

SUMMARY
Centriole duplication occurs once per cell cycle, ensuring that each cell contains two centrosomes, each containing a mother-daughter pair of tightly engaged centrioles at mitotic entry. Loss of the tight engagement between mother and daughter centrioles appears to license the next round of centriole duplication. However, the molecular mechanisms regulating this process remain largely unknown. Mutations in CDK5RAP2, which encodes a centrosomal protein, cause autosomal recessive primary microcephaly (MCPH) in humans. Here we show that CDK5RAP2 loss of function in mice causes centriole amplification with a preponderance of single, unpaired centrioles and increased numbers of daughter-daughter centriole pairs. These results indicate that CDK5RAP2 is required to maintain centriole engagement and cohesion, thereby restricting centriole replication. Early in mitosis, amplified centrosomes assemble multipolar spindles in CDK5RAP2 mutant cells. Moreover, both mother and daughter centrioles are amplified, and the excess mother centrioles template multiple primary cilia in CDK5RAP2 mutant cells.
INTRODUCTION
Centrosomes perform diverse and critical functions. Perturbations in centrosome function form the etiological basis for a growing number of human diseases (Nigg and Raff, 2009). The mammalian centrosome consists of a centriole pair surrounded by pericentriolar material (PCM), a proteinaceous matrix that supports microtubule nucleation, polymerization and stability (Doxsey, 2001; Bornens, 2002; Luders and Stearns, 2007). Centrosomes contain two structurally distinct centrioles, a mature “mother” centriole, distinguished by distal and subdistal appendages, and a “daughter” centriole that lacks these appendages (Vorobjev and Chentsov Yu, 1982; Paintrand et al., 1992). In the absence of centrioles, the PCM becomes unstable and dispersed (Bobinnec et al., 1998). Although typical centrosomes consist of centriole pairs, a single centriole can also assemble PCM. Therefore, centriole numbers define centrosome numbers within cells.
The centriole duplication cycle is tightly regulated to ensure that duplication occurs only once per cell cycle (Tsou and Stearns, 2006a; Azimzadeh and Bornens, 2007; Bettencourt-Dias and Glover, 2007; Nigg, 2007). Deregulation of the duplication cycle can lead to centrosome amplification, thereby increasing the frequency of multipolar spindles and likely contributing to aneuploidy due to errors in chromosome segregation (Brinkley, 2001; Pihan and Doxsey, 2003; Nigg, 2006; Ganem et al., 2009). The formation of multipolar spindles when centrosomes amplify is offset by a centrosome clustering mechanism that suppresses such occurrences (Sluder et al., 1997; Quintyne et al., 2005; Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008). The mitotic checkpoint is activated in cells with multiple centrosomes (Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008; Ganem et al., 2009), yet aberrant microtubule attachments to kinetochores can still occur prior to clustering (Ganem et al., 2009).
Centriole biogenesis is a highly orchestrated process that culminates in the organization of triplet microtubule blades into an elegant 9-fold rotationally symmetric cylinder (Bettencourt-Dias and Glover, 2007; Strnad and Gonczy, 2008). The proteins and mechanisms involved in restricting centriole duplication to once per cell cycle have recently begun to be elucidated (Tsou and Stearns, 2006a; Nigg, 2007; Strnad and Gonczy, 2008). After cell division, a cell inherits a pair of disengaged but cohered centrioles (Nigg, 2007). Centriole cohesion is the tethering of centriole pairs by cohesion fibers during interphase (Bahe et al., 2005; Yang et al., 2006). In G1, the mother centriole initiates formation of the primary cilium (Ishikawa et al., 2005; Bettencourt-Dias and Glover, 2007). In S-phase, each centriole templates the assembly of a single daughter centriole, which grows from its proximal base and remains tightly bound, or “engaged” until disengagement occurs at mitosis. During G2, the engaged centriole pairs remain tethered by cohesion. Cohesion is lost at mitotic onset coincident with centrosome separation in preparation for mitotic spindle assembly (Faragher and Fry, 2003). The mother-daughter centriole pairs, however, remain engaged until anaphase of the ensuing mitosis. At the metaphase to anaphase transition, centriole disengagement requires the activation of the protease separase (Tsou and Stearns, 2006b). According to current models, the loss of centriole engagement is the licensing step that allows centrioles to undergo a single round of replication (Tsou and Stearns, 2006b, a; Nigg, 2007).
CDK5RAP2 is a resident centrosome protein and an ortholog of Drosophila centrosomin (CNN) (Megraw et al., 2001). Homozygous mutations in CDK5RAP2, microcephalin (MCPH1), abnormal spindle-like microcephaly associated (ASPM), centromere associated protein J (SAS4/CPAP/CENPJ), and SCL/TAL1 interrupting locus (STIL) cause autosomal recessive primary microcephaly (MCPH, [MIM 251200]), a condition characterized by the overall reduction of brain size (Bond et al., 2002; Jackson et al., 2002; Trimborn et al., 2004; Bond et al., 2005; Kumar et al., 2009). All five of the mapped MCPH genes encode centrosomal proteins, implicating a critical role for the centrosome in brain development (Bond et al., 2005; Zhong et al., 2005; Zhong et al., 2006; Pfaff et al., 2007). In addition, the essential centrosomal protein pericentrin is linked to Seckel syndrome [MIM 210600] and microcephalic osteodysplastic primordial dwarfism type II (MOPD2, [MIM 210720]) (Griffith et al., 2008; Rauch et al., 2008). Like MCPH, Seckel syndrome and MOPD2 are associated with reduced brain size, suggesting a common role for these centrosome proteins in related processes during development.
In this study we show that centrioles are amplified in loss-of-function CDK5RAP2 mutant cells. Consequently, CDK5RAP2 mutant mouse embryonic fibroblasts (MEFs) frequently displayed multipolar spindles and were delayed in mitosis. In addition, the excess mother centrioles in CDK5RAP2 mutant MEFs promoted assembly of multiple primary cilia. In strong loss-of-function CDK5RAP2 mutant MEFs, centrioles were disengaged and lost the normal paired configuration. We propose that CDK5RAP2 is required to maintain centriole engagement and cohesion. As centriole engagement is a key step in licensing centriole replication, CDK5RAP2 is therefore a negative regulator of centriole licensing. Thus, CDK5RAP2 restricts centriole duplication by maintaining centriole engagement.
RESULTS
Centrosomal levels of CDK5RAP2 change with the cell cycle
CDK5RAP2 is a member of the centrosomin family of proteins, conserved among eukaryotes from yeast to humans. The founding member of this family, centrosomin (CNN) is required for mitotic centrosome function in Drosophila melanogaster (Megraw et al., 2001; Mahoney et al., 2006). The yeast S. pombe ortholog, Mto1p, is similarly required for MTOC functions (Sawin et al., 2004; Venkatram et al., 2004; Zimmerman and Chang, 2005). CDK5RAP2 shares homology with two domains in Drosophila CNN, CNN Motifs 1 and 2 (Figure 1A). In flies, CNN is essential for the formation of the PCM; it is recruited to centrosomes at mitotic onset and maintains centrosomal localization throughout mitosis until its dissociation during cytokinesis (Li and Kaufman, 1996). We examined the subcellular localization and dynamics of CDK5RAP2 to investigate its relationship with the centrosome.
Figure 1.

CDK5RAP2 is a centrosomal protein whose levels change with the cell cycle (A) Schematic comparing Drosophila centrosomin (CNN), with human and mouse CDK5RAP2 proteins. Two conserved domains, each about 60 amino acids in length, are designated CNN motifs 1 (CM1) and 2 (CM2). The black line indicates the region used to raise antibodies. The identities and similarities between CNN and mCDK5RAP2 within CM1 and CM2 are shown. In addition to these conserved blocks, CNN family members contain extensive coiled-coil regions. (B) Immunofluorescence images of NIH-3T3 mouse fibroblasts stained for CDK5RAP2 (red), the microtubule marker α-tubulin (green), and DNA (blue) at different stages of the cell cycle. Insets are enlargements of CDK5RAP2 signal to highlight changes in CDK5RAP2 levels at centrosomes. Scale bar = 10 μm. See also Figure S1.
