

Abstract
Oxalate oxidase (E.C. 1.2.3.4) catalyzes the oxygen-dependent oxidation of oxalate to carbon dioxide in a reaction that is coupled with the formation of hydrogen peroxide. Although there is currently no structural information available for oxalate oxidase from Ceriporiopsis subvermispora (CsOxOx), sequence data and homology modeling indicate that it is the first manganese-containing bicupin enzyme identified that catalyzes this reaction. Interestingly, CsOxOx shares greatest sequence homology with bicupin microbial oxalate decarboxylases (OxDC). We show that CsOxOx activity directly correlates with Mn content and other metals do not appear to be able to support catalysis. EPR spectra indicate that the Mn is present as Mn(II), and are consistent with the coordination environment expected from homology modeling with known X-ray crystal structures of OxDC from Bacillus subtilis. EPR spin-trapping experiments support the existence of an oxalate-derived radical species formed during turnover. Acetate and a number of other small molecule carboxylic acids are competitive inhibitors for oxalate in the CsOxOx catalyzed reaction. The pH dependence of this reaction suggests that the dominant contribution to catalysis comes from the monoprotonated form of oxalate binding to a form of the enzyme in which an active site carboxylic acid residue must be unprotonated.
Keywords: Oxalate Oxidase, Mn(II), EPR Spectroscopy, Pichia pastoris, cupin, pH dependence
Introduction
Commercial applications of oxalate degrading enzymes include their use in clinical assays of oxalate in blood and urine [1, 2], the production of transgenic plants as a means of protection against pathogens and to reduce the amount of oxalate present [3, 4], the bioremediation of oxalate waste, the production of hydrogen peroxide, and pulping in the paper industry [3, 5–7]. Oxalate oxidase (OxOx) catalyzes the difficult carbon-carbon bond cleavage of oxalate to yield carbon dioxide and hydrogen peroxide [8–10] (Scheme 1). OxOx was first described in a study of wheat flour and has since been found in numerous microbes and plants [11]. OxOx activity has been detected in barley [8, 12, 13], beet [14, 15], sorghum [16, 17], maize, oats, rice, and rye [10, 18]. Hydrogen peroxide formation is believed to serve as a defense mechanism against infection by pathogens and to contribute to cell wall crosslinking in these species. The best characterized oxalate oxidases are from barley and wheat. These plant enzymes contain a single β-barrel fold characteristic to the cupin superfamily of proteins and are therefore classified as monocupins.
Scheme 1.

The reaction catalyzed by Ceriporiopsis subvermispora oxalate oxidase.
Fungal OxOx activity was first reported in Ceriporiopsis subvermispora, a white rot basidiomycete fungus able to degrade lignin, in 1999 [19]. In 2005, the gene encoding CsOxOx was cloned and sequenced [20]. Although there is currently no structural information available for oxalate oxidase from Ceriporiopsis subvermispora (CsOxOx), sequence data and homology modeling indicate that it is the first manganese-containing bicupin enzyme identified that catalyzes this reaction. Interestingly, CsOxOx shares greatest sequence homology with bicupin microbial oxalate decarboxylases (OxDC). OxDC catalyzes the carbon-carbon bond cleavage of oxalate to yield carbon dioxide and formate in a reaction in which there is no net oxidation or reduction [20]. UV-visible spectroscopy, spin trapping studies, and structural studies of barley oxalate oxidase have led to a proposed mechanism that involves the binding of oxalate directly to Mn(II), the formation of Mn(III), and a radical intermediate species (Scheme 2) [9, 10, 21, 22]. The first step after oxalate and dioxygen bind to the Mn(II) ion is a reversible proton-coupled electron transfer that facilitates decarboxylation to yield a manganese-bound formyl radical. In OxDC an active site general acid (Glu162 in the N-terminal domain or alternatively, Glu333 in the C-terminal domain) is proposed to protonate the manganese-bound formyl radical and formate is released.
Scheme 2.

Model of free-radical mechanism for oxalate oxidation. Modified from [20].
Initial characterization of the native CsOxOx was done by Aguilar et al., who purified 0.38 mg of native enzyme from 30 g of mycelium. The pH optimum reported for catalysis was 3.5, the specific activity 10.4 U/mg, the Km 0.1 mM. Histochemical studies of mycelium slices showed reaction products in both membrane vesicles connected to the outer membrane and peroxisome-like structures [19]. The fact that enzyme activity could be localized to multivesicular membrane bodies suggests that the enzyme is transported and secreted to the periplasmic space. The role of OxOx in C. subvermispora is still not well understood, but it has been proposed that CsOxOx may provide H2O2 to enable manganese peroxidase to oxidize extracellular Mn(II) to Mn(III). Mn(III), a diffusible and strong oxidant, is thought to facilitate lignin degradation [23].
Previous attempts to purify recombinant CsOxOx were not successful. Recombinant CsOxOx could only be expressed in E. coli with the assistance of co-transformed DnaK and DnaJ chaperone proteins, and attempts to purify the recombinant enzyme resulted in protein of only 40% purity, which precluded characterization of this novel enzyme [20]. We describe herein the first purification to homogeneity and characterization of recombinant bicupin oxalate oxidase. By characterizing oxalate oxidase from Ceriporiopsis subvermispora we can enhance the understanding of how subtle structural changes effect remarkable functional variation in evolutionarily related proteins.
Materials and methods
Materials
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich and were of the highest available purity. All DNA primers were obtained from Integrated DNA Technologies, Inc., and DNA sequencing was performed by the core facility in the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. Restriction enzymes and other molecular biology reagents were purchased from New England Biolabs.
Expression and purification of recombinant bicupin oxalate oxidase
The coding sequence for CsOxOx isoform-G (GenBank accession number: AJ746412) was amplified by PCR with primers that included an XbaI restriction site followed by a Kex2 protease cleavage site at the 5' and an XhoI restriction site at the 3'. The PCR product was digested with XbaI and XhoI and this fragment was cloned into the pPICZαA vector (Invitrogen) cut with the same restriction enzymes. CsOxOx was transformed into Pichia pastoris X-33 strain using the EasyComp transformation procedure (Invitrogen). Recombinants were screened for Zeocin resistance and the ability to grow using methanol as a carbon source. Putative clones were screened on minimal media plates containing 40 mM oxalate, pH 4.2. Bromophenol blue was included in the media for color selection.
