provides the following advantages for deep tissue imaging: 1) high transmittance in tissues and blood, 2) minimal interference from light scattering and autofluorescensce, and 3) the use of nonionizing photons as a source of fluorescence excitation. Optical probes can also be combined with MRI and CT for combined anatomical imaging (61).
For imaging apoptosis, NIR probes target annexin, a calcium-binding protein on the cell surface that has a high affinity for externalized aminophospholipids which are early markers of apoptosis (61). Annexin V, however, cannot distinguish between externalized aminophospholipids on cells with intact membranes (true apoptotic cells) from those with damaged cell membranes (necrotic cells) (62). Alternatively, the NIR probe can target apoptosis-associated caspase activity in vivo. These enzymes are cysteine proteases that are critical for the initiation and execution of apoptosis. Thus far, these probes have been used mainly for cancer imaging, but these techniques can be potentially applied to detect apoptosis after stem cell delivery (63). Interestingly, a recent study demonstrated that conjugation of annexin to stem cells improved homing to apoptotic cells in vitro (64). Further investigation is needed to better define the utility of these techniques for stem cell therapy.
4. Functional Measurements
The goal of stem cell therapy is to replace injured tissue so that the function of the damaged organ may be restored. Functional measurements, however, should not be restricted to cells in vitro but should be extended to include in vivo measurements. The evaluation of improvement in organ function is specific to the organ and often specific to the disease.
4.1. Cardiac function
Heart failure is a leading cause of morbidity and mortality worldwide, yet current medical therapy at best only delays disease progression. Ultimately, most patients will require cardiac transplantation or will succumb to the disease. Regenerative therapy with stem cells is a promising alternative strategy to replace damaged cardiomyocytes with new cells to restore heart function. Improvement in heart function after stem cell delivery can be measured by assessing the changes in left ventricular size and systolic function, myocardial perfusion, and myocardial viability.
4.1.1. LV size and systolic function
LV size and systolic function can be measured non-invasively by various imaging modalities, including MRI, echocardiography, computed tomography (CT), and nuclear techniques. Compared to MRI and echocardiography, both CT and nuclear techniques are not preferred due to their lower temporal resolution and exposure to radiation, which may be harmful to both the patient and the injected stem cells. MRI is often selected over echocardiography because of its high spatial resolution, allowing detection of very small changes in LV size and function. Previous clinical trials using adult progenitor cells performed in the setting of acute myocardial infarction and chronic ischemic heart disease have shown that following stem cell injection, improvement in LV function could range from as little as 3% to as much as 18% (65, 66), with the most benefit noted in the area of tissue damage (29). Some have suggested that improvement in LV function may be time-dependent (29). In a recent trial using bone marrow cells, treated patients had a significant increase of 6.7% in LV function compared to controls at 6 months. However, at 18 months, there was no significant difference (67). Analysis of a time course of recovery revealed that treated patients had a faster recovery of LV function.
4.1.2. Myocardial perfusion
Evaluation of myocardial blood perfusion can be performed non-invasively using nuclear techniques or MRI. Most clinical studies have used SPECT (29, 30) and have shown that adult progenitor cells improve myocardial perfusion, suggesting that these cells may promote growth of new small blood vessels (29, 30). Reduction in myocardial perfusion defect size has been shown in both acute and chronic heart ischemia. For example, in a recent clinical study using bone marrow progenitor cells, SPECT demonstrated a decrease in resting perfusion defect size at 4 months (68).
4.1.3. Viability
The final measure of functional outcome is myocardial viability, which can be assessed by either nuclear imaging with PET (mainly using [18F]-FDG) and SPECT (99mTc-labeled agents), or low-dose dobutamine echocardiography (assessing contractile reserve). Clinical studies using nuclear imaging techniques have shown increased tracer uptake ranging from 15–55% post-therapy with adult progenitor cells. Contractile reserve as measured by echocardiography, however, has not shown significant enhancement (30). These inconsistent results may be due to the fact that contractile reserve might be lost early relative to glucose utilization, which is used to measure viability in nuclear studies. However, it is also possible that echocardiography lacks adequate spatial resolution to detect changes in contractile reserve. A preliminary pilot, clinical study using MRI have shown that manganese, an intracellular contrast agent, may provide additional information on myocardial viability (69).
