

Abstract
Cytokinesis and abscission are complicated events that involve changes in membrane transport and cytoskeleton organization. We have used the combination of time-lapse microscopy and correlative high-resolution 3D tomography to analyze the regulation and spatio-temporal remodeling of endosomes and microtubules during abscission. We show that abscission is driven by the formation of a secondary ingression within the intracellular bridge connecting two daughter cells. The initiation and expansion of this secondary ingression requires recycling endosome fusion with the furrow plasma membrane and nested central spindle microtubule severing. These changes in endosome fusion and microtubule reorganization result in increased intracellular bridge plasma membrane dynamics and abscission. Finally, we show that central spindle microtubule reorganization is driven by localized microtubule buckling and breaking, rather than by spastin-dependent severing. Our results provide a new mechanism for mediation and regulation of the abscission step of cytokinesis.
Key words: Abscission, Cytokinesis, Membrane Trafficking, Microtubules, RAB11, RAB11-FIP3
Introduction
Once mammalian cells have successfully replicated and segregated chromosomes into their respective destinations, they undergo a process of physical separation, called cytokinesis. Cytokinesis begins with the formation of an actomyosin contractile ring, which constricts the plasma membrane (PM) and compacts the overlapping central spindle microtubules at the midzone to form the midbody, which is located within an intracellular bridge (ICB) (Eggert et al., 2006). At this point, the cell must proceed to the final cytokinetic event, abscission, leading to the resolution of the ICB. Recent work has demonstrated that remarkable changes occur inside the ICB, such as the reorganization of post-Golgi vesicle trafficking (Chua et al., 2010; Gromley et al., 2005; Wilson et al., 2005) and increases in ICB microtubule dynamics (Connell et al., 2009; Fanarraga et al., 2010). However, the molecular machinery and the mechanisms that mediate abscission during cytokinesis remain to be fully understood.
Crosstalk between membrane transport and the cytoskeleton play a key role during abscission. With knowledge of an ever-increasing number of membrane trafficking proteins that affect late telophase during cytokinesis, the challenge has become to incorporate all the data into models that can explain the machinery of cell abscission. Recycling endosomes (RE) and secretory vesicles are required for the final steps of cell division, and accumulate within the ICB just before abscission (Fig. 1A) (Gromley et al., 2005). Furthermore, RE resident proteins such as Rab11 and its interacting proteins FIP3 and the Exocyst complex, are all required for the completion of cytokinesis (Fielding et al., 2005). Finally, several SNAREs such as syntaxin 2, syntaxin 4, syntaxin 5, syntaxin 16 and vesicle-associated membrane protein 8 (VAMP8) are all required for abscission in mammalian cells (Jantsch-Plunger and Glotzer, 1999; Low et al., 2003; Song et al., 2009; Xu et al., 2002). Taken together, it was proposed that simultaneous fusion of these furrow endosomes and secretory vesicles with each other and the furrow PM leads to the separation of the ICB PM (Gromley et al., 2005). In addition to RE proteins, the ESCRT (endosomal sorting complex required for transport) complex might play a role in cytokinesis, on the basis of the premise that inward budding of intraluminal vesicles during the maturation of multivesicular bodies (MVB) is topologically similar to abscission (Fig. 1A). Because ESCRT proteins were shown to be involved in MVB formation (Hurley and Hanson, 2010), it was suggested that they mediate the resolution of the ICB. This occurs presumably by the formation and contraction of an ESCRT-III protein ring located within the inside of the ICB PM (Wollert et al., 2009). Consistent with this, several ESCRT proteins are found to localize to the midbody and are required for cytokinesis (McDonald and Martin-Serrano, 2008; Morita et al., 2007).
Fig. 1.

FIP3–GFP dynamics during cell progression through telophase. (A) Proposed abscission models. (B–D) HeLa cells expressing FIP3–GFP (green) were fixed and stained with anti-acetylated tubulin (red) antibodies: (B) cell in early telophase; (C) cell in mid-telophase; (D) cell in late telophase. (E) Staging of telophase cells based on FIP3–GFP localization. Scale bars: 5 μm.
In addition to changes in endocytic membrane transport, the microtubule cytoskeleton undergoes a dramatic reorganization during telophase. During furrow ingression, the central spindle is compacted to overlapping microtubule bundles, which then form a structure known as the midbody, or Flemming's body, located within the ICB between the two daughter cells. The central spindle and the midbody contain several distinct microtubule-associated proteins, such as the PRC1/KIF4 and the centralspindlin complexes (Eggert et al., 2006), each with bundling activities required to properly organize the microtubule cytoskeleton within the ICB. During abscission, these densely packed ICB microtubules need to be removed to allow for fusion of the opposing PM. The spatial regulation of microtubule disassembly during late telophase remains to be fully understood. It has been suggested that disassembly of microtubules is mediated by the microtubule-severing protein spastin (Connell et al., 2009), although the mechanisms regulating spastin targeting and activation within the ICB remain unclear.
In this study, we have investigated the changes in organelle and PM dynamics, as well as microtubule organization during abscission. We used a combination of correlative high-resolution 3D tomography and pHluorin-dependent time-lapse imaging to monitor endosome fusion and microtubule disassembly during the final stages of cytokinesis. By using rapid, high-pressure freezing, followed by freeze-substitution fixation and plastic embedding, we were able to preserve cellular structure for investigation by tomography. This approach previously has been shown to retain organelle structure with low artifacts, and provide high-resolution images of organelles and cytoskeleton (Morphew and McIntosh, 2003). We demonstrate that RE, rather than secretory organelles, accumulate at the ICB during late cytokinesis. We also show that localized microtubule reorganization and RE fusion with the furrow PM precedes formation of a ‘secondary ingression’, which drives abscission. For the first time in mammalian cells, we investigated changes in microtubule organization during abscission by correlative high-resolution 3D tomography and showed that localized microtubule clearance is mediated by buckling-induced severing. Thus, we hypothesize that fusion of REs containing FIP3 and VAMP8 and buckling-induced microtubule severing establish the site of secondary ingression and drive abscission within the ICB.
Results
Dynamics of FIP3- and VAMP8-containing endosomes reflect the progressive stages of abscission
The lack of understanding about the mechanisms of abscission is partly due to difficulties in identifying the stage of cell progression through telophase. Although cells progress from metaphase to telophase in 20–30 minutes, telophase often lasts up to 2–3 hours before daughter cells undergo their final separation. Traditionally, after the formation of a contractile ring, progression through cytokinesis has been studied using light microscopy by visualizing tubulin or proteins known to act during the late stages of cytokinesis, such as Aurora B (Murata-Hori et al., 2002; Murata-Hori and Wang, 2002). However, once the cleavage furrow fully ingresses to form the ICB, the localization of tubulin and Aurora B do not change dramatically over the time that it takes to resolve the ICB. A better marker is needed to monitor the progression of the late stages of cytokinesis. FIP3 and fluorescently tagged FIP3–GFP have previously been shown (Wilson et al., 2005) to change localization during the progression of cytokinesis, and we show that during early telophase FIP3-associated endosomes are localized around the centrosomes (Fig. 1B). As telophase progresses to mid-telophase, the FIP3-associated endosomes move away from the centrosomes and start accumulating just outside the ICB connecting two daughter cells (Fig. 1C). Finally, during late telophase, the FIP3-associated endosomes move inside the ICB, where abscission occurs shortly thereafter (Fig. 1D). By contrast, the localization of tubulin and Aurora B do not change appreciably at different stages of telophase (Fig. 1B–D and data not shown). In this study, FIP3–GFP was used as a marker for the cell progression through telophase and we are referring to the different stages as early or late telophase (Fig. 1E).
The delivery and fusion of post-Golgi organelles are known to be required for abscission (Baluska et al., 2006). Because membrane fusion is mediated by SNAREs (Chen and Scheller, 2001), we set out to identify the SNAREs mediating FIP3-endosome fusion with the furrow PM. We examined the localization of known post-Golgi v-SNAREs (VAMP4, VAMP7 and VAMP8) and show that only VAMP8 co-localizes with FIP3-endosomes during telophase (supplementary material Fig. S1A–I, and data not shown). Consistently, VAMP8 has been implicated in mediating cytokinesis (Low et al., 2003), and VAMP8 knockdown in HeLa cells resulted in an increase in the number of multinucleate cells from 2.12±1.12 in mock to 8.86±2.18 in knockdown cells (data shown are the means and s.d. of three independent experiments, P<0.01). Furthermore, VAMP8 knockdown increased the number of cells in cytokinesis from 12.07% (n=323) in mock to 17.71% (n=271) in knockdown cells, presumably due to the delay in cytokinesis. Taken together, these data are consistent with the involvement of VAMP8 in the late stages of cell division. Although it was suggested that VAMP8 mediates fusion of secretory vesicles (Gromley et al., 2005), we show that VAMP8 also localizes with FIP3-endosomes. Thus, in addition to its role in secretory vesicles, VAMP8 also appears to mediate FIP3-endosome fusion with furrow PM (see VAMP8–pHluorin data below).