To examine subcellular localization and dynamics of CDK5RAP2 during the cell cycle, antibodies were produced against the amino-terminal end of mouse CDK5RAP2 (Figure 1A). Immunostaining showed that CDK5RAP2 is a centrosomal protein (Figure 1B), consistent with previous reports (Bond et al., 2005; Graser et al., 2007; Fong et al., 2008), and localizes to centrosomes independent of microtubules (Figure 2E). GFP-CDK5RAP2 also localized to centrosomes in transiently transfected NIH-3T3 mouse fibroblasts (Figure S1). Next, changes in CDK5RAP2 levels at centrosomes during the cell cycle were examined in NIH-3T3 cells (Figure 1B). Levels of CDK5RAP2 were relatively low at interphase centrosomes, increased at mitotic prophase, and remained high throughout mitosis until telophase, when signal dropped to interphase levels (Figure 1B). While CDK5RAP2 is a centriolar resident (Graser et al., 2007; Fong et al., 2008), the results here show that CDK5RAP2 accumulates at centrosomes in mitosis, consistent with CDK5RAP2 localization to the PCM, which grows at mitosis (Palazzo et al., 2000). Thus, CDK5RAP2 is a centrosomal protein whose centrosomal levels are regulated in a cell cycle-dependent manner. Because CDK5RAP2 impacts human health, we next sought to examine the function of CDK5RAP2 in mammalian cells by generating two mouse models for CDK5RAP2 deficiency.
Figure 2.

Centrosome disruption in CDK5RAP2 mutant MEFs
(A,C) The truncated CDK5RAP2 products expressed in CDK5RAP2RRF465/RRF465 (A) and CDK5RAP2RRU031/RRU031 (C) mutant mice are similar to the two mapped human mutations. Schematics show the protein fragments predicted to result from the human mutations, E385fsX4 and S81X (Bond et al., 2005), and the CDK5RAP2/β-GEO mutant fusion proteins expressed in CDK5RAP2RRF465/RRF465 and CDK5RAP2RRU031/RRU031 mice.
(B,D) Western blots of CDK5RAP2 from whole cell lysates collected from sibling MEF cultures of the indicated genotypes. The positions of the wild type and fusion proteins expressed in the RRF465 mutant (B), and the relative expression level of wild type CDK5RAP2 in the RRU031 mutant (D) are indicated. α-tubulin was probed as a loading control.
(E) Nocodazole-treated CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs stained for CDK5RAP2 (red), α-tubulin (green), and DNA (blue).
(F) CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs stained for CDK5RAP2 (red), the cohesion fiber protein rootletin (green), and DNA (blue).
(G) Images of wild type and CDK5RAP2 mutant MEFs stained for γ-tubulin (red), the cohesion fiber protein rootletin (green), and DNA (blue).
(H) Comparison between strong PCM fibers (CDK5RAP2+/+: 72% ± vs. CDK5RAP2RRU031/RRU031: 34% ± 2%, p<0.05) and weak PCM fibers (CDK5RAP2+/+: 28% ± vs. CDK5RAP2RRU031/RRU031: 66% ± , p<0.05). The CDK5RAP2RRU031/RRU031 MEF in (G) is an example of weak PCM fibers. n = 50 cells total from two independent cell lines.
(I). Centrosome splitting was scored when centrosomes were >2μm apart (CDK5RAP2+/+: 27.4% ± 3.6% vs. CDK5RAP2RRU031/RRU031: 47.2% ± 6.4%, p<0.05). n = 110 cells total from five independent experiments.
Insets are enlargements of the centrosome region and PCM fibers. Scale bar = 10 μm. See also Figure S2.
Generation of CDK5RAP2 mutant mice
CDK5RAP2 mutant mice were derived using two Bay Genomics embryonic stem cell clones, RRU031 and RRF465, harboring transposon splice-trap insertions within introns 3 and 12 of the CDK5RAP2 locus, respectively. The splice-trap vector used to generate the CDK5RAP2 mutations contains a superior splice site upstream of a “β-Geo” fusion cassette, a fusion between β-galactosidase and neomycin phosphotransferase (Stryke et al., 2003).
In CDK5RAP2Gt(RRU031)Byg/Gt(RRU031)Byg and CDK5RAP2Gt(RRF465)Byg/Gt(RRF465)Byg mutant mice (hereafter referred to as CDK5RAP2RRU031/RRU031 and CDK5RAP2RRF465/RRF465), the splice traps result in translation of the first 64 and 435 amino acids of CDK5RAP2, respectively, fused to β-Geo (Figure 2A-D). The truncated proteins expressed from these CDK5RAP2 alleles are similar to the predicted protein products of the human CDK5RAP2 mutations, S81X and E385fsX4, which result in truncations of 81 and 389 amino acids, respectively (Figures 2A and C) (Bond et al., 2005). Homozygous mutant CDK5RAP2 mice (RRU031 and RRF465) were viable and born at expected Mendelian ratios. Female CDK5RAP2RRF465/RRF465 mice were fertile (n=5). Males, however, showed variable fertility, with 2 out of 6 mice showing infertility (n=6). Neither group of CDK5RAP2 mutant mice showed evidence of reduced brain size (Figure S2A and data not shown).
We characterized and compared CDK5RAP2 expression between the two mutants using affinity-purified CDK5RAP2 antibody. The region used to raise antibodies was retained in the CDK5RAP2RRF465/RRF465 mutant fusion protein (Figures 1A and 2B). Immunoblots of whole cell lysates showed that CDK5RAP2+/+ MEFs expressed full-length protein, heterozygous CDK5RAP2+/RRF465 MEFs expressed full-length and mutant fusion proteins, and homozygous CDK5RAP2RRF465/RRF465 MEFs expressed only the CDK5RAP2 mutant fusion protein (Figure 2B). On the other hand, homozygous CDK5RAP2RRU031/RRU031 MEFs expressed full-length CDK5RAP2 at ~7% of wild type levels (Figure 2D). Therefore, the RRU031 mutation is leaky and hypomorphic. These results reveal differences in the relative strengths of the two splice trap mutations, and establish a mouse mutant model of MCPH and of the centrosomin family of centrosomal proteins.
The centrosome phenotypes described below support the observations that the CDK5RAP2RRU031/RRU031 mutation is hypomorphic, expressing a low amount of full-length CDK5RAP2, and the CDK5RAP2RRF465/RRF465 mutation is more severe.
Centrosome disruptions in CDK5RAP2 mutant cells
To investigate CDK5RAP2 functions in centrosome structure and regulation we established MEF primary cell cultures from embryonic day E14.5 embryos. The CDK5RAP2RRF465/RRF465 protein was stable (Figure 2B), and retained centrosome localization (Figure 2E,F). From these data we conclude that the first 435 amino acids of CDK5RAP2 are sufficient for centrosomal localization. However, full-length CDK5RAP2 and the mutant fusion protein in CDK5RAP2RRF465/RRF465 MEFs exhibited different patterns of localization at centrosomes. Full-length CDK5RAP2 localized to centrioles and also to fibrous projections that emanated from the centrosome (Figure 2E,F). These PCM fibers were not microtubules or microtubule-dependent, as localization of CDK5RAP2 to these structures persisted upon microtubule disassembly with nocodazole (Figures 2E and S2B). The CDK5RAP2RRF465/RRF465 protein failed to localize to PCM fibers, yet retained centriole localization (Figures 2E,F). Thus, the normal PCM architecture of centrosomes was disrupted in CDK5RAP2RRF465/RRF465 MEFs.