4.2. Neurological function
Neurological disorders including Parkinson’s disease and stroke are caused by the loss of neurons and glial cells in the brain and spinal cord. Studies on cell replacement therapy and gene therapy have been promising, but limited by the availability of suitable cells for transplant. Because of their pluripotent potential, stem cells can differentiate into the three major cell types (i.e., neurons, astrocytes and oligodendrocytes) of the nervous system to repair these areas of neurological damage. Assessment of the functional effectiveness of therapy depends on the cell type affected as well as the disease process.
4.2.1. Parkinson’s disease
Parkinson’s disease (PD) affects more than 500,000 people in the United States. It is characterized by extensive loss of dopamine neurons in the substantia nigra pars compacta and their terminals in the striatum. Patients have been given L-dihydroxyphenyl alanine (L-DOPA) for the treatment of Parkinson’s disease, but long-term administration has resulted in side effects. Since the late 1980’s, successful therapy has been achieved with transplantation of human fetal ventral mesencephalic tissues into the striatum of patients with Parkinson’s disease (70); however, treatment with fetal tissue has remained controversial. Treatment with ESCs and adult progenitor cell derived neurons with dopamine phenotype is a practical and effective alternative and has been shown to attenuate the functional deficits associated with the disease in preclinical trials.
Dopamine neurons have been generated from murine ESCs (71) and human adult progenitor stem cells (72),(73). Behavioral improvement has been noted in rats and monkeys who develop signs of Parkinson’s disease after administration of toxins (6-hydroxy dopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, respectively), which cause selective permanent destruction of dopamine neurons in the striatum (72, 74). Recently, transplantation of human neural stem cells carrying the genes of two rate limiting enzymes for dopamine production (e.g., guanosine triphosphate cyclohydroxylase and tyrosine hydroxylase) resulted in long-term recovery of function in a rat model of Parkinson’s disease (74). A trial in humans has not yet been performed.
4.2.2. Stroke
Stroke is the second highest cause of death in Asia and the third in the United States. Stroke can be caused by ischemia or intracerebral hemorrhage, both of which result in neuronal loss. Unfortunately, once damage from a stroke occurs, little can be done despite the best efforts to protect neural function. Other than cell replacement therapy, there is no current treatment to restore neural function. Like Parkinson’s disease, the effects of therapy can be assessed using behavioral tests including the rotarod test, the modified limb placing test, and Neuro score (75). The Neuro score is a well-validated scoring method for the assessment of stroke deficits in animals. It consists of 10 different motor, coordinative, and sensory items to evaluate the effect of stroke on the levels of consciousness, hearing, neglect, visual field loss, extra-ocular movement, motor strength, ataxia, and sensory loss. Promising results have been shown in animal models after treatment with stem cells but, thus far, no trials in humans have been performed. Preclinical trials in a rat model of intracerebral hemorrhage have shown that human neural stem cells and mesenchymal stem cells cross the blood-brain barrier, migrate to the lesion area, differentiate into neurons and astrocytes, and induce functional recovery (76–78)
4.3. Pancreatic function
Diabetes is a devastating chronic disease that affects hundreds of millions of children and adults worldwide. Currently, there is no cure for diabetes, and despite great improvement in therapy, the morbidity and mortality associated with diabetic complications remain high. Although transplantable pancreatic islets containing insulin-secreting β cells have been used successfully to treat the disease, there remains a critical shortage of donor transplants. Stem cell therapy is thus a promising alternative. To evaluate the functional effectiveness of stem cell therapy in the treatment of diabetes, researchers measure C-peptide, which is produced upon splitting of pro-insulin into insulin. Although this is an indirect measure of viable β cell function, it can be performed in vivo and only requires a blood test. After years of failed or unconvincing clinical trial data (2), the potential for stem cell therapy for diabetes has recently been shown in a recent phase I/II study in 23 patients with Type I diabetes who received autologous non-myeloablative hematopoietic stem cell transplantation (79). After mean follow-up of 29.8 months, 20 of 23 patients became insulin free (12 continuously and 8 transiently) with good glucose control for up to 4 years. C-peptide levels increased significantly, indicating that the majority of glycemic control was due to endogenous insulin secretion.