Increased plasma membrane dynamics precedes abscission
We show that VAMP8 is involved in FIP3-endosome fusion during cytokinesis, yet the spatial and temporal fusion of FIP3-endosomes within the ICB is unknown. To determine the events leading to abscission, we investigated the ICB PM dynamics through the stages of telophase. Using FIP3-dependent staging, we imaged cells by brightfield microscopy in early telophase, as well as during later stages of telophase. During early telophase, the ICB PM was relatively stable, showing only subtle changes in the width of the PM (Fig. 2A). By contrast, as cells progressed toward abscission and FIP3-endosomes accumulated in the ICB, there was a dramatic increase in the ICB PM dynamics (Fig. 2B). Numerous PM waves were observed on both sides of the midbody (Fig. 2Bc, arrows). These membranous waves were filled with FIP3-endosomes (Fig. 2Ba,Bb, arrows) and appeared to originate in close proximity to the midbody (Fig. 2Bc). These observations are consistent with previous studies that have shown PM waves within the ICB (Byers and Abramson, 1968), although the function of these waves was not determined. Our data indicate that these PM waves precede a dramatic thinning of ICB, that we term a secondary ingression (Fig. 2C and supplementary material Movie 1). Once the thickness of the secondary ingression decreased to ~100 nm, the ICB eventually broke and each side of the ICB retracted into the cell body (Fig. 2C). Interestingly, once the ICB broke, the FIP3-endosomes could only be observed on the intact side of the midbody (Fig. 2Ce), suggesting that the resolution of the ICB might coincide with the fusion of the FIP3-endosomes with the PM.
Fig. 2.

Changes in the ICB PM dynamics during telophase. (A,B) HeLa cells expressing FIP3–GFP at different stages of cytokinesis were analyzed by time-lapse microscopy. (Ac,Bc) Kymographs of the ICB (marked by lines in Aa and Ba); arrows mark membrane waves. (C) Time-lapse microscopy analysis of the cell undergoing secondary ingression and abscission. (Ca,Ce) FIP3–GFP localization before (Ca) and after (Ce) abscission. (Cb–Cd) Brightfield images of the cell undergoing a secondary ingression (arrows) and abscission. ICB is marked by the line in Cb. (Cf,Cg) Kymographs of membrane waves (Cf) and abscission (Cg). (D) Time-lapse microscopy analysis of the ‘wave’ formation in cells treated with mock siRNA or with siRNA targeting FIP3 or VAMP8. Images shown are kymographs from the ICB of the late telophase cells. (E) Quantitation of wave formation in cells treated mock siRNA or with siRNA targeting FIP3 or VAMP8. Data shown are means ± s.d. of the number of waves formed within the ICB during each time-lapse series (2.5 minutes long). Asterisks mark the means that are significantly different from mock at P<0.05; n is the number of cells analyzed. Scale bars: 5 μm.
Because the membranous waves were enriched in FIP3 endosomes (Fig. 2Ba,Bb, arrows) it raised an interesting possibility that the waves, at least in part, might be generated by the localized fusion of FIP3-endosomes with the ICB plasma membrane. To test that, cellular VAMP8 or FIP3 was depleted using RNA interference and the formation of the ICB waves was visualized by time-lapse microscopy. Knockdown of either VAMP8 or FIP3 significantly reduced the number of waves formed during late telophase (Fig. 2D,E), suggesting that the fusion of FIP3-endosomes with the ICB plasma membrane is required for increased plasma membrane dynamics during late telophase.
Changes in FIP3-containing RE dynamics during cell progression from early and late telophase
It has been proposed that the fusion of organelles with the furrow PM is important during the resolution of the ICB (Baluska et al., 2006). However, the function and timing of these fusion events remain controversial. Some studies suggest that asymmetric and synchronous fusion of secretory organelles during late telophase mediates abscission (Gromley et al., 2005). By contrast, secretory organelles can be transported to and fuse with the furrow PM early in telophase, and do not appear to undergo asymmetric and synchronous fusion events (Goss and Toomre, 2008). However, RE accumulate at the furrow (Baluska et al., 2006) and it remains unclear whether they undergo fusion with the furrow PM. Therefore, we investigated the properties and function of FIP3-endosome fusion during progression from early to late telophase.
To determine whether FIP3- and VAMP8-endosomes actually fuse with the PM during late telophase when PM waves occur, we incubated HeLa cells, transduced with VAMP8–GFP and FIP3–mCherry, with an anti-GFP antibody and the uptake of the anti-GFP antibody was visualized by fluorescence microscopy. FIP3- and VAMP8-endosomes undergo dynamic membrane fusion and uptake events during telophase, as indicated by the extensive co-localization between anti-GFP antibodies, FIP3–mCherry, and VAMP8–GFP during early and late telophase (supplementary material Fig. S1J). Furthermore, anti-GFP antibody uptake is mediated by the dynamin pathway, as treatment with a dynamin inhibitor, dynasore, blocks anti-GFP uptake (data not shown). Our data demonstrate that FIP3-endosomes can fuse with the PM during telophase, yet the timing and location of these fusion events remain unclear. To investigate the spatio-temporal properties of FIP3-endosome fusion, we attached a pH-sensitive GFP tag, pHluorin, to the C-terminus of VAMP8. Because VAMP2–pHluorin was successfully used to monitor synaptic vesicle fusion with pre-synaptic PM (Granseth et al., 2006; Miesenbock et al., 1998), we speculated that VAMP8–pHluorin could be used to analyze the spatio-temporal dynamics of FIP3-endosome fusion with the PM. Indeed, we established that VAMP8–pHluorin co-localizes with FIP3–mCherry and that its fluorescence is pH-dependent (supplementary material Fig. S2D–H). Furthermore, we demonstrated that during interphase we can use VAMP8–pHluorin to visualize the fusion of a single endosome with the PM (supplementary material Fig. S2A–C; supplementary material Movie 2).
To establish whether FIP3-endosomes fuse with ICB PM during early stages of cell division, we imaged early telophase cells coexpressing FIP3–mCherry and VAMP8–pHluorin (supplementary material Fig. S3A–C). As in interphase, we could detect multiple VAMP8–pHluorin-endosome fusion events (supplementary material Fig. S3E, arrows). Interestingly, these fusion events always occurred outside the ICB (supplementary material Fig. S3E, arrowhead). Whereas endosomes containing VAMP8–pHluorin could be seen entering and exiting the ICB (supplementary material Fig. S3E, arrows), we did not observe any fusion events with the ICB PM (supplementary material Fig. S3E, supplementary material Movie 3) suggesting that FIP3-endosomes during early telophase can enter the ICB but usually do not fuse with the ICB PM.
Because the formation of the secondary ingression is preceded by the generation of ICB PM waves, we hypothesized that the progression to late telophase might be associated with an increase in FIP3-endosome fusion. To test this, we investigated the dynamics and localization of FIP3–mCherry and VAMP8–GFP during the formation of the secondary ingression (Fig. 3A,B). Consistent with the possible involvement of FIP3-endosomes in the abscission, FIP3 and VAMP8 accumulated at the site of the formation of the secondary ingression (Fig. 3A,B; asterisk in B marks the forming secondary ingression) and, as the secondary ingression elongated and thinned the ICB, FIP3 and VAMP8 stayed at the leading edge of the expanding secondary ingression (Fig. 3C; asterisks mark both sides of the secondary ingression; supplementary material Movie 4). The accumulation of FIP3 at the ICB and formation of the secondary ingression was observed in every case when the abscission event was imaged in FIP3–GFP-expressing HeLa cells (from five randomly chosen cells).
Fig. 3.