We examined the impact of CDK5RAP2RRF465/RRF465 on the localization of other centrosome proteins, and found that rootletin, a cohesion fiber protein (Bahe et al., 2005), co-localized with CDK5RAP2 at PCM fibers in CDK5RAP2+/+ MEFs. Again, nocodazole treatment demonstrated that co-localization of CDK5RAP2 and rootletin at PCM fibers was not microtubule-dependent (Figure S2B). However, rootletin localization to PCM fibers was disrupted in CDK5RAP2RRF465/RRF465 MEFs and, to a lesser and variable degree, also in CDK5RAP2RRU031/RRU031 MEFs (Figure 2F,G). While rootletin localization to PCM fibers was completely disrupted in CDK5RAP2RRF465/RRF465 MEFs, the incidence of weak rootletin signal at PCM fibers increased more than 2-fold in CDK5RAP2RRU031/RRU031 MEFs (CDK5RAP2+/+: 28% vs. CDK5RAP2RRU031/RRU031: 66%) (Figure 2G,H). These results show that CDK5RAP2 regulates assembly of a cohesion fiber protein, consistent with the requirement for CDK5RAP2 in centrosome cohesion (Figures 2I and S3B and (Graser et al., 2007)). Interestingly, PCM fibers were not prominent in all mouse cells; centrosomes in NIH-3T3 cells did not display these structures as elaborately (Figure 1B). Nevertheless, disruption of rootletin localization shows that centrosome structure is altered in CDK5RAP2 mutant MEFs. In contrast, CDK5RAP2 mutant MEFs showed normal centrosomal localization of cenexin/ODF2, centrobin, centrin, γ-tubulin, pericentrin, TOG, aurora-A, and EB1 (Figures 3A, 5A, 7B and S5A; others not shown).
Figure 3.

CDK5RAP2 restricts centriole duplication
(A) CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs were stained for the PCM marker γ-tubulin (red), the centriole marker centrin (green), and DNA (blue). Paired centrioles in G1 and G2 CDK5RAP2+/+ MEFs are shown. The example CDK5RAP2RRF465/RRF465 MEF shows three singlet centrioles, one apparent pair, and a cluster of three. Three enlargements are shown on the right. Scale bar = 10 μm.
(B) Quantification of cells with 2, 3, 4, or greater than 4 centrioles in CDK5RAP2+/+ (green bars) and CDK5RAP2RRF465/RRF465 (red bars) cells. MEFs containing 2 centrioles (CDK5RAP2+/+: 85.0% ± 1.3% vs. CDK5RAP2RRF465/RRF465: 49.8% ± 2.5%, p<0.01), 3 centrioles (CDK5RAP2+/+: 2.5% ± 0.3% vs. CDK5RAP2RRF465/RRF465: 11.2% ± 0.6%, p<0.05), 4 centrioles (CDK5RAP2+/+: 8.3% ± 0.3% vs. CDK5RAP2RRF465/RRF465: 17% ± 2.8%, p>0.05), and greater than 4 centrioles (CDK5RAP2+/+: 4.2% ± 0.7% vs. CDK5RAP2RRF465/RRF465: 22.0% ± 1.5%, p<0.005).
(C) Quantification of cells showing singlet centrioles in CDK5RAP2+/+ (9.7% ± 1.4%, green bars) and CDK5RAP2RRF465/RRF465 (40.8% ± 0.6%, p<0.005, red bars) cultures. Data were collected from 3 independent experiments, n = 200 cells per experiment.
(D) E14.5 coronal brain sections from CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 embryonic mice were stained for pericentrin and DNA and the numbers of nuclei and centrosomes counted in multiple fields. Three or more fields were counted in three separate brain sections. CDK5RAP2+/+ brains had a centrosome to nuclei ratio of 0.96 ± 0.01 compared to 1.62 ± 0.05 in CDK5RAP2RRF465/RRF465 brains (p=0.0051).
(E) Model of daughter centriole amplification in CDK5RAP2RRF465/RRF465 cells. In wild type cells, disengaged mother (with red subdistal appendages) and daughter centrioles template assembly of a new centriole in S phase. The two centriole pairs remain engaged and at G2 the older daughter centriole is decorated with mother centriole vestments. We propose that in CDK5RAP2RRF465/RRF465 cells the parent centrioles fail to remain engaged with the nascent centrioles they have fostered, leading to re-licensing and reduplication of the disengaged centrioles, thereby increasing the pool of “daughter-daughter” pairs transiting from S to G2. See also Table 1 and Figure S3.
Figure 5.

Supernumerary mother centrioles and primary cilia in CDK5RAP2RRF465/RRF465 MEFs
(A) CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs stained with the daughter centriole-specific marker centrobin (left, red in merged panel), the mother centriole-specific marker cenexin/ODF2 (middle, green in merge), and DNA (blue in merge). Insets show enlargements of the centriole region.
(B) Percentages of cells with 0, 1, 2, or greater than 2 mother centrioles in CDK5RAP2+/+ (green bars) and CDK5RAP2RRF465/RRF465 (red bars) cells. MEFs containing 0 mother centrioles (CDK5RAP2+/+: 0.2% ± 0.2% vs. CDK5RAP2RRF465/RRF465: 1.2% ± 0.7%, p>0.05), 1 mother centriole (CDK5RAP2+/+: 89.3% ± 0.7% vs. CDK5RAP2RRF465/RRF465: 62.5% ± 0.5%, p<0.001), 2 mother centrioles (CDK5RAP2+/+: 8.2% ± 0.6% vs. CDK5RAP2RRF465/RRF465: 26.3% ± 0.9%, p<0.0005), or more than 2 mother centrioles (CDK5RAP2+/+: 2.3% ± 0.7% vs. CDK5RAP2RRF465/RRF465: 10.0% ± 1.0%, p<0.005).
(C) Dot plot of the average number of mother (CDK5RAP2+/+: 1.3 ± 0.05 vs. CDK5RAP2RRF465/RRF465: 1.6 ± 0.2) and daughter (CDK5RAP2+/+: 1.3 ± 0.1 vs. CDK5RAP2RRF465/RRF465: 2.4 ± 0.15) centrioles per cell. Data were collected from three independent experiments, 90 cells total.
(D) CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs stained for γ-tubulin (left, red in merged panel), the cilium axoneme marker acetylated α-tubulin (middle, green in merge), and DNA (blue in merge). Image in bottom panel shows four cilia in a cell with eight centrioles.
(E) The percent of cells that formed primary cilia in CDK5RAP2+/+ (green bars) and CDK5RAP2RRF465/RRF465 (red bars) MEFs (CDK5RAP2+/+: 71.2% ± 3.6% vs. CDK5RAP2RRF465/RRF465: 74.0% ± 0.9%, p>0.05).
(F) The percent of MEFs forming a single primary cilium (CDK5RAP2+/+: 95.3% ± 0.8% vs. CDK5RAP2RRF465/RRF465: 66.7% ± 2.4%, p<0.005) or more than one primary cilium (CDK5RAP2+/+, 4.7% ± 0.8% vs. CDK5RAP2RRF465/RRF465, 33.3% ± 2.4%, p<0.005). For (B), (E) and (F), data were collected from three independent experiments, n = 200 cells per experiment (except (F), where n = 150 cells with primary cilia per experiment). Scale bar = 10 μm. MEFs in (C-F) were blocked in G1 by serum starvation. See also Figure S5
CDK5RAP2 restricts centriole duplication
Centrosome amplification was a prominent feature of CDK5RAP2RRF465/RRF465 MEFs. Co-staining for γ-tubulin to label PCM and centrin to label centrioles showed that CDK5RAP2+/+ MEFs contained one or two pairs of centrin puncta, representing normal centriole complements in G1 or G2, respectively (Figure 3A). In contrast, a high percentage of CDK5RAP2RRF465/RRF465 MEFs had greater than two centrin and γ-tubulin puncta, showing that these cells had amplified centrioles (Figure 3A and see below).
