

Abstract
Immunohistochemical characterization of the distribution of GABAA receptor subunits γ 1/3 and 2 in the hippocampus relative to neurofibrillary tangle (NFT) pathology staging was performed in cognitively normal subjects (Braak stage I/II, n=4) and two groups of Alzheimer’s disease (AD) patients (Braak stage III/IV, n=4; Braak stage V/VI, n=8). In both Braak groups of AD patients, neuronal γ1/3 and γ2 immunoreactivity was preserved in all hippocampal subfields. However, compared to normal controls neuronal γ1/3 immunoreactivity was more intense in several end-stage AD subjects. Despite increased NFT pathology in the Braak V/VI AD group, GABAAγ 1/3 and γ2 immunoreactivitydid not co-localize with markers of NFT. These results suggest that upregulating or preserving GABAAγ 1/3 and γ2 receptors may protect neurons against neurofibrillary pathology in AD.
Keywords: Alzheimer’s disease, GABA, excitotoxicity, neurodegeneration, tau
Introduction
Alzheimer’s disease (AD) is characterized pathologically by the presence of neurofibrillary tangles (NFT) and neuritic plaques, and loss of neurons and synapses in selected brain regions, including the hippocampus. Cell loss in AD is in part due to glutamate-mediated excitotoxicity1,2, which could be countered by compensatory changes in the γ-aminobutyric acid (GABA) inhibitory neurotransmitter system1. There are two major classes of GABA receptors, GABAA and GABAB. GABAA receptors are composed of multiple subunits grouped according to sequence similarity into α (α1–6), β (β1–4), γ (γ1–4), δ (δ1–2), ρ(1–3), ε, π, and θ subunits3. We previously examined alterations in the expression of GABAAα4 and β5,6 and GABAB7 receptor subunits in the hippocampus in AD groups with different Braak stages of NFT severity8. These studies reported that with increased NFT severity by Braak staging, the β2/3 subunit protein (but not β3 mRNA) is stable while α1 subunit decreases in the CA1- CA2 hippocampus. These findings indicated that in AD hippocampus, there are complex changes in GABAA receptor subunit composition relative to NFT pathology. This warrants continued investigations, as changes in GABAA receptors could provide protection against glutamate-induced excitotoxicity, and guide development of therapies targeting specific GABAA receptor subunits1,9. The present immunohistochemical study tested the hypothesis that GABAA receptor γ subunits are altered during the progression of NFT pathology in AD hippocampus.
Materials and methods
Postmortem hippocampal tissue was obtained from 16 elderly subjects: 12 with a clinical and neuropathological diagnosis of AD (mean age ± SD = 77.8 ± 13.9 years) and 4 age-matched controls with no history of dementia (ages 73.3 ± 16.5 years). The mean postmortem delay to tissue collection was not significantly different between control and AD groups (Table 1). All AD subjects were participants in a longitudinal research program maintained by the University of Pittsburgh’s Alzheimer’s Disease Research Center (ADRC). Clinical diagnosis of AD was based on a standardized ADRC evaluation at a Consensus Conference, utilizing DSM-IV10 and National Institute of Neurological and Communicative Disorders an Stroke/Alzheimer’s Disease and Related Diseases Association NINCDS/ADRDA11 criteria. AD cases number 14 and 15 had old infarcts restricted to cortical and thalamic regions. Controls were cognitively normal subjects received for regular autopsy and showed no evidence of neurological disorders. Neuropathological diagnosis was determined by a certified neuropathologist, and all AD subjects fulfilled CERAD criteria for the diagnosis of “definite” AD12. All brains (controls and AD) had NFTs and were assigned Braak scores using neuropathological staging for NFT by Braak and Braak8. The four non demented controls were Braak stage I/II with only isolated hippocampal NFT. Four of the 12 AD cases were Braak stage III/IV with moderate hippocampal NFT pathology, and eight AD cases were Braak stage V/VI with severe hippocampal NFT pathology. The three Braak groups are referred to as mildly, moderately or severely affected, referring to the extent of NFT pathology in the hippocampus.
Table 1.