FIP3- and VAMP8-containing endosomes fuse with the ICB at the site of the secondary ingression. (A–C) Late telophase HeLa cell expressing FIP3–mCherry and VAMP8–GFP were imaged by time-lapse microscopy. (A) Localization of FIP3–mCherry (left panel) or VAMP8–GFP (right panel) at the beginning of time-lapse analysis. (B,C) Bright field and fluorescence images at the formation (B, marked with asterisk) and expansion (C, marked with asterisks) of the secondary ingression. Scale bar: 5 μm. (D–F) Late telophase cells expressing FIP3–mCherry (E) and VAMP8–pHluorin (F) were imaged by time-lapse microscopy. Only cells with forming secondary ingression were chosen for analysis. (D) Bright field images of the same cell during the initiation (left image) and expansion (middle and right images) of the secondary ingression. Asterisks mark the midbody; arrows mark leading edges of expanding secondary ingression; LE (leading edge) and C (control) show the areas quantified. (G) Quantification of the ratio between VAMP8–pHluorin and FIP3–mCherry fluorescence in cells with the secondary ingression. In each image, two sections within the ICB (each 2 μm long) were quantified (for example, see D). One section is at the leading edge of expanding secondary ingression (LE), and the second control section (C) is on the same side of the ICB close to the cell body. Data shown are individual ratios derived from three cells with secondary ingression at different time points. Lines mark the mean (1.56 for LE and 1.18 for C); n is the number of cells analyzed.
To test whether FIP3-endosomes fuse with the ICB PM during late telophase, we imaged secondary ingression formation and elongation in HeLa cells co-transduced with FIP3–mCherry and VAMP8–pHluorin (Fig. 3D–G). FIP3–mCherry accumulated at the expansion site of the secondary furrow (Fig. 3D–F; arrows mark the leading edge of the secondary ingression) and, as the secondary ingression formed and elongated, we saw an increase in the amount of VAMP8–pHluorin fluorescence at the leading edge of the secondary ingression (Fig. 3D; compare pHluorin fluorescence at time point 0 and 8 minutes; supplementary material Movie 5). Due to florescent microscopy resolution limits, we cannot resolve the fusion of the individual FIP3-endosomes. However, quantification of the fluorescence intensity showed a higher VAMP8–pHluorin to FIP3–mCherry ratio at the leading edge of the secondary ingression, as compared to the other parts of the ICB (Fig. 3G). Note that the means in Fig. 3G are not statistically different, due to the high variation in expression levels of FIP3–mCherry and VAMP8–pHluorin in different cells. However, in every cell and at every time point after the formation of the secondary ingression, the ratio in leading edge was higher (~132%, n=6) as compared to the fluorescence in other parts of the ICB.
Spatial analysis of organelles within the ICB during early telophase
A problem when imaging small, compact structures such as the ICB and midbody under the light microscope is that you reach the limits of the diffraction of light, which confines the resolution of the image to approximately 200 nm. To further analyze the organelle distribution within the ICB during early telophase, we investigated the ultrastructure of the ICB by using high-resolution electron tomography. Tomograms of HeLa cells expressing FIP3–GFP during early telophase displayed a 1.5–2.0 μm wide ICB, with the midbody region containing overlapping microtubule bundles (Fig. 4A). The midbody region was more electron dense than the surrounding cytoplasm (Fig. 4C, also see Fig. 5A,C). In addition to the overlapping microtubules, it is known to contain several proteins (Skop et al., 2004) so we will refer to it as the midbody matrix. In early telophase, the ICB is inhabited by three distinct populations of membrane-bound organelles, localized predominately outside the midbody matrix (Fig. 4B,C). The first class of organelles are tubulo-vesicular organelles, which often assume a ‘dumbbell’ shape (Fig. 4B, blue) and are modeled on the basis of their morphology (supplementary material Fig. S4F). These tubulo-vesicular organelles are FIP3-endosomes because they contain FIP3–GFP, as determined by via immunoelectron microscopy using anti-GFP antibodies (supplementary material Fig. S4A–E). FIP3-endosomes are positioned parallel to the spindle microtubules inside the ICB, but were rarely observed within midbody matrix (Fig. 4B). The second class of organelles are spherical vesicles of about 53±9 nm diameter (n=36) (supplementary material Fig. S4G) and are localized throughout the ICB (Fig. 3B, green). The identity of these organelles is not clear because the majority of them do not contain FIP3–GFP (supplementary material Fig. S4A–E). It is possible that they represent secretory organelles because they are abundant in the ICB during early telophase. Consistent with that, the time-lapse analysis of secretory–GFP (a marker for secretory vesicles) transport during cytokinesis showed accumulation of secretory vesicles at the ICB before the delivery of FIP3–mCherry-containing endosomes (data not shown). The third class of organelles are lytic endosomes, which were identified on the basis of the presence of at least two intraluminal vesicles (supplementary material Fig. S4H) and were mainly located just outside the ICB (Fig. 4B,C, red).
Fig. 4.

Spatial distribution of organelles within the ICB during early telophase. (A–C) Tomograms (A,C) and reconstructed model (B) of a HeLa cell expressing FIP3–GFP in early telophase. Asterisks and magenta lines mark midbody matrix; blue marks FIP3-endosomes; red marks lytic organelles; green marks 53-nm spherical vesicles; yellow marks plasma membrane. Scale bars: 200 nm. (D,E) Sequential high magnification images (D) and reconstructed model (E) of the boxed region in C. Asterisks mark different organelles that are embedded within microtubule bundle. Side (Ea) or end (Eb) views of modeled organelles and microtubules are shown; orange represents microtubules. (F) HeLa cells expressing FIP3–GFP in early telophase were treated with nocodazole (noc) for 10 minutes and then imaged by time-lapse microscopy. Bright field (Fb,Fd) or fluorescence (Fa,Fc) images before (Fa,Fb) or after (Fc,Fd) nocodazole treatment. (Fe) Kymograph of the ICB PM before (top panel) or after (bottom panel) treatment with nocodazole. Scale bars: 5 μm.
Fig. 5.

Spatial distribution of organelles within the ICB during late telophase. (A,B) Tomogram (A) and reconstructed model (B) of late telophase cell. Blue represents FIP3-endosomes. Green represents 53-nm spherical vesicles. (C,D) Tomogram (C) and reconstructed model (D) of late telophase cell forming a secondary ingression. Blue represents FIP3-endosomes; green represents 53-nm spherical vesicles; red represents lytic endosomes. (E,F) Consecutive images from tomograms showing exo/endo events (marked by asterisks). (Ea–Ed) Higher magnification images of the area boxed in C. Note that all exo/endo events in D are located in PM protrusion/wave. Scale bars: 200 nm.
Although during early telophase the ICB contains multiple organelles, including FIP3-endosomes, our VAMP8–pHluoring fusion data suggest that these organelles rarely fuse with the ICB PM. To further confirm this observation, we modeled the PM of the ICB (Fig. 4B,C, yellow) and counted all organelles that maintained continuity with the PM (Fig. 5E,F). This type of analysis does not allow us to separate organelles that are in the process of fusion from organelles that represent budding events, therefore we will refer to them as exo/endo events. Consistent with our VAMP8–pHluorin data, exo/endo events were rarely observed within the ICB during early telophase (1.7 events/μm2, data obtained from two different early telophase tomograms).
Closer examination of the various organelles and their surrounding environment yielded the observation that the majority of the organelles are tightly surrounded by microtubule bundles (Fig. 4D,E). As a result, we hypothesized that these microtubules might restrict lateral movement of the organelles to the PM, whereas still allowing the movement of organelles in and out of the ICB. We have previously shown that FIP3-endosomes move bi-directionally to and from the ICB, while exhibiting little lateral movement (Simon et al., 2008). To test whether organized microtubule bundles prevent FIP3-endosome fusion and ICB PM dynamics, we imaged HeLa cells expressing FIP3–GFP during early telophase before and after addition of the microtubule depolymerization agent nocodazole. Incubation of HeLa cells expressing FIP3–GFP for 10 minutes in 7.5 μM nocodazole resulted in an almost complete depolymerization of microtubules, leaving only microtubules associated with the midbody (data not shown). Unfortunately, we could not detect any rapid nocodazole-induced increases in VAMP8–pHluorin fluorescence, probably due to the fact that nocodazole is a slow-acting microtubule depolymerizing agent, as compared to the speed of vesicle fusion (it takes at least 10 minutes to depolymerize the microtubules within the ICB). However, consistent with the involvement of microtubules in inhibiting FIP3-endosome fusion, after nocodazole treatment, FIP3–GFP endosomes became diffuse. Punctuate FIP3-endosomes could no longer be observed within the ICB, which might represent the fusion of FIP3-endosomes with the PM (Fig. 4F and data not shown). Furthermore, nocodazole also induced an increase in the ICB PM dynamics (Fig. 4Fe). Together, our data suggest that depolymerization of the ICB microtubules induces PM dynamics, possibly by increasing fusion of FIP3-endosomes.