CDK5RAP2RRF465/RRF465 cells had a greater than 4-fold increase in the occurrence of three centrioles (CDK5RAP2+/+: 2.5% vs. CDK5RAP2RRF465/RRF465: 11.2%), and a greater than 5-fold increase in cells with more than four centrioles (CDK5RAP2+/+: 4.2% vs. CDK5RAP2RRF465/RRF465: 22.0%). A reciprocal decrease in cells with two centrioles occurred in CDK5RAP2RRF465/RRF465 MEFs (CDK5RAP2+/+: 85.0% vs. CDK5RAP2RRF465/RRF465: 49.8%) (Figure 3B). Heterozygous RRF465 MEFs were similar to CDK5RAP2+/+ MEFs; only 2% of cells had greater than four centrioles (data not shown). Therefore, the RRF465 gene trap allele is not a dominant mutation in CDK5RAP2. Overall, 33.2% of CDK5RAP2RRF465/RRF465 MEFs, exhibited amplified centrioles, compared to only 6.7% of CDK5RAP2+/+ MEFs. Thus, CDK5RAP2RRF465/RRF465 MEFs have an altered centriole replication cycle that results in centriole amplification. In contrast, no centrosome amplification was observed in CDK5RAP2RRU031/RRU031 MEFs, an indication that only a small fraction of full-length CDK5RAP2 is sufficient to restrict centriole replication (Figure S3A).
Centrosome amplification was also observed in vivo in fetal CDK5RAP2RRF465/RRF465 mice. We counted centrosome numbers in sections of E14.5 fetal brains by immunofluorescence staining for pericentrin and DNA to identify centrosomes and nuclei, respectively. Centrosomes were amplified in cells of the fetal frontal cortex in vivo compared to wild-type siblings (Figure 3C), indicating that centrosome amplification occurs in vivo in the developing cerebral cortex of CDK5RAP2RRF465/RRF465 mice.
In CDK5RAP2RRF465/RRF465 MEFs centriole amplification could arise as an indirect effect on the cell cycle. To assess cell cycle block or delay, MEF cell cycle profiles were analyzed using flow cytometry. Cell cycle profiles from CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 cultures were indistinguishable (Figure S3C,D), indicating that centriole amplification in CDK5RAP2RRF465/RRF465 MEFs is not due to an overt indirect effect on the cell cycle. Nevertheless, wild type and CDK5RAP2 mutant sibling MEFs had different growth capacities. Whereas CDK5RAP2+/+ MEFs proliferated to passage 15 (P15), in contrast, CDK5RAP2RRF465/RRF465 MEFs slowed or arrested between P8 and P10, and after P4 a higher percentage of cells were needed to seed a culture. Following P8-P10, CDK5RAP2RRF465/RRF465 MEFs remained viable but would no longer grow to confluence. The underlying cause of this premature senescence and its link to centriole amplification is unclear. However, overall, these data show that CDK5RAP2RRF465/RRF465 MEFs have an altered centriole replication cycle, uncoupled from the cell cycle, which results in centriole amplification.
Centriole amplification is associated with increased nuclear size
Another salient phenotype of CDK5RAP2RRF465/RRF465 MEFs was the presence of enlarged nuclei (Figure 3A). Frequently, the nuclear diameter was increased approximately 3-fold (Figure S4). Quantification of nuclear size revealed a small but significant increase in the average nuclear area of CDK5RAP2RRF465/RRF465 cells (CDK5RAP2+/+: 230.3 μm2 vs. CDK5RAP2RRF465/RRF465: 262 μm2). However, the mean nuclear area was significantly larger, 528 μm2, among CDK5RAP2RRF465/RRF465 MEFs with amplified centrioles (Figure 4A). Since centriole amplification and increased DNA content would coincide if cytokinesis failed, we measured DNA content between CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs. DNA analysis by flow cytometry revealed no increase of polyploid cells in CDK5RAP2RRF465/RRF465 MEFs, nor did we observe an increase of binucleate cells to indicate cytokinesis defects. In addition, we compared the fluorescence signal of the nuclear stain, DAPI, between CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs with amplified centrosomes, and compared this to wild-type binucleate cells as a polyploid control. Binucleate cells had approximately 2-fold increase in DAPI signal compared to CDK5RAP2+/+ or CDK5RAP2RRF465/RRF465 MEFs with amplified centrosomes (Figure 4B). Therefore, we conclude that CDK5RAP2RRF465/RRF465 MEFs exhibit nuclear enlargement without increased DNA content, and that increased nuclear size correlates with centriole amplification. While nuclear size correlated with centriole amplification, this was not specific to CDK5RAP2 mutant cells and was seen in the wild type MEFs upon the relatively rare centriole amplification event (Figure 4B).
Figure 4.

CDK5RAP2RRF465/RRF465 MEFs with amplified centrioles have enlarged nuclei (A) Nuclei were stained with DAPI, and nuclear area (μm2) was measured from micrographs (CDK5RAP2+/+: 230.3 μm2 ± 5.6 μm2 vs. CDK5RAP2RRF465/RRF465: 262.0 μm2 ± 9.5 μm2, p<0.05); n ≥ 200 cells per experiment. CDK5RAP2RRF465/RRF465 MEFs with amplified centrioles predominate this phenotype, with an area of 528.0 μm2 ± 13.4 μm2; n = 50 cells per experiment. Data were collected from 3 independent experiments.
(B) DAPI signal (arbitrary units) was compared between CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs with one or two centrosomes (CDK5RAP2+/+: 3.9 × 106 ± 0.1 × 106, filled square vs. CDK5RAP2RRF465/RRF465: 4.0 × 106 ± 0.3 × 106, filled triangle) and MEFs with greater than two centrosomes (CDK5RAP2+/+: 7.2 × 106 ± 1.2 × 106, open circle vs. CDK5RAP2RRF465/RRF465: 8.5 × 106 ± 0.8 × 106, open triangle) and these were then compared to wild type binucleate cells (15.0 × 106 ± 0.7 × 106, open square); n = 10 and 11 for wild type binucleate cells and CDK5RAP2+/+ MEFs with one or two centrosomes, respectively and n ≥20 for all other samples. See also Figure S4.
Centrioles lose engagement and cohesion in CDK5RAP2 mutant MEFs
In addition to centriole amplification, CDK5RAP2RRF465/RRF465 MEFs showed a significantly higher incidence of single centrioles (Figure 3A,D and Table 1). This is in contrast to wild type cells where centrioles are predominantly configured as pairs. Centrosome structure within MEFs was examined with immunofluorescence microscopy, using antibodies against the centriole marker centrin and PCM marker γ-tubulin. Typical of normal cells, CDK5RAP2+/+ MEFs had two centrioles in close proximity that each assembled PCM in G1, and two engaged pairs of centrioles, where each pair assembled PCM in G2 (Figure 3A). In contrast, CDK5RAP2RRF465/RRF465 MEFs contained several different centriole configurations including paired centrioles, single centrioles and clusters of three or more centrioles (Figure 3A).
Table 1. Centriole configurations.
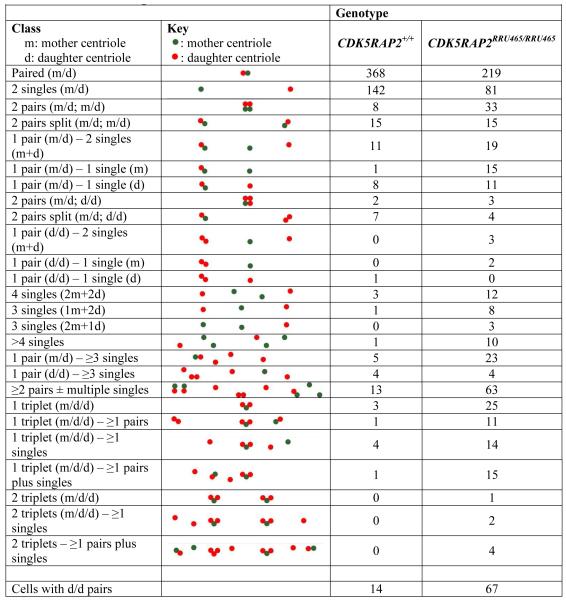
|
We quantified these effects by counting mother and daughter centrioles immunostained for the mother centriole marker cenexin/ODF2 and γ-tubulin or centrin as general centriole markers, and with cenexin plus centrobin as a mother and daughter centriole combination (Figure 5A and S5A). In this analysis, the paired configuration, including one mother and one daughter centriole, was used to describe “normal” centrosomes. The incidence of MEFs containing one or more singlet centrioles increased more than 4-fold in the CDK5RAP2RRF465/RRF465 mutant relative to wild type (CDK5RAP2+/+: 9.7% vs. CDK5RAP2RRF465/RRF465: 40.8%) (Figure 3D) (Table 1 summarizes all centriole configurations). The presence of singlet centrioles indicated that both centriole engagement and cohesion failed. Normally, late in G2, loss of centrosome cohesion occurs when centrosomes separate prior to mitotic spindle formation.