Case demographics
| NFT severity (Braak stage) |
Total | Kruskal Wallis test |
||||
|---|---|---|---|---|---|---|
| Braak I/II | Braak III/IV | Braak V/VI | ||||
| (N=4) | (N=4) | (N=8) | (N=16) | |||
| Age | Mean ± SD (Range) | 73.3 ± 16.5 (57–88) | 79.8 ± 8.4 (72–91) | 76.9 ± 16.5 (48–100) | 76.7 ± 14.2 (48–100) | NS |
| PMI | Mean ± SD (Range) | 7.38 ± 1.3 (5.5–8) | 4.5 ± 1.7 (3–7) | 4.6 ± 1.7 (2–8) | 5.3 ± 2.0 (2–8) | NS |
| BW | Mean ± SD (Range) | 1217 ± 196 (990–1400) | 1217 ± 74 (1150–1300) | 1091 ± 116 (960–1330) | 1154 ± 139 (960–1330) | NS |
| Gender | M/F | 0/4 | 3/1 | 5/3 | 8/8 | |
NFT, neurofibrillary tangle; PMI, postmortem interval, BW, brain weight; NS, not significant; P-value cutoff was 0.01
Brain tissue was processed according to procedures described previously5,7. Blocks of tissue containing hippocampus were cut in the coronal plane at the level of the lateral geniculate body and placed in 0.1 mol/L phosphate buffer (PB, pH = 7.4) containing 4 % paraformaldehyde for 48 h at 4 C. Fixed tissue blocks were cryoprotected by immersion into 30% sucrose in PB for several days, and cut at 40 μm thickness on a sliding, freezing microtome. For each case, at least one tissue section was stained for Nissl substance to delineate the cytoarchitectural boundaries of the hippocampal fields as defined by Duvernoy13.
GABAA receptor subunit immunohistochemistry was performed on free-floating tissue sections as described previously5,7. At least 3 randomly chosen hippocampal tissue sections from each case were immunohistochemically labeled using polyclonal antisera against the γ3 subunits (Sc-7371, Santa Cruz, Santa Cruz CA, 1: 25), which recognizes both γ1 and γ3 (γ1/3), and against the γ2 subunit (345231, Calbiochem, 1: 100) (Fig. 1). As a control for nonspecific staining, sections were incubated with initial incubation media minus the primary antibody, and otherwise processed as described. No positive staining was detectable in these control sections. Sections from all three groups of Braak staging were processed together. In each case, one immunostained section was photographed and then counterstained with the X-34, a fluorescent amyloid binding dye that is a sensitive marker of amyloid-containing structures in AD brain, including senile plaques, NFT (both intracellular and extracellular “ghost” tangles), dystrophic neurites and neuropil threads, as previously described14.
Fig. 1.
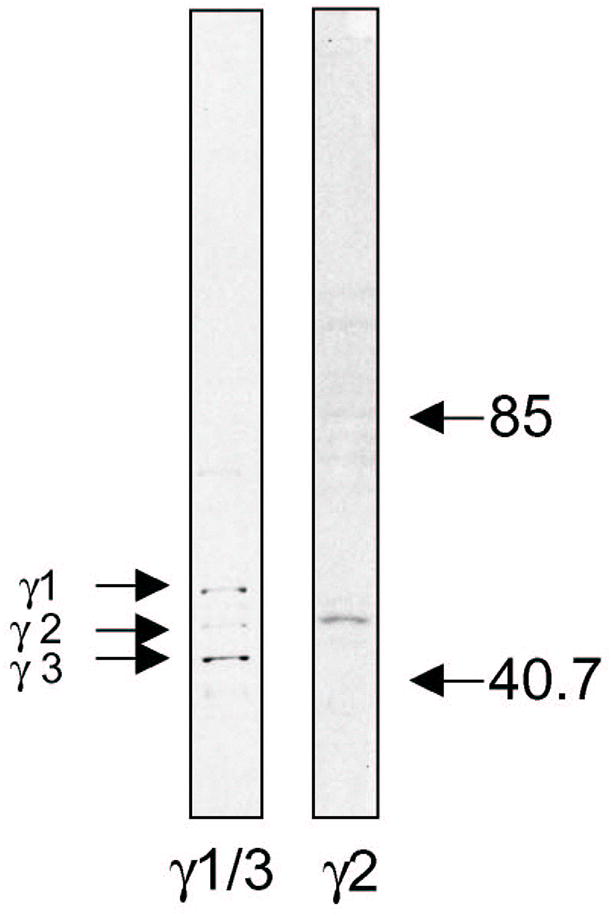
Western blotting shows that Sc-7371 (Santa Cruz) recognizes both γ1 and γ3, and 345231 (Calbiochem) recognizes γ2 subunit.
