

Abstract
Heme serves as a co-factor in proteins involved in fundamental biological processes including oxidative metabolism, oxygen storage and transport, signal transduction and drug metabolism. In addition, heme is important for systemic iron homeostasis in mammals. Heme has important regulatory roles in cell biology, yet excessive levels of intracellular heme are toxic; thus, mechanisms have evolved to control the acquisition, synthesis, catabolism and expulsion of cellular heme. Recently, a number of transporters of heme and heme synthesis intermediates have been described. Here we review aspects of heme metabolism and discuss our current understanding of heme transporters, with emphasis on the function of the cell-surface heme exporter, FLVCR. Knockdown of Flvcr in mice leads to both defective erythropoiesis and disturbed systemic iron homeostasis, underscoring the critical role of heme transporters in mammalian physiology.
Keywords: Heme, Heme oxygenase, FLVCR, Transporter, Transmembrane proteins, Anemia, Porphyria, Erythrophagocytosis
1. Introduction
Porphyrins are heterocyclic organic rings composed of four pyrrole subunits that are usually linked by methine bridges; their conjugation to diverse divalent metal ions such as Mg2+, Co+ and Fe2+ gives rise to the “pigments of life”, i.e., chlorophyll, vitamin B12 and heme, respectively. Heme, a complex of iron with protoporphyrin IX, is ubiquitous in aerobic cells and essential for cellular oxidation-reduction reactions. It serves as a critical component of hemoproteins, a large group of proteins that includes cytochromes (for mitochondrial respiratory chain electron transfer and drug metabolism), oxidases (e.g., NADPH oxidase) and peroxidases, catalases and synthases (e.g., nitric oxide synthase, NOS), as well as the oxygen storage and transport molecules, myoglobin and hemoglobin (Hb) (for reviews see [1, 2]). In addition, two-thirds of Western dietary iron intake is derived from dietary heme [3], while the majority of iron in the body is present in the form of heme (as Hb, myoglobin and cytochromes [4]).
In single-cell organisms the primary importance of heme derives from its role as a source of iron and as a critical component of enzymes of the respiratory chain. Its regulated production and use appears to be intimately related to the presence of oxygen, providing the cell with the ability to exploit the benefits of oxidative metabolism [5, 6]. Thus it is not surprising that heme (like Mg2+-protoporphyrin in plants [7]) has itself evolved to serve as a fundamental cell regulatory molecule in single-cell and more complex organisms. In mammals heme has pleiotropic functions, but its underlying relationship to oxygen and oxidative metabolism remains central. For example it is intimately involved in the regulation of erythropoiesis, regulating erythroid gene transcription—e.g., nuclear heme inhibits repression of the globin locus by the transcription factor Bach1, likely through direct binding [8]; erythroid gene translation, through inhibition of an erythroid-specific eIF-2α kinase (HRI) that regulates protein, especially globin translation [9, 10]; and heme synthesis protein targeting and stability [11, 12]. Recently, the role of heme in other fundamental cell processes—e.g., the regulation of microRNA processing [13], circadian rhythm [14] and ion-channel functions [15]—has been described. In addition, heme appears to promote specific gene expression patterns in mammalian adipose, neuronal and erythroid cells, inducing their differentiation [16–20]. Finally, in addition to its redox ability in electron transfer (e.g., the respiratory chain hemoproteins), iron compartmentalization function and aforementioned regulatory roles, heme also binds NO, CO and O2, allowing hemoproteins to serve as gas sensors and signal transducers [21, 22].
The observations outlined above would seem to argue for a physiological role for a “free” or “uncommitted” heme pool, i.e., intracellular heme that is not a component of hemoproteins. However, free heme is lipophilic and toxic to cells, promoting lipid peroxidation [2, 23] and the production of reactive oxygen species (ROS) [24], resulting in membrane injury and cell apoptosis [2]. Therefore, both from the viewpoint of its toxicity and its regulatory function, the intracellular levels of free heme must be tightly controlled (estimated at <0.1 μM; reviewed in [2, 25]). Like zinc in prokaryotes [26], “free heme” has been proposed to be chaperoned by proteins within cells [27]. Putative intracellular heme carrier proteins include glutathione S-transferase B (ligandin, [28]), Z-type–fatty acid–binding protein [29], heme-binding protein 22 (p22HBP, [30]) and HBP23, [31], and, more recently, biliverdin reductase (BVR) [32]. However, the specific proteins responsible for the safe carriage of heme within cells remain for the most part unidentified.
Control of intracellular heme levels was previously thought to occur through a balance among its mitochondrial biosynthesis, utilization by hemoproteins, and catabolism by heme oxygenases (HO) (predominantly by the heme-inducible HO-1) [1]. However, studies describing transporters of heme and heme synthesis intermediates indicate further layers of complexity in heme homeostasis. Being lipophilic, heme readily associates with cell and organelle membranes; yet, the presence of anionic carboxylate side chains, responsible for heme binding to heme carrier proteins, such as albumin and hemopexin (Hpx), limits transmembrane diffusion [33], indicating the need for specific transporters, as supported by a number of studies [34–38] (see also [39, 40] for recent reviews).
In recent years a number of these heme transport proteins have been identified. The purpose of this review is to describe relevant aspects of heme synthesis and catabolism and then outline our current understanding of heme transporters. We discuss heme transporters that function at different sites within the cell, with emphasis on the role of the plasma membrane heme exporter FLVCR. Table 1 summarizes our current knowledge of these transporters.
Table 1.
Transmembrane proteins implicated in the transport or biosynthesis of heme.
| Protein | Description | Function | Localization | Disorders | Knockout | Reference |
|---|---|---|---|---|---|---|
| FLVCR | MFS transporter. FeLV-C viral receptor. Virus cause anemia in cats. | Exports heme (and PPIX). Does not transport bilirubin. | Plasma membrane. | Diamond Blackfan anemia? Posterior column ataxia and retinitis pigmentosa | Mouse KO is embryonic lethal. Early erythroid progenitor arrest and systemic iron overload in conditional KO. | 36, 37, 189 |
| ABCG2/BCRP | ABC family member. Confers multidrug resistance in breast cancer cell line. | Potentially exports heme. Exports (conjugated) PPIX, pheophorbide A, urate. | Plasma membrane. | Increased serum urate level, resulting in gout. | Mouse KO have 10-fold elevated PPIX in RBCs and in plasma. No hematologic abnormality, no evidence of iron overload. | 229, 235, 236, 238 |
| ABCB6 | ABC family member. Functionally complements yeast Atm1p mutant which accumulates iron in mitochondria. | Unknown, suggested may transport mitochondrial CPgen III. | Mitochondrial OM, plasma membrane, golgi, endosome/lysosome. | Mouse KO generated by Schuetz group (unpublished). | 47, 58 | |
| ABCB10 | ABC family member. GATA-1 regulated. | Transport substrate unknown. Interacts with Fech and Mfrn1. Stabilizes Mfrn1 to allow increased mitochondrial iron uptake during erythropoiesis. | Mitochondrial IM. | Mouse KO embryonic lethal. Arrested erythroid differentiation, increased mitochondrial oxidative stress. | 67, 70, 71 | |
| ABCB7 | ABC family member. Human ortholog of yeast Atm1p which accumulates iron in mitochondria. | Interacts with Fech. Cytosolic Fe-S protein maturation, iron homeostasis. | Mitochondrial IM. | Hereditary X-linked sideroblastic anemia and cerebellar ataxia. | Mouse conditional KO has defective hematopoiesis, sideroblastic-like anemia, ataxia and hepatocellular iron deposits. | 75, 76, 77, 79 |
| PCFT/HCP1 | MFS transporter. Proton coupled folate transporter/Heme carrier protein isolated from hypotransferrinemic mouse duodenal tissue. | Primarily a folate transporter. Physiological role in duodenal heme import unclear. | Plasma membrane. Endosome? | Hereditary familial folate malabsorption, folate-responsive anemia in newborns | Mouse KO has increased N-homocysteine protein levels. | 139, 241, 249 |
| HRG-1 | Transporter with 4 predicted transmembrane domains. Heme regulated gene in C. elegans. Ortholog present in humans. | Exports heme from lysosome/endosome into cytoplasm. | Endosome/Lysosome. | Transient knockdown of zebrafish ortholog results in hydrocephalus, yolk-tube malformations and a profound defect in erytropoiesis. | 150, 248 | |
| HRG-4 | Transporter with either 3 or 5 predicted transmembrane domains. Heme regulated gene in C. elegans, no mammalian orthologs. | Imports heme. | Plasma membrane. | 150, 248 |
2. Heme synthesis and transport of heme synthesis intermediates
The first of eight steps in heme synthesis (Fig. 1; for comprehensive reviews see [41–43]), mediated by the enzyme ALA synthase 2 (ALAS2) in erythroid cells or by the heme-repressible enzyme ALAS1 in non-erythroid cells, is the condensation of succinyl co-enzyme A and glycine to form 5-amino-levulinic acid (ALA), which occurs within the mitochondrial matrix. ALA is then transported across both mitochondrial inner and outer membranes (IM and OM) into the cytosol. Patients with mutations of ALAS2 develop sideroblastic anemia, a hematological disorder characterized by mitochondrial iron overload and impaired heme synthesis [44], while accumulation of substrates related to defects in the remaining pathway enzymes results in porphyrias, disorders characterized by either acute neurovisceral attacks or photosensitivity or sometimes both (reviews, [43, 45]). Intriguingly, a recent study identified mutations of the solute carrier SLC25A38, a mitochondrial IM transporter, as a frequent cause of inherited sideroblastic anemia [46]. Deletion of the yeast ortholog decreases heme synthesis, which is rescued by supplementing the culture media with glycine or ALA. Yeast cytosolic levels of ALA were reduced 6.7-fold compared to controls, leading the authors to propose that SLC25A38 functions to facilitate ALA production by transporting glycine into the matrix or exchanging glycine for ALA across the IM.
Fig. 1. The heme biosynthesis pathway and putative mitochondrial transporters.

The first step of heme synthesis occurs in the mitochondrial matrix with the condensation of succinyl-CoA and glycine by ALAS to generate ALA. ALA may be exported in exchange for glycine by an IM transporter SLC25A38. The mechanism of ALA export across the OM into the cytoplasm is unknown. ALA is converted to CPgenIII by four enzymatic reactions. CPgenIII is then transported back into the mitochondrial intermembrane space (IMS), possibly via ABCB6, where it is converted to PPgenIX by CPO. The conversion of PPgenIX to PPIX by PPO, its transport into the matrix and the addition of Fe+2 by ferrochelatase (FECH) to generate heme are coupled processes, with transport of PPIX likely occurring via a proposed direct channeling through a complex of PPO-FECH (likely part of a larger complex with the mitochondrial iron importer, Mitoferrin, ABCB7 and ABCB10). The transfer of heme from the matrix to the IMS, for respiratory chain cytochrome assembly, may be mediated by ABCB10; the transporter responsible for its transfer across the outer mitochondrial membrane remains unidentified. See text for details.
In the cytosol the cyclic tetrapyrrole, coproporphyrinogen III (CPgenIII), is formed from ALA in a series of four enzymatic reactions. How the heme synthesis intermediates are transferred from one cytosolic enzyme to the next in the pathway is at present unknown, but a macrocomplex comprised of all four cytosolic enzymes, as has been proposed for the terminal pathway enzymes (see below), may occur. CPgenIII is then transported back across the OM into the mitochondrial intermembrane space (IMS). Krishnamurthy et al. suggest that an ATP-binding cassette transporter ABCB6 is involved in porphyrin transport across the OM [47] (reviewed in [48]).
Cell surface ABC transporters serve to efflux a variety of compounds, including xenobiotics in eukaryotic cells [49, 50]. In addition, family members are involved in mitochondrial transport [51]. These transporters have a characteristic structure consisting of two transmembrane domains (TMD) and two intracellular nucleotide-binding domains (NBD). Energy derived from hydrolysis of ATP by the NBD provides energy for the uphill movement of substrates across membranes. Approximately 50 ABC transporters have been identified in the human genome and classified into seven subfamilies (A–G) based on the structure of their ATP-binding domains.
ABCB6 was initially identified as a mitochondrial protein that functionally complements the Saccharomyces cerevisiae atm1-1 mutant [52] that encodes Atm1p, a yeast mitochondrial IM protein believed to be involved in the biogenesis of cytosolic Fe-S clusters [53]. Fe-S clusters are modular protein cofactors consisting of iron and sulfur, which are usually linked by bonds joining the cysteine sulfur atoms of a polypeptide protein to iron atoms of the cluster. Fe-S clusters function as part of enzyme catalytic centers (e.g., aconitase, succinate dehydrogenase), in chemical sensing, electron transfer and generation of radicals [54–56]. Similar to mutations of other genes involved in eukaryotic FeS cluster biosynthesis (e.g., human fxn and glrx5; see [56] for review) Atm1 mutants accumulate free iron in the mitochondria and have depressed mitochondrial respiratory function, which is almost completely reversed by expression of human ABCB6 [52, 57]. However, unlike its mitochondrial IM paralog ABCB7, ABCB6 is localized to the OM in the human erythroid cell line K562 [47, 58] (although exclusive mitochondrial localization of ABCB6 in other cell types has been disputed; see [59–61]), and studies demonstrate conclusively that ABCB7 is the true Atm1 ortholog [62]. Nonetheless, ABCB6 is upregulated by hemin or DMSO-induced differentiation of the murine erythroid cell line MEL cells [47] and a recent large-scale expression analysis of genes associated with heme biosynthesis also identified ABCB6 [63]. ATP- and temperature-dependent inward transport of 55Fe-hemin into mitochondria isolated from K562 cells overexpressing ABCB6 has been demonstrated [47]; inward transport (although non-physiological) is consistent with the presence of the nucleotide-binding domain in the cytosol. The oxidized form of the physiological heme synthesis intermediate CPgenIII, coproporphyrin III (CPIII) competes with mitochondrial 55Fe-hemin uptake and dissociates ABCB6 from hemin-agarose [47]. However CPIII is not a heme synthesis pathway intermediate and it remains to be established whether ABCB6 functions in CPgenIII uptake.