A role for CDK5RAP2 in cohesion was previously reported by siRNA knockdown, yet no centriole amplification or disengagement was observed (Graser et al., 2007). We wondered whether CDK5RAP2RRU031/RRU031 MEFs also displayed loss of centriole cohesion since the low level of full-length protein in this mutant may mimic siRNA knockdown. Indeed, centrosome splitting increased by more than 70% in CDK5RAP2RRU031/RRU031 MEFs (Figures 2I and S3B). We conclude that RRU031 is a weak loss of function CDK5RAP2 mutation due to the low expression level of full-length CDK5RAP2. Moreover, we conclude that the hypomorphic phenotype of CDK5RAP2 is loss of cohesion, whereas a strong loss of function results in loss of cohesion and also loss of centriole engagement.
CDK5RAP2RRF465/RRF465 MEFs possessed multiple mother centrioles and, since CDK5RAP2RRF465/RRF465 MEFs exhibited no irregularities in cell cycle profile (Figure S3C,D), we attributed this to the accumulation of mother centrioles following centriole amplification (Figure 5A,B). The majority of CDK5RAP2+/+ MEFs contained one (89.3%) or two (8.2%) mother centrioles, reflecting the G1/S and G2 stages of the cell cycle, respectively. In contrast, fewer CDK5RAP2RRF465/RRF465 MEFs contained one mother centriole (62.5%), while significantly more cells contained two mother centrioles (26.3%). In addition, mother centriole numbers in excess of two were increased more than 4-fold in CDK5RAP2RRF465/RRF465 MEFs (CDK5RAP2+/+: 2.3% vs. CDK5RAP2RRF465/RRF465: 10.0%) (Figure 5B). Since the cell cycle was unaffected, the increased number of CDK5RAP2RRF465/RRF465 cells with two mother centrioles must represent a substantial fraction of cells in G1 with excess mother centrioles. With this consideration, we infer that approximately 28.1% of CDK5RAP2RRF465/RRF465 MEFs have amplified mother centrioles. Daughter centriole numbers exceeded mother centrioles (Figure 5C), consistent with our model that centriole engagement is lost and multiple rounds of daughter synthesis occur in each cell cycle (Figure 3E, see below). Thus, the prevalence of single centrioles and amplified daughter centrioles in CDK5RAP2RRF465/RRF465 cells indicates that CDK5RAP2 restricts centriole duplication by maintaining centriole engagement.
Another frequent centriole configuration seen in CDK5RAP2RRF465/RRF465 MEFs was “daughter-daughter” pairs (Table 1). Centriole maturation, monitored here by recruitment of cenexin/ODF2 to mother centrioles, occurs late in G2 phase (Lange and Gull, 1995). Therefore, the population of “daughter-daughter” centriole pairs, normally seen only in cells transiting between S and G2, will increase if centriole engagement fails and daughter centrioles reduplicate (Figure 3E). CDK5RAP2RRF465/RRF465 MEFs showed an approximately 5-fold increase in daughter-daughter pairs (CDK5RAP2+/+: 2.3% ± 0.4% vs. CDK5RAP2RRF465/RRF465: 11.2% ± 1.2%, mean ± SEM, p<0.05). Thus, CDK5RAP2RRF465/RRF465 centrioles lose engagement and become re-licensed to duplicate, as evidenced not only by the presence of singlet centrioles but also by the increased numbers of daughter-daughter pairs (see model, Figure 3E).
Amplified mother centrioles template ectopic primary cilia in CDK5RAP2RRF465/RRF465 MEFs
New centrioles form in S-phase and mature into mother centrioles in G2 phase of the following cell cycle. Most vertebrate cells contain a single non-motile primary cilium (Bettencourt-Dias and Glover, 2007; Nigg and Raff, 2009). In G1 the mother centriole migrates to the plasma membrane, becomes a basal body, and templates assembly of the primary cilium (Ishikawa et al., 2005). Centriole amplification led us to hypothesize that the supernumerary mother centrioles in CDK5RAP2RRF465/RRF465 MEFs (Figure 5A-C) could result in the assembly of ectopic primary cilia.
We asked whether the amplification of mother centrioles in CDK5RAP2RRF465/RRF465 MEFs was associated with changes in the proficiency and frequency of primary cilium assembly. When cultured in serum-deprived medium, many cells assemble a primary cilium upon entering G0, a prolonged resting state. We subjected MEFs to serum starvation and immunostained with anti-acetylated α-tubulin antibody to label axoneme microtubules, and with antibodies to γ-tubulin to label the centrioles/basal bodies (Figure 5D). CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs were equally proficient at primary cilium formation (CDK5RAP2+/+: 71.2% vs. CDK5RAP2RRF465/RRF465: 74.0%) (Figure 5E). CDK5RAP2+/+ MEFs formed a single primary cilium templated from one of two interphase centrioles (Figure 5D,F). In contrast, CDK5RAP2RRF465/RRF465 MEFs with amplified centrioles formed multiple primary cilia (Figure 5D,F). The incidence of multiple primary cilia increased more than 7-fold in CDK5RAP2RRF465/RRF465 MEFs (CDK5RAP2+/+: 4.7% vs. CDK5RAP2RRF465/RRF465: 33.3%; Figure 5F), and was consistent with the frequency of centriole amplification (Figure 3B). Immunostaining for additional cilium markers, polyglutamylated tubulin and polaris/IFT88, axonemal and intraflagellar transport components of primary cilia, respectively, confirmed the assembly of multiple cilia in mutant cells (Figure S5B,C). These data reveal a propensity for mammalian cells to form more than one primary cilium.
CDK5RAP2RRF465/RRF465 MEFs assemble multipolar spindles
The presence of supernumerary centrosomes is a potentially dangerous condition at mitosis because centrosomes are dominant MTOCs, and therefore have the potential to organize microtubules aberrantly into the spindle apparatus, make inappropriate kinetochore attachments, and assemble multipolar spindles. The transient occurrence of multipolar spindles in cells with amplified centrosomes can cause chromosomal instability by the formation in inappropriate merotelic kinetochore-microtubule attachments prior to centrosome clustering (Ganem et al., 2009). Thus, spindle multipolarity results in the inappropriate segregation of chromosomes, leading to aneuploidy with increased cell death, but also a heightened propensity for tumorigenesis (Brinkley, 2001; Pihan and Doxsey, 2003; Nigg, 2006; Ganem et al., 2009). Centrosome clustering at spindle poles assures spindle bipolarity in cells with more than two centrosomes (Quintyne et al., 2005; Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008). Multipolar spindles were defined here as one or more MTOC’s positioned greater than 45° away from the major axis of the spindle, as determined from spindle microtubules and the configuration of aligned chromosomes (Figure 6A). We found that CDK5RAP2RRF465/RRF465 MEFs with two centrosomes formed bipolar spindles normally, as did CDK5RAP2+/+ MEFs. However, 51% of mitotic CDK5RAP2RRF465/RRF465 MEFs with supernumerary centrosomes formed multipolar spindles, while the remaining 49% assembled a bipolar, or pseudo-bipolar, spindle (Figure 6B). Since ~30% of mutant MEFs had amplified centrioles, this amounts to approximately 15% of total mitotic CDK5RAP2RRF465/RRF465 MEFs with multipolar spindles. Thus, centriole amplification in CDK5RAP2RRF465/RRF465 MEFs can lead to multipolar spindle formation. However, aberrant anaphase or telophase stage cells were not observed, an indication that multipolar spindles are resolved into bipolar spindles before anaphase onset.
Figure 6.

Multipolar spindles form in CDK5RAP2RRF465/RRF465 MEFs.