Spatial analysis of organelles within the ICB during late telophase
To investigate the spatial distribution of organelles during late telophase, we completed tomographic reconstructions of HeLa cells expressing FIP3–GFP during late stages of cytokinesis (Fig. 5; images are representative of three tomographic reconstructions of late telophase cells). The composition of organelles populating the ICB dramatically changed during the progression from early to late telophase (Fig. 5A,B). In late telophase, FIP3-endosomes predominately filled the ICB (Fig. 5B), and the 53-nm vesicles were rarely observed. Because it was previously suggested that the simultaneous fusion of organelles might mediate the abscission event, we calculated the combined membrane surface of all organelles present in the ICB. Our data suggest that even if all these organelles fused simultaneously at one plane of the ICB, it would only provide ~25% of the membrane needed to fully bridge the ICB. Thus, it is unlikely that fusion-mediated, rapid formation of separating membrane drives the abscission event. Instead, our data are more consistent with endosome fusion being required for the formation and gradual contraction of the secondary ingression.
To further confirm that FIP3-endosomes accumulate at the site of the secondary ingression, we performed tomographic reconstructions of the cells during the initiation of the secondary ingression (Fig. 5C,D; supplementary material Movie 6). Once again, we observed the accumulation of FIP3-endosomes at the site of the forming secondary ingression. Surprisingly, we observed a large number of lytic endosomes within close proximity of the secondary furrow (Fig. 5C,D; supplementary material Movie 6). Consistent with the increase in fusion of the endosomes during secondary ingression, we observed numerous exo/endo events (4.36 events/μm2; calculated from four different tomographs derived from two different cells) at the ICB during late telophase (Fig. 5E,F). Furthermore, numerous exo/endo events were situated inside the membranous projections (possibly ‘waves’) in the ICB (Fig. 5F; supplementary material Movie 7). Thus, our data suggest that the progression of cells from early to late telophase and the formation of secondary ingression coincide with an increase in RE endosome fusion with the ICB PM.
Spatial organization of central spindle microtubules changes during early to late telophase
It has been widely accepted that central spindle microtubules need to be disassembled for abscission to occur, although the mechanisms and regulation of microtubule disassembly remains elusive. To better understand the changes in the microtubule organization at different stages of telophase, we completed large volume tomographic reconstructions of the microtubules within the ICB. During early telophase, the ICB microtubules were organized into bundles in a characteristic overlapping manner (Fig. 6A,B) with 64% of the microtubule plus-ends localized within the midbody matrix (Fig. 6A and Fig. 7F). Consistent with our previous observations, most of the organelles were imbedded within the microtubule bundles (data not shown). In addition, the status of microtubule ends can affect microtubule organization, where flared ends of microtubules form during microtubule catastrophe and depolymerization, and this flaring of microtubule ends can be visualized by tomography (Austin et al., 2005). Interestingly, 18% of microtubules within the midbody matrix contained flared ends (supplementary material Fig. S4I,J). Thus, remodeling of the microtubules inside the midbody matrix might occur during early telophase (Fig. 6B and Fig. 7F).
Fig. 6.

Changes in microtubule organization during progression from early to late telophase. (A–D) Reconstructed models of microtubules in early (A,B) and late (C,D) telophase. (A,C) Microtubule with plus-ends (white spheres) and minus-ends (red spheres), defined on the basis of microtubule orientation. Ends facing cell body were labeled as minus-ends whereas ends facing midbody were termed plus-ends. (B,D) Microtubule with non-flared (white spheres) and flared (red spheres) ends. Arrowheads and inset in D show localized microtubule buckling and severing. Scale bar: 200 nm. (E,F) X-, Y- and Z-projections of dividing RK cells expressing tubulin–GFP. Arrowhead identifies region of tubulin–GFP depletion. Scale bar: 5 μm.
Fig. 7.

Microtubule severing during late telophase. (A–E) Tomograms of HeLa cells expressing FIP3–GFP in late telophase. (A) Microtubules severed or broken at one side of the midbody. (B) Higher magnification tomogram of the boxed area in A. (C,D) Bent and severed microtubules with the breaking point marked with asterisks. (E) Microtubules severed or broken at both sides of the midbody. (Ea,Eb) Higher magnification tomograms of the areas boxed in E. (F) Quantification of the number of microtubule plus-ends contained within the midbody matrix and total number of microtubules with flared ends in early and late telophase. Numbers within bars represents total number of microtubules analyzed.
Next, we analyzed microtubule organization during late telophase. Most of the microtubules were still organized in anti-parallel overlapping bundles, resembling early telophase (Fig. 6C,D). Overall, the mass of microtubules did not seem to change, suggesting that microtubules do not undergo complete depolymerization, but instead are remodeled at localized sites within the ICB. However, late telophase cells, prior to abscission, exhibited localized regions of severed microtubules (Fig. 6C,D; Fig. 7A–D), and the regions of severed microtubules were associated with an increased number of flared microtubule ends (Fig. 6D and Fig. 7F). These data suggest that microtubule severing results in increased depolymerization, presumably due to the formation of new uncapped plus-ends outside the midbody matrix (Fig. 6C). Although in many cases the severed microtubule regions are localized asymmetrically, in some cells we could observe severed microtubules on both sides of the midbody (Fig. 7E). Taken together, our results indicate that as a cell begins to progress into the later stages of cytokinesis, the overlapping microtubule network of the two daughter cells begins to break down in nested regions of the ICB. This localized microtubule severing and depolymerization might lead to formation of the secondary ingression. Consistent with that possibility, within the secondary ingression, as determined by narrowing of the ICB and increased PM protrusions (supplementary material Fig. S5A,B, marked by arrow), all microtubules had been cleared out and a region of depolymerizing and flared microtubule ends was found adjacent to the secondary ingression (supplementary material Fig. S5B). Whereas the microtubules adjacent to the secondary ingression did not display buckling within the plane of the tomogram, their altered orientation suggests that buckling had occurred either above or below this section.
Central spindle buckling mediates localized microtubule depolymerization during late telophase
Our tomography data show that during the progression of dividing cells from early to late telophase, the ICB microtubules undergo localized buckling, severing and depolymerization. To confirm that this localized microtubule reorganization occurs in live cells, we used rat kidney (RK) cells stably expressing GFP–tubulin to visualize microtubule dynamics during cell progression through cytokinesis (supplementary material Fig. S5C). As cells form the furrow, we saw the characteristic bundled microtubules within the ICB (supplementary material Fig. S5C). Localized GFP–tubulin depletion could be detected as early as 15 minutes after initial ICB formation, suggestive of microtubule thinning and depolymerization (supplementary material Fig. S5C, see arrowheads). During late telophase, almost complete loss of microtubules on one side of the ICB could be observed (supplementary material Fig. S5C, see arrowheads at 30 minutes), leading to abscission and retraction of the midbody (marked by arrow) to one daughter cell (supplementary material Fig. S5C, 45 minutes). Thus, the microtubule dynamics in live cells confirm that during late telophase microtubules undergo localized depolymerization usually (although not always) on one side of the midbody.
Our tomography analysis of the ICB microtubules also demonstrate that during late telophase microtubules undergo coordinated buckling (Fig. 6C, Fig. 7C,D). To confirm this, we imaged tubulin–GFP-expressing RK cells at different stages of telophase and analyzed the angle of the microtubules across the ICB. As shown in Fig. 6E,F, over 81% of late telophase cells displayed bent central spindle microtubules. By contrast, 92% of the central spindles during early telophase were straight (Fig. 6F, imaged at early telophase, 0 minutes, to late telophase, 45 minutes). Interestingly, in all cases, the sites of localized microtubule depolymerization coincided with the point of microtubule bending (Fig. 6F). These observations raise the interesting possibility that central spindle microtubule severing might be caused by microtubule bending and buckling. Although the role of microtubule buckling during cytokinesis has never been investigated, microtubule buckling was shown to play an important role in regulation of microtubule severing and length during interphase (Dogterom et al., 2005; Guo et al., 2007; Liu et al., 2006). Furthermore, it was shown that during interphase microtubule buckling and breakage usually leads to a rapid depolymerization and retraction of the plus-ends (Dogterom et al., 2005; Guo et al., 2007; Liu et al., 2006).
