

Abstract
Non-selective cyclooxygenase (COX) inhibitors target many of the same cancer-associated molecular pathways as COX-2 specific inhibitors. Although these non-steroidal anti-inflammatory drugs (NSAIDs) are often associated with gastrointestinal toxicity, there is renewed interest in their use as colorectal cancer (CRC) chemopreventive agents due to the adverse side effects associated with long-term use of selective COX-2 inhibitors. In this study, we investigated the effects of long-term use (up to 25 years) of NSAIDs (ibuprofen or aspirin) on adenoma pathology and β-catenin-mediated signaling in sporadic human colon adenomas. Although NSAID use did not impact overall adenoma size or degree of dysplasia, it did cause a significant inhibition of nuclear β-catenin localization, which correlated with suppression of cyclin D1 expression. In order to further elucidate the effect of these agents in regulating β-catenin, we treated SW480 colon cancer cells with a panel of NSAIDs and determined their effects on β-catenin levels and cellular localization. In agreement with our in vivo results, both S-ibuprofen and aspirin were found to decrease total levels of β-catenin, while increasing its phosphorylation. In addition, S-ibuprofen induced both degradation of IκBα and nuclear localization of NF-κB. Despite its nuclear localization, however, the activation of the NF-κB target genes, Bcl-2, survivin and cyclin D1, was suppressed. This reduction in NF-κB transcriptional activity may be due to increased phosphorylation of GSK-3β following S-ibuprofen treatment. These data suggest that ibuprofen can effectively target both the Wnt/β-catenin and NF-κB pathways, and potentially uncovers a novel mechanism through which NSAIDS may exert their chemopreventive efficacy.
Keywords: colon cancer, chemoprevention, NSAID, ibuprofen, β-catenin, NF-κB, GSK-3β
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit the cyclooxygenase-1 and -2 (COX-1 and-2) enzymes, have an impressive history as effective chemopreventive agents for colorectal cancer (CRC) (1, 2). These effects are largely dependent on their ability to inhibit the production of proliferative and inflammatory prostaglandins (PGs), most notably PGE2 (3). More recently, specific COX-2 inhibitors were developed as a means to reduce the gastrointestinal toxicity associated with inhibiting COX-1. However, treatment with this drug class is associated with increased incidence of adverse cardiovascular events, even after short-term exposures (4, 5). Consequently, these negative side effects have limited their clinical application in chemoprevention (6–8). Therefore, there is renewed interest in developing non-selective NSAIDs for clinical use in chemoprevention, especially those chemically modified to circumvent gastrointestinal side effects.
Several double-blinded, placebo-controlled and case-controlled studies have reported that both selective COX-2 inhibitors, such as celecoxib and rofecoxib, and non-selective inhibitors, such as sulindac and aspirin, are effective in reducing adenoma recurrence, as well as the risk for CRC (9–16). Apart from their abilities to inhibit COX activity and prostaglandin production, both selective and non-selective COX-2 inhibitors can target β-catenin, a key mediator of colon tumorigenesis. NSAIDs, including celecoxib, sulindac and aspirin, can increase the phosphorylation of β-catenin, thus decreasing its nuclear accumulation and transcription of Wnt/β-catenin target genes, such as cyclin D1, c-myc, and PPARδ (17–22). Sulindac has also been shown to inhibit β-catenin expression in FAP-related adenomas (21, 23).
In addition to their effects on Wnt/β-catenin signaling, a number of NSAIDs, including aspirin, diclofenac, sulindac, sulindac sulfone, indomethacin, celecoxib and NS-398, have been shown to induce the degradation of the NF-κB inhibitor, IκBα, resulting in the direct nuclear translocation of the p65 subunit of NF-κB in the absence of additional NF-κB activators (24–29). In fact, additional studies in human CRC cell lines have demonstrated that aspirin and non-aspirin NSAIDs induce the translocation of NF-κB-p65 into the nucleolus, resulting in inhibition of NF-κB related gene expression. (25, 29, 30). It has thus been proposed that NSAID-mediated suppression of NF-κB target gene expression may contribute to the COX-2-independent growth inhibition and apoptosis induced by these drugs (26, 31, 32). Thus, the identification of additional chemopreventive agents that target both β-catenin and NF-κB-mediated signaling in human tumors may lead to more effective therapeutic approaches.
In the following study, we evaluated the effects of the non-specific COX-2 inhibitor, ibuprofen, on the Wnt/β-catenin pathway in human sporadic colon adenomas. Notably, it was found that ibuprofen treatment inhibited the accumulation of nuclear β-catenin expression, an outcome that correlated with reduced levels of cyclin D1. In colon cancer cells in vitro, S-ibuprofen, the active form in cell culture, along with a panel of other NSAIDs, increased phosphorylation-dependent inactivation of β-catenin. In addition, S-ibuprofen induced degradation of IκBα together with phosphorylation and nuclear localization of NF-κB-p65. Despite its nuclear localization, however, the activation of NF-κB target genes was suppressed, possibly related to the observed increase in phosphorylation of GSK-3β. These data suggest that ibuprofen may provide an effective alternative, or useful adjunct, to selective COX-2 inhibitors for use in chemoprevention.
Materials and methods
Human Subjects
Formalin-fixed, paraffin embedded (FFPE) sections of adenomas from 37 patients taking daily NSAIDs (ibuprofen or aspirin) for up to 25 years and from 39 patients not taking daily NSAIDs were obtained from the Mayo Clinic (Rochester, MN). In total there were 76 samples comprised of 32 tubular adenomas and 5 tubulovillous adenomas from patients taking NSAIDs as well as 27 tubular adenomas and 12 tubulovillous adenomas from patients not taking NSAIDs. There was also a separate group of 17 adenomas from FAP patients given sulindac for one year. Samples were obtained in accordance with University of Connecticut Health Center Institutional Review Board (IRB) guidelines and use of these samples was approved by the IRB at the Mayo Clinic (Rochester, MN).
Immunohistochemistry
Staining was performed as described in our previous study (33). Briefly, FFPE tissues were de-paraffinized and incubated with 1% hydrogen peroxide for 20 min at room temperature. Tissues were subjected to antigen retrieval and blocked with 10% normal goat serum (Vector Laboratories, Inc.) in PBS-Brij solution. Tissues were then incubated overnight at 4°C with anti-β-catenin (1:2000; Sigma-Aldrich), anti-cyclin D1 (1:100; Immuno-Biological Laboratories; Gunma, Japan, a kind gift from Dr. A. Arnold), anti-proliferating cell nuclear antigen (PCNA; 1:150; Novacastra Laboratories) or anti-p21 (1:100; Neomarkers) antibodies. Tissues were washed and incubated with biotinylated anti-mouse or anti-rabbit secondary antibodies for 30 min at room temperature followed by incubation with avidin-biotin complex reagent (Vector Laboratories, Inc.) for 30 min at room temperature. Signal was detected with 3,3’-diaminobenzidine (DAB) solution (Vector Laboratories) and tissues were counterstained with hematoxylin. For scoring of β-catenin and cyclin D1 nuclear immunostaining, the following scale was used: grade 1, 1–10% positive nuclei; grade 2, 10–20% positive nuclei, grade 3, 20–50% positive nuclei; grade 4, >50% positive nuclei. Samples were considered negative if there were no positively staining nuclei.