A further two oxygen-dependent enzymatic steps, mediated by the mitochondrial IMS enzyme, coproporphyrinogen oxidase (CPO) and protoporphyrinogen oxidase (PPO), are required to convert CPgenIII into protoporphyrin IX (PPIX); the latter was proposed to be transported across the IM into the matrix by adenine nucleotide translocator (ANT), without requiring ATP energy [64]. However, because the enzymes PPO and ferrochelatase (Fech), catalyzing respectively the penultimate and final steps of heme synthesis, are both localized at the mitochondrial IM—PPO is an IMS-facing protein while Fech is exposed to the matrix—a channeling of PPgenIX and PPIX through a complex formed by PPO and Fech within the IM is more likely, based on biochemical studies [65] and the crystal structure of the two enzymes [66]. The final step, the addition of one atom of Fe2+ to PPIX by Fech that results in the formation of protoheme or heme b occurs on the matrix side of the IM, necessitating its transfer, by as yet unidentified transporters, across both the IM and OM again in order to reach the cytosol.
Recent studies indicate that Fech interacts with mitoferrin-1 (Mfrn1)–a mitochondrial IM protein involved in mitochondrial iron uptake that is upregulated during erythroid differentiation [67]. Like ALAS, there are two forms of Mfrn in zebrafish and mammals, Mfrn1 and Mfrn2, respectively, with Mfrn1 uniquely important for iron import in developing erythroid cells [68]. Elegant studies subsequently showed that Mfrn1 but not Mfrn2 was stabilized in a post-translational manner during erythroid differentiation [69]. Chen et al. then isolated an IM ABC transporter, ABCB10 (formerly ABC-me; [70]), among a number of proteins that were precipitated with Mfrn1 isolated from mitochondria derived from MEL cells undergoing erythroid differentiation. Their studies demonstrate that ABCB10 stabilizes Mfrn1 during erythroid differentiation, to allow for high efficiency iron uptake into mitochondria for optimal heme synthesis [71]. Notably, in undifferentiated MEL cells, forced expression of Abcb10 and Mfrn1 enhances Mfrn1-mediated 59Fe uptake by mitochondria approximately 2-fold, but has no effect on mitochondrial 59Fe-heme levels. It was proposed that this lack of heme synthesis was due to an inadequate supply of protoporphyrin precursors [71]; however an accompanying commentary speculated that Fech levels may be limiting and that the enzyme formed part of a macromolecular complex with Mfrn1 and ABCB10 [72]. In fact, further studies have shown that the expression of all three proteins is co-induced during MEL cell erythroid differentiation, whereas analysis of the higher order protein complexes of Mfrn1 during differentiation indicate that Mfrn1 and ABCB10 transiently interact with Fech [73]. Previous studies had shown that MEL cells transfected with Abcb10 undergoing erythroid differentiation had enhanced hemoglobin synthesis (2–4-fold; [70, 74]). Yet overexpression of ABCB10 alone does not initiate erythroid differentiation or alter the timing of hemoglobin synthesis upon differentiation. It would be interesting to discern whether the structural role of ABCB10 in stabilizing Mfrn1 is sufficient to produce enhanced Hb levels or whether ABCB10 transport function is required. The potential substrate of ABCB10 has not been identified, but heme is a possible candidate. Mfrn1 is found in higher-order protein complexes than those predicted from a macromolecular complex of Mfrn1-ABCB10-Fech suggesting that yet unidentified proteins are also part of a multi-protein complex at the IM. Chen et al. speculate that PPO and perhaps CPO are also present in this complex to allow coordinated supply of PPIX and Fe2+ to Fech for heme synthesis [73] (and perhaps coordinated heme transport by ABCB10).
In addition to its use for heme synthesis, mitochondrial iron is also utilized for mitochondrial Fe-S cluster biogenesis. Fech possesses a [2Fe-2S] cluster that is not required for catalytic function, but the enzyme is sensitive to the availability of Fe-S clusters. Rouault’s group has recently shown that when Fe-S cluster synthesis is impaired, due to deficiency of proteins required for Fe-S cluster biosynthesis or iron deficiency, apo-Fech is rapidly degraded in mitochondria, potentially providing a direct link between Fe-S cluster and heme synthesis [75]. As mentioned, the mammalian IM transporter ABCB7 is the yeast Atm1 ortholog and appears to be important for cytosolic but not mitochondrial Fe-S cluster biogenesis [76]. Mutations in the human Abcb7 gene result in defective cytosolic Fe-S–containing protein maturation and mitochondrial iron overload, leading to an X-linked congenital sideroblastic anemia with ataxia [77, 78]. Prior to the identification of cytosolic Fe-S cluster biogenesis (review; [79]) it was believed that ABCB7 was involved in mitochondrial Fe-S cluster export. More recent studies suggest that it exports a substrate important for cytosolic iron homeostasis or a signal of mitochondrial iron status, with deficiency of this signal indicating depletion of mitochondrial iron and resulting in diversion of cytosolic iron to the mitochondria [76]. The specific reason for the impaired heme synthesis observed in Abcb7 knockout mice remains unclear. The physical interaction of ABCB7 with the C-terminal region (Fe-S ligation site) of Fech has been proposed to function in mitochondrial iron homeostasis [80]. The knockout mouse model shows elevated mitochondrial iron levels without an increase in PPIX levels, yet zinc PPIX levels are increased (which requires functional Fech) suggesting that ABCB7 plays a role in the availability of iron destined for incorporation into PPIX by Fech [78].
Nilsson and colleagues recently performed a large-scale gene expression analysis to identify potential mitochondrial transporters of heme and heme synthesis intermediates, seeking genes co-expressed consistently and specifically with genes encoding enzymes of the heme synthesis pathway [63]. Reassuringly, ABCB6, ABCB10 and Mfrn-1 were identified, as expected. The transporters SLC25A39, TMEM14C, and SLC22A4 and the Fe-S cluster biogenesis proteins ISCA1 and C1orf69 were identified as potential candidates involved in mitochondrial heme/iron metabolism. Silencing of each candidate gene in zebrafish results in an anemic phenotype. SLC25A39 was the only gene characterized in detail. Silencing of this gene in differentiating MEL cells results in a 4-fold decrease in the 59Fe-heme content of mitochondria, without the accumulation of 59Fe. The authors predict a role for SLC25A39 in Fe-S assembly that affects heme synthesis.
3. Heme Catabolism and the need for heme transport
Although chemical degradation of heme occurs in mammals through the action of H2O2 or ascorbic acid [81, 82], the HO system is the major physiologically relevant mechanism of heme catabolism (for review see [83]). In humans, HO is present in two functionally active isoforms, HO-1 and HO-2, encoded by different genes and, apart from their heme degradative function, dissimilar in molecular and biochemical properties. A pseudogene variant of HO-2, HO-3 has also been described in the rat brain [84–86].
HO is a mixed function oxidase, i.e., it utilizes molecular oxygen, generating oxidized products and H2O. The oxidase catalyzes the regio-specific cleavage of the α-methine bridge of heme—producing iron, CO, and biliverdin IXα (BV)—and is the rate-limiting step in the degradation sequence. The concerted activity of NADPH-cytochrome P450 reductase is also necessary, both to supply electrons and to activate oxygen. BV, a physiological inhibitor of HO function, is rapidly converted to bilirubin IXα (BR) by BVR. HO and NADPH-cytochrome P450 reductase are located in the endoplasmic reticulum (ER), forming a complex with each other and, at least in vitro, with cytosolic BVR [87]. Incidentally, HO-1 has also been localized to plasma membrane caveolae (with NOS and BVR [88]), mitochondria, and the nucleus [89, 90]. BR is, in turn, conjugated in the liver by UDP-glucuronyl transferase and eventually excreted into the bile. The daily production of BR in man is ~400 mg, of which ~300 mg is derived from Hb breakdown [91].
In bacteria, breakdown of imported heme serves primarily as a source of iron, and early studies in mammals likewise focused for the most part on the importance of HO for heme catabolism during Hb breakdown. However, it is now well recognized that HO-mediated heme degradation has multiple roles in mammals, including antioxidative and iron reutilization functions.
3.1. Antioxidative function of HO-1 and HO-2
HO-1 is activated by a greater number of stimuli than that of any other gene [83]. It is inducible by increased heme levels; various metals (including sodium arsenite, cobalt and selenium); hypoxia in certain cell types; hyperoxia; environmental chemicals; H2O2; NO; depletion of intracellular glutathione; UV light; and heat shock (and thus known as HSP32). Notably, basic leucine zipper (bzip) “stress-responsive” transcription factors, including members of the ATF family, c-Fos, c-Jun and Nrf2, maf family members and Bach-1, all regulate HO-1 expression. These transcription factors function as homo-dimeric transcriptional complexes or, with other bzip members, as hetero-dimeric transcriptional complexes, vastly expanding the scope of HO-1 regulation [92, 93]. More recently, BVR has been identified as a key regulator of HO-1 expression. The enzyme has kinase, transcription factor and intracellular heme transport activities. Thus BVR can serve as a dimeric bzip transcription factor; or through its kinase function, directly phosphorylate and regulate other bzip members; or via transport of heme into the nucleus enable derepression of Bach-1–mediated repression of HO-1 [92–94]. An overriding principle of HO-1 expression appears to be its rapid induction in response to an increase in cellular oxidative stress and then subsequent repression to low levels.
In contrast, HO-2, the predominant form of HO protein in the brain and testis, is constitutively expressed in all cells, with the promoter responding solely to glucocorticoids (for review see [83]). The N-terminus of HO-2 protein contains heme-binding Heme Regulatory Motifs (HRMs) consisting of CP-based short amino acid sequences [95], an ancient motif found in proteins that function as heme-oxygen sensors in bacteria [27], yeast (HAP-1 [41]) and mammals (ALAS2, Bach-1 and HRI [8, 9, 11]). The presence of HRMs thus suggested a further function for this enzyme—that of an intracellular sink, binding and regulating (or “sensing”) the heme gaseous ligands NO, CO and O2. Subsequently, HO-2 has been shown to co-localize with NOS, binding the vasodilator NO with high affinity [96] and, with less affinity, the potential vasodilator CO, or O2. Furthermore, HO-2 interacts with and, through proximal production of CO, stimulates the calcium-sensitive potassium (BK) channels that mediate the excitatory responses of the carotid body to hypoxia [97]. HO-2−/− mice have mild hypoxemia and their carotid bodies are enlarged (as also seen in patients with chronic hypoxemia [98]); however, their carotid bodies respond normally to acute hypoxia [99], and the mice have only partially impaired acute ventilatory responses and (proposed) chronic pulmonary ventilation/perfusion mismatches, perhaps related to changes in CO signaling [100]. Thus it is postulated that redundant mechanisms, including CO, NADPH-oxidase and AMP:ATP ratios, regulate the acute response of both central and peripheral chemoreceptors to hypoxia through inhibition of ion channels [101]. In addition, HO-1 appears to be upregulated in the lungs of HO-2−/− mice, perhaps functioning as an alternate source of CO production. Recent studies indicate an additional role for the HRMs of HRM-containing proteins like HO-2; that of a thiol/disulfide redox switch that responds to the cellular redox state. Thus, during oxidative stress, heme binds with a much higher affinity to the disulfide cysteine residues, increasing its function as a sink for NO and derivatives, CO and O2, while, under reducing conditions, the cysteines would become thiols and thus have a lower affinity for heme [102].
The reason for the reduction of the heme catabolite BV to BR has been questioned, as BV is water soluble and thus easily excreted, whereas BR is lipid soluble and believed to be toxic. However, studies by Stocker et al. [103], extended by Snyder’s laboratory [104–106] and others, have clearly shown that BR at intracellular nanomolar (i.e., physiological) concentrations is a potent antioxidant that protects cells from up to a 10,000-fold excess of H2O2; in so doing, it is itself oxidized to BV, which is then recycled back to the reducing agent BR through the action of PKC-dependent signaling that activates BVR [105]. In neurons this reaction depends on the substrate BV being supplied by HO-2, rather than by HO-1 [106]. Recent studies indicate that BR serves mostly to defend lipid membranes from ROS, whereas the other major intracellular antioxidant, the water-soluble glutathione system (GSH/GSSG), protects water-soluble proteins [104].