Microtubule-severing enzyme spastin regulates central spindle organization, but is not required for ICB microtubule severing
In addition to microtubule buckling, several severing enzymes were shown to mediate microtubule cutting and to play a role during cytokinesis (Roll-Mecak and McNally, 2010). Specifically, spastin was shown to have a role in regulating microtubule branching and organization (Sherwood et al., 2004; Yu et al., 2008) and in the release of microtubule minus ends from the centrosomes during cytokinesis (Zhang et al., 2007). Because spastin is known to localize to the ICB during telophase (Connell et al., 2009), it was postulated that it might also mediate microtubule cutting during abscission. Consistent with that, spastin knockdown was found to delay abscission (Connell et al., 2009). To further test whether spastin might be required for localized microtubule cutting, we treated HeLa cells expressing FIP3–GFP with spastin-specific siRNA and analyzed tubulin distribution during late telophase. Consistent with previous studies (Connell et al., 2009), spastin knockdown increased the number of cells in telophase (supplementary material Fig. S6D), although it had no effect on the number of binucleated cells (supplementary material Fig. S6A–C), suggesting a delay but not complete block of cytokinesis. Surprisingly, a lack of spastin did not result in an increased mass of central spindle microtubules during late telophase, as might have been expected if spastin mediates microtubule clearance from the ICB (Fig. 8). Instead, in over 72% of spastin knockdown cells, central spindle microtubules were not well organized and were still connected to the centrosomes (Fig. 8A,B and supplementary material Fig. S6E). Because central spindle microtubules are known to mediate organelle transport to the ICB, we wanted to investigate the effect of spastin knockdown on the transport of FIP3-endosomes to the ICB during cytokinesis. Cells treated with spastin-specific siRNA showed diffuse FIP3-endosome localization (Fig. 8C,D and supplementary material Fig. S6E), as compared with mock cells, in which during late telophase FIP3-endosomes were largely concentrated at the ICB during late stages of cytokinesis. This, presumably, was due to the unorganized nature of the microtubule network during spastin knockdown (Fig. 8B), making it difficult for the FIP3-endosomes to traffic efficiently. Eventually, some of the FIP3-endosomes did arrive at the ICB, consistent with spastin knockdown only affecting the efficiency of cytokinesis, rather than blocking it.
Fig. 8.

Spastin depletion alters microtubule organization and FIP3-endosome trafficking. (A–D) Fluorescent microscopy of HeLa cells treated with mock siRNA (A,C) or spastin-specific siRNA (B, D) and stained for tubulin (A,B) or FIP3–GFP localization (C,D). Scale bars: 5 μm. (E,F) Tomograms and reconstructed models of cells treated with spastin-specific siRNA. Asterisks mark midbody matrix. Arrowheads point to the areas of disorganized midbody matrix. (Ea) Higher magnification image of area boxed in E. (Fa) Modeled area of box in F. (G) Complete model of tomogram shown in F. Left microtubules, orange; right microtubules, blue; midbody matrix, purple; lytic organelles, red; recycling endosomes, blue; 53-nm vesicles, green; microtubule minus ends, white; microtubule plus ends, red. Scale bars: 200 nm.
Recent work suggests that microtubule-severing enzymes might preferentially bind to and cut bent microtubules (Baas et al., 2005). Thus, it is possible that spastin might be required for buckling-induced microtubule severing. To test that, we analyzed spastin-depleted cells during late telophase using large volume tomographic reconstructions to properly visualize the effect of spastin knockdown on microtubule organization. As shown in spastin knockdown tomograms, the central spindle microtubules overlap at the midbody matrix, yet they were much longer and appeared to extend and possibly still be connected to centrosomes (Fig. 8E). Furthermore, spastin knockdown cells exhibited an overall decrease in the organization of the central spindle microtubules, consistent with our previous observations (Fig. 8F,G). It appears that the overall mass of the central spindle microtubules remained consistent, or might be even decreased, in the various spastin knockdown tomograms, compared to untreated cells. However, even in spastin-depleted cells, there were still areas of localized microtubule bending and severing (Fig. 8F,G; supplementary material Fig. S5). Taken together, our data suggest that spastin plays a role in the release of central spindle microtubules from centrosomes, as well as in the organization of microtubule bundles within the ICB. Although we cannot fully discount some role of spastin in severing central spindle microtubules during secondary furrow ingression, spastin does not seem be absolutely required for the severing and localized depolymerization of microtubules within the ICB during abscission.
Discussion
Whereas different models have been proposed to explain the mechanisms behind abscission (Steigemann and Gerlich, 2009) there is little data defining the events leading up to abscission and the mechanisms behind the physical separation of the daughter cells. It is known that various organelles and the cytoskeleton participate in cytokinesis, yet how each component interacts with each other remains to be fully understood. Here, for the first time, we have used a combination of time-lapse microscopy and high-resolution tomography of high-pressure frozen and freeze-substituted fixed HeLa cells to analyze the changes in endosomal and microtubule spatio-temporal organization and dynamics as a cell progresses through telophase and abscission. Our data allow us to better define the mechanisms of abscission, as well as the factors regulating microtubule cytoskeleton remodeling and endosomal fusion during the terminal steps of cytokinesis.
It has been suggested that secretory vesicles and/or recycling endosomes fill the ICB at the midbody and mediate abscission, presumably by a series of fusion events between organelles and the PM, leading to a separation in the PM at the abscission site (Baluska et al., 2006). Interestingly, our tomography data suggest that during late telophase FIP3-endosomes are the dominant organelles present inside the ICB. Although we cannot completely rule out the contributions from other organelles, it is unlikely that secretory organelles play a major role in abscission. We also saw little evidence for the ‘plant-like’ formation (the construction of a cell plate in the middle of the two daughter cells) of the separating PM. Furthermore, our tomographic reconstructions of the ICB at late telophase demonstrate that accumulating organelles would not provide enough membrane to create a separating membrane that would cross-bridge the ICB PM. Instead, we have demonstrated that abscission appears to be a two-step process. First, during late telophase, we observed the formation of the secondary ingression, which decreases the thickness of the ICB from 1.5–2 μm to about 100–200 nm. Second, this secondary ingression expands to form a thin bridge, which then rapidly undergoes scission, leading to the separation of daughter cells. In addition, the initiation of the secondary ingression is usually preceded by an increase in dynamics of the PM, resulting in the formation of PM waves. The role of these waves in the formation of the secondary ingression remains to be fully understood, but it is tempting to hypothesize that increased dynamic instability of the ICB plasma membrane might be required for the initiation and expansion of the secondary ingression. Consistent with that, the knockdown of FIP3 and VAMP8 results in both a decrease in PM wave formation and inhibition of the abscission.
What is the source of the ICB PM waves that precede the secondary ingression? Several pieces of evidence suggest that these waves and the secondary ingression are probably induced by FIP3-endosome fusion with the ICB PM. First, we show that FIP3-endosomes accumulate at the ICB before the initiation of secondary ingression. Second, we show that the origin sites of PM waves co-localize with accumulations of FIP3-endosomes. Third, our VAMP8–pHluorin data show that FIP3-endosomes fuse at the site of forming secondary ingression. Fourth, FIP3 or VAMP8 depletion using RNA interference leads to a decrease in PM wave formation. Finally, our tomography data demonstrate that the frequency of exo/endo events increases at the site of secondary furrow formation. Taken together, our data strongly support the idea that fusion of endosomes with the ICB PM is required for the wave formation and increase in the ICB PM dynamic instability.
Interestingly, whereas FIP3 knockdown has a very dramatic effect on the ICB wave formation, VAMP8 knockdown cells exhibited a much milder phenotype. Similarly, VAMP8 knockdown also resulted in only a moderate increase in multinucleated cells as well as cells delayed in late telophase. It is likely that other VAMPs can partially substitute for the loss of VAMP8. For example, VAMP3, also known as cellubrevin, is also known to be present on recycling endosomes (McMahon et al., 1993). Furthermore, post-Golgi VAMPs have been shown to be functionally quite promiscuous and capable of substituting for each other in many different membrane transport pathways (Gordon et al., 2010).
Whether and how endosome fusion mediates the initiation and expansion of the secondary ingression remains to be fully understood. It is possible that rapid fusion of endosomes provide the necessary membrane to generate the PM waves and/or to allow the secondary ingression to narrow the ICB to 100–200 nm. In addition, the fusion of endosomes with the PM probably results in a change of the lipid composition at the site of the secondary ingression. Localized changes in lipid composition were shown to lead to changes in membrane shape and can result in scission via the mechanism known as ‘phase separation’ (Roux et al., 2005). Thus, it is tempting to speculate that localized endosome fusion might cause phase separation leading to the formation of the secondary ingression.