Cell culture
SW480 and DLD-1 cells were maintained in Dulbecco’s Modified Eagle medium supplemented with 10% (v/v) fetal bovine serum and 1% penicillin/streptomycin. Cells were treated with 1 mM S-ibuprofen, 5 mM aspirin, 100 µM sulindac sulfide, 600 µM sulindac sulfone, 600 µM indomethacin (all Sigma-Aldrich) or 1 µM PGE2 (Cayman Chemical) for 24 hours. Control cells were treated with vehicle (DMSO or 100% ethanol) alone.
PGE2 Quantification
The amount of PGE2 present in cells was quantified using the PGE2 Monoclonal EIA Kit (Cayman Chemical) according to the manufacturer’s instructions. Briefly, cells were grown in 24-well plates to 80% confluence. Following drug treatments (as above for 24 hours), cell culture supernatants were collected and immediately incubated for 18 hours at 4°C with a PGE2-acetylcholinesterase conjugate (PGE2 Tracer) and a PGE2 monoclonal antibody in plates pre-coated with goat anti-mouse IgG. Following washing to remove unbound reagents, the plates were developed with Ellman’s Reagent, which contains the substrate for acetylcholinesterase, and read at 405 nm. The absorbance readings were proportional to the amount of bound PGE2 Tracer and inversely proportional to the amount of PGE2 in the samples. A standard curve was also measured using known concentrations of PGE2 in order to calculate the absolute amounts of PGE2 in the samples.
Immunoblotting
Following drug treatment (as above for 24 hours), cells were lysed in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA and 1% Triton X-100 supplemented with protease and phosphatase inhibitors (50 mM NaF, 10 mM Na β-glycerol PO4, 5 mM Na pyroPO4, 1 mM sodium vanadate, 1 µg/ml leupeptin and 1 mM PMSF). Following centrifugation at 14000 rpm for 15 minutes at 4°C, the supernatant was removed and quantified for total protein. 30 µg of protein was incubated at 95°C for 5 minutes and 4× sample loading buffer containing 8% β-mercaptoethanol and loaded onto a 10–15% polyacrylamide gel. Proteins were transferred to an Immobilon-P membrane (Millipore Corp.) and blocked in 5% non-fat dry milk in TBS-T (1× TBS, 0.1% Tween-20) for 1 hour. Blots were incubated overnight at 4°C with phospho-β-catenin (1:2000), phospho-NF-κB-p65 (1:1000), PARP (1:1000), total NF-κB-p65 (1:1000), phospho-GSK-3β (1:1000), total GSK-3β (1:1000), total IκBα (1:1000), phospho-AKT (1:1000), or total AKT (1:1000) antibodies (Cell Signaling), as well as β-catenin (1:2000) (Sigma-Aldrich), cyclin D1 (1:1000) (IBL), Bcl-2 (1:1000) (BD Transduction Laboratories), Survivin (1:1000) (Lab Vision Corp.), β-tubulin (1:2000) (Sigma-Aldrich), or β-actin (1:6000; Santa Cruz Biotechnology) antibodies. Blots were then washed multiple times and incubated with goat anti-mouse IgG HRP (1:10,000; Upstate Biotechnology) or donkey anti-rabbit IgG HRP (1:4000; Santa Cruz Biotechnology) for 45 minutes at room temperature. HRP was visualized with enhanced luminal reagent (Millipore Corp.).
Immunofluorescence
Cells were grown on 22 × 22 mm glass coverslips in 6-well plates to approximately 50% confluence and then treated with drugs (as above, for 24 hours). Cells were washed with cold 1× PBS and then fixed in 4% para-formaldehyde for 10 min followed by permeabilization in 0.5% Triton X-100 in PBS for 5 minutes. Cells were then blocked in 3% BSA (10 mg/ml) in PBS for 1 hour at room temperature followed by incubation with anti-β-catenin antibody (1:100; Sigma-Aldrich) in 1% BSA in PBS for 1 hour at room temperature. Cells were washed in PBS and then incubated with secondary antibody (goat anti-mouse IgG Alexa 568; 1:200 in 1% BSA in PBS; Molecular Probes) for 1 hour at room temperature in the dark. Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI; 1:10,000). Cells on coverslips were mounted onto glass slides using Prolong Gold Antifade Reagent (Molecular Probes) and visualized using an Olympus IX50 fluorescence microscope (Olympus Corp.).
Statistical analyses
For associations comparing adenoma phenotype and NSAID use, statistical analyses were performed using a two-tailed Fisher’s exact test. To determine statistical significance of PGE2 levels, analyses were performed using ANOVA between groups. P values were considered to be statistically significant at P < 0.05.
Results
Long-term NSAID use does not significantly affect adenoma size or morphology
The sample set of human adenomas was comprised of 76 sporadic adenomas, 37 from patients taking daily ibuprofen or aspirin for one year or more and 39 from patients with no history of NSAID use. In addition, there were 17 adenomas from FAP patients given sulindac for one year. To evaluate the effects of one year or more of ibuprofen or aspirin use on adenoma phenotype, we performed statistical analyses comparing dysplasia or size with NSAID use. As shown in Table 1, there was no significant correlation found between degree of dysplasia and NSAID use (P = 0.12, two-tailed Fisher’s exact test) or between size and NSAID use (P = 0.21, two-tailed Fisher’s exact test). However, given the limited number of dysplastic adenomas observed in the present study (5 out of a total of 76), a conclusive determination of the effects of NSAID use on histological tumor grade is not possible. This indicates that in this sample set, NSAID use did not significantly impact the pathological features of the adenomas.
Table 1.