In closing this section on the antioxidant function of HO, we should note that there have been reports suggesting that HO-1 induction could be harmful to the cell under certain circumstances (see [92]). In particular, the release of heme-chelated ferrous iron that could potentially interact with H2O2, producing the highly toxic hydroxide radical (the Fenton reaction), is seen as problematic. Yet, provided the iron released is either sequestered by storage within ferritin or exported out of the cell by the cell membrane iron exporter ferroportin (FPN1) [107, 108], then an increase in HO-1 activity should be beneficial to the cell. One group found that transfection of human cells with catalytically inactive rat HO-1 (mHO-1) provided greater protection from (very high levels of) H2O2 than transfection with wild-type HO-1, an effect reversed by treatment of the cells with a HO inhibitor (zinc protoporphyrin) or iron chelators. They also noted an inverse correlation between the levels of glutathione and catalase and HO-1 activity [109]. One could argue that these are not physiologic studies; HO-1 is never induced in cells for long periods of time, as heme levels may fall precipitously, perhaps leading to decreases in catalase and other heme-containing antioxidant proteins (see [58]). Ferris et al. noted equivalent levels of ferritin in HO-1−/− cells and cells overexpressing HO-1 [110]. Similarly, a study published recently demonstrates a lack of ferritin synthesis, despite HO-1 induction, in a cell-line in response to oxidizing reagents (as opposed to an appropriate response to extracellular heme) [111]. As the known intracellular level of free heme is very low, it was suggested that there is insufficient substrate for HO-1, or that it perhaps acts through other, non-enzymatic mechanisms [111]. However, one could also speculate that under oxidative stress all available iron is shunted directly to the mitochondria for heme synthesis for heme-containing antioxidant proteins, without a concomitant increase in ferritin synthesis. Moreover, as these investigators acknowledge, they may be unable to measure the small quantity of heme degradation that occurs during exposure to oxidative stress. In sum, there is a large body of literature that supports a seminal role for the HO system, in particular the inducible HO-1 enzyme, in protecting cells from oxidative stress, provided the result is limited heme degradation, which might otherwise limit the function of other critical cell hemoproteins.
3.2. Iron reutilization function of HO-1 and HO-2
Under normal physiological conditions, ~25 mg of iron is consumed daily by immature erythrocytes in the bone marrow (BM) for heme biosynthesis. As the daily dietary iron intake is 1–2 mg, recycling of heme-iron from senescent erythrocytes constitutes the main source of iron for erythropoiesis. Erythrophagocytic macrophages in the liver, spleen and BM are responsible for this recycling. Hb-derived heme is catabolized by HO-1 to release iron, which is subsequently exported into circulation by the sole cell iron exporter FPN1 (reviews [112, 113]). Hemoglobin also appears in the plasma under physiological conditions due to intra-vascular hemolysis of senescent erythrocytes (~10% [114]) and during enucleation of erythroblasts in the BM. Hemoglobin is bound by haptoglobin for delivery to CD163-receptor–bearing macrophages, whereas any free heme is rapidly bound to albumin and lipoproteins before being passed on to the high-affinity heme binding plasma protein Hpx (review [115]). Hpx-heme complexes undergo (LRP1/CD91) receptor-mediated endocytosis by hepatic parenchymal cells; however, they can also be taken up by macrophages, neurons and syncytio-trophoblasts [116].
In approaches to determining the relative contributions of HO-1 and HO-2 to erythrocyte heme-iron recycling, both HO-1−/− and HO-2−/− mice were generated [117, 118]. In contrast to HO-1−/− mice, HO-2−/− mice have no evidence of disturbed iron homeostasis, apart from increased pulmonary iron in the setting of hyperoxia [119]. As mentioned, it is likely that the inducible enzyme HO-1 is upregulated in HO-2−/− mice. A patient with homozygous HO-1 deficiency has been described [120]; the child, who died at age 6 years, had evidence of severe hemolytic anemia with high serum heme levels, low serum bilirubin, absent Hpx, increased haptoglobin, and hypoferremia. Very high von Willebrand Factor and thrombo-modulin levels and disseminated intravascular coagulation were observed, indicating endothelial injury. The child also had hyperlipidemia, and, at autopsy, aortic atheroma were noted. There was iron overload of the liver (involving both the hepatic reticulo-endothelial system [RES] Kuppfer cells and parenchymal cells) and kidney (proximal tubule epithelium). The findings are consistent with an important role for heme catabolism both in protection of the vascular endothelium from potentially high levels of exposure to heme and for heme-iron reutilization. HO-1−/− mice develop a similar iron overload phenotype [110, 117] that becomes evident between 20 and 40 weeks, with twice the amount of iron in the liver at 10 weeks, when compared with controls. The erythrocytophagocytes of the liver (Kuppfer cells) showed evidence of iron overload, but no excess iron deposition was observed within splenic erythrocytophagic macrophages; the spleen however, was enlarged due to increased hematopoiesis (the child had asplenia). More recently, the murine model of HO-1 deficiency has been re-examined at various ages between 1.5 and 22 months [121]. The animals develop asplenia over time, due to fibrosis and loss of the red pulp. Notably, there is a marked loss of erythrocytophagocytes in the liver spleen and BM, which is shown to be due to the toxicity of heme released within macrophages during ingestion of senescent erythrocytes. The authors suggest that the high levels of haptoglobin observed in the human subject with HO-1 deficiency are explained by the specific loss of erythrophagocytic macrophages (which also bear the haptoglobin receptor CD163). Iron loading of the proximal tubules of the mouse kidney was seen as previously, and splenic tissue iron levels were low (due to a lack of macrophages). Notably, liver tissue non-heme iron levels were similar to controls, however the distribution was altered, with iron found in hepatocytes, perhaps from Hpx-heme delivery, rather than in (the reduced numbers of) Kuppfer cells. Although HO-2 knockdown in mice is not associated with disordered systemic iron homeostasis, it has been postulated to contribute to iron homeostasis in liver parenchymal cells [122]. Finally, when HO-1−/− BM cells are transplanted into irradiated HO-1+/+ mice, the animals show no evidence of hepatic or renal iron overload at 20 weeks, suggesting a role for endothelial, hepatic and renal parenchymal HO-1 in heme-iron recycling [117]. In sum, the analysis by Kovtunovych et al. indicates that the loss of erythrophagocytic macrophages from heme toxicity, resulting in a large increase in circulatory heme and Hb is the key to the pathogenesis of HO-1 deficiency [121].
Hepcidin is the hormone that regulates systemic iron homeostasis through binding to and thus decreasing the cell-surface expression of FPN1 [123]. Hence, it regulates both enterocyte iron absorption and the release of iron from erythrocytophagic macrophages. Hepcidin is itself regulated by iron, erythropoiesis, tissue hypoxia and inflammation (review [124]). Of interest, FPN1 and HO-1 are co-induced in macrophages upon ingesting erythrocytes, whereas macrophages from HO-1−/− mice have chronically high levels of FPN1 expression [121], likely related to the presence of a heme-inducible MARE/ARE element in the enhancer region of the gene [125]. Recent studies have shown decreased hepcidin levels in an HO-1–deficient patient [126], despite evidence of chronic inflammation, which normally increases hepcidin levels, whereas studies of the murine HO-1−/− animal demonstrate normal hepcidin levels [121]. These findings reiterate the predominance of the proposed erythroid iron regulator [127] and serum hypoferremia in suppressing hepcidin to allow for the flow of iron from the RES to the BM for erythropoiesis.
As expected, human and murine HO-1−/− cells rapidly undergo apoptosis upon exposure to heme [120], and HO-1−/− mice also have evidence of increased oxidative stress, with increased free radical expression in response to heme, H2O2, paraquat or CdCl2 [128]. The mice are extremely sensitive to endotoxemia, dying upon exposure to low doses of lipopolysaccharide. Notably, these deaths are due to massive parenchymal hepatocyte iron loading, resulting in liver necrosis. A similar hepatocyte injury pattern is seen when Hpx-null mice are given moderate intravenous doses of hemin; that is, the majority of intravascular heme ends up in the liver parenchyma (presumably bound to albumin and LDL [115]), resulting in hepatic inflammation and congestion, likely secondary to sinusoidal injury and thrombosis. This inflammation is prevented by prior induction of HO [129]. Hershko et al. reported hepatic and RES iron loading in a rat model of inflammation more than three decades ago [130]. After injection of heme in the form of damaged erythrocytes or Hb, there was increased catabolism of heme to ferritin, as well as a decrease in ferritin secretion. It is now acknowledged that the iron loading of RES and hepatic parenchymal cells in response to inflammation is, for the most part, secondary to a lack of release of iron from these cells due to cytokine-mediated upregulation of hepcidin, decreasing FPN1-mediated iron export [131]. Presumably HO-1 is upregulated to cope with the heme load. It is unknown why or how mobilization of iron and heme to the liver occurs with inflammation/infection. It may be a protective mechanism to decrease the levels of both iron and heme-iron in the systemic circulation, thus preventing bacterial access to iron (iron in ferritin is likely inaccessible to bacteria) or, perhaps, as Poss and Tonegawa propose, to reduce oxidative stress in the tissues [128].
In summary, both HO-1 and, to a lesser extent, HO-2 are important for heme-iron re-utilization. What is striking, however, is the longevity of the HO-1 knockout animals, given the low-level expression of HO-2 (e.g., there appears to be no upregulation of HO-2 in PBMCs isolated from the patient with HO-1 deficiency [120]), and the enormous iron requirement for continued high level erythrocyte production in the face of severe hemolysis. This observation would suggest that there are other methods of recycling heme-iron. Poss and Tonegawa suggest contributions from a microsomal cytochrome P450 reductase and a cytosolic xanthine oxidase [117]. In the following sections we will discuss the potential importance of FLVCR-mediated heme export.
4. Heme Absorption
Ingested heme serves as the most important source of dietary iron in meat-eating animals [3, 132], comprising one-third of ingested iron in Western diets but up to two-thirds of absorbed body iron. This is likely related to the increased bioavailability of heme relative to iron, especially when bound to proteins [133–135]. Unlike expression of the duodenal iron importer (DMT1, [136]), which increases approximately 10-fold during iron deficiency, intestinal heme uptake increases 3-fold, suggesting a separate, carrier-mediated process [137]. Although the molecule(s) mediating duodenal brush-border enterocyte heme uptake has not been identified, considerable evidence suggests uptake occurs via a receptor-mediated endocytotic pathway (see [138] and [134] and references therein) or by heme importer(s) (possibly HCP1 [139]; see below). Early studies demonstrated the presence of a mammalian enterocyte heme-binding protein that binds heme with high affinity (Kdiss up to 10−9 M [140]), with binding subsequently lost upon treatment of the cells with trypsin. More recent studies indicate heme uptake is temperature dependent and saturable, decreased by inhibition of oxidative phosphorylation, but unaffected by ATP depletion [141–143]. Similar findings in mammalian erythroid and hepatic tissue cell lines suggest heme import is a generalized cell process [141, 144]. Two separate electron microscopy studies of closed mammalian duodenal loop tissues indicate that heme is taken up intact by enterocytes—subsequently appearing first in apical tubulo-vesicular structures within the cytosol and then in secondary lysosomes [145, 146]. Notably, heme disappeared from these vesicles within 3 h and was not present in the basolateral regions of the cells, the extracellular space or the portal vein, indicating intracellular catabolism, consistent with findings in mammals, including humans, where the majority of ingested radioactive Hb iron is detected in the portal vein as non-heme iron within 2–3 h [133, 147, 148]. In this scenario, either the endosome/lysosome possesses HO activity and an iron transporter for ferrous iron, such as DMT1 [136] or TRPML1 (see review; [149]), or perhaps the lysosomal heme transporter HRG-1 delivers heme to the cytosol ([150]; discussed below). HO is upregulated 2.5-fold in rat enterocytes in response to iron deficiency, and enzyme activity is highest in the proximal small intestine, the site of maximal heme absorption [151–153]. Studies in dogs confirm that HO activity is limiting for heme absorption [154], while HO inhibitors—e.g., Sn mesoporphyrin (po or iv)—inhibit intestinal heme absorption and result in increased biliary excretion of heme [155]. Surprisingly, recent confocal microscopy studies of rat enterocytes after heme dosing show that, while HO-1 is distributed throughout the cell, HO-2 is the enzyme found exclusively in the subapical region, where it co-localizes extensively with endosomes [156].
In contrast to these studies on the importance of HO-mediated heme catabolism for heme-iron absorption, an older study of Hb absorption in guinea pigs suggested the absorption of intact heme [157]. In addition, in the human enterocyte–like cell line, Caco2, which has very high expression of the heme exporter FLVCR, heme appears to be exported across both the basolateral and apical surfaces [158]. In addition, haptoglobin gene knockout in mice causes fpn1 upregulation in intestinal cells, which may also suggest the uptake of heme from the circulation into intestinal cells [159].
In summary, in most mammals, dietary heme is transported intact into intestinal mucosal cells, where it is mostly catabolized, with the subsequent appearance of inorganic iron in the portal vein. Systemic heme may also be taken up by hepatic (or perhaps intestinal) cells and transferred, if necessary, into the gastrointestinal lumen [160].
5. Heme Trafficking and Transporters
Both cell surface and organelle-associated heme transporters are likely involved in the sequestering and trafficking of intracellular heme (Fig. 2; see also reviews [39, 48, 138, 161–164]). Because the last step of endogenous heme synthesis occurs in the mitochondrial matrix, heme must traverse both mitochondrial membranes to enter the cytoplasm (Fig. 1). Although the identity of the mitochondrial heme exporter is unknown, ABC transporter family members may channel heme synthesis intermediates across both mitochondrial and plasma membranes (see sections 2 and 5.1.2) [47, 165]. Heme is also known to function as a transcriptional regulator; therefore, it must enter the nucleus, probably through binding to a carrier protein such as BVR [32]. Approximately 85% of synthesized heme is utilized for Hb production in the BM. The other 15% is produced in the liver, primarily for cytochrome (especially P450) synthesis in the ER and for catalase synthesis in peroxisomes, suggesting the presence of heme transporters in these organelle membranes [39, 166]. Thus, heme needs to be transferred to various compartments within the cell, presumably chaperoned by cytosolic heme–binding proteins (reviewed in [39]) However, the identity of the intracellular heme–binding proteins and many of the heme transporters have not yet been identified. In the next section, the current understanding of various established and potential heme transporters is discussed.
Fig. 2. A generalized depiction of known and putative cell-surface and organelle-associated heme transporters.