Whereas the secondary ingression is caused by localized endosome fusion within the ICB, we see no PM waves and little endosome fusion during early telophase. Our work suggests that, at least in part, this lack of endosome fusion might be caused by dense microtubule bundles, which restrict endosome lateral movement and fusion with the surrounding ICB PM. Consistent with that, our tomography data show that the majority of organelles during early telophase are localized within microtubule bundles. Previously, it was shown that during early telophase FIP3-endosomes move in and out of the ICB, without actually fusing with the PM (Simon et al., 2008). In addition, here we show that nocodazole-induced depolymerization of microtubules during early telophase results in FIP3-endosome fusion and increased PM dynamics within the ICB. Finally, our tomographic reconstructions demonstrate that at late telophase, the sites of microtubule depolymerization coincide with the sites of increased endosome fusion and formation of the secondary ingression. Thus, we propose that localized depolymerization of microtubules can serve as a cue to establish the site of endosome fusion, secondary ingression and eventual separation of daughter cells.
Central spindle microtubules traditionally are seen as non-kinetochore microtubules that, in late telophase, form microtubule bundles in the ICB and need to be depolymerized for abscission to take place. Although the mechanisms leading to microtubule depolymerization are not fully understood, it has been proposed that microtubule severing proteins, such as spastin, cut microtubules within the ICB resulting in the creation of free plus-ends and rapid depolymerization of microtubule bundles. Indeed, it was shown that spastin knockdown delays cytokinesis (Connell et al., 2009), although the mechanism for this delay remains unclear. Here we tested the role of spastin in the ICB microtubule clearance by analyzing the spatio-temporal organization of the ICB microtubules in spastin knockdown cells. Spastin knockdown did not result in decreased microtubule severing or increased microtubule mass within the ICB. Instead, spastin depletion resulted in disorganization of the central spindle microtubules, which appeared to still be attached to centrosomes even at late telophase. These data are consistent with the studies performed in neuronal axons and plant root cells, where spastin was implicated in microtubule organization by decreasing microtubule branching and crossing, thus enabling tight packing of microtubule bundles (Roll-Mecak and McNally, 2010). Similarly, spastin was shown to play a role in bundling non-centrosomal microtubule arrays in synaptic boutons, where loss of spastin leads to a thinning of microtubule arrays (McNally et al., 2006). Perhaps during cytokinesis, spastin is needed to release microtubules from the centrosomes and compact the central spindle bundles, but plays no direct role in the ICB microtubule severing and depolymerization. Our data show that spastin knockdown also results in the delay of FIP3-endosome delivery to the ICB, probably due to the disorganization of microtubule tracks leading to the ICB. The observations that spastin is required for efficient central spindle organization and delivery of endosomes is consistent with previous reports that cell division is still occurs after spastin depletion, but at a slower pace.
Surprisingly, our data show that localized microtubule severing and depolymerization coincides with increased microtubule buckling. Because buckling-induced microtubule severing has been shown to play an important role in microtubule remodeling during interphase (Guo et al., 2007; Liu et al., 2006), it is tempting to speculate that buckling might also induce microtubule breaking during late telophase. Indeed, it was shown that buckling-induced microtubule severing causes microtubules to undergo rapid depolymerization (Waterman-Storer and Salmon, 1997). Similarly, we show a dramatic increase in flared-end (indication of rapid depolymerization) microtubules at the sites of microtubule buckling. It is worth noting that microtubule severing enzymes were suggested to prefer bent or buckled microtubules (Baas et al., 2005). Thus, it is possible that spastin might increase the efficiency of buckled microtubule severing, although spastin does not appear to be absolutely required for it. Despite the fact that the cause of microtubule buckling remains elusive, previous studies suggested mechanisms mediating microtubule bending and buckling. It has been shown that kinesin and dynein motors might result in microtubule buckling and breaking during telophase (Bicek et al., 2009). Because several microtubule motors are known to be necessary for the completion of cytokinesis (Zhu et al., 2005), perhaps these microtubule motors might also contribute to the central spindle buckling by either anchoring themselves to the PM or by anchoring to another microtubule to create a bending force. In addition, it has been shown in vitro, that microtubule bundles can form wave-like structures through coordinated buckling by polymerization-induced forces (Guo et al., 2007; Liu et al., 2006). Additional studies will be needed to determine the mechanisms mediating central spindle microtubule buckling and severing.
On the basis of our analysis of endosome and microtubule dynamics during telophase, we propose a new model for the mechanisms involved in the abscission of mammalian cells (Fig. 9). During early telophase, microtubules form parallel bundles that overlap at the midbody and fill most of the ICB, preventing the Model for mammalian cell abscission.lateral transport and fusion of FIP3-endosomes with the PM. As cells progress to late telophase, microtubule buckling and severing lead to localized depolymerization of microtubule bundles and allow for increased fusion of FIP3-endosomes to the ICB PM. Organelle fusion increases the dynamics of the ICB membrane, leading to the induction and expansion of the secondary ingression. This secondary ingression then leads to the thinning of the ICB, eventually resulting in the scission and separation of daughter cells. The scission mechanism remains to be determined. One possible candidate is the ESCRT complex. It was previously suggested that the ESCRT complex proteins might form a ring around the ICB and induce scission of the ICB membrane. However, it is unclear how the ESCRT complex, which normally produces intraluminal vesicles and viral particles ranging in size from 25 to 50 nm, can actually sever the ICB measuring 1.5 to 2 μm. Furthermore, we found little evidence of the existence of gradual, ESCRT-dependent, constriction of the ICB during late telophase, as well as the involvement of ESCRT-dependent recruitment of spastin in microtubule severing during abscission. Further research is needed to determine the exact role of the ESCRT complex in cytokinesis. Perhaps the ESCRT complex might mediate the scission step after the formation of the secondary ingression. Indeed, the 100–200 nm thickness of the secondary ICB is much more consistent with the size of the membrane tubes that the ESCRT complex is known to sever.
Fig. 9.
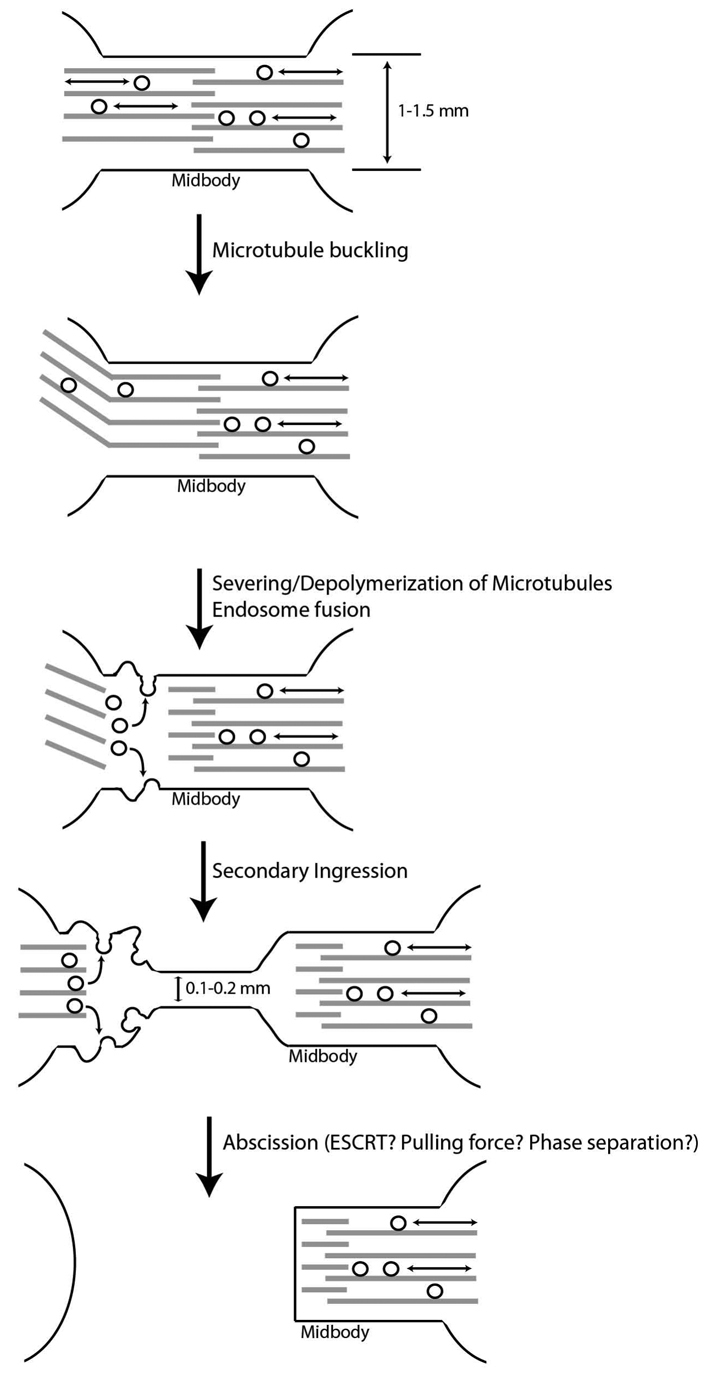
Model of secondary ingression formation and abscission.