25 mL cultures of MGY media (1.34% yeast nitrogen base, 1% glycerol, 4 ×10−5 % biotin) were inoculated with glycerol stocks of a single CsOxOx colony in a 250 mL baffled flask and grown overnight at 28–30 °C shaking at 250 rpm until the culture reaches log phase growth (OD600 = 2 – 6). Each of these cultures was used to inoculate 500 mL of MGY media in a 2 liter baffled flask at 28–30 °C shaking at 250 rpm until the culture reaches log phase growth. Cells were harvested by centrifugation at 2500 × g for five minutes at room temperature. Induction of expression of CsOxOx was accomplished by resuspending cells to OD600 = 1.0 in 500 mL aliquots of MM media (1.34% yeast nitrogen base, 4 ×10−5 % biotin, 0.5% methanol) with 5 mM MnCl2 at 28–30 °C shaking at 250 rpm. Methanol was added to 0.5 % every 24 hours for 4–5 days. Cells were removed by centrifuging 5000 × g for 45 minutes at 15 °C. The media containing secreted CsOxOx was concentrated to approximately 100 mL using a Biomax-30 tangential flow cassette and stirred cell ultrafiltration (Millipore). The concentrated sample was dialyzed against 50 mM Imidazole-Cl, pH 7.0 (buffer A) and applied to a DEAE-Sepharose Fast Flow column (2.5 × 25 cm) equilibrated with buffer A. Elution was performed using a 500 mL linear gradient from buffer A to buffer A containing 1 M NaCl. Fractions containing OxOx activity were pooled, and solid (NH4)2SO4 added to a final concentration of 1.7 M. The resulting solution was applied to a butyl-Sepharose Hi-Performance column (2.5 × 18 cm) (GE Healthcare, Piscataway, NJ) equilibrated with 50 mM Imidazole-Cl, pH 7.0, containing 1.7 M (NH4)2SO4 (buffer B). Bound proteins were eluted using a 500 mL linear gradient from buffer B to buffer A, and fractions containing purified CsOxOx were pooled, and concentrated by stirred cell ultrafiltration (Millipore) to a final volume of 10 mL before being exhaustively dialyzed against 25 mM Imidazole-Cl, pH 7.0. The dialyzed enzyme was then concentrated to approximately 10 mg/mL, and stored in aliquots at −80 °C. Expression and purification of heterologously expressed CsOxOx was monitored by SDS-PAGE carried out using a Mini Protean chamber system (Bio-Rad). Gels were stained for proteins using Coomassie stain and for carbohydrates using a Gel-Code Glycoprotein Staining Kit (Pierce). Protein concentrations were determined using a modified Lowry assay (Pierce, Rockford, IL) for which standard curves were constructed with bovine serum albumin [24].
Steady-state kinetic assays
The level of oxalate oxidase activity was determined using a continuous assay in which H2O2 production is coupled to the horseradish peroxidase (HRP) catalyzed oxidation of 2,2'-azinobis-(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) [13]. Reaction mixtures contained 25 U HRP, 5 mM ABTS, 50 mM potassium oxalate, CsOxOx (at concentrations up to 0.050 mg/mL) dissolved in 50 mM sodium acetate, sodium succinate, or sodium citrate, pH 4.0 (total volume 1.0 mL). Assays were monitored at 650 nm and an extinction coefficient of 10,000 M−1 cm−1 for the ABTS radical product was assumed in these experiments. Control samples omitted HRP in order to distinguish between H2O2 production and any oxalate-dependent dye oxidation activity by CsOxOx. Measurements were made at specific substrate and enzyme concentrations in duplicate, and data were analyzed to obtain the values of Vmax and Vmax/Km by standard computer-based methods [25]. Assay mixtures for the construction of the pH profile with acetate present contained a final concentration of 50 mM glycine, 50 mM acetate, and 50 mM MES.
The continuous coupled assay described above (using succinate buffer) was used to measure the inhibition of acetate, malonate, malate, glycolate, glyoxylate, and pyruvate. The mode of inhibition for all compounds tested was determined by measuring the enzyme velocity at a variety of substrate concentrations in the presence and absence of the possible inhibitor. All of the compounds that showed inhibition diminished enzyme velocity at low concentrations of substrate but the velocity reached uninhibited maximal levels at high concentrations of substrate. Since, therefore, the compounds were competitive inhibitors, the inhibition constants were determined by observing the apparent Km (Km,obs) at two concentrations of inhibitor and calculating Ki by equation 1 which defines Ki as the inhibitor concentration that doubles the apparent Km of the competing substrate.
| (1) |
The pH dependence of Vmax/Km was determined by measuring the initial rate at varying concentrations of oxalate. Assay mixtures in these experiments each contained a final concentration of 50 mM glycine, 50 mM succinate, and 50 mM MES adjusted to specific pH values. Substrate solutions were pH adjusted also. The log(Vmax) and log(Vmax/Km) were plotted versus pH, and the data were fit to equations 2 and 3 using the program GraphPad Prism.
| (2) |
| (3) |
In addition to measuring the rate at which hydrogen peroxide was produced by the enzyme-catalyzed oxidation of oxalate, we determined the level of oxalate decarboxylase activity of CsOxOx. Assay mixtures consisted of 50 mM NaOAc, pH 4.2, 0.2% Triton X-100, 0.5 mM o-phenylenediamine, 1–50 mM potassium oxalate, and CsOxOx (50 μM) (100 μL total volume). Reactions were initiated by the addition of substrate, incubated at ambient temperature (21–23°C), and quenched by the addition of 1 N NaOH (10 μL). The amount of formate produced was determined by an end-point assay [26] consisting of 50 mM potassium phosphate, pH 7.8, 0.09 mM NAD+, and 0.4–1.0 U/mg of formate dehydrogenase (1 mL final volume). The absorbance at 340 nm was measured after overnight incubation at 37 °C, and formate was quantified by comparison to a standard curve generated by spiking pre-quenched assay mixtures with known amounts of sodium formate.