FLVCR is a cell-surface heme exporter. ABCG2 is a potential cell-surface heme exporter; HCP1 a putative heme importer. CD163 receptor-mediated endocytosis of the Hp:Hb complex suggests the existence of an endosomal/lysosomal heme exporter; HCP1 or HRG1 may fulfill these role(s). The transporters that mediate import of heme into peroxisomes and lysosomes for assembly of catalases and peroxidases are unknown (it is also unclear at present where final assembly of hemoproteins occurs). Heme is transferred into the nucleus in association with BVR, while cytoplasmic heme appears to be chaperoned by proteins such as glutathione S-transferase B and HBP23. See text for further details. Putative transporters of mitochondrial heme and heme biosynthesis intermediates are shown in detail in Fig. 1.
5.1. Heme Exporters
5.1.1. FLVCR, a cell surface heme exporter
Infection of cats with Feline Leukemia Virus subgroup C (FeLV-C) invariably causes a severe non-regenerative anemia (hematocrit 4%–15%), with an absence of reticulocytes in the circulation and hemoglobinized cells in the BM, despite high erythropoietin levels. Studies have subsequently shown evidence of an arrest in erythropoiesis at the colony-forming unit–erythroid progenitor (CFU-E)/pro-erythroblast stage [167, 168], where heme synthesis begins [169, 170]. The disease bears a resemblance to human pure red cell aplasia because, despite viral infection of all BM cells, there is normal development of the myeloid and megakaryocytic lineages [171]. FeLV subgroups (A, B and C) were defined by host cell range, neutralization and interference assays that indicate their viral envelopes exploit different cell receptors [172, 173]. The nonpathogenic FeLV-A has a narrow host range, while both FeLV-B and FeLV-C have broad host ranges. The genomes of FeLV A, B and C are most divergent in the segment that encodes for variable region 1 (VR1) of their envelope proteins. In studies using chimeric envelope genes of FeLV-A and FeLV-C, an 11-aa sequence of VR1 of FeLV-C envelope protein was sufficient to transform FeLV-A into a pathogenic feline virus causing anemia [174]. This suggests that the anemia is caused by a dominant negative effect of the FeLV-C envelope protein interfering with the display and/or function of its receptor on CFU-E/pro-erythroblasts. As the function of the receptor appeared to be critical for the development of erythroid progenitors we and others functionally cloned the gene, termed Flvcr, Feline Leukemia Virus subgroup C Receptor [175, 176].
5.1.1.1. Gene, protein structure and expression
FLVCR is highly conserved throughout evolution with orthologs present in animals, plants and bacteria, suggesting an important role in cell biology [177]. The human gene (also known as Mfsd7b) is located at chromosome 1q32, contains 10 exons and spans approximately 40 Kb. The promoter region contains consensus GATA-1, GATA-2 and STAT5a binding sites, providing potential mechanisms for upregulation of the gene during early erythroid development [36]. Of interest, both FLVCR and one of its 3 paralogs (on chromosome 3q, GI:74732717) have an adjacent gene upstream that shares the same promoter; LQK-1, 177bps upstream of FLVCR, is of unknown function, whereas the 3q gene product HSPBAP1, binds HSP27, an antioxidant protein [178]. In addition, there is marked homology between a group of genes that includes FLVCR on chromosome 1q and a paralogous cluster that includes FLVCR2 on chromosome 14q, a duplication that occurred over 500 million years ago [177]. Such clustering of genes has been previously associated with functional linkage—e.g., the murine surfeit locus [179].
The major mature FLVCR transcript encodes a protein of 555 aa with a predicted molecular weight of 60 kDa and a pI of 5.72 (Fig. 3). The protein belongs to the major facilitator superfamily (MFS) of secondary permeases, which transport small solutes such as glucose (e.g., GLUT-1), amino acids, and ions across cell membranes in response to chemico-osmotic gradients. Topology predictions indicate that FLVCR is a 12-transmembrane domain protein with both N- and C-termini inside the cell. Both the N- and C-termini of MFS transporters generally contain domains important for transporter trafficking and proper localization—e.g., in the polarized epithelial cells of the intestine and kidney [180]. FLVCR has both a proximal tyrosine-based motif YXXØ and a more distal di-leucine motif in the N-terminal intracytosolic region. These signals in mammalian proteins mediate rapid internalization and targeting to endosomal-lysosomal compartments, suggesting that FLVCR may be targeted to these locales. Sequences conforming to this motif have also been implicated in baso-lateral targeting in polarized epithelial cells [181, 182]. In addition, the C-terminus contains a PDZ domain–binding motif that likely promotes protein-protein interactions (see below and reviews [183, 184]). Both the putative human heme importer PCFT/HCP1 and the C. elegans lysosomal heme transporter HRG-1 (see below) have di-leucine motifs, whereas FPN has a predicted PDZ domain–binding motif.
Fig. 3. FLVCR membrane topology.
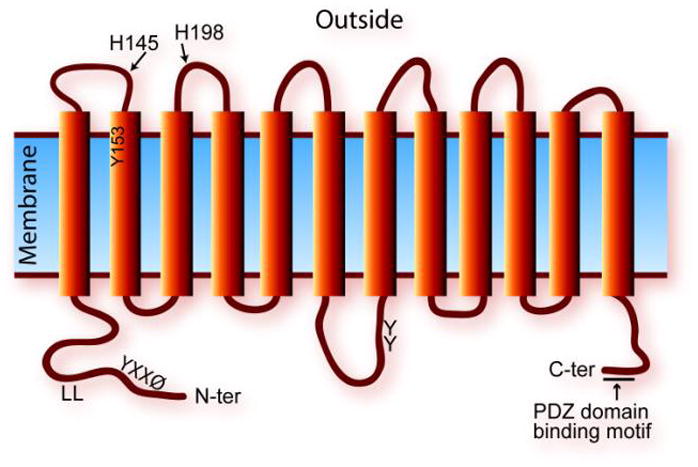
FLVCR is predicted to contain 12-transmembrane domains, with both N- and C-termini within the cell. It has YXXØ and di-leucine (LL) motifs in the N-terminal region that may allow proper membrane domain targeting and recycling. His and Tyr residues in the extracellular loops are predicted to be important for heme binding/transfer, whereas Tyr residues in the large intracellular loop may serve as targets of kinases and phosphatases. The 4-aa PDZ domain–binding motif at the C-terminal region is important for its interaction with the PDZ domain of the nonreceptor tyrosine phosphatase PTPN3.
Human FLVCR mRNA is expressed ubiquitously with highest expression observed in the liver, duodenum, kidney, lung, spleen, brain, placenta and BM (including CD34+ stem/progenitor cells) [36, 37]. Notably, in studies of human erythropoiesis, specific upregulation of FLVCR protein expression is observed at the CFU-E stage; FLVCR is subsequently downregulated with further differentiation [36]. There are wide disparities between mRNA levels and cell surface protein expression (see [36, 37]) suggesting that posttranslational control mechanisms, such as protein trafficking, regulate FLVCR expression. This is not unexpected as regulation of transporter cycling is a control mechanism important in the cell surface expression of other MFS members, such as glucose and iron transporters (e.g., GLUT-4 [185]; Arn1p [186]). More recently, alternative splicing of FLCVR mRNA has been described, with enhanced alternative splicing resulting in reduced levels of the normal protein observed in erythroid cells from patients with a congenital form of red cell aplasia, Diamond-Blackfan anemia (DBA) [187]. In earlier studies we found no evidence of FLVCR gene mutations in a subset of DBA patients with linkage to the FLVCR locus [188].
Of interest, a recent study describes homozygous missense mutations in FLVCR in affected individuals from three families with a syndrome of posterior column ataxia and retinitis pigmentosa [189]. Analysis of murine posterior column neurons and retinal tissues shows high expression of FLVCR. The authors hypothesize that FLVCR mutations abrogate the known neuro-protective effects of a hemoglobin-related protein, neuroglobin. However, FLVCR may also serve to protect these tissues from heme toxicity related to capillary leaks.
5.1.1.2. FLVCR trafficking and localization
Interacting proteins regulate trafficking of MFS members like FLVCR to and from the plasma membrane. For example, the glucose transporter–4 (GLUT4) resides in intracellular storage compartments, but rapidly translocates to the plasma membrane in response to a signaling cascade initiated by insulin [190, 191]. Studies of FLVCR expression demonstrate a marked discordance between protein expression and mRNA levels [36, 37], while analysis of mammalian FLVCR protein sequences suggest they contain a PDZ domain–binding motif at the extreme C-terminus, suggesting PDZ domain–containing proteins may regulate FLVCR trafficking and localization. A PDZ domain is a protein structural motif of 80–90 aa that helps anchor transmembrane proteins to the cytoskeleton [192, 193]. Using a yeast-two-hybrid screen, we recently identified a PDZ domain–containing protein, the nonreceptor protein tyrosine phosphatase, PTPN3 that interacts with FLVCR. Both proteins co-immunoprecipitate in cell expression and pull-down assays. Importantly, their interaction is abolished when either the PDZ domain of PTPN3 or the 4-aa PDZ–domain binding motif of FLVCR is deleted [194]. A role for PTPN3 in anchoring FLVCR to the plasma membrane may thus have important implications for trafficking and/or membrane retention of FLVCR in for example, the polarized epithelial cells of the intestine and kidney.
5.1.1.3. FLVCR-mediated heme export
Heme synthesis (for Hb production) initiates at the CFU-E/pro-erythroblast stage of erythroid development [169, 170]; thus, a potential role for FLVCR in heme transport was examined. Using rat kidney epithelial (NRK) cells overexpressing FLVCR, we demonstrated that FLVCR exports 55Fe-hemin or zinc mesoporphyrin (ZnMP, a heme analog that also inhibits HO) over a 90 minute washout period, after initial loading [36]. Approximately 50% of the heme taken up by the cells is exported. Using HPLC methodology, the export rate of exogenously supplied heme by NRK/FLVCR cells was quantified as approximately 2-fold that of control NRK cells (which presumably express the rat ortholog), provided that HO function is first blocked by a short incubation with ZnMP. In K562 cells, interference with endogenous FLVCR expression by FeLV-C envelope protein dramatically reduces heme export, to 3% versus 54% in naive K562 cells. FLVCR-mediated heme export in K562 cells can also be blocked using a polyclonal antibody specific to FLVCR (10% export vs. 54% in control cells), confirming that cell-surface FLVCR mediates heme export. The heme export activity of FLVCR has also been demonstrated using macrophages from wild-type and Flvcr-deleted mice [37]. There is no evidence that FLVCR imports heme [36]. Recently, endogenous or exogenously supplied heme was demonstrated to be the primary physiological substrate for FLVCR. In addition, under (non-physiological) conditions that increase intracellular PPIX or coproporphyrin levels, FLVCR exports these porphyrins [38]. However FLVCR does not export unconjugated bilirubin.
Heme export by FLVCR requires an extracellular heme-binding protein such as albumin or Hpx, as export is not observed in the absence of a carrier protein [38]. Thus heme may be channeled through FLVCR, docked at extracellular amino acid residues, and then released to a heme carrier protein. By analogy to the bacterial hemophore HasA [195, 196], CssBA [197], and other heme-binding proteins, the residues His 145, Tyr 153 and His 198 of FLVCR are likely to be involved in the heme transport route and/or heme transfer to extracellular heme-binding proteins (the common predicted motif is: H-x(7)-Y-x(44)-H, using the Prosite system [198] pattern syntax; see http://ca.expasy.org/tools/scanprosite/scanprosite-doc.html#pattern_syntax). The histidine residues, conserved in mammalian FLVCR proteins, but absent in the closely related paralogs that do not export heme, are predicted to be on extracellular loops 1 and 2, and thus optimally located to coordinate heme docking and export. Recent studies suggest that Hpx—with a similar arrangement of His residues—functions physiologically to bind heme that is bound to FLVCR, therefore facilitating for example, the uptake of heme via FLVCR from macrophages [38]. Export of heme by NRK/FLVCR cells is 100-fold more efficient when the media contains Hpx rather than albumin [38], in keeping with the relative affinities of Hpx and albumin for heme (Kd <1 pM vs. Kd = 5 nM; [199, 200]). The model is also supported by the finding that apo-Hpx directly binds to a 70 aa FLVCR peptide that encompasses His 145, Tyr 153, and His 198. The in vitro binding of Hpx to the peptide can be inhibited by incubation of the (heme-binding) peptide with heme: presumably Hpx removes heme from the peptide and is then released into the media [38]. Notably, Hpx is present in high concentration in human serum (6.7–25 μM [201]), a concentration similar to that of the iron transport protein transferrin (22–31 μM [202]).
5.1.1.4. FLVCR heme export function in erythropoiesis
As discussed, cats infected with FeLV-C lack reticulocytes in the circulation and hemoglobinized cells in the marrow, indicating a block in erythroid differentiation. Similarly, inducible deletion of its murine receptor, Flvcr, in one-week-old mice rapidly results in a severe hyperchromic macrocytic anemia, reticulocytopenia, splenomegaly and a lack of hemoglobinized cells, again caused by a block in erythropoiesis at the CFU-E/pro-erythroblast stage, as demonstrated by colony assays or FACS analysis [37]. In contrast to this purported role for FLVCR in heme export during early erythropoiesis, previous studies have shown that exogenous heme promotes erythroid differentiation of K652 cells and of the early erythroid progenitor cells, burst-forming units–erythroid [203–205]. In addition, increasing levels of heme in the nucleus directly represses a transcriptional repressor of globin genes, Bach1, during erythropoiesis [8], presumably to allow globin expression for maximal Hb production. Thus, the requirement for a heme exporter during erythropoiesis appears counterintuitive.