Materials and Methods
Materials, antibodies and expression constructs
Various reagents were obtained from Invitrogen (Carlsbad, CA), Sigma-Aldrich (St Louis, MO) or Fisher (Pitsburg, PA), unless otherwise specified. Mouse monoclonal anti-acetylated tubulin antibody was purchased from Sigma-Aldrich. Mouse monoclonal anti-transferrin receptor antibody was purchased from Invitrogen (Carlsbad, CA). Rabbit polyclonal anti-VAMP8 antibody was a generous gift from Andrew Peden (Cambridge University, Cambridge, UK) (Gordon et al., 2010). Polyclonal rabbit anti-GFP antibody was generated using recombinant GFP as described previously (Wilson et al., 2005). Secondary antibodies conjugated with FITC, Cy-5 or Texas Red were purchased from Jackson ImmunoResearch (West Grove, PA).
FIP3–GFP, VAMP8–GFP and GFP–tubulin constructs are previously described (Wilson et al., 2005). Superecliptic pHluorin cDNA was a generous gift from Gero Miesenbock (University of Oxford, Oxford, UK) and was previously characterized (Miesenbock et al., 1998). Vamp8–pHluorin was created by using NotI and BamHI sites in Vamp8–GFP to excise GFP replacing it with superecliptic pHluorin. FIP3–mCherry was created by cloning FIP3 cDNA into pmCherry-N1 (Clontech, Mountain View, CA).
Light microscopy
For immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde, permeabilized in either PBS containing 0.4% saponin, 0.2% BSA and 1% fetal bovine serum, or PBS containing 0.1% Triton X-100, 0.2% BSA and 1% fetal bovine serum. Cells were stained by standard immunofluorescence procedures and imaged with an inverted Zeiss Axiovert 200M deconvolution microscope. Images were acquired and processed using Intelligent Imaging Innovations (Denver, CO) three-dimensional rendering and exploration software. Where indicated, Z-axis mini-stack (step 0.1 μm) was taken and maximum projection image generated.
For time-lapse microscopy, cells were plated on collagen-coated coverslips for 24 hours. Cells were then mounted on PH2-heated platform fitted with a TC-344B dual automatic temperature controller (Warner Instruments), and imaged at 37°C using a 63× oil-immersion lens. Ministacks (step size 0.2 μm) for GFP–tubulin expressing cells were acquired every 15 minutes for two hours.VAMP8–pHluorin expressing HeLa cells were imaged continuously at 0.1 second exposure for 100 or 200 frames.
Transfection and RNA interference
To knockdown spastin, HeLa cells were transfected with 2.5 nM spastin-specific siRNA using Lipofectamine 2000 (Invitrogen) according to manufacturer's protocol. Transfected cells were incubated for 48 hours, trypsinized and plated on collagen-coated coverslips for 24 hours, then processed for imaging, western blotting or tomography. siRNAs targeting spastin were previously characterized (Connell et al., 2009).
Correlative high-resolution tomography
HeLa cells expressing FIP3–GFP were plated on sapphire discs and imaged by brightfield and fluorescence microscopy to stage FIP3–GFP HeLa cells. Discs and cells were transferred to aluminium freezing planchettes (Wohlwend, Switzerland) containing a layer of hexadecane (Sigma) and were high-pressure frozen on HPM-010 from BAL-TEC AG (now Leica Microsystems, Wetzlar, Germany).
Frozen discs and cells were transferred to cryo-tubes (Nalgene-Nunc) containing freeze substitution media and moved to an automatic freeze-substitution system (AFS) (Leica Microsystems) and cooled to −85°C. Freeze-substitution protocols were as follows. Procedures for electron tomography were as described previously (McIntosh et al., 2005). Briefly, the samples were placed in 0.5% glutaraldehyde (EMS, Fort Washington, PA) and 0.1% tannic acid (Mallinckrodt, St Louis, MO) in acetone and a second substitution bath, containing 1% OsO4 (EMS) and 0.1% uranyl acetate (EMS) in acetone. The tubes were then warmed to 0°C, rinsed in acetone, and infiltrated with epoxy resin (EPOX-Araldite) (EMS) and flat-embedded following a method previously described (Reymond and Pickett-Heaps, 1983). After polymerization of the resin, the sapphire disc was released from the epoxy, and designated cells were mounted and sectioned en face. Serial sections (250 nm) were cut on an Ultracut microtome (Leica, Wetzlar, Germany), collected on formvar-coated, copper slot grids and post-stained with uranyl acetate and lead citrate. For immunolocalization, samples were placed in 0.1% glutaraldehyde and 0.05% uranyl acetate in acetone, rinsed in acetone, and infiltrated with Lowicryl HM20 resin (EMS) as previously described (Morphew, 2007). Discs were flat-embedded between Thermanox coverslips (Nunc, Naperville, IL) and polymerized with UV light in the AFS. Designated cells were remounted as previously described. Serial sections (75 nm) were collected as above and immunostained.
Cellular regions were selected and imaged with a Tecnai F-30 electron microscope (FEI, Eindhoven, NL), using Gatan CCD cameras to record serial tilts from ±60° in increments of 1° using image acquisition software Serial EM (Mastronarde, 2005). Each section was imaged in two tilt series around orthogonal axes and then assembled into a single reconstruction using the IMOD software (Mastronarde, 1997). Tomographic reconstructions were modeled by manual contour tracing using IMOD (Kremer et al., 1996). FIP3-associated endosomes were traced in blue, lytic organelles in red, spherical organelles in green, midbody matrix in purple, and microtubules were rendered by one quarter of their actual size. Microtubules left and right of the midbody matrix were colored orange and blue, respectively.
Immunoelectron microscopy
Serial sections were treated for 30 minutes in phosphate buffer containing 0.8% BSA and 0.1% fish gelatin (block buffer), blotted and incubated in primary antibodies for 2 hours at room temperature. GFP primary antibody was used as previously described (Zeng et al., 1999). Grids were rinsed in phosphate buffer and incubated in secondary antibodies conjugated to colloidal gold (BB International, Cardiff, UK) for 2 hours at room temperature. After rinsing, grids were post-fixed in 0.5% glutaraldehyde for 5 minutes, rinsed with water and post-stained as above. Cells were examined using Philips CM-100 electron microscope operating at 80 kV and images recorded using an AMT V600 camera (Advanced Microscopy Techniques, Danvers, MA).