Patient demographics and adenoma classification
| Group | Overall | − NSAID | + NSAIDa |
|---|---|---|---|
| A. Sex | |||
| Male | 50 | 25 (50%) | 25 (50%) |
| Female | 26 | 14 (54%) | 12 (46%) |
| B. Age (yrs) | |||
| <40 | 2 | 2 (100%) | 0 (0%) |
| 40–65 | 43 | 26 (60%) | 17 (40%) |
| >65 | 31 | 11 (35%) | 20 (65%) |
| C. Polyp Type | |||
| TAb | 61 | 29 (48%) | 32 (52%) |
| TVc | 15 | 10 (67%) | 5 (33%) |
| FAPd | 17 | 0 (0%) | 17 (100%) |
| D. Dysplasiae | |||
| low grade | 71 | 35 (49%) | 36 (51%) |
| high grade | 4 | 4 (100%) | 0 (0%) |
| invasive | 1 | 0 (0%) | 1 (100%) |
| E. Size (mm)f | |||
| <10 | 23 | 9 (39%) | 14 (61%) |
| 10–30 | 46 | 26 (57%) | 20 (43%) |
| >30 | 7 | 4 (57%) | 3 (43%) |
NSAID treatment is aspirin or ibuprofen for sporadic patients and sulinac for FAP patients, both for 1 year
TA, tubular adenoma
TV, tubulovillous adenoma; sporadic patients only
Familial adenomatous polyposis (FAP) patients, no patient demographic or adenoma classification data available
Dysplasia, P=0.12 between groups (two-tailed Fisher’s exact test)
Size, P=0.21 between groups (two-tailed Fisher’s exact test)
Long-term NSAID use affects β-catenin cellular localization
NSAIDs, including aspirin, sulindac and indomethacin, have been shown to decrease nuclear levels of β-catenin in colon cancer cells (17–19). In order to determine if long-term NSAID use in this adenoma sample set was able to affect cellular localization of β-catenin, immunohistochemical analysis of β-catenin was performed in the 76 sporadic adenomas and 17 FAP adenomas. As shown in the representative examples in Figure 1A, in adenomas from non-NSAID patients there was strong nuclear staining of β-catenin (a, b), whereas NSAID use significantly decreased nuclear β-catenin expression (c, d). Nuclear β-catenin was quantified and these data are shown in Table 2. Overall 14/36 (39%) of adenomas in the non-NSAID group had nuclear β-catenin staining, compared to only 3/34 (9%) in adenomas from patients with a history of NSAID intake (P = 0.0047). In addition, the three adenomas from the NSAID group with positive nuclear β-catenin staining were all from patients taking daily aspirin, indicating that daily ibuprofen use completely inhibited nuclear β-catenin. Neither ibuprofen nor aspirin use had an effect on cell proliferation, as assessed by PCNA immunostaining, nor p21 expression. In the adenomas from FAP patients treated with sulindac, 9/17 (53%) had nuclear β-catenin staining, indicating that sulindac was only variably effective in inhibiting translocation of β-catenin in these patients. However, it was not possible to determine if the percentage of positive nuclear β-catenin cells in the adenomas was suppressed by sulindac because there was no non-treated group for comparison.
Figure 1. β-catenin and cyclin D1 expression in human sporadic adenomas.

A, representative examples of β-catenin immunohistochemical analyses in untreated (a,b) and NSAID-treated (c, d) adenomas at 200× (a,c) and 400× (b,d) magnification, performed as described under Materials and methods. B, representative examples of cyclin D1 immunohistochemical analysis in untreated (a,b) and NSAID treated (c,d) adenomas at 200× (a,c) and 400× (b,d) magnification, performed as described under Materials and Methods. C, representative examples of β-catenin (a,b) and cyclin D1 (c,d) co-localized immunostaining in serial sections of an untreated adenoma at 100× (a,c) and 400× (b,d) magnification.
Table 2.
Association of beta-catenin/cyclin D1 nuclear localization with NSAID intake
| grade | − NSAID | + NSAID | FAP sulindac | + nuclear beta-catenin | |
|---|---|---|---|---|---|
| + nuclear beta-catenina | 1 | 8 | 1 | N/A | |
| 2 | 5 | 1 | |||
| 3 | 1 | 1 | |||
| 4 | 0 | 0 | |||
| total positive | 14 (39%) | 3 (9%) | 9 (53%) | ||
| total | 36 | 34 | 17 | N/A | |
| + nuclear cyclin D1b | 1 | 14 | 10 | 12 (75%) | |
| 2 | 4 | 1 | |||
| 3 | 0 | 0 | |||
| 4 | 0 | 0 | |||
| total positive | 18 (50%) | 11 (31%) | N/A | ||
| total | 36 | 35 | N/A | 16 |
There is a significant association between nuclear β-catenin and NSAID use, P = 0.0047 (two-tailed Fisher’s exact test).
There is a significant association between nuclear cyclin D1 and nuclear β-catenin in adenomas, P = 0.022 (two-tailed Fisher’s exact test) but not with NSAID use, P = 0.149.
Nuclear β-catenin correlates with nuclear cyclin D1 expression in human adenomas
When β-catenin translocates to the nucleus and binds to TCF/LEF transcription factors, it drives the transcription of a panel of Wnt target genes including cyclin D1, c-myc, COX-2, and PPARδ (34–36). Thus, we wanted to determine if nuclear β-catenin localization in the adenoma samples might have downstream effects on the expression of cyclin D1. As shown in the representative photomicrographs (Figure 1B), immunohistochemical analysis revealed strong nuclear expression of cyclin D1 in non-NSAID adenomas that had nuclear β-catenin staining (a, b); however, in adenomas from patients in the NSAID exposure group, the absence of nuclear β-catenin was associated with diminished nuclear cyclin D1 immunostaining (c, d). As shown in Table 2, however, there was no significant correlation between nuclear cyclin D1 expression and NSAID use (P > 0.05); however 12/16 (75%) adenomas with nuclear β-catenin expression also had nuclear expression of cyclin D1 (P = 0.022). This finding is illustrated in greater detail in Figure 1C, which shows a representative example of β-catenin (a–c) and cyclin D1 (d–f) co-localization in serial sections of a representative non-NSAID adenoma. This indicates that nuclear β-catenin does affect Wnt target gene expression.
Ibuprofen affects β-catenin localization in human colon cancer cells
To further investigate the effect of ibuprofen on β-catenin cellular localization, we treated human colon cancer cells with S-ibuprofen and a panel of other NSAIDs for comparison. Treatment of SW480 or DLD-1 cells with either 1 mM S-ibuprofen, 5 mM aspirin, 100 µM sulindac sulfide, 600 µM sulindac sulfone or 600 µM indomethacin for 24 hours significantly inhibited PGE2 production compared to vehicle alone, indicating that COX-1/2 enzymes were being effectively targeted by drug treatment (Figure 2A & B).
Figure 2. NSAIDs inhibit PGE2 production in human colon cancer cells.

SW480 (A) and DLD-1 (B) human colon cancer cell lines were treated with a panel of NSAIDs (S-ibuprofen, aspirin, sulindac sulfide, sulindac sulfone and indomethacin) for 24 hours as described under Materials and Methods. At the end of drug treatment, supernatants were collected and PGE2 levels were determined by ELISA. Columns, mean of three samples per group; bars, SE. *, P = 0.0008 for A and 0.033 for B, ANOVA between groups.