Determination of Mn content
The metal content of CsOxOx samples was quantified at the University of Wisconsin Soil and Plant Analysis Laboratory on the basis of inductively-coupled plasma mass spectroscopy (ICPMS) [27] and by Flame Atomic Absorption Spectroscopy (FAAS) [28] using a Varian SpectrAA-55 and comparing measurements to a standard curve constructed using dilutions of a standard solution of MnCl2 (Fisher). To prepare samples and blanks for determination of metal content, divalent cations were removed from the final enzyme storage buffer by passing through a 1.5 × 16 cm column containing Chelex 100 (Bio-Rad) in the Na+ form. Purified protein samples were exchanged into the resulting buffer by washing 2.5 mg samples three times with 10-fold volumes of the “scrubbed” buffer in Centricon or Centriprep 30 (Amicon) concentrators [29]. The final filtrates recovered were used as blanks, which possessed insignificant metal content.
Preparation of Apoenzyme
This procedure was modified from that used to prepare manganese-deficient oxalate decarboxylase [30]. Recombinant CsOxOx was dialyzed against 3.5 M guanidinium hydrochloride (GuHCl), 20 mM Tris-HCl, and 10 mM EDTA, pH 3.1, for 8 h at 4 °C. A second round of dialysis against 2.5 M GuHCl, 20 mM Tris-HCl, and 10 mM EDTA, pH 7.0, was performed, and excess EDTA removed in a third dialysis against 2.5 M GuHCl containing 20 mM Tris-HCl, pH 7.0 again for 8 h at 4 °C. CsOxOx was then re-folded by dialysis against 25 mM Imidazole-Cl, pH 7.0, over 8 h at 4 °C, to yield a Mn-deficient form of OxDC (“apoenzyme”) that exhibited no catalytic activity when incubated with oxalate.
EPR spectroscopy
EPR spectra were determined using CsOxOx (16 mg/mL) in 25 mM Imidazole-Cl, pH 7.0 (100 μL total volume). For low temperature EPR spectra the samples were taken as prepared and added to a 3×4 mm (IDxOD) quartz tube (approx. 100 μL per sample) then quickly frozen in pre-cooled isopentane. A test tube of liquid isopentane was inserted into a dewar filled with liquid nitrogen. After 1 to 2 min as the isopentane started to freeze at the walls of the test tube the EPR sample tube was plunged into the cold isopentane which resulted in immediate freezing of the sample inside. When preparing samples at lower pH in acetate buffer a 1.0 M acetate buffer stock solution was prepared and adjusted to pH 4.0 using HCl. For oxalate addition we prepared a 0.5 M oxalic acid solution using KOH to adjust the pH to 4.0. 15 μL of the acetate buffer stock solution was added to 150 μL of CsOxOx enzyme solution for a sample at low pH (measured as 4.6) and a final acetate concentration of about 91 mM. To this sample 18 μL of the oxalate stock solution was added for a final oxalate concentration of about 50 mM. The sample was then frozen as described above in pre-cooled isopentane after a reaction time of about 5 seconds. EPR experiments at cryogenic temperatures were conducted on a Bruker Elexsys E580 spectrometer equipped with an Oxford ESR900 helium cryostat. Pre-frozen samples were inserted into the pre-cooled cryostat. The instrumental settings for the spectra shown here were: sweep width, 7,000 G; number of data points/sweep, 2048; microwave frequency, 9.454 GHz; microwave power, 630 μW; modulation amplitude, 20 G; modulation frequency, 100 kHz; receiver gain, 70 dB; time constant, 40 ms; conversion time, 40 ms, temperature, 5 K, and each spectrum was the average of four field sweeps.
EPR spin-trapping experiments
The reaction mixtures, conducted in 0.2 M potassium acetate buffer pH 4.1, contained: 100 mM oxalate (or 1,2-13C-oxalate), 100 mM KCl, 20 mM PBN or 50 mM DMPO, and 50 μM CsOxOx. A small aliquot of the reaction mixture was transferred into a quartz capillary of approximately 1×3 mm ID×OD for EPR measurement. The EPR spectrum was recorded at room temperature, using a Bruker Elexsys E580 spectrometer, employing the high-Q cavity (ER 4123SHQE). EPR parameters were typically: 100 KHz modulation frequency, 1 G modulation amplitude, 20.18 ms time constant, 81.92 ms conversion time/point, 70 G sweep width, 9.87 GHz microwave frequency, and 2 mW microwave power.
Results and discussion
Enzyme expression and purification
Previous work with the native CsOxOx suggested that this enzyme is secreted. We considered this an important aspect that should be taken into account in the expression of the recombinant enzyme and might explain why previous attempts to express and purify this protein in E. coli failed. In order to produce recombinant secreted protein, CsOxOx was amplified by PCR with an engineered Kex2 protease cleavage site at the 5' and this fragment was cloned into the pPICZαA vector (Invitrogen) as described in Materials and Methods. This cloning strategy led to the translation of untagged, full-length CsOxOx preceded by the α-mating factor signal secretion sequence. The engineered Kex2 protease cleavage site ensures that the α-factor signal sequence is removed from the expressed recombinant CsOxOx. CsOxOx was transformed into Pichia pastoris X-33 strain and recombinant colonies were screened for Zeocin resistance and the ability to grow using methanol as a carbon source.
In order to ensure that the selected colonies were expressing functional recombinant enzyme that retained OxOx activity, we screened candidate colonies on minimal media plates containing 40 mM oxalate, pH 4.2 and bromophenol blue. Changes in pH affect the color of the dye, which allowed us to monitor oxalate degradation. Colonies grown under non-inducing conditions presented a yellow tint (Figure 1A). When methanol was used to induce recombinant protein expression (Figure 1B), colonies expressing functional CsOxOx (first row) were able to degrade oxalate and presented a blue tint due to the rise in pH, while empty vector (second row) and untransformed control (third row) did not show blue color. The Mut− strain KM71H is unable to grow in the presence of methanol; it is used as a control for the presence of methanol in the plates and as a negative control for the Mut+ mutation. The blue color noticeable in the fourth row (Figure 1B) is due to changes in pH resulting from cell death. Therefore, viable, recombinant colonies that were able to produce blue color on oxalate selection plates were selected for large scale protein expression and purification.