Results
GABA γ1/3 immunostaining intensity in Braak stage I/II cases was light to moderate in the cytoplasm and in the neuropil of CA2-4 and DG neurons (Fig. 2A,D). Particularly in CA2-4, cellular γ1/3 immunostaining was more intense at the junction of the primary dendrite and cell soma (Fig. 2A,D). In the CA1, γ1/3 immunostaining was relatively weak in pyramidal neurons and smaller multipolar neurons (Fig. 2G), and lightly-immunolabeled dendrites were observed traversing the pyramidal cell layer. In the Braak III/IV cases, γ1/3 immunoreactivity was indistinguishable from the Braak stage I/II cases (Fig. 2B,E,H). In four of the eight Braak V/VI cases, increased intensity ofγ1/3 immunoreaction in the cell soma was observed in all CA regions (Fig. 2C,F,I).
Fig. 2.
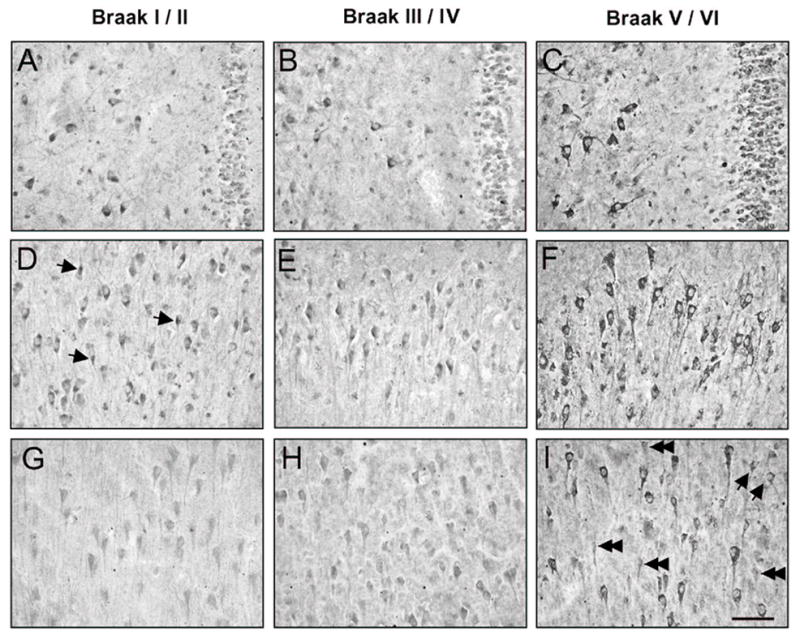
GABAAγ1/3 immunoreactivity in CA4/DG (A-C), CA2/3 (D-F) and CA1 (G-I) hippocampus from a representative Braak stage I/II control case (A,D,G), and AD cases in Braak stages III/IV (B,E,H) and V/VI (C,F,I). Arrows in (D) point to concentrations of γ1/3 immunoreactivity in CA2/3 neurons. In the Braak V/VI AD case, increased immunostaining of neuronal cells is evident in all hippocampal regions, filling the entire cytoplasm of large pyramidal cells and smaller multipolar neurons (arrows in I). A subset of CA1 cells (double arrowheads in I) shows γ1/3 immunoreactivity that is comparable to controls. Scale bar = 100 μm.
We next examined the pattern of GABAA γ2 immunoreactivity in the same three Braak staged groups of subjects. In Braak stage I/II (controls) group, moderately intenseγ2 immunoreactivity was detected in neurons and neuropil throughout the hippocampus (Fig. 3A,D,G,J). In all hippocampal fields, neuropil immunostaining was diffuse, while more prominent cellular immunostaining was punctate and filled the entire cell soma, extending into the proximal dendrites. In the dentate gyrus (DG),γ2 immunoreactivity labeled cell soma and proximal dendrites in the granular cell layer. In the CA fields, γ2 immunoreactivity was found primarily in pyramidal neurons; a subset of smaller, multipolar neurons was also observed. The γ2 staining pattern of the Braak III-IV and V-VI severity groups was comparable to that of control Braak I/II cases, except that labeling of dendrites, but not cell bodies, in the CA1 was less prominent in Braak stage V/VI AD cases relative to Braak stage I/II control cases (Fig. 3).