5. Problems and Perspectives
Stem cell therapy has the potential to treat a myriad of diseases that have limited options for cure. Challenges, however, still remain before cell based therapy can be used routinely to treat disorders of the heart, nervous system, and pancreas. An ongoing debate is the identification of the most optimal cell type. The advantage of using hESCs is their ability to self-renew and to differentiate into any cell; thus, these cells can potentially regenerate the entire organ. Their disadvantage is a potential for immunologic rejection and need for immunosuppressive therapy. Use of cells from embryos has also met with several regulatory hurdles. Only recently has the FDA approved the first Phase 1 clinical trial using hESCs for the neural regeneration in patients with severe spinal cord injury.
Adult progenitor cells, on the other hand, are limited to differentiating into cell types from the tissue of origin. Adult progenitor cells are rare in mature organs and can be difficult to expand in culture. However, they are considered less immunogenic than hESCs and their therapeutic application has been less controversial. Thus, the majority of clinical trials have used adult progenitor cells. Results, however, have been mixed, leading many to question their regeneration potential and ability to differentiate, survive and engraft (29).
Some of these challenges can be addressed by using iPSCs. iPSCs are derived from the patients’ adult cells; thus, there is an unlimited supply and rejection is not an issue. These cells can be cultured, expanded and differentiated in vitro and transplanted into injured tissue. However, the efficiency of reprogramming and differentiation remains low. Whether these cells truly differentiate into target tissue or retain some features of their tissue of origin has also been questioned (80, 81). Finally, efficient derivation of iPSCs still require viral transfection for reprogramming, presenting obstacles for regulatory approval. A clinical trial has not been yet been initiated using iPSCs.
For the heart, challenges facing stem cell therapy involve each step of treatment, from cell isolation to long-term safety (82). For example, determining the sufficient cell number and the mode of cell delivery remain unclear. In addition, survival and proliferation in the inflammatory environment of the infarcted myocardium has been challenging as >90% of cells die within a week. Addressing this issue will require better understanding of the acute donor cell death phenomenon. Electromechanical integration of cells is also required for improvement of cardiac function. Transplanted cells must have long-term electromechanical stability to prevent the development of dangerous arrhythmias. Currently, these challenges have not been adequately met as most clinical studies have focused on the safety of the transplantation procedure and the improvement in LV systolic function or perfusion after stem cell therapy.
The application of stem cells for neurological disorders may be even more complex than other diseases (83). Stem cells need to integrate into a sophisticated system of interconnected cells that extend over great distances, which may be challenging in the absence of stimuli to guide the development of neural networks. In addition to the poor survival rate, transplanted cells are also subject to the same progressive and recurrent pathological processes that caused the initial neurological injury, making it necessary to implant large grafts. Controlling stem cell proliferation and differentiation into appropriate cellular phenotypes is also necessary for organ specific function and to prevent tumor formation. Finally, it is unclear whether findings in animal models can translate into human studies because of the differences in species plasticity and an incomplete understanding of disease processes. In Parkinson’s disease (83), for example, there has not yet been a clear demonstration that neurons generated in vitro can efficiently reinnervate the striatum, release dopamine in vivo, or result in functional recovery from deficits resembling human symptoms, despite promising results in preclinical studies.
For treatment of diabetes, a recent study has demonstrated the potential of autologous non-myeloablative hematopoietic stem cell transplantation; however, almost half the patients had adverse effects associated with the procedure, including late endocrine dysfunction, autoimmune polyendocrine syndrome, and cyclophosphamide-related oligospermia (79). The use of hESCs or iPSCs may minimize these complications, but early protocols have failed to generate functional β-islet cells. More recently, protocols, which included all stages of β-cell development, have successfully produced glucose-responsive insulin secreting cells in vivo (16). Although promising, communication between transplanted cells and different islet cells and the extracellular matrix, which together provide normal glycemic control, has not yet been demonstrated. The function, survival, and replication of these cells ultimately rely on their ability to exist in the highly specialized microenvironment of the islet (2). Finally, replacement of the cells does not address the cause for disease, which not only destroys native cells but also transplanted cells (2). Specifically, in the case of Type I diabetes, autoimmune antibodies can also destroy stem cells, leading to the need for repeat transplantations.
Although the future for patients with heretofore incurable diseases of the heart, nervous system, and pancreas appears brighter with the advent of stem cell based therapy, more research is needed before stem cells can effectively repair damaged tissue. By performing a complete evaluation of the stem cell lineage, fate, and function in preclinical and clinical trials, we will ensure that one day patients will routinely receive this novel therapy.