It is likely however that regulation of potentially toxic heme in erythroid cells is markedly different from that in non-erythroid cells. For example, the first step in heme synthesis is initiated by erythroid ALAS2, which is not repressed by increasing heme levels, but rather regulated by iron supply (see [1]), in contrast to heme repression of the ubiquitous enzyme ALAS1 in non-erythroid cells [161]. There is also no evidence to date that HO-1 is induced at this stage of erythropoiesis—studies, for example, of differentiation of a murine erythroid (MEL) cell line observed downregulation of HO-1 [206, 207]. There is therefore a paucity of mechanisms in erythroid progenitors to cope with any excess free heme that may occur as heme and then globin synthesis ramps up. We hypothesize that FLVCR is specifically required during this initial phase of heme synthesis, serving as an overflow valve to protect CFU-E/pro-erythroblasts from fluctuating heme levels and potential heme toxicity until sufficient globin becomes available to bind heme. In accordance with this hypothesis, FLVCR expression is downregulated with further erythroid differentiation [36].
Inhibition of FLVCR function during erythroid differentiation of K562 cells leads to apoptosis, which suggests apoptotic pathway(s) may be stimulated by direct cellular heme toxicity or as a secondary effect of impaired erythroid differentiation [36]. It appears that FLVCR, through rapid modulation of its surface expression by trafficking or regulatory factors, is well suited to augment regulation of the heme content of erythroid progenitors, as opposed to the alternative: heme degradation by induction of HO-1, which, as an enzyme with a long half-life (~20 h) may prevent optimal erythroid hemoglobinization.
Of interest, a potential imbalance between heme and globin synthesis during erythropoiesis may be key to a human congenital form of red cell aplasia, DBA. Mutations of ribosomal proteins (notably RPS19, 24 and 17) are seen in approximately 45% of patients with DBA (reviews, [208, 209]). Mutation of RPS19 results in defective maturation of the 40S (translation initiation) ribosomal subunit [210], whereas RPS24 and 17 are also associated with the 40S subunit [202]. These mutations may result in defects in ribosome biogenesis and, in developing erythroid cells thus delay or decrease initiation of protein, predominantly globin, translation. Therefore a transient excess in erythroid progenitor intracellular free heme may occur, analogous to that predicted for Flvcr-deleted mice where heme export is impaired. Although a clear link between ribosome biogenesis defects and the development of the erythroid and myriad non-erythroid tissue defects seen in DBA is still unclear, Flvcr-null mice embryos do exhibit not only erythroid tissue but also morphologic abnormalities similar to those observed in patients with DBA—specifically, flattened faces, hypertelorism, upper limb and hand/digit abnormalities (see [211]), suggesting that FLVCR dysfunction and/or heme excess may be important in the pathogenesis of this disease.
5.1.1.5. FLVCR heme export function in macrophages and systemic iron homeostasis
Macrophage processing of senescent erythrocytes, ~360 billion per day [212], generates 25 mg or ~85% of our daily iron requirements. The iron derived from heme catabolism is either exported by the phagocytic cell-specific divalent metal transporter (Nramp1) [213] and FPN1, or stored as ferritin [214]. But, during erythrocytophagocytosis not all heme appears to be broken down by HO-1 [215, 216], and FLVCR provides an important heme (iron)–export function that impacts systemic iron homeostasis [37]. When compared to control macrophages, Flvcr-deleted murine macrophages have reduced heme export. Upon phagocytosis of opsonized erythrocytes they accumulate more ferritin (from erythroid heme catabolism) than controls; with their ferritin content increasing further when FPN1-mediated iron export is blocked by prior incubation with hepcidin [37]. These findings confirm studies by Knutson et al. indicating that approximately 25% 30% of heme is re-exported intact from macrophages following erythrophagocytosis [215]. This fraction may increase during anemia of chronic disease, when hepcidin levels are high and FPN1 expression downregulated.
Hpx, a high affinity heme-binding protein, is expressed in the liver, CNS, retina, skeletal muscle and kidney (review; [217]). It preferentially transports heme in the plasma to CD91-expressing hepatocytes, where it is subsequently catabolized by heme oxygenases. Hpx interacts with FLVCR in heme transfer studies and is present in the circulation at levels similar to that of the iron transporter transferrin (see above; [38]), suggesting that the systemic circulation of heme bound to Hpx (or the more abundant plasma protein albumin) may be of relevance to iron homeostasis. Although it is difficult to find measurements of circulating heme in the literature, a review [201] indicates the heme-binding capacity of Hpx is approximately 600 μg/dL (in agreement with an older study [218]), which would contain approximately 60 μg/dL of iron. In comparison, plasma transferrin-bound iron lies in a similar range, 50–180 μg/dL. Hpx knockout mice have been generated [219]; they show no evidence of anemia, with plasma iron levels and blood parameters (e.g., hematological parameters, BR, and albumin) comparable to those of control mice. In addition there was no evidence of oxidative damage to tissues or abnormal iron deposits. The lack of a phenotype under physiological conditions is likely related to transport of circulating heme by albumin, present in the plasma at a concentration approximately 100 times that of Hpx (30–55 mg/mL vs. 0.5–1 mg/mL). However it may be possible to dissect the role of systemic heme circulation further in that the Hpx knockout mouse is available while the heme-binding sites on albumin are known; in particular, Tyr 138 and Tyr 161 appear critical [220].
FLVCR is also highly expressed at other potential sites of heme flux—e.g., in the liver, kidney, duodenum, placenta and uterus–suggesting a role for cell heme export at these sites. Furthermore, inducible deletion of Flvcr in one-week old mice results in iron overload in hepatocytes within 5 weeks and subsequently in enterocytes and splenic macrophages [37], while FLVCR appears to traffic to the apical membrane of both hepatic and duodenal cells in vitro when cellular iron levels are high. Notably, by 7 months there is swelling of the hepatocytes that line bile canaliculi and biliary stasis, suggesting that heme export from the liver into bile may serve as a mechanism to ameliorate systemic iron overload. This mechanism may be especially important for example, during episodes of hemolysis (likely occurring in Flvcr-deleted mice) when there is increased heme delivery to the liver. Kappas et al. found that there was little or no excretion of heme into the biliary system of rats under physiological circumstances, but infusion of heme or heat-damaged erythrocytes lead to excretion of small amounts of biliary heme (e.g., 14% of the dose of injected heme) [160]. The amount excreted into the bile increased 3–4-fold when Sn-protoporphyrin, a HO inhibitor, was infused simultaneously with either heme or heat-damaged erythrocytes, this latter result suggesting that tissue macrophages can export heme to hepatocytes when HO function is impaired. Similar to studies where HO-1−/− BM cells were transplanted into irradiated HO-1+/+ mice (see section 3), when Flvcr-deleted BM cells were transplanted into irradiated Flvcr+/+ mice the transplanted mice showed no evidence of hepatic iron loading at 5–6 weeks, suggesting that liver excretion (or, more speculatively, enterocyte secretion [158]) of heme may be important in conditions of heme overload.
A related question is why or under what circumstances cells would export rather than catabolize heme. We have no clear understanding of this at present. However, one could speculate that (i) when cells are overwhelmed by heme and/or iron, for example, when endothelial cells are exposed to large amounts of heme during intravascular hemolysis, both pathways are operative; (ii) under hypoxic conditions cells may preferentially export excess heme, rather than utilize molecular oxygen to degrade heme using HO; (iii) as intact heme may also serve as an intercellular signaling molecule, macrophages may export heme as a signal of increased erythroid cell turnover; and (iv) more speculatively, cells may export heme as a form of redox reservoir for adjacent cells.
5.1.1.6. Anopheles FLVCR-like proteins export heme—relevance to malaria
Hematophagous mosquitoes (females) ingest large amounts of blood (up to 3x their bodyweight). The responses triggered include activation of genes for blood digestion, peritrophic matrix formation, and egg development (vitellogenesis). The rewards are iron (98% from Hb-heme) and amino acid absorption (60% from Hb-globin), nutrients critical for vitellogenesis. Hb degradation, however, also releases large amounts of heme into the mosquito midgut lumen, with a resultant increase in epithelial exposure to toxic ROS [221–223]. Thus, multiple systems exist to detoxify heme, potentially including mosquito FLVCR-like transporters that may export heme from the apical surface of midgut epithelial cells back into the lumen and/or via the basolateral surfaces into the hemolymph. Using bioinformatics, orthologs of human Flvcr in the genomes of the most prevalent malaria transmission vectors, Anopheles gambiae and An. stephensi were identified and cloned. Similar to human FLVCR, overexpression of these proteins in NRK (or Sf9 (insect)) cells leads to increased export of ZnMP, which is decreased by treatment of the cells with specific siRNA. In addition, mosquito midgut expression of Flvcr mRNA and protein is upregulated within 24 h of hematophagy, as expected for a heme exporter during assimilation of a heme load, while confocal microscopy of midgut cryo-sections reveals epithelial cell-surface expression of AsFLVCR following a blood meal [224].
Notably, hematophagy represents the major bottleneck for Plasmodium transmission from mosquitoes to humans. Fertilization of P. falciparum gametocytes in infected ingested blood produces <100 ookinetes in the midgut lumen, with susceptible strains of An. gambiae killing >80% of them within the midgut [225]. Surviving ookinetes are susceptible to ROS, and mosquitoes with increased midgut oxidative stress are resistant to Plasmodium transmission [226]. Recent studies emphasize the importance of oxidative stress during Plasmodium ookinete invasion of the mosquito gut epithelium; P. berghei carrying a deletion of an antioxidant enzyme are arrested at the oocyst stage [227]. We hypothesize that dysregulation of midgut epithelium FLVCR-mediated heme export will increase epithelial cell oxidative stress, potentially impacting Plasmodium transmission at its weakest point.
5.1.2. The role of ABCG2 in cell heme export
The proposed role of the mitochondrial transporters ABCB6, ABCB7 and ABCB10 in transport of heme biosynthesis intermediates has been discussed. Here we review the role of ABCG2/BCRP, a potential cell surface heme exporter.
ABCG2 or Breast Cancer Resistance Protein (BCRP) is a ~70 kDa reverse (i.e., NBD is at the N-terminus) half transporter that requires dimerization to form a functional transporter [228]. It is expressed in multiple tissues including the intestine, kidney, liver, blood-brain barrier and placenta, where it can modulate the absorption and excretion of multiple xenobiotic compounds (reviewed in [229, 230]). It is also expressed in stem cells, including hematopoietic stem cells [231]. ABCG2 expression is upregulated during erythroid differentiation and the transporter is present on the cell surface of erythrocytes [232]. Like Flvcr, the Abcg2 promoter region contains consensus GATA-1 and GATA-2 sites, providing potential mechanisms for upregulation of the gene during erythroid development.
Knockout mice were generated to investigate the physiological role of Abcg2 [233, 234]. The mice are normal with no apparent phenotypic (including hematologic) abnormalities. However, knockout mice serendipitously housed close to a light source while feeding an alfalfa-containing chow, developed skin photosensitivity with lesions on their ears, tails, snouts and eye rims [233]. The photosensitivity was traced to an accumulation of pheophorbide a, a degradation product of chlorophyll structurally related to PPIX. Compared to wild type mice, knockout mice are 100-fold more photosensitive to orally administered pheophorbide a, and have 20-fold higher plasma pheophorbide a levels. Importantly, erythrocytes and the plasma of Bcrp1−/− mice contain higher levels of PPIX relative to controls, regardless of their diet [233]. Control murine erythrocytes accumulate similar levels of PPIX when ABCG2 function is inhibited either by a general decrease in cell ATP synthesis (using 2-deoxy-D-glucose) or by a specific ABCG2 inhibitor, Ko143. Conversely, K562 cells engineered to overexpress ABCG2 have decreased retention of exogenously supplied PPIX and an 11-fold decrease in endogenous PPIX accumulation after ALA treatment. This increase in PPIX export is specifically blocked by Ko143 [232]. Jonker et al. observed significantly increased levels of conjugated PPIX in the porphyrin-producing harderian gland and liver of Bcrp1−/− mice, with increased PPIX in the biliary system. They suggest that ABCG2 transports conjugated PPIX, and has a potential role in murine hepatic excretion of excess PPIX [235].
A recent report indicates that human ABCG2 exports the heme analog ZnMP when overexpressed in K562 cells [236], an effect abrogated by Ko143. Unfortunately only a single (60-minute export) time point was presented, precluding assessment of initial loading. In addition, examination of transport of 55Fe-hemin or HPLC analysis of the cell media to rigorously demonstrate export [36] was not performed. Using biophysical methods, the binding of heme to ABCG2 or its large extracellular loop 3 was demonstrated (Kd 0.2 μM and Kd 0.5–1 μM, respectively). PPIX, ZnPPIX (present in iron-deficient maturing erythroid cells) and pheophorbide a also bind to ABCG2. As albumin can remove ABCG2-bound hemin, a heme export and transfer mechanism similar to mechanism of transfer of heme from FLVCR to Hpx is proposed [236]. Based on these studies, ABCG2 appears to have functional redundancy with FLVCR.
Both FLVCR [36, 237] and ABCG2 are expressed in hematopoietic stem cells and erythroid progenitors. However, the Abcg2 knockout phenotype suggests that ABCG2 heme export function is not critical during erythropoiesis or for systemic iron homeostasis. ABCG2 has broad substrate specificity including xenobiotics, porphyrins, and, as a recent study indicates, urate [238]. A physiological role for ABCG2 in heme transport therefore requires further investigation.
5.2. Heme Importers
Although older studies have suggested the presence of cell-surface heme importers (e.g., [140]), they have not been identified to date. Recently two groups of investigators have used novel approaches in attempts to identify these proteins in mice and the heme auxotroph, C. elegans.