(A) Schematic outlining the criteria used to designate bipolar and multipolar spindle formation. To qualify as multipolar, spindles had an MTOC offset by greater than 45° from the dominant spindle axis as determined from spindle microtubules and DNA alignment.
(B) Mitotic figures immunostained for γ-tubulin (1st row, red in merged panel), α-tubulin (2nd row, green in merge), and DNA (3rd row, blue in merge). In CDK5RAP2RRF465/RRF465 mitotic MEFs with multiple centrosomes, 51% of mitotic spindles were multipolar (n = 43). Arrows indicate examples of the excess spindle poles, and arrowheads indicate chromosomes configured improperly on the metaphase plate. Scale bar = 10 μm.
(C) Scatter plot showing the timing from nuclear envelope breakdown (NEBD) to anaphase onset in CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 MEFs. In a small population of CDK5RAP2RRF465/RRF465 MEFs, the time between NEBD and anaphase onset was prolonged, with an approximately 8 min, or 47.7%, increase in the time spent to reach anaphase (CDK5RAP2+/+: 17.2 min. ± 0.7 min. vs. CDK5RAP2RRF465/RRF465: 25.4 min. ± 2.1 min., p<0.0001); n ≥ 25.
Centrosome clustering is accomplished during a delay in mitosis due to activation of the spindle checkpoint by amplified centrosomes, allowing the cell time to correct spindle assembly defects (Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008). Using live cell imaging, we observed that the timing of anaphase onset was prolonged by approximately 48% in CDK5RAP2RRF465/RRF465 MEFs compared to CDK5RAP2+/+ MEFs (Figure 6C). These results are consistent with the idea that CDK5RAP2RRF465/RRF465 MEFs accomplish spindle bipolarity by prolonging their stay in metaphase through checkpoint activation, providing time for supernumerary centrosomes to cluster.
Discussion
These data show that CDK5RAP2 maintains centriole engagement and cohesion. Since centriole disengagement is the licensing step that initiates centriole replication (Tsou and Stearns, 2006b, a; Nigg, 2007), the loss of CDK5RAP2 activity results in centriole amplification due to the failure to maintain engagement of mother-daughter pairs. CDK5RAP2 is therefore a negative regulator of centriole licensing. The amplified centrioles in CDK5RAP2RRF465/RRF465 MEFs impact not only mitotic spindle assembly but also promote the ectopic assembly of multiple primary cilia, templated by the excess mother centrioles that accumulate in these cells.
CDK5RAP2’s role in centriole engagement and cohesion
The presence of single centrioles is an uncommon occurrence in wild type cells because mother-daughter centriole pairs remain engaged following duplication until anaphase (Tsou and Stearns, 2006b, a; Nigg, 2007). Following disengagement, the centriole pair normally remains in close proximity through cohesion, which persists from G1 through the G2/M transition. The scattering of singlet centrioles and the centriole amplification in CDK5RAP2RRF465/RRF465 MEFs show that engagement and cohesion both require CDK5RAP2 function. Moreover, from the two mutants we have generated, one hypomorphic and one strong, it is evident that centrosome cohesion is more sensitive to disruption of CDK5RAP2 than is engagement. Thus, even the low (approximately 7% of endogenous) level of CDK5RAP2 expression from the leaky allele was sufficient to maintain centriole engagement.
The prevailing model for control of centriole replication underscores disengagement as the initiating step for centriole licensing: the step in the cell cycle that authorizes a single round of centriole replication (Tsou and Stearns, 2006b, a; Nigg, 2007). Disengagement is activated by separase at the metaphase-to-anaphase transition (Tsou and Stearns, 2006b). The molecules required to maintain engagement of centrioles, thus preventing re-licensing and restricting duplication, are not known. Our data indicate that CDK5RAP2 is required to restrict centriole replication by maintaining mother-daughter centrioles in the engaged state. The prevalence of singlet centrioles and excess daughter-daughter pairs strongly implicates disengagement combined with re-licensing as mechanisms underlying centriole amplification in CDK5RAP2RRF465/RRF465 MEFs (see model, Figure 3E).
Since separase initiates disengagement, it is possible that CDK5RAP2 is a target of this protease or that its localization or activity is inhibited by separase. We tested CDK5RAP2 as a separase substrate using in vitro translated human CDK5RAP2 with purified human separase, but detected no apparent cleavage of CDK5RAP2 (data not shown). Despite this negative result we cannot rule out the possibility that additional factors, post-translational modifications, or its centriolar context are needed for separase to target CDK5RAP2. The extent of the relationship between these two engagement regulators remains to be determined.
An alternative hypothesis for the amplification of centrioles in CDK5RAP2RRF465/RRF465 MEFs is unrestricted de novo centriole biogenesis. In contrast to the canonical “templated” assembly of daughters from mothers, centrioles can form in cells that lack any centrioles by de novo assembly (Marshall et al., 2001; Khodjakov et al., 2002; La Terra et al., 2005; Uetake et al., 2007). However, the presence of even one centriole in a cell suppresses de novo centriole assembly (La Terra et al., 2005). Since centrioles are present in CDK5RAP2RRF465/RRF465 MEFs during amplification we favor the loss of engagement/re-licensing model as proposed above. However, we cannot exclude the possibility that CDK5RAP2 suppresses de novo centriole formation.
How CDK5RAP2 regulates engagement and cohesion is unclear, but it is likely that these two centriole-binding processes are related at the molecular level, as demonstrated by the CDK5RAP2 mutant phenotypes. The displacement of rootletin, a structural component of cohesion fibers (Bahe et al., 2005), from its normal localization pattern at CDK5RAP2 mutant centrosomes is consistent with the role for CDK5RAP2 in maintaining cohesion, as presented here and reported recently by RNAi knockdown in cell culture (Graser et al., 2007). That the RNAi depletion of CDK5RAP2 did not produce the single centrioles and centriole amplification phenotypes reported here is likely due to the partial depletion of CDK5RAP2 by siRNAs. This supposition is strongly supported by our results with the weak CDK5RAP2 mutation in CDK5RAP2RRU031/RRU031 MEFs. Thus, partial loss of CDK5RAP2 function results in loss of cohesion, while strong loss of function additionally results in loss of engagement.
Another recent study, again targeting CDK5RAP2 by RNAi, reported that depletion of CDK5RAP2 disrupted mitotic centrosome MTOC activity and γ-tubulin recruitment (Fong et al., 2008), similar to the phenotype of cnn mutant Drosophila cells (Megraw et al., 2001). In contrast, we observed no apparent effect on mitotic centrosome MTOC activity or γ-tubulin recruitment to centrosomes in CDK5RAP2RRF465/RRF465 MEFs. Nevertheless, the possibility remains that such a function is retained by the first 435 amino acids of CDK5RAP2, which are still present in the RRF465 truncation mutant. It is also possible that off-target affects contributed to the phenotypes seen by Fong et al. Myomegalin/PDE4DIP, the other centrosomin ortholog in mammals (Verde et al., 2001), might work redundantly with CDK5RAP2 in mitotic centrosome assembly and could have been an off-target in those experiments. Since antibodies against mouse myomegalin were unavailable, we were unable to examine its expression or the effect, if any, of CDK5RAP2 mutations on myomegalin localization. It remains possible that myomegalin shares redundant functions with CDK5RAP2 in mitotic centrosome function, similar to the defined role for centrosomin in Drosophila.
CDK5RAP2 mutant mice
The CDK5RAP2 mutant mice described here showed no overt defects in somatic growth, body weight, adult behavior, or female fertility. Male fertility was mixed, with about 33% of males showing reduced fertility in CDK5RAP2RRF465/RRF465 mice. Male infertility might be a genetic background effect, a possibility that will be tested via backcrosses to the C57BL/6 and 129Ola strains. Brain size and gross morphology appeared to be normal, indicating that disruption of CDK5RAP2 does not cause microcephaly in mice. It is possible that in mice, unlike in humans, the development of the cerebral cortex is not CDK5RAP2-dependent. In one scenario, mice may have redundancy in CDK5RAP2 function, and compensatory mechanism(s) might account for the lack of an obvious brain phenotype in these mutants. Myomegalin might play this role. Alternatively, CDK5RAP2 may be required in humans to populate the cerebral cortex with the significantly larger number of neurons (1011) than exist in the mouse (107); human brain development was proposed to involve an extra set of neural progenitors (Fish et al., 2008). Alternatively, other differences in cerebral cortex development between mouse and human may account for the different requirements for CDK5RAP2. Yet another possibility is that strain background effects masked phenotypic penetrance in the mouse mutants reported here.