To determine the effect of this panel of NSAIDs on β-catenin expression, immunofluorescence microscopy was performed on SW480 cells, which have mutant APC and consequent high levels of nuclear β-catenin. As shown in Figure 3A, strong membrane, cytoplasmic and nuclear staining of β-catenin was present in untreated cells. However, treatment with NSAIDs greatly decreased the overall intensity of β-catenin staining, most notably with S-ibuprofen, sulindac sulfide and indomethacin treatment. These data indicate that ibuprofen is able to inhibit the accumulation of nuclear β-catenin, as well as suppress the levels of total cellular β-catenin despite the loss of APC protein. Figure 3B shows that in cytoplasmic extracts from SW480 cells, S-ibuprofen, aspirin, sulindac sulfide and indomethacin were able to increase the accumulation of the phosphorylated form of β-catenin, suggesting that these drugs decrease nuclear levels of β-catenin by increasing its cytoplasmic degradation.
Figure 3. S-ibuprofen inhibits β-catenin expression and nuclear localization in SW480 colon cancer cells.

A, Immunofluorescence microscopy of β-catenin (Alexa Fluor 568, red) in SW480 cells following treatment with a panel of NSAIDs (1 mM S-ibuprofen, 5 mM aspirin, 100 µM sulindac sulfide, 600 µM sulindac sulfone and 600 µM indomethacin) for 24 hours as described in Materials & Methods. Nuclei were counter-stained with 4',6-diamidino-2-phenylindole (DAPI, blue). Merged images represent the overlay of the image at 603 nm (β-catenin, Alexa Fluor 568) with the image at 488 nm (DAPI). B, Western blot analysis of phospho-β-catenin and total β-catenin in the cytoplasmic fraction of SW480 cells following 24-hour treatment with a panel of NSAIDs (S-ibuprofen, aspirin, sulindac sulfide, sulindac sulfone and indomethacin). PGE2 was added as a negative control. Total cell lysates from SW480 cells containing both nuclear and cytoplasmic fractions is also included as a negative control. Blots were re-probed with anti-β-actin as a loading control.
Ibuprofen treatment results in NF-κB-p65 phosphorylation and nuclear localization
To explore the possibility that ibuprofen may affect other signaling pathways in addition to the Wnt/β-catenin cascade, NF-κB signaling was examined in human SW480 colon cancer cells. NSAIDs, including aspirin, sulindac and diclofenac, have been shown to activate NF-κB in a variety of CRC cells (24, 26). As shown in Figure 4A, 24 hour treatment of SW480 cells with increasing concentrations of S-ibuprofen resulted in a dose-dependent decrease in the levels of the NF-κB inhibitor protein, IκBα. Furthermore, a dose-dependent increase in the phosphorylated form (serine-536) of the p65 subunit of the NF-κB transcription factor complex was observed (Fig. 4A). While phosphorylation of NF-κB-p65 at this serine residue has been demonstrated to be important in the kinetics of its nuclear import (37), further direct evidence of NF-κB-p65 nuclear localization was required. As shown in Figure 4B, 24-hour treatment with S-ibuprofen resulted in nuclear translocation and accumulation of NF-κB-p65 in SW480 cells, as assessed by immunofluorescence staining.
Figure 4. Ibuprofen treatment relieves IκBα-mediated repression of NF-κB-p65 and induces its phosphorylation and nuclear localization.
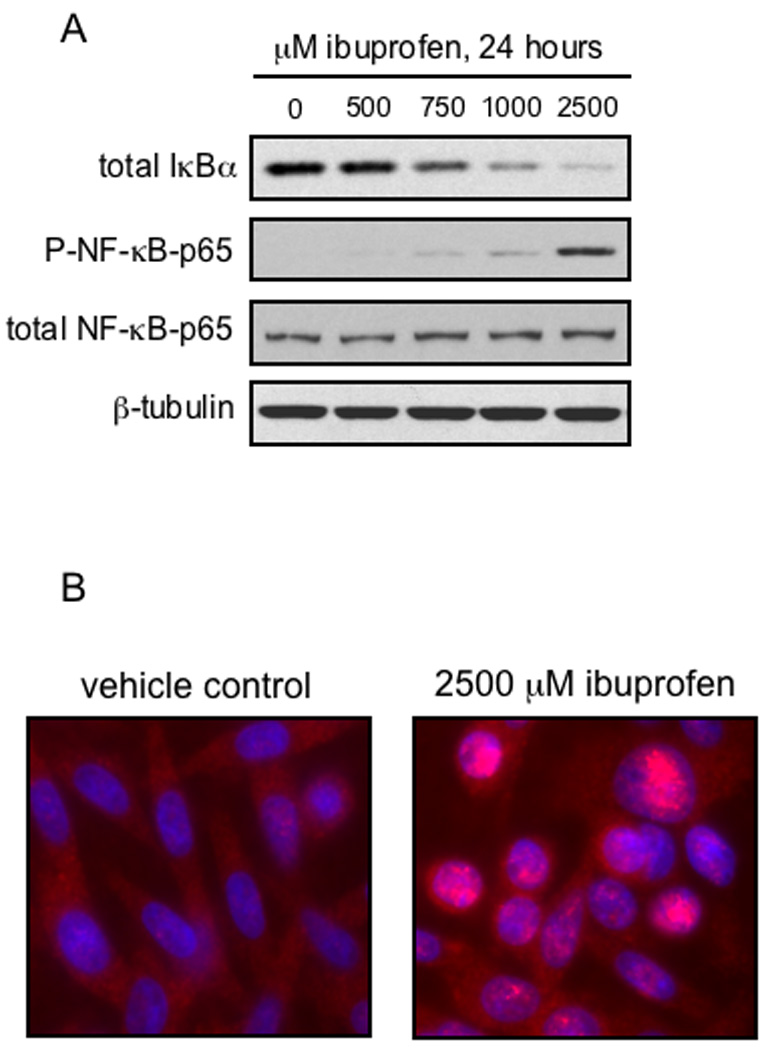
A, SW480 cells were treated for 24 hours with increasing doses of S-ibuprofen and protein lysates were collected as described under Materials and methods. Proteins were resolved by SDS-PAGE and blotted for IκBα, phospho-NF-κB-p65 (serine-536) and β-tubulin (loading control). The phospho-NF-κB-p65 blot was stripped and blotted for total NF-κB-p65. B, SW480 cells were grown overnight on cover-slips and then treated for 24 hours with either vehicle control or 2500 µM S-ibuprofen. Immunofluorescence microscopy was performed to visualize NF-κB-p65 (Alexa Fluor 568, red). Nuclei were counterstained with DAPI (blue). Localization of NF-κB-p65 in the nucleus is represented by magenta staining after merging of the two images.