Acknowledgments
We thank Andrew Lee, Blake Wu, and Ian Chen for assistance with preparing the manuscript. This work was supported in part by ACC-GE Cardiovascular Imaging Award (PKN), Stanford VPUE (DN), NIH HL099117 (JCW), and NIH EB009689 (JCW).
References
- 1.Beeres SL, Bax JJ, Dibbets-Schneider P, et al. Sustained effect of autologous bone marrow mononuclear cell injection in patients with refractory angina pectoris and chronic myocardial ischemia: twelve-month follow-up results. Am Heart J. 2006;152(684):e11–6. doi: 10.1016/j.ahj.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Halban PA, German MS, Kahn SE, Weir GC. Current status of islet cell replacement and regeneration therapy. J Clin Endocrinol Metab. 2010;95:1034–43. doi: 10.1210/jc.2009-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104:398–402. doi: 10.1161/CIRCRESAHA.108.187724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 5.van der Bogt KE, Sheikh AY, Schrepfer S, et al. Comparison of different adult stem cell types for treatment of myocardial ischemia. Circulation. 2008;118:S121–9. doi: 10.1161/CIRCULATIONAHA.107.759480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428:668–73. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Wilson GF, Soerens AG, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu BY, Weick JP, Yu J, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107:4335–40. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hori Y, Rulifson IC, Tsai BC, Heit JJ, Cahoy JD, Kim SK. Growth inhibitors promote differentiation of insulin-producing tissue from embryonic stem cells. Proc Natl Acad Sci U S A. 2002;99:16105–10. doi: 10.1073/pnas.252618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science. 2001;292:1389–94. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 11.Rajagopal J, Anderson WJ, Kume S, Martinez OI, Melton DA. Insulin staining of ES cell progeny from insulin uptake. Science. 2003;299:363. doi: 10.1126/science.1077838. [DOI] [PubMed] [Google Scholar]
- 12.Hansson M, Tonning A, Frandsen U, et al. Artifactual insulin release from differentiated embryonic stem cells. Diabetes. 2004;53:2603–9. doi: 10.2337/diabetes.53.10.2603. [DOI] [PubMed] [Google Scholar]
- 13.Cao F, Wagner RA, Wilson KD, et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS One. 2008;3:e3474. doi: 10.1371/journal.pone.0003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boheler KR, Czyz J, Tweedie D, Yang HT, Anisimov SV, Wobus AM. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ Res. 2002;91:189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- 15.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–41. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 17.Regenerative Medicine, Appendix E: Stem Cell Markers. Department of Health and Human Services; 2006. (Accessed at http://stemcells.nih.gov/info/scireport/appendixe.asp.) [Google Scholar]
- 18.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–73. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 19.Nygren JM, Jovinge S, Breitbach M, et al. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 20.Synnergren J, Akesson K, Dahlenborg K, et al. Molecular signature of cardiomyocyte clusters derived from human embryonic stem cells. Stem Cells. 2008;26:1831–40. doi: 10.1634/stemcells.2007-1033. [DOI] [PubMed] [Google Scholar]
- 21.Gurok U, Steinhoff C, Lipkowitz B, Ropers HH, Scharff C, Nuber UA. Gene expression changes in the course of neural progenitor cell differentiation. J Neurosci. 2004;24:5982–6002. doi: 10.1523/JNEUROSCI.0809-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eiges R, Schuldiner M, Drukker M, Yanuka O, Itskovitz-Eldor J, Benvenisty N. Establishment of human embryonic stem cell-transfected clones carrying a marker for undifferentiated cells. Curr Biol. 2001;11:514–8. doi: 10.1016/s0960-9822(01)00144-0. [DOI] [PubMed] [Google Scholar]