The physiological consequences of the plethora of cellular defects we describe, including multiple centrosomes in interphase and mitotic cells, and excess numbers of primary cilia in CDK5RAP2RRF465/RRF465 mice are at present unknown. The establishment of these mouse mutants will enable deeper investigations into the mechanisms by which the organism “copes” with these centrosome-based aberrations during development and in the adult. It is an intriguing possibility that centrosome amplification underlies the etiology of human MCPH. Since the primary cilium, once thought to be a cell vestige with no function, has emerged as a key signaling “antenna” for the cell (Singla and Reiter, 2006), the amplified cilia could produce altered responses to intercellular signaling the affect cell division, cell fates or cell migration. An alternative mechanism might be a primary defect in neural stem cell divisions caused by the presence of multipolar spindles. Early in the development of the cerebral cortex, ventricular neural stem cells divide symmetrically to expand the progenitor pool; neurons arise somewhat later from asymmetric cell divisions of ventricular stem cells and from symmetric divisions of intermediate cortical progenitor cells (Fish et al., 2006; Buchman and Tsai, 2007; Fish et al., 2008). The formation of multipolar spindles, even transiently prior to anaphase, could alter the control of symmetric and asymmetric divisions, thereby affecting neural cell fates.
Materials and Methods
Mouse Genetics
Chimeras were generated by injection of embryonic stem cells from clones RRF465 and RRU031 (Bay Genomics, San Francisco, CA, USA) into host blastocysts of C57BL/6J mice and then transferred into uteri of pseudo-pregnant C57BL/6J females. Male chimeras were mated with C57BL/6J females and F1 progeny were subsequently intercrossed. The β-Geo insertion site was mapped by Southern blotting and PCR analysis. Splice trap transposon sites were mapped to sites approximately 8.8 and 11.1 kbp from the 5′ ends of exons 3 and 12 in CDK5RAP2RRU031 and CDK5RAP2RRF465 mutant mouse lines. Genotyping was done by PCR. Sequencing of these alleles revealed that, while the CDK5RAP2RRF465 line had an intact transposon, the CDK5RAP2RRU031 line had a 133 bp deletion at the 5′ end of the transposon. Mouse protocols were approved by IACUC committees at UT Southwestern and Florida State University.
MEF Derivation and culture
A heterozygous cross was observed for the copulatory plug (day E0.5). MEFs were then derived from gestation day E14.5. After removal of the head and internal organs, embryos were rinsed in phosphate-buffered saline (PBS) and incubated in 0.05% trypsin at 4°C overnight. Embryos were then mechanically dissociated with a 1ml pipette and allowed to settle. Cells from single embryos were plated onto a 100 mm culture dish (Grainer Bio-One, Frickenhausen, Germany) with DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin and incubated at 37°C in a 5% CO2 and 5% O2-humidifier chamber. Cell culture reagents were purchased from GIBCO-BRL (Invitrogen, Carlsbad, CA, USA) except where noted. Plating after dissociation was considered passage 0 (P0) and the first replating 2 days later was P1. All experiments were performed using cells between passages P4 and P8, on sibling MEFs cultured side by side. MEF genotypes were confirmed by PCR.
Cell Culture and Treatments
NIH-3T3 cells were cultured in DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin and incubated at 37°C in a 5% CO2-humidifier chamber. Cell culture reagents were purchased from GIBCO-BRL (Invitrogen, Carlsbad, CA, USA) except where noted. Serum starvation to induce cilium formation was accomplished by culturing cells in DMEM containing 0.5% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin and incubated for 36 hours. Cells were cultured in complete medium containing 30 μM Nocodazole (Sigma, St. Louis, MO, USA) for 2.5 hours to achieve microtubule depolymerization. MEFs were synchronized by double-thymidine block by incubating freshly plated cells for 12 hours in complete medium containing 2 mM thymidine (Sigma), released for 16 hours in complete medium, and then replated and incubated 12 hours in complete medium containing 2 mM thymidine. Cells were finally released for 8 hours before fixation to examine mitotic cells.
Immunostaining and Microscopy
Cells were seeded onto 4 well slides and incubated overnight. Next, cells were rinsed briefly (2 seconds) in PBS and fixed in −20°C methanol for 10 minutes. After fixation cells were washed 5 min in PBS and primary antibodies (see Supplemental Experimental Methods) added in staining solution (5 mg/ml BSA, 0.1% Saponin, and 0.1% Triton X-100) for 1 hour, thereafter cells were washed 3 times 5 min in PBS and secondary antibody conjugates to Alexa 488 or 546 (Invitrogen, Carlsbad, CA, USA) were applied at 1:500 dilution for 1 hour. Three 5 min washes in PBS preceded the mounting of slides. Cells were imaged with a Leica TCS SP2 confocal microscope (Leica, Wetzlar, Germany) with a 63X/NA1.4 oil immersion objective, or a Zeiss Axioskop microscope (Carl Zeiss, Inc., Oberkochen, Germany) with a 63X/NA1.4 oil immersion objective and a Coolsnap FX CCD camera (Photometrics, Tucson, AZ, USA) with Metamorph software (Molecular Devices, Downingtown, PA, USA). Where noted, imageJ (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA) was used for data analysis.
Tissue Sectioning and Microscopy
A CDK5RAP2+/RRF465 timed pregnant female that was mated with a heterozygous male was deeply anesthetized (12.5 mg/ml Tribromoethanol [Avertin], administered at 250 mg/kg, i.p.) followed by cervical dislocation. Embryonic brains were recovered, post-fixed overnight in 4% Paraformaldehyde in 0.1M phosphate buffer, cryoprotected in 30% sucrose in 0.1M phosphate buffer, and frozen in −80°C 2-methylbutane. Frozen 30-μm thick sections were immunostained on-slide. The tissue was incubated in primary antibodies overnight, washed, and then incubated with secondary antibodies for two hours. Sections were imaged as described above. For adult brain sections, CDK5RAP2+/+ and CDK5RAP2RRF465/RRF465 mice and brains were processed as above, fixed overnight in 4% Paraformaldehyde in PBS and embedded in paraffin. Mounted 20μm thick coronal sections were stained 1 minute in Harris Hematoxylin (American Master Tech Inc., Lodi, CA, USA), washed 1 minute in water, rinsed in Scott’s Solution, stained 1 minute in Eosin Y stain (American Master Tech Inc.), and dehydrated in ethanol (95% followed by 100% ethanol), quickly rinsed in Xylene and coverslip mounted with Permount (Fisher Scientific, Fairlawn, NJ, USA)
Statistical Analysis
Statistical analyses were performed using Prism 5 (GraphPad Software, Inc., San Diego, CA). All statistical tests were two-tailed and were considered to be statistically significant at p<0.05. An * denotes p<0.05 and ** denotes p<0.01. In all cases, error bars represent the standard error of the means (SEM).