Ibuprofen treatment decreases expression of NF-κB target genes, inhibits GSK-3β and induces apoptosis
After demonstrating that ibuprofen treatment (in the absence of additional NFκB activation) resulted in nuclear localization of NF-κB-p65, we next examined the possible effects of prolonged ibuprofen treatment on NF-κB target gene expression. As demonstrated in Figure 5A, despite the observed nuclear localization, expression of the NF-κB target genes, Bcl-2, survivin and cyclin-D1, were decreased in a time-dependent manner following treatment of SW480 cells with S-ibuprofen, an effect that occurred through 72 hours. Additionally, S-ibuprofen caused a time-dependent increase in apoptosis in SW480 cells treated, assessed by PARP cleavage (Figure 5A). Recent studies have indicated that the serine/threonine kinase, GSK-3β, positively regulates NF-κB-mediated gene expression (38, 39). Thus we investigated the possibility that ibuprofen treatment may inhibit the activity of GSK-3β. GSK-3β is present constitutively in an active form, and phosphorylation of its serine-9 residue by Akt results in the inhibition of its signaling activity (40). As shown in Figure 5A, there was a time-dependent increase in the inhibitory phosphorylation of GSK-3β at serine-9. Additionally, as shown in Figure 5B, we observed a rapid (within minutes) increase in the phosphorylation of GSK-3β at serine-9 in SW480 cells following treatment with S-ibuprofen. Interestingly, there was a concomitant increase in the active, phosphorylated form of Akt (serine-476), consistent with its established role in GSK-3β inhibition (40–42).
Figure 5. Ibuprofen decreases selective NF-κB target gene expression and induces inhibitory phosphorylation of GSK-3β.
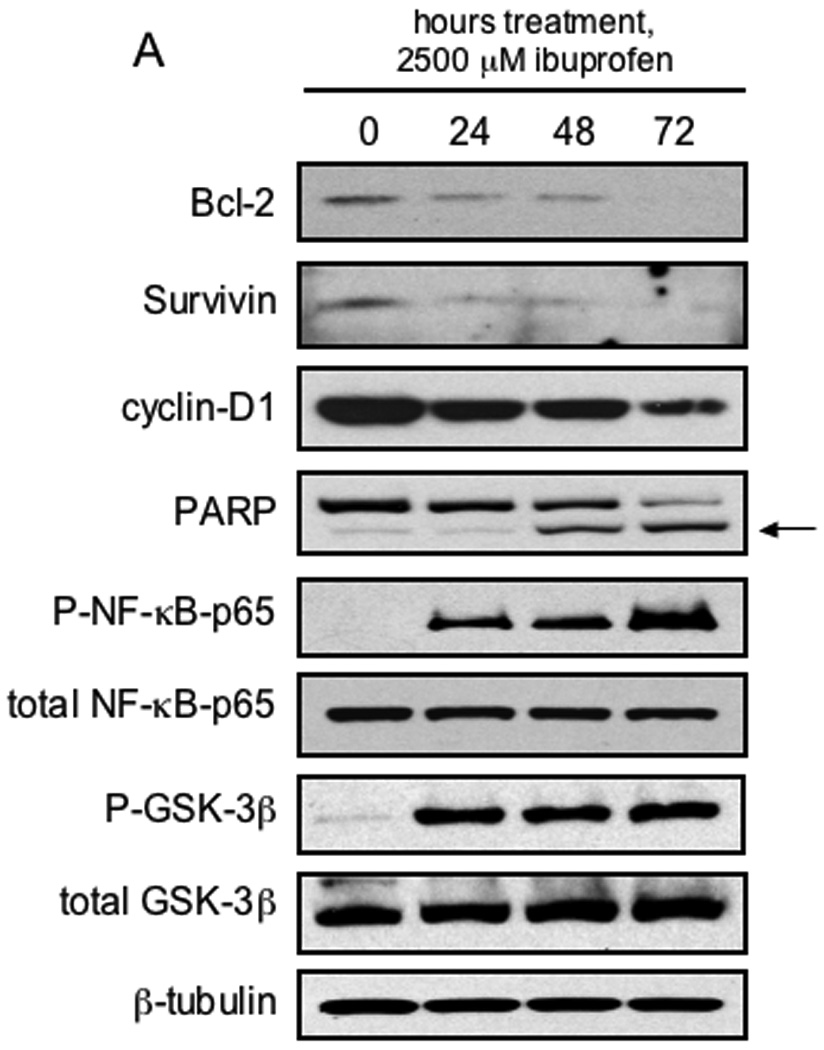
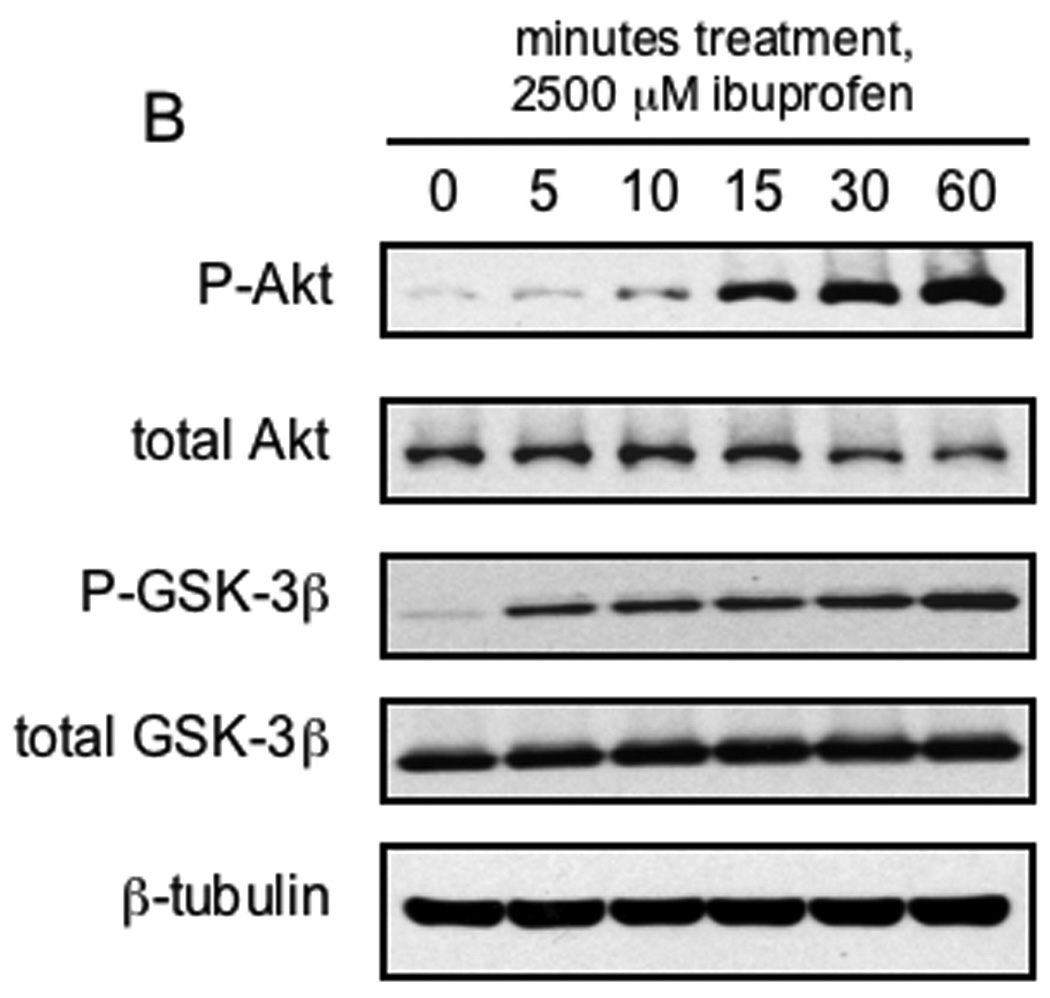
A, SW480 cells were treated with 2500 µM S-ibuprofen for either 24, 48 or 72 hours and protein lysates were collected. Additional SW480 cells were treated for 24 hours with vehicle only and served as a baseline control. Proteins were resolved by SDS-PAGE and initially blotted for the NF-κB target genes Bcl-2, survivin and cyclin-D1. Protein lysates were also blotted for PARP, phospho-NF-κB-p65 (serine-536), phospho-GSK-3β (serine-9) and β-tubulin. Blots were stripped and blotted for total NF-κB-p65 and total GSK-3β. Arrow indicates the apoptosis-dependent cleavage product of PARP. B, SW480 cells were treated with 2500 µM S-ibuprofen for various time points up to 60 minutes and protein lysates were collected. Proteins were resolved by SDS-PAGE and initially blotted for phospho-Akt (serine-473), phospho-GSK-3β (serine-9) and β-tubulin. Blots were stripped and re-blotted for total Akt and total GSK-3β.
Discussion
The adverse cardiovascular effects associated with specific COX-2 inhibitors have limited their application in cancer chemoprevention, especially in population-risk individuals (43–46). Therefore, a renewed interest in the development of less toxic non-selective NSAIDs has emerged. Our study found significant inhibition of nuclear β-catenin expression in sporadic colon adenomas from patients with a history of daily intake of ibuprofen or aspirin. One important distinguishing feature of our study is related to the patient population, comprised primarily of sporadic adenoma cases. Currently, the only clinical human data demonstrating that NSAIDs can suppress β-catenin expression are in FAP patients (21, 23). Of note, no adenomas from subjects in our study who reported daily ibuprofen use exhibited the presence of nuclear β-catenin, indicating that this drug was able to completely suppress its nuclear translocation. Surprisingly, adenomas from FAP patients maintained on sulindac for up to one year showed only a modest reduction in the levels of nuclear β-catenin. However, pre-treatment adenomas were not available from either population group, and thus it is difficult to establish a definitive comparison in response between these groups. It remains a possibility that sulindac treatment suppressed nuclear β-catenin translocation if compared to pre-treatment adenomas.