- 23.Cheng L, Du C, Murray D, et al. A GFP reporter system to assess gene transfer and expression in human hematopoietic progenitor cells. Gene Ther. 1997;4:1013–22. doi: 10.1038/sj.gt.3300507. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Wu JC, Sheikh AY, et al. Differentiation, survival, and function of embryonic stem cell derived endothelial cells for ischemic heart disease. Circulation. 2007;116:I46–54. doi: 10.1161/CIRCULATIONAHA.106.680561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spees JL, Olson SD, Ylostalo J, et al. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci U S A. 2003;100:2397–402. doi: 10.1073/pnas.0437997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzmenkin A, Liang H, Xu G, et al. Functional characterization of cardiomyocytes derived from murine induced pluripotent stem cells in vitro. Faseb J. 2009;23:4168–80. doi: 10.1096/fj.08-128546. [DOI] [PubMed] [Google Scholar]
- 27.Biella G, Di Febo F, Goffredo D, et al. Differentiating embryonic stem-derived neural stem cells show a maturation-dependent pattern of voltage-gated sodium current expression and graded action potentials. Neuroscience. 2007;149:38–52. doi: 10.1016/j.neuroscience.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Schroeder IS, Rolletschek A, Blyszczuk P, Kania G, Wobus AM. Differentiation of mouse embryonic stem cells to insulin-producing cells. Nat Protoc. 2006;1:495–507. doi: 10.1038/nprot.2006.71. [DOI] [PubMed] [Google Scholar]
- 29.Beeres SL, Bengel FM, Bartunek J, et al. Role of imaging in cardiac stem cell therapy. J Am Coll Cardiol. 2007;49:1137–48. doi: 10.1016/j.jacc.2006.10.072. [DOI] [PubMed] [Google Scholar]
- 30.Zhang SJ, Wu JC. Comparison of imaging techniques for tracking cardiac stem cell therapy. J Nucl Med. 2007;48:1916–9. doi: 10.2967/jnumed.107.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ransohoff KJ, Wu JC. Advances in cardiovascular molecular imaging for tracking stem cell therapy. Thromb Haemost. 104:13–22. doi: 10.1160/TH09-08-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Hong KS, Song J. The present status of cell tracking methods in animal models using magnetic resonance imaging technology. Mol Cells. 2007;23:132–7. [PubMed] [Google Scholar]
- 33.Rogers WJ, Meyer CH, Kramer CM. Technology insight: in vivo cell tracking by use of MRI. Nat Clin Pract Cardiovasc Med. 2006;3:554–62. doi: 10.1038/ncpcardio0659. [DOI] [PubMed] [Google Scholar]
- 34.Acton PD, Kung HF. Small animal imaging with high resolution single photon emission tomography. Nucl Med Biol. 2003;30:889–95. doi: 10.1016/s0969-8051(03)00112-4. [DOI] [PubMed] [Google Scholar]
- 35.Wu JC, Chen IY, Sundaresan G, et al. Molecular imaging of cardiac cell transplantation in living animals using optical bioluminescence and positron emission tomography. Circulation. 2003;108:1302–5. doi: 10.1161/01.CIR.0000091252.20010.6E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao F, Lin S, Xie X, et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation. 2006;113:1005–14. doi: 10.1161/CIRCULATIONAHA.105.588954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17:545–80. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 38.Chen IY, Greve JM, Gheysens O, et al. Comparison of optical bioluminescence reporter gene and superparamagnetic iron oxide MR contrast agent as cell markers for noninvasive imaging of cardiac cell transplantation. Mol Imaging Biol. 2009;11:178–87. doi: 10.1007/s11307-008-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Himes N, Min JY, Lee R, et al. In vivo MRI of embryonic stem cells in a mouse model of myocardial infarction. Magn Reson Med. 2004;52:1214–9. doi: 10.1002/mrm.20220. [DOI] [PubMed] [Google Scholar]
- 40.Hoehn M, Kustermann E, Blunk J, et al. Monitoring of implanted stem cell migration in vivo: a highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc Natl Acad Sci U S A. 2002;99:16267–72. doi: 10.1073/pnas.242435499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daadi MM, Li Z, Arac A, et al. Molecular and magnetic resonance imaging of human embryonic stem cell-derived neural stem cell grafts in ischemic rat brain. Mol Ther. 2009;17:1282–91. doi: 10.1038/mt.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evgenov NV, Medarova Z, Pratt J, et al. In vivo imaging of immune rejection in transplanted pancreatic islets. Diabetes. 2006;55:2419–28. doi: 10.2337/db06-0484. [DOI] [PubMed] [Google Scholar]
- 43.Lewin M, Carlesso N, Tung CH, et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–4. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 44.Kostura L, Kraitchman DL, Mackay AM, Pittenger MF, Bulte JW. Feridex labeling of mesenchymal stem cells inhibits chondrogenesis but not adipogenesis or osteogenesis. NMR Biomed. 2004;17:513–7. doi: 10.1002/nbm.925. [DOI] [PubMed] [Google Scholar]