5.2.1. Proton Coupled Folate Transporter/Heme Carrier Protein 1 (PCFT/HCP1)
Hypotransferrinemic mice have anemia related to low levels of the iron-binding protein transferrin, which results in low serum iron levels, repression of hepcidin and a marked increase in intestinal iron absorption. Since dietary heme represents the most abundant pool of bio-available iron, it was postulated that heme uptake would also be increased in this mouse model. Therefore, Shayeghi et al. used a subtractive RNA hybridization approach on duodenal (the site of heme import) versus ileal (“zero” heme import) tissues derived from hypotransferrinemic mice to isolate a putative duodenal heme importer, heme carrier protein 1 (HCP1, SLC46A1) [139]. The protein (~50 kDa), like FLVCR, is an MFS member, conserved among species and predicted to have 12 transmembrane domains [239]. The role of HCP1 in heme/iron biology [240] was later overshadowed by the finding that HCP1 primarily functions as an intestinal proton-coupled folate transporter (PCFT) [241]. Several patients with hereditary familial folate malabsorption have been identified as having mutations of the PCFT/HCP1 gene; one mutation, an exon 3 deletion, was shown to eliminate membrane localization of the protein and (folate) transport activity [241, 242]. Although a consequence of the resultant folate deficiency in newborns is anemia, this is fully corrected by oral doses of 5-formyltetrahydrofolate. Furthermore, PCFT/HCP1 gene variants are infrequent in hemochromatosis and iron overload screening (HEIRS) study participants [243].
The heme import activity of PCFT/HCP1 was demonstrated in Xenopus laevis oocytes; injection of PCFT/HCP1 cRNA increases the uptake of L-arginine–conjugated 55Fe-heme 2.5-fold in a temperature-dependent manner. In comparison, PCFT/HCP1-mediated 3H-folic acid uptake was 200-fold that of controls, demonstrating PCFT’s preference for folate [241]. Competition studies with an antibody against PCFT/HCP1 inhibited uptake of both heme and folate, and each substrate competed with the other to some extent [244]. However, the greater affinity of PCFT/HCP1 for folate (Km 1 μM) as compared to hemin (Km >100 μM) suggests that folate is the primary and physiologically more relevant substrate for this transporter [244, 245]. However it should be mentioned that the concentration of heme in the gastrointestinal tract following a meal is likely many times higher than that of folate.
Ectopically expressed PCFT/HCP1 in HEK293 cells co-localizes with Hb:Hp complexes and transferrin in early endosomes, and has thus been hypothesized to cycle back and forth to the plasma membrane, possibly due to its di-leucine motif [246]. These investigators speculate that PCFT/HCP1 functions in endosomes to export heme released from Hb:Hp into the cytosol for degradation by microsomal HO. A bona fide physiological role for PCFT/HCP1 in heme metabolism awaits further clarification.
5.2.2. Heme Responsive Genes in Caenorhabditis elegans
The nematode, C. elegans is a heme auxotroph, that is, it lacks endogenous heme synthesis, depending on environmental heme for survival, thus providing a “clean” genetic background to study intracellular heme trafficking pathways. C. elegans has been studied as a model organism for more than 40 years. The genome has been sequenced, and many human genes are conserved in C. elegans. In addition, the ease of genetic manipulation makes it a unique model system for the identification of heme transporters [162, 247]. Heme uptake is a regulated process in C. elegans; when worms are grown in optimal-to-low heme conditions (<20 μM) and then exposed to ZnMP, upregulation of heme transporters allows rapid internalization of ZnMP, however ZnMP uptake is reduced significantly when worms are grown in medium containing 100 μM heme [150]. Hamza’s group performed microarray analyses to identify heme-regulated genes (hrg) in C. elegans, during growth under low heme (4 μM) versus optimal (20 μM) versus high heme conditions (500 μM), identifying a total of 288 genes (now available, [248]). Initially focusing on genes upregulated in low heme conditions, they further sorted them based on the predicted presence of transmembrane domains, known transporter function and/or heme- or metal-binding domains. A heme-responsive gene fulfilling these criteria, hrg-4, and three of its putative paralogs (hrg-1, hrg-5 and hrg-6) were subjected to further studies [150]. The expression of both hrg-4 and hrg-1 is modulated by the heme concentration in the growth media in contrast to hrg-5 and hrg-6, which are expressed constitutively. RNAi-based knockdown of CeHRG-4 abolishes uptake of ZnMP in worms grown in low heme, and their progeny are resistant to feeding with the cytotoxic heme analog gallium protoporphyrin IX (GaPP). In addition, in voltage clamp experiments on Xenopus oocytes in the presence of hemin, injection of the Cehrg-4 cRNA into the oocytes resulted in significant inward currents. These findings combined with localization studies indicate that CeHRG-4 is a plasma membrane–associated heme importer, expressed in the worm intestine. Unfortunately, mammalian orthologs are lacking [150].
In contrast, hrg-1 has orthologs in vertebrates. CeHRG-1 (19 kDa) has four predicted transmembrane domains and, like FLVCR, YXXØ and distal di-leucine motifs; these motifs lie adjacent to a C-terminal heme-binding motif. CeHRG-1 knockdown paradoxically increases ZnMP uptake and does not protect C. elegans from GaPP toxicity. The human ortholog hHRG-1 is widely expressed, but not regulated by heme or iron, at least at the transcriptional level. Both CeHRG-1 and hHRG-1 extensively co-localize with acidic endosome-lysosome organelles in HEK293 cells, and their affinity for heme decreases with increasing pH. Xenopus oocytes studies (as above) were also consistent with heme transport across a membrane. Thus the available evidence indicates that these proteins transport heme across the endosome-lysosomes into the cytoplasm. Notably, knockdown of the zebrafish ortholog results in marked anemia and developmental defects suggesting the importance of lysosomal heme-recycling from organelle hemoproteins.
In a more recent study [248], Hamza’s group have systematically knocked down all 288 worm genes in a transgenic worm that responds to changes in heme levels by altering GFP expression. Genes causing alterations of ±2-fold were further examined using a cytotoxicity assay (resistance to GaPP toxicity). In addition to identification of HRG-4, an ABC transporter, MRP-1, which appears to be a cell membrane permease that exports heme, and F22B5.4, a single TMD protein of unknown function, were identified. In sum, their studies vindicate the use of C. elegans to rapidly identify both cell surface and cell organelle heme transporters. Further analysis may uncover yet other heme transporters. Approximately 40% of the 288 worm genes have orthologs in the human genome and thus it is likely that vertebrate transporters will also be identified.
6. Conclusion
There is a large body of literature that supports a seminal role for the HO system, predominantly HO-1, in protecting cells from oxidative stress. Although both HO-1 and, to a lesser extent, HO-2 are important for heme-iron reutilization, the murine model of FLVCR deficiency emphasizes the importance of FLVCR-mediated heme export and heme recycling in systemic iron homeostasis. Future studies should delineate the importance of systemic heme recycling, especially in anemia of chronic disease where iron but not heme export is subject to suppression by hepcidin; the relative contributions of the HO system and heme export in maintaining intracellular heme levels; and the physiological conditions whereupon heme export rather than HO-mediated catabolism occurs.
FLVCR is required during erythropoiesis and likely functions as an overflow valve to mitigate fluctuations in heme levels that may occur prior to or at the onset of globin synthesis. Investigation of FLVCR trafficking, protein-protein interactions and internalization/degradation pathways will yield additional insights into the function and regulation of this protein.
Of the mammalian ABC transporters, ABCG2 may have functional redundancy with FLVCR. However, the distinctively different knockout mice phenotypes suggest their physiological roles do not overlap. Notably, both proteins are expressed in K562 cells; thus, it would be of interest to inhibit the function of each in turn and determine their relative contributions to cell heme export.
PCFT/HCP1 was initially identified as a duodenal heme importer, yet subsequent studies indicate that it functions primarily as a folate transporter; thus, its physiological role in heme acquisition requires further clarification. The Pcft knockout mouse has recently been generated. Although it appears to have significant disorders in homocysteine metabolism related to folate deficiency, its potential role in heme/iron metabolism has not been addressed [249].
Both ABCB6 and ABCB10 are mitochondrial proteins consistently co-expressed with heme synthesis pathway enzymes, suggesting roles for these proteins in heme metabolism. A physiological substrate of ABCB6, relevant to heme synthesis, has yet to be identified. The last step of heme synthesis, iron insertion into PPIX by Fech, occurs on the matrix side of mitochondria, thus indicating the presence of unidentified heme exporters on both the mitochondrial IM and OM. As discussed, ABCB10 physically interacts with Fech and stabilizes Mfrn1, but it remains to be established whether it has a transport function (perhaps heme) in addition to its structural role on the mitochondrial IM.
A recent breakthrough in heme transport biology is the identification of HRG proteins in the heme auxotroph C. elegans. HRG-1 and its human ortholog co-localize to the endosome/lysosome. Further analysis of HRG proteins in C. elegans [248] and humans [63] will further our knowledge of heme transport and metabolism.
Increased understanding of cell-surface heme transport proteins will provide insights into disease states characterized by release of free (toxic) heme, including both congenital and acquired hemolytic anemias, sickle cell disease, thalassemias, malaria and the myelodysplastic syndromes. Fundamental questions in heme physiology remain unanswered, including the identity of transporters involved in transfer of heme and heme intermediates across mitochondrial membranes, the sites of heme transfer to apoproteins to form hemoproteins, the identity of intracellular heme chaperones, the comparative contributions of FLVCR and the HO system to cell protection from oxidative stress and/or heme excess, and the role of systemic circulation of heme in iron homeostasis and its other functions.
Acknowledgments
We thank Dr. A. Chishti for critical review of the manuscript and many stimulating discussions. We would also like to acknowledge the help of Ms. E. Quigley and Dr. B. Quigley with editing and Ms. F. Hashemi with figures. Work in the JQ laboratory is funded by grants from the National Institutes of Health and the Roche Foundation for Anemia research (RoFAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ponka P. Blood. 1997;89:1–25. [PubMed] [Google Scholar]
- 2.Ryter SW, Tyrrell RM. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 3.Carpenter CE, Mahoney AW. Crit Rev Food Sci Nutr. 1992;31:333–67. doi: 10.1080/10408399209527576. [DOI] [PubMed] [Google Scholar]
- 4.Andrews NC. N Engl J Med. 1999;341:1986–95. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 5.Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, McKnight SL. Proc Natl Acad Sci U S A. 2007;104:16886–91. doi: 10.1073/pnas.0708365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forsburg SL, Guarente L. Annu Rev Cell Biol. 1989;5:153–80. doi: 10.1146/annurev.cb.05.110189.001101. [DOI] [PubMed] [Google Scholar]
- 7.Strand A, Asami T, Alonso J, Ecker JR, Chory J. Nature. 2003;421:79–83. doi: 10.1038/nature01204. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, Fujita H, Igarashi K. EMBO J. 2001;20:2835–43. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Haro C, Mendez R, Santoyo J. FASEB J. 1996;10:1378–87. doi: 10.1096/fasebj.10.12.8903508. [DOI] [PubMed] [Google Scholar]
- 10.Crosby JS, Chefalo PJ, Yeh I, Ying S, London IM, Leboulch P, Chen JJ. Blood. 2000;96:3241–8. [PubMed] [Google Scholar]