Studies in human colon cancer cells have found that inhibition of nuclear β-catenin by NSAIDs, such as sulindac and indomethacin, results in a dramatic down-regulation of its transcriptional targets, including cyclin D1 and c-myc (22, 47). Consistent with these in vitro findings, our study also found an association between nuclear β-catenin expression and positive cyclin D1 staining in the sporadic colon adenomas. In fact, 75% of adenomas with nuclear β-catenin staining also exhibited nuclear cyclin D1 expression. Furthermore, the suppression of nuclear β-catenin by either ibuprofen or aspirin was associated with a reduction in nuclear cyclin D1 expression. These data indicate that nuclear expression of β-catenin in adenomas can activate proliferative Wnt target genes, which can also be suppressed by NSAID intake.
To further investigate the effects of ibuprofen on β-catenin localization, additional studies were performed using human colon cancer cell lines. As anticipated, it was found that S-ibuprofen, as well as a panel of additional NSAIDs including aspirin, sulindac sulfide, sulindac sulfone and indomethacin, significantly inhibited PGE2 production in two human colon cancer cell lines, SW480 and DLD-1, both of which have mutant APC {Yang, 2006 #27. However, we selected the SW480 cells for further analysis based on their strong nuclear localization of β-catenin (48). In agreement with our in vivo results, S-ibuprofen inhibited nuclear translocation of β-catenin in SW480 cells, as well as stimulated its phosphorylation at Serine-33,37 and Threonine-41, indicating its increased cytoplasmic degradation. NSAIDs have been shown to induce β-catenin phosphorylation by acting as phosphatase inhibitors {Bos, 2006 #16}. Specifically, aspirin has been shown to negatively regulate protein phosphatase 2A (PP2A) enzymatic activity through phosphorylation of the inhibitory Tyr307 in PP2A (17). Thus, ibuprofen may also act as a phosphatase inhibitor to stabilize the phosphorylation of β-catenin.
In addition to its effects on β-catenin, S-ibuprofen treatment also resulted in the degradation of IκBα in SW480 cells. This was accompanied by phosphorylation of the p65 subunit of NF-κB (Figure 4A). This signaling pathway has been established to play a key role in controlling the expression of a myriad of genes involved in inflammation, differentiation, proliferation and apoptotic cell death (49). In resting cells, the heterodimeric NF-κB transcription factor complex is sequestered in the cytoplasm by the inhibitor protein IκBα. Cellular stimulation by various agents can result in the phosphorylation and proteosomal degradation of IκBα, causing the phosphorylation, release and nuclear translocation of the NF-κB complex (49). As shown in Figure 4B, further evaluation of NF-κB-p65 staining using immunofluorescence analysis revealed its punctate, nuclear localization after treatment with S-ibuprofen. Previous studies have shown that nucleolar sequestration of the p65 subunit of NF-κB is essential for aspirin-mediated apoptosis (26, 50, 51). Furthermore, nucleolar sequestration of NF-κB-p65 may contribute to the suppression of NF-κB target gene activation by preventing proper binding of the NF-κB complex to the promoters of these genes (51). Future studies will be aimed at determining whether S-ibuprofen treatment results in nucleolar localization of NF-κB-p65.
Consistent with these findings, we also observed that treatment of SW480 cells in vitro with S-ibuprofen (for up to 72 hours) resulted in decreased expression of a limited set of NF-κB target genes, including Bcl-2, survivin and cyclin-D1 (Figure 5A). Furthermore, the suppression of NF-κB target gene activation in SW480 cells was accompanied by a time-dependent increase in apoptosis. These data, however, do not directly address the possibility that S-ibuprofen directly mediates the nucleolar localization of NF-κB-p65. Thus we sought an alternative mechanism to explain the phosphorylation and nuclear localization of NF-κB-p65 that occurs despite a marked decrease in the expression of this subset of NF-κB target genes.