Fig. 1.

Oxalate degradation assay. Putative clones were screened on minimal media plates containing 40 mM oxalate, pH 4.2 and bromophenol blue was included in the media for color selection. When colonies were grown under non-induced conditions (Figure 1A, dextrose media), the colonies presented a yellow tint. Under induction conditions (Figure 1B, methanol media), colonies expressing CsOxOx were able to degrade oxalate and presented a blue tint due to the raise in pH. Empty vector and untransformed control did not show blue color. KM71H did not grow on methanol-containing plates.
Under methanol-induced expression, CsOxOx was secreted into the growth media as a soluble, functional enzyme. CsOxOx was purified from the expression media as described in Materials and Methods. Approximately 10 mg of purified CsOxOx was obtained from 2.5 L of growth medium (Table 1). The majority of the purification comes from the hydrophobic interaction chromatography step (32-fold). Gel analysis of both the DEAE pools and butyl-sepharose pools indicate each is purified to greater than 90% homogeneity (Fig. 2). This suggests that the butyl-sepharose column is separating inactive from active enzyme given the increase in activity after the latter step.
Table 1.
Summary of the Purification of CsOxOx.
| Fraction | Vol. mL | Protein mg | Total Act. U | Spe. Act. U/mg | Purification fold | Yield % |
|---|---|---|---|---|---|---|
| Unconcentrated media | 2300 | 268 | 188 | 0.4 | 1.0 | 100 |
| Concentrated dialyzed media | 52 | 240 | 159 | 0.7 | 1.8 | 85 |
| DEAE pool | 50 | 147 | 132 | 0.9 | 2.3 | 70 |
| Butyl Sepharose pool | 32 | 9.5 | 121 | 12.7 | 32.0 | 64 |
| Concentrated CsOxOx | 1.2 | 8.9 | 114 | 12.8 | 32.0 | 61 |
Fig. 2.
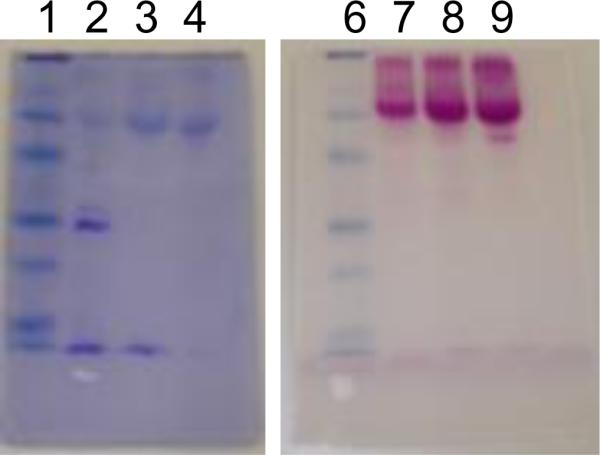
12% SDS PAGE stained for protein and carbohydrate. Samples were applied twice (lanes 1–4 and lanes 6–9). After electrophoresis, the gel was cut in half and one half stained with Coomassie (lanes 1–4) and the other half stained for carbohydrate (lanes 6–9) as described in Materials and Methods. Lanes 1 and 6: prestained molecular weight markers (97.4 kD, 66.2 kD, 45.0 kD, 31.0 kD, 21.5 kD, 14.4 kD), lanes 2 and 7 are 7 ng of concentrated media, lanes 3 and 8 are 7 ng of DEAE pool, and lanes 4 and 9 are 7 ng butyl sepharose pool.
The recombinant enzyme is highly glycosylated, as shown by carbohydrate staining (Fig.2). This is consistent with the presence of three possible N-glycosylation sites previously identified in its sequence [20]. Due to its carbohydrate content, the purified protein runs as a 66 kD protein. The calculated molecular weight of CsOxOx based on amino acid sequence is 45 kD implying that carbohydrates make up approximately 30% of its mass. This high level of glycosylation makes staining with Coomassie a challenge. Gels require long stain times and will eventually destain completely if left in destain solution for long periods of time. Hence, visualizing the Coomassie stained CsOxOx band is a balance between staining and destaining.
Competitive inhibition of CsOxOx by acetate and other carboxylic acids
As shown in Fig.3, the pH optimum of CsOxOx is 4.0. Steady-state experiments carried out in acetate buffer, pH 4.0 yielded a Vmax value of 21.2 U/mg that compares very favorably with the value obtained previously for the native enzyme [19]. The Km for oxalate, determined in acetate buffer, was 14.9 mM, which was, however, significantly larger than the 0.1 mM value reported for the native enzyme. Table 2 shows that the steady-state kinetic parameters are very sensitive to the buffer in which the assay is performed. When succinate buffer, pH 4.0 is used the Km for oxalate is 1.5 mM and when citrate buffer, pH 4.0 is used the Km is 0.1 mM. The apparent Km value of 0.1 mM in citrate suggests that succinate might be a competitive inhibitor. The Vmax in citrate is, however, reduced and the addition of succinate increases the activity of the citrate inhibited enzyme. This suggests that citrate is a weak uncompetitive inhibitor of CsOxOx. Citrate inhibited enzyme still turns over at about 40% of the uninhibited rate. Succinate appears to displace citrate without inhibiting the enzyme. There is no evidence for succinate inhibition. Since the pH optimum for this enzyme catalyzed reaction is 4.0, it is customary (and necessary) to use carboxylic acids as buffers at this pH. We have determined that acetate and a number of other carboxylic acids (malonate, malate, glycolate, glyoxylate, and pyruvate) diminished enzyme velocity at low concentrations of substrate but the velocity reached uninhibited maximal levels at high concentrations of substrate and are, therefore, competitive inhibitors. This is consistent with the structural similarities between acetate and oxalate. The inhibition constants are given in Table 3. None of the inhibitors tested served as alternative substrates.