- 45.Li Z, Suzuki Y, Huang M, et al. Comparison of reporter gene and iron particle labeling for tracking fate of human embryonic stem cells and differentiated endothelial cells in living subjects. Stem Cells. 2008;26:864–73. doi: 10.1634/stemcells.2007-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tai YC, Chatziioannou AF, Yang Y, et al. MicroPET II: design, development and initial performance of an improved microPET scanner for small-animal imaging. Phys Med Biol. 2003;48:1519–37. doi: 10.1088/0031-9155/48/11/303. [DOI] [PubMed] [Google Scholar]
- 47.Wu JC, Tseng JR, Gambhir SS. Molecular imaging of cardiovascular gene products. J Nucl Cardiol. 2004;11:491–505. doi: 10.1016/j.nuclcard.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Love C, Palestro CJ. Radionuclide imaging of infection. J Nucl Med Technol. 2004;32:47–57. quiz 8–9. [PubMed] [Google Scholar]
- 49.Aicher A, Brenner W, Zuhayra M, et al. Assessment of the tissue distribution of transplanted human endothelial progenitor cells by radioactive labeling. Circulation. 2003;107:2134–9. doi: 10.1161/01.CIR.0000062649.63838.C9. [DOI] [PubMed] [Google Scholar]
- 50.Brenner W, Aicher A, Eckey T, et al. 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. J Nucl Med. 2004;45:512–8. [PubMed] [Google Scholar]
- 51.Bindslev L, Haack-Sorensen M, Bisgaard K, et al. Labelling of human mesenchymal stem cells with indium-111 for SPECT imaging: effect on cell proliferation and differentiation. Eur J Nucl Med Mol Imaging. 2006;33:1171–7. doi: 10.1007/s00259-006-0093-7. [DOI] [PubMed] [Google Scholar]
- 52.Barbash IM, Chouraqui P, Baron J, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863–8. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 53.Yaghoubi SS, Gambhir SS. PET imaging of herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk reporter gene expression in mice and humans using [18F]FHBG. Nat Protoc. 2006;1:3069–75. doi: 10.1038/nprot.2006.459. [DOI] [PubMed] [Google Scholar]
- 54.Sun N, Lee A, Wu JC. Long term non-invasive imaging of embryonic stem cells using reporter genes. Nat Protoc. 2009;4:1192–201. doi: 10.1038/nprot.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu JC. Can radionuclide imaging predict future response to stem cell therapy? J Nucl Cardiol. 2008;15:308–10. doi: 10.1016/j.nuclcard.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 56.Sheikh AY, Lin SA, Cao F, et al. Molecular imaging of bone marrow mononuclear cell homing and engraftment in ischemic myocardium. Stem Cells. 2007;25:2677–84. doi: 10.1634/stemcells.2007-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gyongyosi M, Blanco J, Marian T, et al. Serial noninvasive in vivo positron emission tomographic tracking of percutaneously intramyocardially injected autologous porcine mesenchymal stem cells modified for transgene reporter gene expression. Circ Cardiovasc Imaging. 2008;1:94–103. doi: 10.1161/CIRCIMAGING.108.797449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu Y, Dang H, Middleton B, et al. Noninvasive imaging of islet grafts using positron-emission tomography. Proc Natl Acad Sci U S A. 2006;103:11294–9. doi: 10.1073/pnas.0603909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu JC, Spin JM, Cao F, et al. Transcriptional profiling of reporter genes used for molecular imaging of embryonic stem cell transplantation. Physiol Genomics. 2006;25:29–38. doi: 10.1152/physiolgenomics.00254.2005. [DOI] [PubMed] [Google Scholar]
- 60.Wu JC, Cao F, Dutta S, et al. Proteomic analysis of reporter genes for molecular imaging of transplanted embryonic stem cells. Proteomics. 2006;6:6234–49. doi: 10.1002/pmic.200600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr Opin Chem Biol. 2010;14:71–9. doi: 10.1016/j.cbpa.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 62.Maxwell D, Chang Q, Zhang X, Barnett EM, Piwnica-Worms D. An improved cell-penetrating, caspase-activatable, near-infrared fluorescent peptide for apoptosis imaging. Bioconjug Chem. 2009;20:702–9. doi: 10.1021/bc800516n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petrovsky A, Schellenberger E, Josephson L, Weissleder R, Bogdanov A., Jr Near-infrared fluorescent imaging of tumor apoptosis. Cancer Res. 2003;63:1936–42. [PubMed] [Google Scholar]