Further examination of protein lysates from SW480 cells treated for 72 hours with S-ibuprofen (Figure 5A) revealed a time-dependent increase in inhibitory phosphorylation of GSK-3β at serine-9, an effect that occurred within minutes of treatment (Figure 5B). GSK-3β is a multifunctional serine/threonine kinase that was described initially as a regulator of glycogen synthase activity (52, 53). Elevated levels of GSK-3β have been found in human colon cancer cell lines and in primary colon tumors (54). Importantly, there is accumulating evidence for a direct relationship between GSK-3β activity and the NF-κB signaling pathway (55–57). For example, several in vitro studies in MEFs and in pancreatic cancer cell lines have shown that GSK-3β regulates NF-κB signaling downstream of IκBα degradation and nuclear translocation of NF-κB (39, 55). Additionally, GSK-3β is required for the expression of certain NF-κB target genes (58) and NF-κB-mediated cell survival (39). An earlier study also provided evidence that treatment of SW480 cells with aspirin resulted in increased phosphorylation of β-catenin, as well as phosphorylation of GSK-3β at serine-9 (59). As noted above, it has also been shown that aspirin inhibits the phosphatases PP2A (17, 60). In the present study, we examined whether treatment of SW480 cells with S-ibuprofen may lead to an increase in inhibitory PP2A phosphorylation levels. However, we were unable to establish time- or dose-dependent differences in PP2A phosphorylation after S-ibuprofen treatment (data not shown). Although we did not establish a direct association between GSK-3β and PP2A, there are many other phosphatase enzymes that may possibly affect S-ibuprofen-induced GSK-3β inhibitory phosphorylation (61). Future studies will be directed at examining the possibility that other phosphatases may affect GSK-3β signaling with respect to S-ibuprofen treatment.
As noted above, the long-term use of traditional NSAIDs has been associated with gastrointestinal side effects. This toxicity is generally attributed to the key role that COX-1 plays in maintaining the integrity of the intestinal mucosa. The use of modified NSAIDs, including those containing nitric oxide (NO) that purportedly maintain mucosal integrity in a similar manner to that of endogenous prostaglandins, may circumvent gastrointestinal toxicity (5). Other approaches include the use of dual COX and 5-lipoxygenase inhibitors, and NSAIDs that are capable of releasing hydrogen sulfide (62). Thus, continued development of these novel strategies to circumvent gastrointestinal toxicity while still affording chemopreventive benefit are clearly needed to expand the clinical safety and efficacy of this important drug class.
To our knowledge, our study is the first to demonstrate that ibuprofen is effective at inhibiting β-catenin nuclear translocation in both human adenomas and in colon cancer cells in vitro. It is also the first to demonstrate that an NSAID can suppress β-catenin in sporadic adenomas following long-term treatment in patients. This indicates that the Wnt pathway in colon adenomas is sensitive to NSAID exposure, regardless of whether the lesions are from FAP patients, or are sporadic in origin. In addition, our current data suggest that S-ibuprofen treatment results in the phosphorylation and nuclear translocation of NF-κB. However, this effect is associated with a concomitant loss in transcriptional activity. This reduced transcriptional activity of NF-κB may be due to the novel finding of increased S-ibuprofen-dependent phosphorylation of GSK-3β, leading to inhibition of NF-κB signaling. Future studies will establish the precise role that ibuprofen treatment plays in augmenting the GSK-3β-NF-κB signaling axis. Additionally, since the Wnt/β-catenin and NF-κB signaling pathways share overlapping target genes, it will be important to establish the relative contribution of these pathway(s) to the transcriptional changes observed after ibuprofen treatment. In summary, we believe that these findings warrant the development of case-controlled studies for CRC chemoprevention using ibuprofen.
Acknowledgments
This work was supported by National Institute of Health Grants 5RO1CA125691 and 5R01CA114635 (to D.W.R.)
References
- 1.Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 2.Williams CS, Smalley W, DuBois RN. Aspirin use and potential mechanisms for colorectal cancer prevention. J Clin Invest. 1997;100(6):1325–1329. doi: 10.1172/JCI119651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiff SJ, Rigas B. Nonsteroidal anti-inflammatory drugs and colorectal cancer: evolving concepts of their chemopreventive actions. Gastroenterology. 1997;113(6):1992–1998. doi: 10.1016/s0016-5085(97)99999-6. [DOI] [PubMed] [Google Scholar]
- 4.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340(24):1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 6.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial. Cancer Prev Res (Phila Pa) 2009;2(4):310–321. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 8.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 9.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 10.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348(10):891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 11.Baron JA, Sandler RS, Bresalier RS, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131(6):1674–1682. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 12.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 13.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346(14):1054–1059. doi: 10.1056/NEJMoa012015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris RE, Beebe-Donk J, Alshafie GA. Similar reductions in the risk of human colon cancer by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer. 2008;8:237. doi: 10.1186/1471-2407-8-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348(10):883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 16.Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade (review) Oncol Rep. 2005;13(4):559–583. [PubMed] [Google Scholar]
- 17.Bos CL, Kodach LL, van den Brink GR, et al. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25(49):6447–6456. doi: 10.1038/sj.onc.1209658. [DOI] [PubMed] [Google Scholar]
- 18.Hawcroft G, D'Amico M, Albanese C, Markham AF, Pestell RG, Hull MA. Indomethacin induces differential expression of beta-catenin, gamma-catenin and T-cell factor target genes in human colorectal cancer cells. Carcinogenesis. 2002;23(1):107–114. doi: 10.1093/carcin/23.1.107. [DOI] [PubMed] [Google Scholar]
- 19.Rice PL, Kelloff J, Sullivan H, et al. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol Cancer Ther. 2003;2(9):885–892. [PubMed] [Google Scholar]
- 20.Tuynman JB, Vermeulen L, Boon EM, et al. Cyclooxygenase-2 inhibition inhibits c-Met kinase activity and Wnt activity in colon cancer. Cancer Res. 2008;68(4):1213–1220. doi: 10.1158/0008-5472.CAN-07-5172. [DOI] [PubMed] [Google Scholar]