Fig. 3.
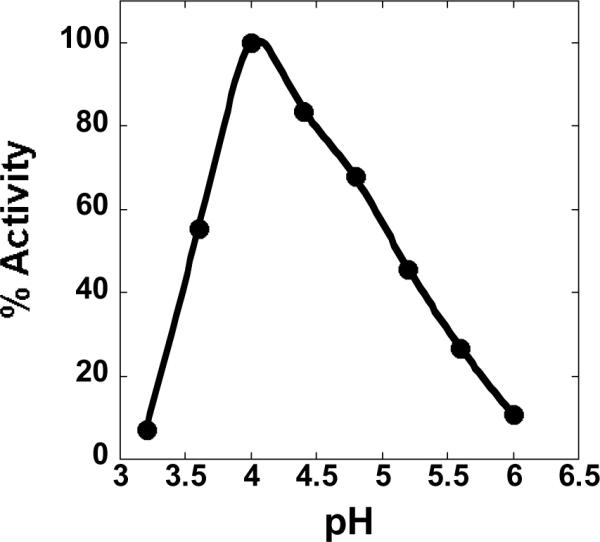
pH profile of recombinant oxalate oxidase activity with acetate buffer present in the reaction mixtures.
Table 2.
Km for oxalate varies with assay buffera.
| Buffer | Km mM | Vmax U/mg |
|---|---|---|
| Acetate, pH 4.0 | 14.9 ± 0.1 | 21.1 ± 0.5 |
| Succinate, pH 4.0 | 1.5 ± 0.1 | 18.2 ± 0.4 |
| Citrate, pH 4.0 | 0.1 ± 0.02 | 8.1 ± 0.4 |
Initial velocities were measured spectrophotometrically using the coupled assay described in Material and Methods with the final concentrations of oxalate ranging from 0.01 mM to 90 mM.
Table 3.
Ki values for a variety of inhibitorsa.
| Ki, mM | |
|---|---|
| acetate | 3.9 ± 0.6 |
| malonate | 3.0 ± 0.3 |
| malate | 52 ± 6 |
| glycolate | 28 ± 5 |
| glyoxylate | 15 ± 3 |
| pyruvate | 17 ± 3 |
Initial velocities were determined spectrophotometrically in succinate buffer, pH 4.0 using the coupled assay described in Material and Methods.
OxDC activity assay of CsOxOx
Previously characterized OxDC's and OxOx's possess between 2% and 4% of the other respective activity [20, 31]. An E162Q mutant of OxDC exhibited an approximately 200-fold decrease in OxDC activity with an approximately 10-fold increase in OxOx activity [32]. A theoretical study suggested that the intrinsic reactivity of the N-terminal Mn-binding site of OxDC is to oxidize oxalate and that decarboxylation is a result of the protein environment modulating this reactivity through the addition of a proton donor as well as through electrostatic changes [33]. Burrell et al. have demonstrated that the decarboxylase can be converted to an oxidase by mutating amino acids of a flexible lid that, when in the “closed” conformation, seals the Mn-binding site from bulk solvent [22]. Mutations of this lid, which contains Glu162, have resulted in specificity switches of up to 282 000 (SEN161-3DSS), 275 000 (SENS161-4DSSN), and 225 000 (SENS161-4DASN). The structure of the SENS161-4DSSN mutant showed that the C-terminal Mn-binding site was not affected. This group's sequence information of oxalate oxidase from Ceriporiopsis subvermispora played a critical role in the recognition that lid peptide sequences can control reaction specificity. It was, therefore, of interest to determine the oxalate decarboxylase activity of recombinant CsOxOx. Recombinant CsOxOx possesses less than 0.1% OxDC activity (data not shown) which is consistent with the observation that the enzyme purified from C. subermispora possesses less than 0.6% OxDC activity [20].
Manganese dependency of CsOxOx
Enzymes in the cupin superfamily appear to be able to employ a variety of metals in catalysis [34–36]. Characterizing the manganese dependence of CsOxOx is significant to place this enzyme in the context of other oxalate degrading enzymes and that of other cupin proteins. Of the 54 unique cupin (double stranded beta helix) proteins with structures deposited in the database 24 contain no metal, 16 contain Fe, 4 contain Mn, 4 contain Zn, 3 contain Ni, 2 Cu and 1 Hg. Two methods were used to determine the manganese content of three independently purified samples: Flame Atomic Absorption Spectroscopy (FAAS) and Inductively-Coupled Plasma Mass Spectroscopy (ICP-MS). As shown in Table 4, the two methods showed comparable results, and manganese levels obtained were between 0.1 and 0.4 mole Mn/mole enzyme. The specific activities of these individual preparations show a direct correlation with manganese content. The specific activity of our preparations is limited by the amount of manganese that we can incorporate using the Pichia expression system.
Table 4.
Comparison of results of FAAS and ICP-MS analyses of CsOxOx samples and specific activity.
| Sample | FAAS mol Mn/mol enz | ICP-MS mol Mn/mol enz | Activity U/mg |
|---|---|---|---|
| OxDC | 1.60 ± 0.08 | 1.60 ± 0.01 | N/A |
| CsOxOx (#1) | 0.29 ± 0.05 | 0.22 ± 0.02 | 6.2 ± 0.3 |
| CsOxOx (#2) | 0.38 ± 0.04 | 0.34 ± 0.01 | 18.2 ± 0.6 |
| CsOxOx (#3) | 0.14 ± 0.03 | 0.18 ± 0.02 | 1.8 ± 0.1 |
To test whether or not other metal ions were able to support catalysis, we prepared “apo-CsOxOx” by unfolding it in the presence of EDTA and refolding it in metal-free buffer in a procedure previously used for oxalate decarboxylase [30]. The apo-CsOxOx enzyme does not possess any oxalate oxidase activity. Incubation of the apoenzyme with a 100-fold molar excess of MgCl2, CoCl2, CuCl2, ZnCl2, NiCl2, FeCl2 or FeCl3, individually, does not result in the return of enzymatic activity. Incubation of the apoenzyme with a 100-fold molar excess of MnCl2 does, however, restore activity. The return of activity by incubation with MnCl2 required four weeks and resulted in only a small but measurable (11%) return of activity. These experiments suggest that CsOxOx activity is highly specific for manganese.