- 11.Lathrop JT, Timko MP. Science. 1993;259:522–5. doi: 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- 12.Qi Z, Hamza I, O’Brian MR. Proc Natl Acad Sci U S A. 1999;96:13056–61. doi: 10.1073/pnas.96.23.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Nat Struct Mol Biol. 2007;14:23–9. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- 14.Imaizumi T, Kay SA, Schroeder JI. Science. 2007;318:1730–1. doi: 10.1126/science.1151360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Publicover S, Gu Y. Proc Natl Acad Sci U S A. 2009;106:2957–62. doi: 10.1073/pnas.0809100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponka P. Am J Med Sci. 1999;318:241–56. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Y, Lee HC, Zhang L. DNA Cell Biol. 2002;21:333–46. doi: 10.1089/104454902753759744. [DOI] [PubMed] [Google Scholar]
- 18.Sengupta A, Hon T, Zhang L. Brain Res Mol Brain Res. 2005;137:23–30. doi: 10.1016/j.molbrainres.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Chen JJ, London IM. Trends Biochem Sci. 1995;20:105–8. doi: 10.1016/s0968-0004(00)88975-6. [DOI] [PubMed] [Google Scholar]
- 20.Sassa S, Nagai T. Int J Hematol. 1996;63:167–78. doi: 10.1016/0925-5710(96)00449-5. [DOI] [PubMed] [Google Scholar]
- 21.Aono S. Dalton Trans. 2008:3137–46. doi: 10.1039/b802070c. [DOI] [PubMed] [Google Scholar]
- 22.Mense SM, Zhang L. Cell Res. 2006;16:681–92. doi: 10.1038/sj.cr.7310086. [DOI] [PubMed] [Google Scholar]
- 23.Balla J, Vercellotti GM, Nath K, Yachie A, Nagy E, Eaton JW, Balla G. Nephrol Dial Transplant. 2003;18(Suppl 5):v8–12. doi: 10.1093/ndt/gfg1034. [DOI] [PubMed] [Google Scholar]
- 24.Halliwell B, Gutteridge JM. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 25.Sassa S. Antioxid Redox Signal. 2004;6:819–24. doi: 10.1089/ars.2004.6.819. [DOI] [PubMed] [Google Scholar]
- 26.Outten CE, O’Halloran TV. Science. 2001;292:2488–92. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 27.Qi Z, O’Brian MR. Mol Cell. 2002;9:155–62. doi: 10.1016/s1097-2765(01)00431-2. [DOI] [PubMed] [Google Scholar]
- 28.Habig WH, Pabst MJ, Fleischner G, Gatmaitan Z, Arias IM, Jakoby WB. Proc Natl Acad Sci U S A. 1974;71:3879–82. doi: 10.1073/pnas.71.10.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincent SH, Muller-Eberhard U. J Biol Chem. 1985;260:14521–8. [PubMed] [Google Scholar]
- 30.Taketani S, Adachi Y, Kohno H, Ikehara S, Tokunaga R, Ishii T. J Biol Chem. 1998;273:31388–94. doi: 10.1074/jbc.273.47.31388. [DOI] [PubMed] [Google Scholar]
- 31.Iwahara S, Satoh H, Song DX, Webb J, Burlingame AL, Nagae Y, Muller-Eberhard U. Biochemistry. 1995;34:13398–406. doi: 10.1021/bi00041a017. [DOI] [PubMed] [Google Scholar]
- 32.Tudor C, Lerner-Marmarosh N, Engelborghs Y, Gibbs PE, Maines MD. Biochem J. 2008;413:405–16. doi: 10.1042/BJ20080018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Light WR, 3rd, Olson JS. J Biol Chem. 1990;265:15623–31. [PubMed] [Google Scholar]
- 34.Ricchelli F, Gobbo S, Jori G, Moreno G, Salet C. Eur J Biochem. 1995;233:159–64. doi: 10.1111/j.1432-1033.1995.159_1.x. [DOI] [PubMed] [Google Scholar]
- 35.Goldman BS, Beck DL, Monika EM, Kranz RG. Proc Natl Acad Sci U S A. 1998;95:5003–8. doi: 10.1073/pnas.95.9.5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Cell. 2004;118:757–66. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. Science. 2008;319:825–8. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z, Philips JD, Doty RT, Giraudi P, Ostrow JD, Tiribelli C, Smith A, Abkowitz JL. J Biol Chem. 2010 doi: 10.1074/jbc.M110.119131. JBC Papers in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Severance S, Hamza I. Chem Rev. 2009;109:4596–616. doi: 10.1021/cr9001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thöny-Meyer L. Tetrapyrroles. 2009:149–159. [Google Scholar]
- 41.Medlock AE, Dailey HA. Tetrapyrroles. 2009:116–127. [Google Scholar]
- 42.Heinemann IU, Jahn M, Jahn D. Arch Biochem Biophys. 2008;474:238–51. doi: 10.1016/j.abb.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 43.Ajioka RS, Phillips JD, Kushner JP. Biochim Biophys Acta. 2006;1763:723–36. doi: 10.1016/j.bbamcr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Sheftel AD, Richardson DR, Prchal J, Ponka P. Acta Haematol. 2009;122:120–33. doi: 10.1159/000243796. [DOI] [PubMed] [Google Scholar]
- 45.Sassa S. Br J Haematol. 2006;135:281–92. doi: 10.1111/j.1365-2141.2006.06289.x. [DOI] [PubMed] [Google Scholar]
- 46.Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, Saint-Amant L, Schmidt PJ, Orr A, Bottomley SS, Fleming MD, Ludman M, Dyack S, Fernandez CV, Samuels ME. Nat Genet. 2009;41:651–3. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 47.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, Schuetz JD. Nature. 2006;443:586–9. doi: 10.1038/nature05125. [DOI] [PubMed] [Google Scholar]
- 48.Krishnamurthy P, Xie T, Schuetz JD. Pharmacol Ther. 2007;114:345–58. doi: 10.1016/j.pharmthera.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Rees DC, Johnson E, Lewinson O. Nat Rev Mol Cell Biol. 2009;10:218–27. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dean M, Rzhetsky A, Allikmets R. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 51.Zutz A, Gompf S, Schagger H, Tampe R. Biochim Biophys Acta. 2009;1787:681–90. doi: 10.1016/j.bbabio.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Mitsuhashi N, Miki T, Senbongi H, Yokoi N, Yano H, Miyazaki M, Nakajima N, Iwanaga T, Yokoyama Y, Shibata T, Seino S. J Biol Chem. 2000;275:17536–40. doi: 10.1074/jbc.275.23.17536. [DOI] [PubMed] [Google Scholar]
- 53.Kispal G, Csere P, Prohl C, Lill R. EMBO J. 1999;18:3981–9. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hausmann A, Aguilar Netz DJ, Balk J, Pierik AJ, Muhlenhoff U, Lill R. Proc Natl Acad Sci U S A. 2005;102:3266–71. doi: 10.1073/pnas.0406447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balk J, Aguilar Netz DJ, Tepper K, Pierik AJ, Lill R. Mol Cell Biol. 2005;25:10833–41. doi: 10.1128/MCB.25.24.10833-10841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye H, Rouault TA. Biochemistry. 2010;49:4945–56. doi: 10.1021/bi1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kispal G, Csere P, Guiard B, Lill R. FEBS Lett. 1997;418:346–50. doi: 10.1016/s0014-5793(97)01414-2. [DOI] [PubMed] [Google Scholar]
- 58.Lynch J, Fukuda Y, Krishnamurthy P, Du G, Schuetz JD. Cancer Res. 2009;69:5560–7. doi: 10.1158/0008-5472.CAN-09-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuchida M, Emi Y, Kida Y, Sakaguchi M. Biochem Biophys Res Commun. 2008;369:369–75. doi: 10.1016/j.bbrc.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 60.Jalil YA, Ritz V, Jakimenko A, Schmitz-Salue C, Siebert H, Awuah D, Kotthaus A, Kietzmann T, Ziemann C, Hirsch-Ernst KI. Am J Physiol Cell Physiol. 2008;294:C579–90. doi: 10.1152/ajpcell.00612.2006. [DOI] [PubMed] [Google Scholar]
- 61.Paterson JK, Shukla S, Black CM, Tachiwada T, Garfield S, Wincovitch S, Ernst DN, Agadir A, Li X, Ambudkar SV, Szakacs G, Akiyama S, Gottesman MM. Biochemistry. 2007;46:9443–52. doi: 10.1021/bi700015m. [DOI] [PubMed] [Google Scholar]
- 62.Csere P, Lill R, Kispal G. FEBS Lett. 1998;441:266–70. doi: 10.1016/s0014-5793(98)01560-9. [DOI] [PubMed] [Google Scholar]
- 63.Nilsson R, Schultz IJ, Pierce EL, Soltis KA, Naranuntarat A, Ward DM, Baughman JM, Paradkar PN, Kingsley PD, Culotta VC, Kaplan J, Palis J, Paw BH, Mootha VK. Cell Metab. 2009;10:119–30. doi: 10.1016/j.cmet.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Azuma M, Kabe Y, Kuramori C, Kondo M, Yamaguchi Y, Handa H. PLoS One. 2008;3:e3070. doi: 10.1371/journal.pone.0003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferreira GC, Dailey HA. J Biol Chem. 1987;262:4407–12. [PubMed] [Google Scholar]
- 66.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. EMBO J. 2004;23:1720–8. doi: 10.1038/sj.emboj.7600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen W, Dailey HA, Paw BH. Blood. :116–628. 30. doi: 10.1182/blood-2009-12-259614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 69.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Mol Cell Biol. 2009;29:1007–16. doi: 10.1128/MCB.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shirihai OS, Gregory T, Yu C, Orkin SH, Weiss MJ. EMBO J. 2000;19:2492–502. doi: 10.1093/emboj/19.11.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, Hyde BB, Shirihai OS, Ward DM, Kaplan J, Paw BH. Proc Natl Acad Sci U S A. 2009;106:16263–8. doi: 10.1073/pnas.0904519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andrews NC. Proc Natl Acad Sci U S A. 2009;106:16012–3. doi: 10.1073/pnas.0909137106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen W, Dailey HA, Paw BH. Blood. 2010;116:628–30. doi: 10.1182/blood-2009-12-259614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graf SA, Haigh SE, Corson ED, Shirihai OS. J Biol Chem. 2004;279:42954–63. doi: 10.1074/jbc.M405040200. [DOI] [PubMed] [Google Scholar]
- 75.Crooks DR, Ghosh MC, Haller RG, Tong WH, Rouault TA. Blood. 2010;115:860–9. doi: 10.1182/blood-2009-09-243105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye H, Rouault TA. Adv Hematol 2010. 2010 doi: 10.1155/2010/329394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF. Blood. 2000;96:3256–64. [PubMed] [Google Scholar]
- 78.Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. Blood. 2007;109:3567–9. doi: 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharma AK, Pallesen LJ, Spang RJ, Walden WE. J Biol Chem. 2010;285:26745–51. doi: 10.1074/jbc.R110.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taketani S, Kakimoto K, Ueta H, Masaki R, Furukawa T. Blood. 2003;101:3274–80. doi: 10.1182/blood-2002-04-1212. [DOI] [PubMed] [Google Scholar]
- 81.Bonnett R, McDonagh AF. J Chem Soc Perkin. 1973;1–9:881–8. doi: 10.1039/p19730000881. [DOI] [PubMed] [Google Scholar]
- 82.Brown SB, Hatzikonstantinou H, Herries DG. Biochem J. 1978;174:901–7. doi: 10.1042/bj1740901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maines MD, Gibbs PE. Biochem Biophys Res Commun. 2005;338:568–77. doi: 10.1016/j.bbrc.2005.08.121. [DOI] [PubMed] [Google Scholar]
- 84.McCoubrey WK, Jr, Huang TJ, Maines MD. Eur J Biochem. 1997;247:725–32. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 85.Scapagnini G, D’Agata V, Calabrese V, Pascale A, Colombrita C, Alkon D, Cavallaro S. Brain Res. 2002;954:51–9. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 86.Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M. Gene. 2004;336:241–50. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 87.Yoshinaga T, Sassa S, Kappas A. J Biol Chem. 1982;257:7786–93. [PubMed] [Google Scholar]
- 88.Kim HP, Wang X, Galbiati F, Ryter SW, Choi AM. FASEB J. 2004;18:1080–9. doi: 10.1096/fj.03-1391com. [DOI] [PubMed] [Google Scholar]
- 89.Converso DP, Taille C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. FASEB J. 2006;20:1236–8. doi: 10.1096/fj.05-4204fje. [DOI] [PubMed] [Google Scholar]
- 90.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, Dennery PA. J Biol Chem. 2007;282:20621–33. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- 91.Berk PD, Howe RB, Bloomer JR, Berlin NI. J Clin Invest. 1969;48:2176–90. doi: 10.1172/JCI106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kapitulnik J, Maines MD. Trends Pharmacol Sci. 2009;30:129–37. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 93.Igarashi K, Sun J. Antioxid Redox Signal. 2006;8:107–18. doi: 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 94.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K. Mol Cell Biol. 2007;27:6962–71. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang L, Guarente L. EMBO J. 1995;14:313–20. doi: 10.1002/j.1460-2075.1995.tb07005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ding Y, McCoubrey WK, Jr, Maines MD. Eur J Biochem. 1999;264:854–61. doi: 10.1046/j.1432-1327.1999.00677.x. [DOI] [PubMed] [Google Scholar]
- 97.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Science. 2004;306:2093–7. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 98.Lack EE. Hum Pathol. 1977;8:39–51. doi: 10.1016/s0046-8177(77)80064-6. [DOI] [PubMed] [Google Scholar]
- 99.Ortega-Saenz P, Pascual A, Gomez-Diaz R, Lopez-Barneo J. J Gen Physiol. 2006;128:405–11. doi: 10.1085/jgp.200609591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shibahara S, Han F, Li B, Takeda K. Antioxid Redox Signal. 2007;9:2209–25. doi: 10.1089/ars.2007.1784. [DOI] [PubMed] [Google Scholar]
- 101.Kemp JP, Peers C. Adv Exp Med Biol. 2009;648:39–48. doi: 10.1007/978-90-481-2259-2_4. [DOI] [PubMed] [Google Scholar]
- 102.Yi L, Jenkins PM, Leichert LI, Jakob U, Martens JR, Ragsdale SW. J Biol Chem. 2009;284:20556–61. doi: 10.1074/jbc.M109.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Science. 1987;235:1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 104.Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH. Proc Natl Acad Sci U S A. 2009;106:5171–6. doi: 10.1073/pnas.0813132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baranano DE, Rao M, Ferris CD, Snyder SH. Proc Natl Acad Sci U S A. 2002;99:16093–8. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dore S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Proc Natl Acad Sci U S A. 1999;96:2445–50. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Nature. 2000;403:776–81. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 108.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 109.Hori R, Kashiba M, Toma T, Yachie A, Goda N, Makino N, Soejima A, Nagasawa T, Nakabayashi K, Suematsu M. J Biol Chem. 2002;277:10712–8. doi: 10.1074/jbc.M107749200. [DOI] [PubMed] [Google Scholar]