- 64.Gerasimou A, Ramella R, Brero A, et al. Homing of annexin-labeled stem cells to apoptotic cells. Cell Mol Biol Lett. 2009;14:100–12. doi: 10.2478/s11658-008-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Assmus B, Honold J, Schachinger V, et al. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006;355:1222–32. doi: 10.1056/NEJMoa051779. [DOI] [PubMed] [Google Scholar]
- 66.Chen SL, Fang WW, Ye F, et al. Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol. 2004;94:92–5. doi: 10.1016/j.amjcard.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 67.Meyer GP, Wollert KC, Lotz J, et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months’ follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006;113:1287–94. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- 68.Bartunek J, Vanderheyden M, Vandekerckhove B, et al. Intracoronary injection of CD133-positive enriched bone marrow progenitor cells promotes cardiac recovery after recent myocardial infarction: feasibility and safety. Circulation. 2005;112:I178–83. doi: 10.1161/CIRCULATIONAHA.104.522292. [DOI] [PubMed] [Google Scholar]
- 69.Skjold A, Amundsen BH, Wiseth R, et al. Manganese dipyridoxyl-diphosphate (MnDPDP) as a viability marker in patients with myocardial infarction. J Magn Reson Imaging. 2007;26:720–7. doi: 10.1002/jmri.21065. [DOI] [PubMed] [Google Scholar]
- 70.Hagell P, Brundin P. Cell survival and clinical outcome following intrastriatal transplantation in Parkinson disease. J Neuropathol Exp Neurol. 2001;60:741–52. doi: 10.1093/jnen/60.8.741. [DOI] [PubMed] [Google Scholar]
- 71.Kim JH, Auerbach JM, Rodriguez-Gomez JA, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418:50–6. doi: 10.1038/nature00900. [DOI] [PubMed] [Google Scholar]
- 72.Redmond DE, Jr, Bjugstad KB, Teng YD, et al. Behavioral improvement in a primate Parkinson’s model is associated with multiple homeostatic effects of human neural stem cells. Proc Natl Acad Sci U S A. 2007;104:12175–80. doi: 10.1073/pnas.0704091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yasuhara T, Matsukawa N, Hara K, et al. Transplantation of human neural stem cells exerts neuroprotection in a rat model of Parkinson’s disease. J Neurosci. 2006;26:12497–511. doi: 10.1523/JNEUROSCI.3719-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim SU, Park IH, Kim TH, et al. Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease. Neuropathology. 2006;26:129–40. doi: 10.1111/j.1440-1789.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- 75.Wilhelm-Schwenkmezger T, Pittermann P, Zajonz K, Kempski O, Dieterich M, Nedelmann M. Therapeutic application of 20-kHz transcranial ultrasound in an embolic middle cerebral artery occlusion model in rats: safety concerns. Stroke. 2007;38:1031–5. doi: 10.1161/01.STR.0000257966.32242.0b. [DOI] [PubMed] [Google Scholar]
- 76.Lee HJ, Kim KS, Kim EJ, et al. Brain transplantation of immortalized human neural stem cells promotes functional recovery in mouse intracerebral hemorrhage stroke model. Stem Cells. 2007;25:1204–12. doi: 10.1634/stemcells.2006-0409. [DOI] [PubMed] [Google Scholar]
- 77.Lee HJ, Kim KS, Park IH, Kim SU. Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS One. 2007;2:e156. doi: 10.1371/journal.pone.0000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagai A, Kim WK, Lee HJ, et al. Multilineage potential of stable human mesenchymal stem cell line derived from fetal marrow. PLoS One. 2007;2:e1272. doi: 10.1371/journal.pone.0001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Couri CE, Oliveira MC, Stracieri AB, et al. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. Jama. 2009;301:1573–9. doi: 10.1001/jama.2009.470. [DOI] [PubMed] [Google Scholar]
- 80.Ghosh Z, Wilson KD, Wu Y, Hu S, Quertermous T, Wu JC. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS One. 2010;5:e8975. doi: 10.1371/journal.pone.0008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010 doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Segers VF, Lee RT. Stem-cell therapy for cardiac disease. Nature. 2008;451:937–42. doi: 10.1038/nature06800. [DOI] [PubMed] [Google Scholar]
- 83.Lindvall O, Kokaia Z. Stem cells for the treatment of neurological disorders. Nature. 2006;441:1094–6. doi: 10.1038/nature04960. [DOI] [PubMed] [Google Scholar]