Supplementary Material
Acknowledgments
We thank Kathryn Howell and Chad Pearson (University of Colorado, Denver) for critical reading of the manuscript. We also thank Andrew Peden (Cambridge University) for VAMP8–GFP construct, anti-VAMP8 antibodies and VAMP8-targeted siRNAs, Gero Miesenbock (University of Oxford) for pHluorin cDNA, Tom Kirchhausen for dynasore (Harvard Medical School) and Bruce Banfield for RK cells stably expressing GFP–tubulin. This work was supported in part by grant from NIH-NIDDK (DK064380 to R.P.). E.R. is a Wellcome Trust Senior Research Fellow in Clinical Science. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/9/1411/DC1
References
- Austin J. R., 2nd, Segui-Simarro J. M., Staehelin L. A. (2005). Quantitative analysis of changes in spatial distribution and plus-end geometry of microtubules involved in plant-cell cytokinesis. J. Cell Sci. 118, 3895-3903 [DOI] [PubMed] [Google Scholar]
- Baas P. W., Karabay A., Qiang L. (2005). Microtubules cut and run. Trends Cell Biol. 15, 518-524 [DOI] [PubMed] [Google Scholar]
- Baluska F., Menzel D., Barlow P. W. (2006). Cytokinesis in plant and animal cells: endosomes ‘shut the door’. Dev. Biol. 294, 1-10 [DOI] [PubMed] [Google Scholar]
- Bicek A. D., Tuzel E., Demtchouk A., Uppalapati M., Hancock W. O., Kroll D. M., Odde D. J. (2009). Anterograde microtubule transport drives microtubule bending in LLC-PK1 epithelial cells. Mol. Biol. Cell 20, 2943-2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B., Abramson D. H. (1968). Cytokinesis in HeLa: post-telophase delay and microtubule-associated motility. Protoplasma 66, 413-435 [DOI] [PubMed] [Google Scholar]
- Chen Y. A., Scheller R. H. (2001). SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98-106 [DOI] [PubMed] [Google Scholar]
- Chua C. E., Lim Y. S., Tang B. L. (2010). Rab35-a vesicular traffic-regulating small GTPase with actin modulating roles. FEBS Lett. 584, 1-6 [DOI] [PubMed] [Google Scholar]
- Connell J. W., Lindon C., Luzio J. P., Reid E. (2009). Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 10, 42-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom M., Kerssemakers J. W., Romet-Lemonne G., Janson M. E. (2005). Force generation by dynamic microtubules. Curr. Opin. Cell Biol. 17, 67-74 [DOI] [PubMed] [Google Scholar]
- Eggert U. S., Mitchison T. J., Field C. M. (2006). Animal cytokinesis: from parts list to mechanisms. Annu. Rev. Biochem. 75, 543-566 [DOI] [PubMed] [Google Scholar]
- Fanarraga M. L., Bellido J., Jaen C., Villegas J. C., Zabala J. C. (2010). TBCD links centriologenesis, spindle microtubule dynamics, and midbody abscission in human cells. PLoS ONE 5, e8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding A. B., Schonteich E., Matheson J., Wilson G., Yu X., Hickson G. R., Srivastava S., Baldwin S. A., Prekeris R., Gould G. W. (2005). Rab11-FIP3 and FIP4 interact with Arf6 and the exocyst to control membrane traffic in cytokinesis. EMBO J. 24, 3389-3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E., Bond L. M., Sahlender D. A., Peden A. A. (2010). A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic 11, 1191-1204 [DOI] [PubMed] [Google Scholar]
- Goss J. W., Toomre D. K. (2008). Both daughter cells traffic and exocytose membrane at the cleavage furrow during mammalian cytokinesis. J. Cell Biol. 181, 1047-1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B., Odermatt B., Royle S. J., Lagnado L. (2006). Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 51, 773-786 [DOI] [PubMed] [Google Scholar]
- Gromley A., Yeaman C., Rosa J., Redick S., Chen C. T., Mirabelle S., Guha M., Sillibourne J., Doxsey S. J. (2005). Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell 123, 75-87 [DOI] [PubMed] [Google Scholar]
- Guo Y., Liu Y., Tang J. X., Valles J. M., Jr (2007). Polymerization force driven buckling of microtubule bundles determines the wavelength of patterns formed in tubulin solutions. Phys. Rev. Lett. 98, 198103 [DOI] [PubMed] [Google Scholar]
- Hurley J. H., Hanson P. I. (2010). Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11, 556-666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch-Plunger V., Glotzer M. (1999). Depletion of syntaxins in the early Caenorhabditis elegans embryo reveals a role for membrane fusion events in cytokinesis. Curr. Biol. 9, 738-745 [DOI] [PubMed] [Google Scholar]
- Kremer J. R., Mastronarde D. N., McIntosh J. R. (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71-76 [DOI] [PubMed] [Google Scholar]
- Liu Y., Guo Y., Valles J. M., Jr, Tang J. X. (2006). Microtubule bundling and nested buckling drive stripe formation in polymerizing tubulin solutions. Proc. Natl. Acad. Sci. USA 103, 10654-10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low S. H., Li X., Miura M., Kudo N., Quinones B., Weimbs T. (2003). Syntaxin 2 and endobrevin are required for the terminal step of cytokinesis in mammalian cells. Dev. Cell 4, 753-759 [DOI] [PubMed] [Google Scholar]
- Mastronarde D. N. (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol. 120, 343-352 [DOI] [PubMed] [Google Scholar]
- Mastronarde D. N. (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36-51 [DOI] [PubMed] [Google Scholar]
- McDonald B., Martin-Serrano J. (2008). Regulation of Tsg101 expression by the steadiness box: a role of Tsg101-associated ligase. Mol. Biol. Cell 19, 754-763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh R., Nicastro D., Mastronarde D. (2005). New views of cells in 3D: an introduction to electron tomography. Trends Cell Biol. 15, 43-51 [DOI] [PubMed] [Google Scholar]
- McMahon H. T., Ushkaryov Y. A., Edelmann L., Link E., Binz T., Niemann H., Jahn R., Sudhof T. C. (1993). Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature 364, 346-349 [DOI] [PubMed] [Google Scholar]
- McNally K., Audhya A., Oegema K., McNally F. J. (2006). Katanin controls mitotic and meiotic spindle length. J. Cell Biol. 175, 881-891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G., De Angelis D. A., Rothman J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192-195 [DOI] [PubMed] [Google Scholar]
- Morita E., Sandrin V., Chung H. Y., Morham S. G., Gygi S. P., Rodesch C. K., Sundquist W. I. (2007). Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26, 4215-4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphew M. K. (2007). 3D immunolocalization with plastic sections. Methods Cell Biol. 79, 493-513 [DOI] [PubMed] [Google Scholar]
- Morphew M. K., McIntosh J. R. (2003). The use of filter membranes for high-pressure freezing of cell monolayers. J. Microsc. 212, 21-25 [DOI] [PubMed] [Google Scholar]
- Murata-Hori M., Wang Y. L. (2002). Both midzone and astral microtubules are involved in the delivery of cytokinesis signals: insights from the mobility of aurora B. J. Cell Biol. 159, 45-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata-Hori M., Tatsuka M., Wang Y. L. (2002). Probing the dynamics and functions of aurora B kinase in living cells during mitosis and cytokinesis. Mol. Biol. Cell 13, 1099-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond O. L., Pickett-Heaps J. D. (1983). A routine flat embedding method for electron microscopy of microorganisms allowing selection and precisely orientated sectioning of single cells by light microscopy. J. Microsc. 130, 79-84 [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A., McNally F. J. (2010). Microtubule-severing enzymes. Curr. Opin. Cell Biol. 22, 96-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A., Cuvelier D., Nassoy P., Prost J., Bassereau P., Goud B. (2005). Role of curvature and phase transition in lipid sorting and fission of membrane tubules. EMBO J. 24, 1537-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood N. T., Sun Q., Xue M., Zhang B., Zinn K. (2004). Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2, e429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon G. C., Schonteich E., Wu C. C., Piekny A., Ekiert D., Yu X., Gould G. W., Glotzer M., Prekeris R. (2008). Sequential Cyk-4 binding to ECT2 and FIP3 regulates cleavage furrow ingression and abscission during cytokinesis. EMBO J. 27, 1791-1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skop A. R., Liu H., Yates J., 3rd, Meyer B. J., Heald R. (2004). Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 305, 61-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S. J., Kim S. J., Song M. S., Lim D. S. (2009). Aurora B-mediated phosphorylation of RASSF1A maintains proper cytokinesis by recruiting Syntaxin16 to the midzone and midbody. Cancer Res. 69, 8540-8544 [DOI] [PubMed] [Google Scholar]
- Steigemann P., Gerlich D. W. (2009). Cytokinetic abscission: cellular dynamics at the midbody. Trends Cell Biol. 19, 606-616 [DOI] [PubMed] [Google Scholar]
- Waterman-Storer C. M., Salmon E. D. (1997). Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J. Cell Biol. 139, 417-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson G. M., Fielding A. B., Simon G. C., Yu X., Andrews P. D., Hames R. S., Frey A. M., Peden A. A., Gould G. W., Prekeris R. (2005). The FIP3-Rab11 protein complex regulates recycling endosome targeting to the cleavage furrow during late cytokinesis. Mol. Biol. Cell 16, 849-860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T., Wunder C., Lippincott-Schwartz J., Hurley J. H. (2009). Membrane scission by the ESCRT-III complex. Nature 458, 172-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Brill J. A., Hsien J., McBride R., Boulianne G. L., Trimble W. S. (2002). Syntaxin 5 is required for cytokinesis and spermatid differentiation in Drosophila. Dev. Biol. 251, 294-306 [DOI] [PubMed] [Google Scholar]
- Yu W., Qiang L., Solowska J. M., Karabay A., Korulu S., Baas P. W. (2008). The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol. Biol. Cell 19, 1485-1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X., Kahana J. A., Silver P. A., Morphew M. K., McIntosh J. R., Fitch I. T., Carbon J., Saunders W. S. (1999). Slk19p is a centromere protein that functions to stabilize mitotic spindles. J. Cell Biol. 146, 415-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Rogers G. C., Buster D. W., Sharp D. J. (2007). Three microtubule severing enzymes contribute to the “Pacman-flux” machinery that moves chromosomes. J. Cell Biol. 177, 231-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C., Zhao J., Bibikova M., Leverson J. D., Bossy-Wetzel E., Fan J. B., Abraham R. T., Jiang W. (2005). Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. Cell 16, 3187-3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.