- 21.Boon EM, Keller JJ, Wormhoudt TA, et al. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 2004;90(1):224–229. doi: 10.1038/sj.bjc.6601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner SH, Hawcroft G, Hull MA. Effect of nonsteroidal anti-inflammatory drugs on beta-catenin protein levels and catenin-related transcription in human colorectal cancer cells. Br J Cancer. 2004;91(1):153–163. doi: 10.1038/sj.bjc.6601901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koornstra JJ, Rijcken FE, Oldenhuis CN, et al. Sulindac inhibits beta-catenin expression in normal-appearing colon of hereditary nonpolyposis colorectal cancer and familial adenomatous polyposis patients. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1608–1612. doi: 10.1158/1055-9965.EPI-05-0112. [DOI] [PubMed] [Google Scholar]
- 24.Cho M, Gwak J, Park S, et al. Diclofenac attenuates Wnt/beta-catenin signaling in colon cancer cells by activation of NF-kappaB. FEBS Lett. 2005;579(20):4213–4218. doi: 10.1016/j.febslet.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 25.Din FV, Dunlop MG, Stark LA. Evidence for colorectal cancer cell specificity of aspirin effects on NF kappa B signalling and apoptosis. Br J Cancer. 2004;91(2):381–388. doi: 10.1038/sj.bjc.6601913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loveridge CJ, MacDonald AD, Thoms HC, Dunlop MG, Stark LA. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NF-kappaB. Oncogene. 2008;27(18):2648–2655. doi: 10.1038/sj.onc.1210891. [DOI] [PubMed] [Google Scholar]
- 27.Niederberger E, Tegeder I, Vetter G, et al. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-kappaB. Faseb J. 2001;15(9):1622–1624. doi: 10.1096/fj.00-0716fje. [DOI] [PubMed] [Google Scholar]
- 28.Smartt HJ, Elder DJ, Hicks DJ, Williams NA, Paraskeva C. Increased NF-kappaB DNA binding but not transcriptional activity during apoptosis induced by the COX-2-selective inhibitor NS-398 in colorectal carcinoma cells. Br J Cancer. 2003;89(7):1358–1365. doi: 10.1038/sj.bjc.6601266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark LA, Din FV, Zwacka RM, Dunlop MG. Aspirin-induced activation of the NF-kappaB signaling pathway: a novel mechanism for aspirin-mediated apoptosis in colon cancer cells. Faseb J. 2001;15(7):1273–1275. [PubMed] [Google Scholar]
- 30.Stark LA, Reid K, Sansom OJ, et al. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28(5):968–976. doi: 10.1093/carcin/bgl220. [DOI] [PubMed] [Google Scholar]
- 31.Shao J, Fujiwara T, Kadowaki Y, et al. Overexpression of the wild-type p53 gene inhibits NF-kappaB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene. 2000;19(6):726–736. doi: 10.1038/sj.onc.1203383. [DOI] [PubMed] [Google Scholar]
- 32.Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene. 2004;23(57):9247–9258. doi: 10.1038/sj.onc.1208169. [DOI] [PubMed] [Google Scholar]
- 33.Nakanishi M, Montrose DC, Clark P, et al. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 34.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11(24):3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 35.Clapper ML, Coudry J, Chang WC. beta-catenin-mediated signaling: a molecular target for early chemopreventive intervention. Mutat Res. 2004;555(1–2):97–105. doi: 10.1016/j.mrfmmm.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303(5663):1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattioli I, Sebald A, Bucher C, et al. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol. 2004;172(10):6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 38.Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood. 2007;110(2):735–742. doi: 10.1182/blood-2006-12-060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65(6):2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- 40.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 41.Chen EY, Mazure NM, Cooper JA, Giaccia AJ. Hypoxia activates a platelet-derived growth factor receptor/phosphatidylinositol 3-kinase/Akt pathway that results in glycogen synthase kinase-3 inactivation. Cancer Res. 2001;61(6):2429–2433. [PubMed] [Google Scholar]
- 42.Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW, Hunninghake GW. Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J Immunol. 2001;166(7):4713–4720. doi: 10.4049/jimmunol.166.7.4713. [DOI] [PubMed] [Google Scholar]
- 43.Abdel-Tawab M, Zettl H, Schubert-Zsilavecz M. Nonsteroidal anti-inflammatory drugs: a critical review on current concepts applied to reduce gastrointestinal toxicity. Curr Med Chem. 2009;16(16):2042–2063. doi: 10.2174/092986709788682209. [DOI] [PubMed] [Google Scholar]
- 44.Half E, Arber N. Colon cancer: preventive agents and the present status of chemoprevention. Expert Opin Pharmacother. 2009;10(2):211–219. doi: 10.1517/14656560802560153. [DOI] [PubMed] [Google Scholar]
- 45.Pereira C, Pimentel-Nunes P, Brandao C, Moreira-Dias L, Medeiros R, Dinis-Ribeiro M. COX-2 polymorphisms and colorectal cancer risk: a strategy for chemoprevention. Eur J Gastroenterol Hepatol. 2010 doi: 10.1097/MEG.0b013e3283352cbb. [DOI] [PubMed] [Google Scholar]
- 46.Rostom A, Dube C, Lewin G, et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146(5):376–389. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 47.Han A, Song Z, Tong C, et al. Sulindac suppresses beta-catenin expression in human cancer cells. Eur J Pharmacol. 2008;583(1):26–31. doi: 10.1016/j.ejphar.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem. 2006;281(26):17751–17757. doi: 10.1074/jbc.M600831200. [DOI] [PubMed] [Google Scholar]
- 49.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 50.Stark LA, Dunlop MG. Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Mol Cell Biol. 2005;25(14):5985–6004. doi: 10.1128/MCB.25.14.5985-6004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007;67(4):1660–1669. doi: 10.1158/0008-5472.CAN-06-1038. [DOI] [PubMed] [Google Scholar]
- 52.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114(2–3):147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- 54.Shakoori A, Ougolkov A, Yu ZW, et al. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334(4):1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 55.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406(6791):86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 56.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376(6536):167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 57.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284(5412):321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 58.Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25(19):8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dihlmann S, Klein S, Doeberitz MvMK. Reduction of beta-catenin/T-cell transcription factor signaling by aspirin and indomethacin is caused by an increased stabilization of phosphorylated beta-catenin. Mol Cancer Ther. 2003;2(6):509–516. [PubMed] [Google Scholar]
- 60.Bos CL, Diks SH, Hardwick JC, Walburg KV, Peppelenbosch MP, Richel DJ. Protein phosphatase 2A is required for mesalazine-dependent inhibition of Wnt/beta-catenin pathway activity. Carcinogenesis. 2006;27(12):2371–2382. doi: 10.1093/carcin/bgl071. [DOI] [PubMed] [Google Scholar]
- 61.Gallego M, Virshup DM. Protein serine/threonine phosphatases: life, death, and sleeping. Curr Opin Cell Biol. 2005;17(2):197–202. doi: 10.1016/j.ceb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Coruzzi G, Venturi N, Spaggiari S. Gastrointestinal safety of novel nonsteroidal antiinflammatory drugs: selective COX-2 inhibitors and beyond. Acta Biomed. 2007;78(2):96–110. [PubMed] [Google Scholar]