X-band EPR spectroscopy of the manganese centers of CsOxOx
Low temperature X-band EPR spectra of the manganese centers of CsOxOx are shown in Fig. 4. All spectra are from the same sample subjected to different conditions. The trace given in Fig. 4A refers to the sample in imidazole buffer at pH 7.0, as taken from the original preparation. The Fig.4B spectrum is for the same sample that had been thawed and then mixed with acetate buffer for a final pH of 4.6 at an acetate buffer concentration of approximately 91 mM, as explained in “Materials and Methods.” Fig. 4C reflects the sample after addition of oxalate and allowed to react for approximately 2 min after which it was flash frozen in pre-cooled isopentane again. The signal to noise ratio is poor due to the low manganese incorporation in this sample but several interesting observations can be made. In contrast to X-band EPR spectra of oxalate decarboxylase from Bacillus subtilis [37], all CsOxOx spectra, whether in imidazole buffer, pH 7.0 (A) or in acetate buffer, pH 4.6 (B), appear very similar. The addition of oxalate (C) does not lead to significant spectral changes either. In these experiments we were looking to see if a tyrosyl radical could be observed with CsOxOx similar to what is seen in OxDC [38]. The absence of the tyrosyl radical does not come as a surprise since it has not been observed in other oxalate oxidases but appears to be specific to oxalate decarboxylase. The decarboxylase activity of the bicupin OxOx is essentially zero. The addition of dithionite did not significantly increase the signal intensity indicating that the majority of manganese in the sample is already in the Mn(II) oxidation state (spectrum not shown). A simulation (Fig. 4D) was able to reproduce quite well the main broad features of the spectra using a single set of magnetic parameters (D = −3000MHz, E = 300 MHz, g = 2.00088, A = 263 MHz with 20% strain associated with the fine structure constants D and E). The simulation, however, does not do such a good job on the narrower features which is not surprising under the low-field conditions of Mn(II) in the X-band. It is of course possible that a second Mn-site gives rise to some of the narrow spectral features but the spectra are too noisy to lend any confidence in simulations with more than one set of magnetic parameters.
Fig. 4.

X-band EPR of the Mn(II) centers of CsOxOx under several different conditions at 5 K. 4A: CsOxOx in imidazole buffer at pH7.0, as taken from the original preparation. 4B: Same sample, thawed and after addition of 91 mM acetate buffer pH4.0 for a final pH of 4.6. 4C: Same sample as in the red trace, thawed and after addition of 50 mM oxalate and allowed to further react for approximately 2 min. 4D: Spectral simulation. The instrumental settings for the spectra were: sweep width, 7,000 G; number of data points/sweep, 2048; microwave frequency, 9.454 GHz; microwave power, 630 μW; modulation amplitude, 20 G; modulation frequency, 100 kHz; receiver gain, 70 dB; time constant, 40 ms; conversion time, 40 ms, temperature, 5 K, and each spectrum was the average of four field sweeps.
X-band spin trapping experiments
The X-band EPR spectra of spin trapped free radicals produced during turnover by CsOxOx is shown in Figure 5. Figure 5A shows the six-line spectrum of the PBN radical adduct formed during turnover. The hyperfine coupling constants given in the figure legend are consistent with those of a carboxylate radical. These data strongly support the formation of the carbon dioxide radical anion which appears in the proposed catalytic mechanism (Scheme 2). Figure 5B shows further splitting of the PBN adduct signal when doubly 13C-labeled oxalate is used confirming that the radical is oxalate derived. Finally, Figure 5C shows the sextet expected of the DMPO radical adduct which has hyperfine coupling constants consistent with a carboxylate radical. Trapping of the same species in oxalate decarboxylase [39] is consistent with a common element between the two enzymes.
Fig. 5.

EPR spectra of PBN and DMPO radical adducts obtained from the incubation of 100 mM oxalate (or 1,2-13C-oxalate), 100 mM KCl, 20 mM PBN or 50 mM DMPO, and 50 μM CsOxOx. (A) PBN-12C oxalate derived radical adduct displayed the hyperfine coupling constants, a(N) = 15.8 G and a(H) = 4.5 G. (B) PBN-13C oxalate derived radical adduct showed the hyperfine coupling constants, a(N) = 15.9 G, a(13C) = 11.6 G, and a(H) = 4.4 G. (C) DMPO-12C oxalate derived radical adduct displayed the hyperfine coupling constants, a(N) = 15.7 G and a(H) = 18.9 G.
The effect of pH on the affinity of CsOxOx for oxalate
Fig. 6B shows the Vmax/Km profile in a three component buffer system containing succinate is asymmetrical, decreasing more rapidly on the acidic side. The slope of the acidic limb of the Vmax/Km profile is +2 and the slope of the basic limb is −1. The Vmax/Km profile was fit with equation 2 which assumes that monoprotonated substrate binds unprotonated enzyme (EAH− in Fig. 6A). The fit (R2=0.95) was obtained with pKa1= 4.5, pKa2= 3.1, pKenz= 3.7, K2= 4.0 and Vmax= 110. This is the simplest model that approximates the data and it is possible that more pKa values are involved. These data could not be fit with the analogous equations assuming the key species to be those defined in Fig 6A as EAH2, EA2−,(EH+)AH2, or (EH+)AH−. We were able, however, to approximate the data equally well (R2=0.95) by the model that assumes fully unprotonated oxalate binds protonated enzyme ((EH+)A2−) (Fig. 6A) with similar values (pKa1= 4.6, pKa2= 3.0, and pKenz= 3.6, K6= 3.7 and Vmax= 90) (fit not shown). This is not surprising since EAH− and (EH+)A2− are formally equivalent and could represent different routes to the same species. The Vmax data was fit using equation 3 (R2=0.96), which assumes again that monoprotonated substrate binds unprotonated enzyme where Vmax =600, K1=12, K2=4, K3=3.2, K4=40, K5 =0.4 and K6=0.04. Again, the experimental data could be fit equally well by the model that assumes fully unprotonated substrate binds protonated enzyme ((EH+)A2−) (Fig. 6A) where Vmax=30, K1=5, K2=0.09, K3=6.5, K4=28, K5=3, and K6=0.4 but could not be fit with the analogous equations assuming the key species to be those defined in Fig 6A as EAH2, EA2, (EH+)AH2, or (EH+)AH−.