Fig. 3.

GABAAγ2 immunoreactivity in DG (A-C), CA4 (D-F), CA2/3 (G-I) and CA1 (J-L) hippocampus from a representative Braak stage I/II control case (A,D,G,J), and AD cases in Braak stages III/IV (B,E,H,K) and V/VI (C,F,I,L). Neuronal and neuropil immunostaining is preserved across severity groups in AD. Arrows in (J) point to the labeled primary dendrite of a large CA1 pyramidal neuron. Such dendritic labeling is less prominent in the Braak stage V/VI case, although the immunostaining intensity of cell bodies is comparable to that seen in the other Braak staged groupsacross all hippocampal regions. Scale bar = 50 μm.
Counterstaining of γ1/3 and γ2 immunostained sections with the X-34 (used here to visualize both NFT and amyloid plaques), revealed that neurons immunoreactive to either of the γ subunits did not contain NFT (Fig. 4C-H). This was the case for the majority of the X-34 positive NFT that were of a classic intracellular type, as well as for extracellular “ghost” NFT that were also X-34 positive but with distinct morphological differences (Figure 4G,H, arrow).
Fig. 4.

Paired low-power composites (A-B) and high-power (C-D boxed area in CA3, and E-H boxed areas CA1) images of successive GABAAγ1/3 (A-F) and GABA Aγ2 (G-H) immunoreactivity and X-34 histofluorescent amyloid staining in a single hippocampal tissue section from a Braak stage V/VI AD case. GABAAγ1/3 immunoreactive cells are present in all hippocampal regions, regardless of the degree of X-34 labeled NFT and plaque pathology, which is particularly abundant throughout the subiculum/CA1(A-B). In the CA3 region (C-D), most neurons show prominent γ1/3 immunoreactivity, while X-34 labeling is limited to a single NFT (arrow in D) and scarce neuropil threads. In CA1 (E-F), GABAAγ1/3 immunostained neurons do not contain NFT, and are present throughout the field despite numerous plaques (arrows in F). Likewise, GABA Aγ2 (G-H) immunostained neurons do not contain NFT (H). Asterisk matches blood vessels in each high-power field. Scale bar = 7.5 mm (A,B); 75 μm (C-H). CA1-3 = Cornu Ammonis fields; dg = dentate gyrus; Sub = subiculum.
Among cases within each Braak (NFT) pathology group, there was substantial variability in the extent and types of hippocampal amyloid plaques, which were frequent in the CA1, CA4, and dentate gyrus, and scarce in the CA2-3; no association of these lesions with either γ1/3 orγ2 immunoreactive cells was observed.
Discussion
This study provides the first immunohistochemical analysis of GABAAγ receptor subunits in the human hippocampus staged by the progression of NFT pathology in AD. The overall distribution of GABAAγ immunoreactive elements was similar to that from studies of rat hippocampus, where γ2 immunoreactivity was detected in neuronal soma and principal dendrites as well as in the surrounding neuropil15, whileγ1 and γ3 immunoreactivity was observed in pyramidal cell layers, and only faintly in the neuropil16.
Studies of AD hippocampus using radio-labeled ligand binding to GABAA receptors containing γ subunits (especially γ2) have produced inconsistent data17–21, and are difficult to compare to the present immunohistochemical study. For example, loss of GABAAγ protein in AD hippocampus could reflect atrophy related loss of neurons expressing these receptors. However, if remaining cells upregulate γ subunits, as suggested in the present study, the total binding level in the hippocampus would show no change, even in cases affected with more extensive hippocampal cell loss. In our previous studies of hippocampal GABA receptor subunits, α1 immunoreactivity and β3 subunit mRNA were reduced in the Braak V/VI cases, while β2 subunit mRNA was relatively preserved4,6. Other studies have reported reduction of α5 protein22 and mRNA23, while no change in β1 protein22. These studies, together with our present observation of γ subunit changes, suggest that each subunit of the GABAA receptor undergoes unique responses during the progression of NFT pathology in AD hippocampus. The results of our experiments that employed dual labeling of tissue sections with γ1/3 orγ2 immunohistochemistry and the amyloid binding dye X-34 demonstrated that neurons immunoreactive to γ subunits did not contain NFT. This indicates either a protective role of neuronal GABAAγ receptor expression, or their loss precedes neurofibrillary degeneration in AD. While we are not aware of a study demonstrating a direct cause-effect link between dysfunction of the hippocampal GABAergic system and NFT formation, it has been hypothesized that the loss of GABAergic inhibitory innervation contributes to excitotoxicity, due to excessive Ca2+ influx, which can lead to abnormal phosphorylation of tau and its aggregation into NFT 24–26. This warrants future multiple-immunolabeling studies examining the relationship between γ receptor subunit changes and specific markers of different stages in the course of NFT formation (e.g., tau hyper-phosphorylation) and neuronal cell death in AD.