Supplementary Material
Acknowledgements
We thank J. L. Salisbury, K. Lee, Eugene Xu, Brad Shuster, C. Janke, and K. Rhee for providing antibodies. We also thank Hui Zou for performing the separase assay against hCDK5RAP2, Bill Snell for helpful discussions, and two anonymous reviewers for comments that led to significant improvements of this work. This work was supported by a grant from the National Institutes of Health (NIH) (GM068756) to T. L. Megraw, and an NIH Pharmacological Sciences Training Grant to J. A. Barrera (GM007062).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120:2139–2142. doi: 10.1242/jcs.005231. [DOI] [PubMed] [Google Scholar]
- Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J Cell Biol. 2005;171:27–33. doi: 10.1083/jcb.200504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol. 2007;8:451–463. doi: 10.1038/nrm2180. [DOI] [PubMed] [Google Scholar]
- Bobinnec Y, Khodjakov A, Mir LM, Rieder CL, Edde B, Bornens M. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J Cell Biol. 1998;143:1575–1589. doi: 10.1083/jcb.143.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- Buchman JJ, Tsai LH. Spindle regulation in neural precursors of flies and mammals. Nature reviews. 2007;8:89–100. doi: 10.1038/nrn2058. [DOI] [PubMed] [Google Scholar]
- Doxsey SJ. Centrosomes as command centres for cellular control. Nat Cell Biol. 2001;3:E105–108. doi: 10.1038/35074618. [DOI] [PubMed] [Google Scholar]
- Faragher AJ, Fry AM. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol Biol Cell. 2003;14:2876–2889. doi: 10.1091/mbc.E03-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JL, Dehay C, Kennedy H, Huttner WB. Making bigger brains-the evolution of neural-progenitor-cell division. J Cell Sci. 2008;121:2783–2793. doi: 10.1242/jcs.023465. [DOI] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci U S A. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong KW, Choi YK, Rattner JB, Qi RZ. CDK5RAP2 Is a Pericentriolar Protein That Functions in Centrosomal Attachment of the {gamma}-Tubulin Ring Complex. Mol Biol Cell. 2008;19:115–125. doi: 10.1091/mbc.E07-04-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Nigg EA. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci. 2007;120:4321–4331. doi: 10.1242/jcs.020248. [DOI] [PubMed] [Google Scholar]
- Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet. 2008;40:232–236. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Kubo A, Tsukita S. Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat Cell Biol. 2005;7:517–524. doi: 10.1038/ncb1251. [DOI] [PubMed] [Google Scholar]
- Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL, Sluder G, Cassels G, Sibon O, Wang CL. De novo formation of centrosomes in vertebrate cells arrested during S phase. J Cell Biol. 2002;158:1171–1181. doi: 10.1083/jcb.200205102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Girimaji SC, Duvvari MR, Blanton SH. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009;84:286–290. doi: 10.1016/j.ajhg.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Terra S, English CN, Hergert P, McEwen BF, Sluder G, Khodjakov A. The de novo centriole assembly pathway in HeLa cells: cell cycle progression and centriole assembly/maturation. J Cell Biol. 2005;168:713–722. doi: 10.1083/jcb.200411126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange BM, Gull K. A molecular marker for centriole maturation in the mammalian cell cycle. J Cell Biol. 1995;130:919–927. doi: 10.1083/jcb.130.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Kaufman TC. The homeotic target gene centrosomin encodes an essential centrosomal component. Cell. 1996;85:585–596. doi: 10.1016/s0092-8674(00)81258-1. [DOI] [PubMed] [Google Scholar]
- Luders J, Stearns T. Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol. 2007;8:161–167. doi: 10.1038/nrm2100. [DOI] [PubMed] [Google Scholar]
- Mahoney NM, Goshima G, Douglass AD, Vale RD. Making microtubules and mitotic spindles in cells without functional centrosomes. Curr Biol. 2006;16:564–569. doi: 10.1016/j.cub.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Marshall WF, Vucica Y, Rosenbaum JL. Kinetics and regulation of de novo centriole assembly. Implications for the mechanism of centriole duplication. Curr Biol. 2001;11:308–317. doi: 10.1016/s0960-9822(01)00094-x. [DOI] [PubMed] [Google Scholar]
- Megraw TL, Kao LR, Kaufman TC. Zygotic development without functional mitotic centrosomes. Curr Biol. 2001;11:116–120. doi: 10.1016/s0960-9822(01)00017-3. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–2723. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215–221. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Paintrand M, Moudjou M, Delacroix H, Bornens M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol. 1992;108:107–128. doi: 10.1016/1047-8477(92)90011-x. [DOI] [PubMed] [Google Scholar]
- Palazzo RE, Vogel JM, Schnackenberg BJ, Hull DR, Wu X. Centrosome maturation. Curr Top Dev Biol. 2000;49:449–470. doi: 10.1016/s0070-2153(99)49021-0. [DOI] [PubMed] [Google Scholar]
- Pfaff KL, Straub CT, Chiang K, Bear DM, Zhou Y, Zon LI. The zebra fish cassiopeia mutant reveals that SIL is required for mitotic spindle organization. Mol Cell Biol. 2007;27:5887–5897. doi: 10.1128/MCB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan G, Doxsey SJ. Mutations and aneuploidy: co-conspirators in cancer? Cancer Cell. 2003;4:89–94. doi: 10.1016/s1535-6108(03)00195-8. [DOI] [PubMed] [Google Scholar]
- Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127–129. doi: 10.1126/science.1104905. [DOI] [PubMed] [Google Scholar]
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- Sawin KE, Lourenco PC, Snaith HA. Microtubule nucleation at non-spindle pole body microtubule-organizing centers requires fission yeast centrosomin-related protein mod20p. Curr Biol. 2004;14:763–775. doi: 10.1016/j.cub.2004.03.042. [DOI] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Sluder G, Thompson EA, Miller FJ, Hayes J, Rieder CL. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J Cell Sci. 1997;110(Pt 4):421–429. doi: 10.1242/jcs.110.4.421. [DOI] [PubMed] [Google Scholar]
- Strnad P, Gonczy P. Mechanisms of procentriole formation. Trends Cell Biol. 2008;18:389–396. doi: 10.1016/j.tcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, Meng EC, Lee RE, Yee A, L’Italien L, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimborn M, Bell SM, Felix C, Rashid Y, Jafri H, Griffiths PD, Neumann LM, Krebs A, Reis A, Sperling K, et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet. 2004;75:261–266. doi: 10.1086/422855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou MF, Stearns T. Controlling centrosome number: licenses and blocks. Curr Opin Cell Biol. 2006a;18:74–78. doi: 10.1016/j.ceb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Tsou MF, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 2006b;442:947–951. doi: 10.1038/nature04985. [DOI] [PubMed] [Google Scholar]
- Uetake Y, Loncarek J, Nordberg JJ, English CN, La Terra S, Khodjakov A, Sluder G. Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J Cell Biol. 2007;176:173–182. doi: 10.1083/jcb.200607073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatram S, Tasto JJ, Feoktistova A, Jennings JL, Link AJ, Gould KL. Identification and characterization of two novel proteins affecting fission yeast gamma-tubulin complex function. Mol Biol Cell. 2004;15:2287–2301. doi: 10.1091/mbc.E03-10-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I, Pahlke G, Salanova M, Zhang G, Wang S, Coletti D, Onuffer J, Jin SL, Conti M. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J Biol Chem. 2001;276:11189–11198. doi: 10.1074/jbc.M006546200. [DOI] [PubMed] [Google Scholar]
- Vorobjev IA, Chentsov Yu S. Centrioles in the cell cycle. I. Epithelial cells. J Cell Biol. 1982;93:938–949. doi: 10.1083/jcb.93.3.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Adamian M, Li T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol Biol Cell. 2006;17:1033–1040. doi: 10.1091/mbc.E05-10-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Loncarek J, Khodjakov A, Rieder CL. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol. 2008;10:748–751. doi: 10.1038/ncb1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Liu L, Zhao A, Pfeifer GP, Xu X. The abnormal spindle-like, microcephaly-associated (ASPM) gene encodes a centrosomal protein. Cell Cycle. 2005;4:1227–1229. doi: 10.4161/cc.4.9.2029. [DOI] [PubMed] [Google Scholar]
- Zhong X, Pfeifer GP, Xu X. Microcephalin encodes a centrosomal protein. Cell Cycle. 2006;5:457–458. doi: 10.4161/cc.5.4.2481. [DOI] [PubMed] [Google Scholar]
- Zimmerman S, Chang F. Effects of {gamma}-tubulin complex proteins on microtubule nucleation and catastrophe in fission yeast. Mol Biol Cell. 2005;16:2719–2733. doi: 10.1091/mbc.E04-08-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.