Fig. 6.
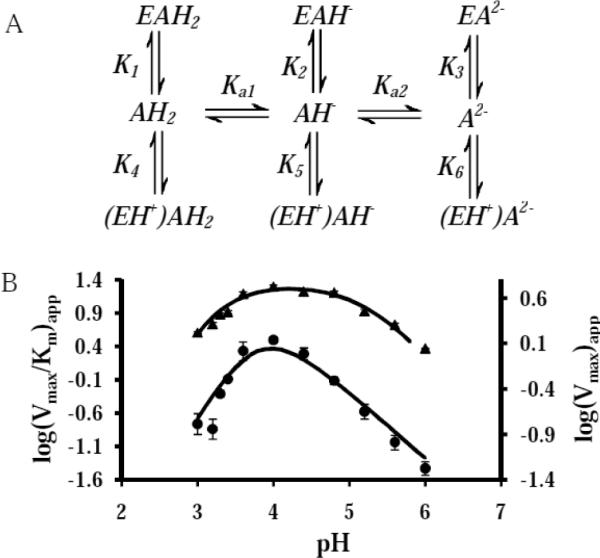
The effect of pH on the affinity of CsOxOx for oxalate. A: Scheme showing the dissociation constants for the three protonation states of oxalate binding to unprotonated enzyme (E) and protonated enzyme (EH+). B: pH dependence on kinetic parameters Vmax/Km (•) and Vmax (▲) for the CsOxOx catalyzed reaction. The curves were generated by a fit to equations 2 and 3, respectively.
The simplest interpretation of these data are that two of the three pKa values required for these models refer to oxalate (pKavalues for species free in solution are 1.3 and 4.2) and that binding to the enzyme alters the more acidic pKa by approximately 2 units. We propose that the third pKa value refers to an active site carboxylate residue. It is not immediately apparent which aspartate or glutamate residue must be unprotonated to bind monoprotonated oxalate (or protonated to bind unprotonated substrate) but it is intriguing to speculate that the aspartate in the DASN sequence analogous to the SENS of oxalate decarboxylase shown to be a specificity switch from decarboxylase activity to oxidase activity by Burrell et al [22] may be implicated. We look forward to carrying out a program of site-directed mutagenesis to test this possibility. In distinguishing between the two modes of substrate binding suggested by the data, we propose that the simplest explanation is that monoprotonated substrate binding to enzyme containing an unprotonated carboxylate residue near the active site is the dominant contributor to catalysis. We base this proposal on the fact that given the closeness of the determined pKa values, at any given pH, the fraction of substrate species that are unprotonated while an active site residue is protonated will be very small.
Conclusion
We have successfully expressed CsOxOx in Pichia pastoris and purified it to homogeneity. This recombinant enzyme is a soluble glycoprotein containing 0.2 to 0.4 mole Mn/mole of enzyme. The specific activity of this recombinant CsOxOx is 12.8 U/mg and its pH optimum is 4.0, which is in good agreement with the values reported for the native enzyme. Enzyme activity directly correlates with Mn content and other metals do not appear to be able to support catalysis. Acetate and other small molecule carboxylic acids have been shown to be competitive inhibitors of the CsOxOx catalyzed reaction. EPR spectra indicate that the Mn is present as Mn(II), and are consistent with the coordination environment expected from homology modeling with known X-ray crystal structures of oxalate decarboxylase from Bacillus subtilis. EPR spin-trapping experiments support the existence of an oxalate-derived radical species formed during turnover. The pH dependence of the CsOxOx catalyzed reaction suggests monoprotonated oxalate binds to a form of CsOxOx in which an important carboxylate residue near the active site must be unprotonated for catalysis.
Acknowledgements
We thank Dr. Stephen Bornemann of the John Innes Center for providing us with the plasmid for oxalate oxidase and for thoughtful discussions of this research. We thank Dr. John Salerno of Kennesaw State University for his help with deriving the equations necessary to model our Vmax and Vmax/Km data. This work was supported by the National Science Foundation (MCB-1041912) to EWM. Partial support was provided by NSF (CHE-0809725) to AA and NIH (DK061666) to NGJR.