Changes in receptor subunit composition during the course of AD could affect regional GABA binding, drug responses and ability to maintain inhibitory tone in the CNS. Currently, the effect of benzodiazepine therapy on psychological and behavioral symptoms in AD patients remains unclear as there have been few controlled clinical trials to date, which have yielded mixed results27. In addition, no study has focused on changes in benzodiazepine sensitivity in AD patients. However, a study by Fastbom et al. (1998) reported a significantly lower incidence of AD in subjects with a history of benzodiazepine use, suggesting a protective role of benzodiazepines in AD, likely through glutamatergic inhibition 28. AD therapies should target the disturbed inhibition-excitation balance, a major driving force for neuronal degenerative changes in AD29.
Conclusions
The results of the present study suggest that neurons expressing GABAAγ receptor subunits resist the progression of neurofibrillary degeneration in AD hippocampus. The potential protective effect of these receptors against NFT pathology warrants their further investigation for the benefit of developing novel therapeutic strategies that target the GABAergic system in AD.
Acknowledgments
This work was supported by NIH grant NIA AG05133 (STDeK) and by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (KM).
References
- 1.Armstrong DM, Sheffield R, Mishizen-Eberz AJ, et al. Plasticity of glutamate and GABAA receptors in the hippocampus of patients with Alzheimer’s disease. Cell Mol Neurobiol. 2003;23:491–505. doi: 10.1023/A:1025063811290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer AM, Gershon S. Is the neuronal basis of Alzheimer’s disease cholinergic or glutamatergic? FASEB J. 1990;4:2745–2752. doi: 10.1096/fasebj.4.10.2165009. [DOI] [PubMed] [Google Scholar]
- 3.Whiting PJ, Bonnert TP, McKernan RM, et al. Molecular and functional diversity of the expanding GABAA receptor gene family. Ann NY Acad Sci. 1999;868:645–653. doi: 10.1111/j.1749-6632.1999.tb11341.x. [DOI] [PubMed] [Google Scholar]
- 4.Mizukami K, Ikonomovic MD, Grayson DR, Sheffield R, Armstrong DM. Immunohistochemical study of GABAA receptor α1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 1998;799:148–155. doi: 10.1016/s0006-8993(98)00437-5. [DOI] [PubMed] [Google Scholar]
- 5.Mizukami K, Ikonomovic MD, Grayson DR, et al. Immunohistochemical study of GABAA receptor β2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp Neurol. 1997;147:333–345. doi: 10.1006/exnr.1997.6591. [DOI] [PubMed] [Google Scholar]
- 6.Mizukami K, Grayson DR, Ikonomovic MD, Sheffield R, Armstrong DM. GABAA receptor β2 and β3 subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology. Mol Brain Res. 1998;56:268–272. doi: 10.1016/s0169-328x(97)00347-1. [DOI] [PubMed] [Google Scholar]
- 7.Iwakiri M, Mizukami K, Ikonomovic MD, et al. Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: association with Braak staging. Acta Neuropathol (Berl) 2005;109:467–474. doi: 10.1007/s00401-005-0985-9. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Eid CN, Jr, Rose GM. Cognition enhancement strategies by ion channel modulation of neurotransmission. Curr Pharm De. 1999;5:345–61. [PubMed] [Google Scholar]
- 10.American Psychiatric Association. Diagnostic and StatisticalManual of Mental Disorders. 4. Washington, DC: AmericanPsychiatric Press; 1994. [Google Scholar]
- 11.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 12.Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 13.Duvernoy HM. The Human Hippocampus. Munchen: JF Bergmann Verlag; 1988. [Google Scholar]