Abbreviations
- OxOx
oxalate oxidase
- OxDC
oxalate decarboxylase
- CsOxOx
OxOx from Ceriporiopsis subvermispora
- HRP
horseradish peroxidase
- ABTS
2,2'-azinobis-(3-ethylbenzthiazoline-6-sulphonic acid)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hesse A, Bongartz H, Heynck H, Berg W. Clin. Biochem. 1996;29:467–472. doi: 10.1016/0009-9120(96)00067-7. [DOI] [PubMed] [Google Scholar]
- [2].Honow R, Bongartz D, Hesse A. Clin. Chim. Acta. 1997;261 doi: 10.1016/s0009-8981(97)06521-2. [DOI] [PubMed] [Google Scholar]
- [3].Dunwell JM, Khuri S, Gane PJ. Microbiol Mol Biol Rev. 2000;64:153–179. doi: 10.1128/mmbr.64.1.153-179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Thompson C, Dunwell JM, Johnstone CE, Lay V, Ray J, Schmitt H, Watson H, Nisbet G. Euphytica. 1995;85:169–172. [Google Scholar]
- [5].Wei Y, Zhang Z, Andersen CH, Schmelzer E, Gregersen PL, Collinge DB, Smedegaard-Petersen V, Thordal-Christensen H. Plant Mol Biol. 1998;36:101–112. doi: 10.1023/a:1005955119326. [DOI] [PubMed] [Google Scholar]
- [6].Strasser H, Burgstaller W, Schinner F. FEMS Microbiol Lett. 1994;119:365–370. doi: 10.1111/j.1574-6968.1994.tb06914.x. [DOI] [PubMed] [Google Scholar]
- [7].Ruttimann-Johnson C, Salas L, Vicuna R, Kirk TK. Appl Environ Microbiol. 1993;59:1792–1797. doi: 10.1128/aem.59.6.1792-1797.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kotsira VP, Clonis YD. Arch Biochem Biophys. 1997;340:239–249. doi: 10.1006/abbi.1997.9896. [DOI] [PubMed] [Google Scholar]
- [9].Whittaker MM, Whittaker JW. J Biol Inorg Chem. 2002;7:136–145. doi: 10.1007/s007750100281. [DOI] [PubMed] [Google Scholar]
- [10].Svedruzic D, Jonsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ. Arch Biochem Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- [11].Dunwell JM. Biotechnol Genet Eng Rev. 1998;15:1–32. doi: 10.1080/02648725.1998.10647950. [DOI] [PubMed] [Google Scholar]
- [12].Chiriboga J. Arch Biochem Biophys. 1966;116:516–523. doi: 10.1016/0003-9861(66)90057-9. [DOI] [PubMed] [Google Scholar]
- [13].Requena L, Bornemann S. Biochem J. 1999;343(Pt 1):185–190. [PMC free article] [PubMed] [Google Scholar]
- [14].Varalakshmi P, Richardson KE. Biochem Int. 1992;26:153–162. [PubMed] [Google Scholar]
- [15].Leek AE, Butt VS. Biochem J. 1972;128:87P. [PMC free article] [PubMed] [Google Scholar]
- [16].Satyapal, Pundir CS. Biochim Biophys Acta. 1993;1161:1–5. doi: 10.1016/0167-4838(93)90188-w. [DOI] [PubMed] [Google Scholar]
- [17].Pundir CS, Kuchhal NK, Satyapal Indian J Biochem Biophys. 1993;30:54–57. [PubMed] [Google Scholar]
- [18].Lane BG. Biochem J. 2000;349:309–321. doi: 10.1042/0264-6021:3490309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aguilar C, Urzua U, Koenig C, Vicuna R. Arch Biochem Biophys. 1999;366:275–282. doi: 10.1006/abbi.1999.1216. [DOI] [PubMed] [Google Scholar]
- [20].Escutia MR, Bowater L, Edwards A, Bottrill AR, Burrell MR, Polanco R, Vicuna R, Bornemann S. Appl Environ Microbiol. 2005;71:3608–3616. doi: 10.1128/AEM.71.7.3608-3616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Opaleye O, Rose RS, Whittaker MM, Woo EJ, Whittaker JW, Pickersgill RW. J Biol Chem. 2006;281:6428–6433. doi: 10.1074/jbc.M510256200. [DOI] [PubMed] [Google Scholar]
- [22].Burrell MR, Just VJ, Bowater L, Fairhurst SA, Requena L, Lawson DM, Bornemann S. Biochemistry. 2007;46:12327–12336. doi: 10.1021/bi700947s. [DOI] [PubMed] [Google Scholar]
- [23].Lobos S, Larrain J, Salas L, Cullen D, Vicuna R. Microbiology. 1994;140(Pt 10):2691–2698. doi: 10.1099/00221287-140-10-2691. [DOI] [PubMed] [Google Scholar]
- [24].Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- [25].Cleland WW. Methods Enzymol. 1979;62:151–160. [Google Scholar]
- [26].Schute H, Flossdorf J, Sahm H, Kula MR. Eur J Biochem. 1976;62:151–160. doi: 10.1111/j.1432-1033.1976.tb10108.x. [DOI] [PubMed] [Google Scholar]
- [27].Olivares JA. Methods Enzymol. 1988;158:205–232. doi: 10.1016/0076-6879(88)58057-6. [DOI] [PubMed] [Google Scholar]
- [28].Slavin W. Methods Enzymol. 1988;158:117–145. doi: 10.1016/0076-6879(88)58052-7. [DOI] [PubMed] [Google Scholar]
- [29].Gonzalez JC, Peariso K, Penner-Hahn JE, Matthews RG. Biochem. 1996;35:12228–12234. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- [30].Moomaw EW, Angerhofer A, Moussatche P, Ozarowski A, Garcia-Rubio I, Richards NG. Biochemistry. 2009;48:6116–6125. doi: 10.1021/bi801856k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tanner A, Bowater L, Fairhurst SA, Bornemann S. J Biol Chem. 2001;276:43627–43634. doi: 10.1074/jbc.M107202200. [DOI] [PubMed] [Google Scholar]
- [32].Svedruzic D, Liu Y, Reinhardt LA, Wroclawska E, Cleland WW, Richards NG. Arch Biochem Biophys. 2007;464:36–47. doi: 10.1016/j.abb.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chang CH, Richards NGJ. J. Chem. Theory Comput. 2005;1:994–1007. doi: 10.1021/ct050063d. [DOI] [PubMed] [Google Scholar]
- [34].Schaab MR, Barney BM, Francisco WA. Biochemistry. 2006;45:1009–1016. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- [35].Gopal B, Madan LL, Betz SF, Kossiakoff AA. Biochem. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- [36].Bowater L, Fairhurst SA, Just VJ, Bornemann S. FEBS Lett. 2004;557:45–48. doi: 10.1016/s0014-5793(03)01439-x. [DOI] [PubMed] [Google Scholar]
- [37].Angerhofer A, Moomaw EW, Garcia-Rubio I, Ozarowski A, Krzystek J, Weber RT, Richards NG. J Phys Chem B. 2007;111:5043–5046. doi: 10.1021/jp0715326. [DOI] [PubMed] [Google Scholar]
- [38].Chang CH, Svedruzic D, Ozarowski A, Walker L, Yeagle G, Britt RD, Angerhofer A, Richards NGJ. J Biol Chem. 2004;279:52840–52849. doi: 10.1074/jbc.M402345200. [DOI] [PubMed] [Google Scholar]
- [39].Imaram W, Centonze CP, Moral M, Richards NGJ, Angerhofer A. Free Radical Biology and Medicine. 2009;47:S188–S188. doi: 10.1016/j.freeradbiomed.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]