- 14.Ikonomovic MD, Abrahamson EE, Isanski BA, et al. X-34 labeling of abnormal protein aggregates during the progression of Alzheimer’s disease. Methods Enzymol. 2006;412:123–44. doi: 10.1016/S0076-6879(06)12009-1. [DOI] [PubMed] [Google Scholar]
- 15.Fritschy JM, Mohjer H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 16.Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W. GABAA receptor subunits in the rat hippocampus. I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80:987–1000. doi: 10.1016/s0306-4522(97)00146-2. [DOI] [PubMed] [Google Scholar]
- 17.Cross AJ, Crow TJ, Johnson JA, et al. Studies on neurotransmitter receptor systems in neocortex and hippocampus in senile dementia of the Alzheimer type. J Neurol Sci. 1984;64:109–117. doi: 10.1016/0022-510x(84)90029-7. [DOI] [PubMed] [Google Scholar]
- 18.Greenamyre JT, Penny JB, D’Amato CJ, Young AB. Dementia of the Alzheimer’s type: changes in hippocampal L-[3H] glutamate binding. J Neurochem. 1987;48:543–551. doi: 10.1111/j.1471-4159.1987.tb04127.x. [DOI] [PubMed] [Google Scholar]
- 19.Shimohama S, Taniguchi T, Fujiwara M, Kameyama M. Changes in benzodiazepine receptors in Alzheimer-type dementia. Ann Neurol. 1988;23:404–406. doi: 10.1002/ana.410230419. [DOI] [PubMed] [Google Scholar]
- 20.Jansen KLR, Faull RLM, Dragunow M, Synek BL. Alzheimer’s disease: changes in hippocampal N-Methyl-D-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors–an autoradiographic study. Neuroscience. 1990;39:613–627. doi: 10.1016/0306-4522(90)90246-z. [DOI] [PubMed] [Google Scholar]
- 21.Penny JB, Maragos WF, Greenamyre JT, Debowey DI, Hollingsworth Z, Young AB. Excitatory amino acid binding sites in the hippocampal region of Alzheimer’s disease and other dementias. J Neurol Neurosurg Psychiatry. 1990;53:314–320. doi: 10.1136/jnnp.53.4.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rissman RA, De Blas AL, Armstrong DM. GABA(A) receptors in aging and Alzheimer’s disease. J Neurochem. 2007;103:1285–92. doi: 10.1111/j.1471-4159.2007.04832.x. [DOI] [PubMed] [Google Scholar]
- 23.Howell O, Atack JR, Dewar D, McKernan RM, Sur C. Density and pharmacology of alpha5 subunit-containing GABA(A) receptors are preserved in hippocampus of Alzheimer’s disease patients. Neuroscience. 2000;98:669–75. doi: 10.1016/s0306-4522(00)00163-9. [DOI] [PubMed] [Google Scholar]
- 24.Smith-Swintosky VL, Mattson MP. Glutamate, beta-amyloid precursor proteins, and calcium mediated neurofibrillary degeneration. J Neural Transm Suppl. 1994;44:29–45. doi: 10.1007/978-3-7091-9350-1_3. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP. Calcium and neuronal injury in Alzheimer’s disease. Contributions of beta-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann N Y Acad Sci. 1994;747:50–76. [PubMed] [Google Scholar]
- 26.Schmitt HP. Neuro-modulation, aminergic neuro-disinhibition and neuro-degeneration. Draft of a comprehensive theory for Alzheimer disease. Med Hypotheses. 2005;65:1106–1119. doi: 10.1016/j.mehy.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Lanctôt KL, Herrmann N, Mazzotta P, Khan LR, Ingber N. GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can J Psychiatry. 2004;49:439–453. doi: 10.1177/070674370404900705. [DOI] [PubMed] [Google Scholar]
- 28.Fastbom J, Forsell Y, Winblad B. Benzodiazepines may have protective effects against Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:14–17. doi: 10.1097/00002093-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt HP. On the paradox of ion channel blockade and its benefits in the treatment of Alzheimer disease. Med Hypotheses. 2005;65:259–265. doi: 10.1016/j.mehy.2005.03.011. [DOI] [PubMed] [Google Scholar]