- 110.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Baranano DE, Dore S, Poss KD, Snyder SH. Nat Cell Biol. 1999;1:152–7. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- 111.Sheftel AD, Kim SF, Ponka P. J Biol Chem. 2007;282:10480–6. doi: 10.1074/jbc.M700240200. [DOI] [PubMed] [Google Scholar]
- 112.Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117:285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 113.Zhang AS, Enns CA. Hematology Am Soc Hematol Educ Program. 2009:207–14. doi: 10.1182/asheducation-2009.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Garby L, Noyes WD. J Clin Invest. 1959;38:1479–83. doi: 10.1172/JCI103925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ascenzi P, Bocedi A, Visca P, Altruda F, Tolosano E, Beringhelli T, Fasano M. IUBMB Life. 2005;57:749–59. doi: 10.1080/15216540500380871. [DOI] [PubMed] [Google Scholar]
- 116.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Blood. 2005;106:2572–9. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 117.Poss KD, Tonegawa S. Proc Natl Acad Sci U S A. 1997;94:10919–24. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poss KD, Thomas MJ, Ebralidze AK, O’Dell TJ, Tonegawa S. Neuron. 1995;15:867–73. doi: 10.1016/0896-6273(95)90177-9. [DOI] [PubMed] [Google Scholar]
- 119.Dennery PA, Spitz DR, Yang G, Tatarov A, Lee CS, Shegog ML, Poss KD. J Clin Invest. 1998;101:1001–11. doi: 10.1172/JCI448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. J Clin Invest. 1999;103:129–35. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Blood. 2010 doi: 10.1182/blood-2010-03-272138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2787. [DOI] [PubMed] [Google Scholar]
- 123.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 124.Nemeth E. Curr Opin Hematol. 2008;15:169–75. doi: 10.1097/MOH.0b013e3282f73335. [DOI] [PubMed] [Google Scholar]
- 125.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, Muckenthaler MU. Haematologica. 2010;95:1261–8. doi: 10.3324/haematol.2009.020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kartikasari AE, Wagener FA, Yachie A, Wiegerinck ET, Kemna EH, Swinkels DW. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Finch C. Blood. 1994;84:1697–702. [PubMed] [Google Scholar]
- 128.Poss KD, Tonegawa S. Proc Natl Acad Sci U S A. 1997;94:10925–30. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vinchi F, Gastaldi S, Silengo L, Altruda F, Tolosano E. Am J Pathol. 2008;173:289–99. doi: 10.2353/ajpath.2008.071130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hershko C, Cook JD, Finch CA. Br J Haematol. 1974;28:67–75. doi: 10.1111/j.1365-2141.1974.tb06640.x. [DOI] [PubMed] [Google Scholar]
- 131.Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. J Biol Chem. 2002;277:39786–91. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- 132.Bezwoda WR, Bothwell TH, Charlton RW, Torrance JD, MacPhail AP, Derman DP, Mayet F. S Afr Med J. 1983;64:552–6. [PubMed] [Google Scholar]
- 133.Turnbull A, Cleton F, Finch CA. J Clin Invest. 1962;41:1897–907. doi: 10.1172/JCI104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Conrad ME, Umbreit JN. Blood Cells Mol Dis. 2002;29:336–55. doi: 10.1006/bcmd.2002.0564. [DOI] [PubMed] [Google Scholar]
- 135.Vaghefi N, Nedjaoum F, Guillochon D, Bureau F, Arhan P, Bougle D. J Agric Food Chem. 2002;50:4969–73. doi: 10.1021/jf0109165. [DOI] [PubMed] [Google Scholar]
- 136.Mims MP, Prchal JT. Hematology. 2005;10:339–45. doi: 10.1080/10245330500093419. [DOI] [PubMed] [Google Scholar]
- 137.Young GP, Rose IS, St John DJ. J Gastroenterol Hepatol. 1989;4:537–45. doi: 10.1111/j.1440-1746.1989.tb00858.x. [DOI] [PubMed] [Google Scholar]
- 138.West AR, Oates PS. World J Gastroenterol. 2008;14:4101–10. doi: 10.3748/wjg.14.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 140.Grasbeck R, Majuri R, Kouvonen I, Tenhunen R. Biochim Biophys Acta. 1982;700:137–42. doi: 10.1016/0167-4838(82)90089-9. [DOI] [PubMed] [Google Scholar]
- 141.Worthington MT, Cohn SM, Miller SK, Luo RQ, Berg CL. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1172–7. doi: 10.1152/ajpgi.2001.280.6.G1172. [DOI] [PubMed] [Google Scholar]
- 142.Vaghefi N, Guillochon D, Bureau F, Neuville D, Lebrun F, Arhan P, Bougle D. J Nutr Biochem. 2000;11:562–567. doi: 10.1016/s0955-2863(00)00120-0. [DOI] [PubMed] [Google Scholar]
- 143.Noyer CM, Immenschuh S, Liem HH, Muller-Eberhard U, Wolkoff AW. Hepatology. 1998;28:150–5. doi: 10.1002/hep.510280120. [DOI] [PubMed] [Google Scholar]
- 144.Galbraith RA, Sassa S, Kappas A. J Biol Chem. 1985;260:12198–202. [PubMed] [Google Scholar]
- 145.Parmley RT, Barton JC, Conrad ME, Austin RL, Holland RM. Exp Mol Pathol. 1981;34:131–44. doi: 10.1016/0014-4800(81)90070-8. [DOI] [PubMed] [Google Scholar]
- 146.Wyllie JC, Kaufman N. Lab Invest. 1982;47:471–6. [PubMed] [Google Scholar]
- 147.Wheby MS, Suttle GE, Ford KT., 3rd Gastroenterology. 1970;58:647–54. [PubMed] [Google Scholar]
- 148.Brown EB, Hwang YF, Nicol S, Ternberg J. J Lab Clin Med. 1968;72:58–64. [PubMed] [Google Scholar]
- 149.Puertollano R, Kiselyov K. Am J Physiol Renal Physiol. 2009;296:F1245–54. doi: 10.1152/ajprenal.90522.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, Krause M, Hamza I. Nature. 2008;453:1127–31. doi: 10.1038/nature06934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Collins JF, Franck CA, Kowdley KV, Ghishan FK. Am J Physiol Gastrointest Liver Physiol. 2005;288:G964–71. doi: 10.1152/ajpgi.00489.2004. [DOI] [PubMed] [Google Scholar]
- 152.Chicault C, Toutain B, Monnier A, Aubry M, Fergelot P, Le Treut A, Galibert MD, Mosser J. Physiol Genomics. 2006;26:55–67. doi: 10.1152/physiolgenomics.00297.2005. [DOI] [PubMed] [Google Scholar]
- 153.Raffin SB, Woo CH, Roost KT, Price DC, Schmid R. J Clin Invest. 1974;54:1344–52. doi: 10.1172/JCI107881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wheby MS, Spyker DA. Am J Clin Nutr. 1981;34:1686–93. doi: 10.1093/ajcn/34.9.1686. [DOI] [PubMed] [Google Scholar]
- 155.Boni RE, Huch Boni RA, Galbraith RA, Drummond GS, Kappas A. Pharmacology. 1993;47:318–29. doi: 10.1159/000139113. [DOI] [PubMed] [Google Scholar]
- 156.West AR, Oates PS. J Gastroenterol Hepatol. 2008;23:150–8. doi: 10.1111/j.1440-1746.2007.05047.x. [DOI] [PubMed] [Google Scholar]
- 157.Conrad ME, Weintraub LR, Sears DA, Crosby WH. Am J Physiol. 1966;211:1123–30. doi: 10.1152/ajplegacy.1966.211.5.1123. [DOI] [PubMed] [Google Scholar]
- 158.Uc A, Stokes JB, Britigan BE. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1150–7. doi: 10.1152/ajpgi.00157.2004. [DOI] [PubMed] [Google Scholar]
- 159.Marro S, Barisani D, Chiabrando D, Fagoonee S, Muckenthaler MU, Stolte J, Meneveri R, Haile D, Silengo L, Altruda F, Tolosano E. Gastroenterology. 2007;133:1261–1271. doi: 10.1053/j.gastro.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 160.Kappas A, Simionatto CS, Drummond GS, Sassa S, Anderson KE. Proc Natl Acad Sci U S A. 1985;82:896–900. doi: 10.1073/pnas.82.3.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Furuyama K, Kaneko K, Vargas PD. Tohoku J Exp Med. 2007;213:1–16. doi: 10.1620/tjem.213.1. [DOI] [PubMed] [Google Scholar]
- 162.Hamza I. ACS Chem Biol. 2006;1:627–9. doi: 10.1021/cb600442b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Latunde-Dada GO, Simpson RJ, McKie AT. Trends Biochem Sci. 2006;31:182–8. doi: 10.1016/j.tibs.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 164.Dunn LL, Rahmanto YS, Richardson DR. Trends Cell Biol. 2007;17:93–100. doi: 10.1016/j.tcb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 165.Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. J Biol Chem. 2004;279:24218–25. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 166.Lazarow PB, de Duve C. J Cell Biol. 1973;59:491–506. doi: 10.1083/jcb.59.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Onions D, Jarrett O, Testa N, Frassoni F, Toth S. Nature. 1982;296:156–8. doi: 10.1038/296156a0. [DOI] [PubMed] [Google Scholar]
- 168.Abkowitz JL, Holly RD, Grant CK. J Clin Invest. 1987;80:1056–63. doi: 10.1172/JCI113160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wickrema A, Krantz SB, Winkelmann JC, Bondurant MC. Blood. 1992;80:1940–9. [PubMed] [Google Scholar]
- 170.Gilmore MM, Bishop TR. Blood Cells Mol Dis. 1999;25:68–77. doi: 10.1006/bcmd.1999.0228. [DOI] [PubMed] [Google Scholar]
- 171.Dean GA, Groshek PM, Mullins JI, Hoover EA. J Virol. 1992;66:5561–8. doi: 10.1128/jvi.66.9.5561-5568.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Riedel N, Hoover EA, Dornsife RE, Mullins JI. Proc Natl Acad Sci U S A. 1988;85:2758–62. doi: 10.1073/pnas.85.8.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Weiss RA, Tailor CS. Cell. 1995;82:531–3. doi: 10.1016/0092-8674(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 174.Rigby MA, Rojko JL, Stewart MA, Kociba GJ, Cheney CM, Rezanka LJ, Mathes LE, Hartke JR, Jarrett O, Neil JC. J Gen Virol. 1992;73(Pt 11):2839–47. doi: 10.1099/0022-1317-73-11-2839. [DOI] [PubMed] [Google Scholar]
- 175.Quigley JG, Burns CC, Anderson MM, Lynch ED, Sabo KM, Overbaugh J, Abkowitz JL. Blood. 2000;95:1093–9. [PubMed] [Google Scholar]
- 176.Tailor CS, Willett BJ, Kabat D. J Virol. 1999;73:6500–5. doi: 10.1128/jvi.73.8.6500-6505.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lipovich L, Hughes AL, King MC, Abkowitz JL, Quigley JG. Gene. 2002;286:203–13. doi: 10.1016/s0378-1119(02)00457-2. [DOI] [PubMed] [Google Scholar]
- 178.Abruzzo PM, diTullio S, Marchionni C, Belia S, Fano G, Zampieri S, Carraro U, Kern H, Sgarbi G, Lenaz G, Marini M. Free Radic Res. 2010;44:563–76. doi: 10.3109/10715761003692487. [DOI] [PubMed] [Google Scholar]
- 179.Colombo P, Yon J, Garson K, Fried M. Proc Natl Acad Sci U S A. 1992;89:6358–62. doi: 10.1073/pnas.89.14.6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.van Beest M, Robben JH, Savelkoul PJ, Hendriks G, Devonald MA, Konings IB, Lagendijk AK, Karet F, Deen PM. Biochim Biophys Acta. 2006;1758:1126–33. doi: 10.1016/j.bbamem.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 181.Matter K, Yamamoto EM, Mellman I. J Cell Biol. 1994;126:991–1004. doi: 10.1083/jcb.126.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Miranda KC, Khromykh T, Christy P, Le TL, Gottardi CJ, Yap AS, Stow JL, Teasdale RD. J Biol Chem. 2001;276:22565–72. doi: 10.1074/jbc.M101907200. [DOI] [PubMed] [Google Scholar]
- 183.Muth TR, Caplan MJ. Annu Rev Cell Dev Biol. 2003;19:333–66. doi: 10.1146/annurev.cellbio.19.110701.161425. [DOI] [PubMed] [Google Scholar]
- 184.Brone B, Eggermont J. Am J Physiol Cell Physiol. 2005;288:C20–9. doi: 10.1152/ajpcell.00368.2004. [DOI] [PubMed] [Google Scholar]
- 185.Bryant NJ, Govers R, James DE. Nat Rev Mol Cell Biol. 2002;3:267–77. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 186.Kim Y, Lampert SM, Philpott CC. EMBO J. 2005;24:952–62. doi: 10.1038/sj.emboj.7600579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rey MA, Duffy SP, Brown JK, Kennedy JA, Dick JE, Dror Y, Tailor CS. Haematologica. 2008;93:1617–26. doi: 10.3324/haematol.13359. [DOI] [PubMed] [Google Scholar]
- 188.Quigley JG, Gazda H, Yang Z, Ball S, Sieff CA, Abkowitz JL. Blood Cells Mol Dis. 2005;35:189–92. doi: 10.1016/j.bcmd.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 189.Rajadhyaksha AM, Elemento O, Puffenberger EG, Schierberl KC, Xiang JZ, Putorti ML, Berciano J, Poulin C, Brais B, Michaelides M, Weleber RG, Higgins JJ. Am J Hum Genet. 2010;87:643–54. doi: 10.1016/j.ajhg.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hou JC, Pessin JE. Curr Opin Cell Biol. 2007;19:466–73. doi: 10.1016/j.ceb.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Muretta JM, Mastick CC. Vitam Horm. 2009;80:245–86. doi: 10.1016/S0083-6729(08)00610-9. [DOI] [PubMed] [Google Scholar]
- 192.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Science. 1997;275:73–7. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 193.Jemth P, Gianni S. Biochemistry. 2007;46:8701–8. doi: 10.1021/bi7008618. [DOI] [PubMed] [Google Scholar]
- 194.Khan AA, Jeong J, Walker MJ, Chishti AH, Quigley JG. Blood. 2009;114:779a. [ http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/779]
- 195.Letoffe S, Debarbieux L, Izadi N, Delepelaire P, Wandersman C. Mol Microbiol. 2003;50:77–88. doi: 10.1046/j.1365-2958.2003.03686.x. [DOI] [PubMed] [Google Scholar]
- 196.Deniau C, Gilli R, Izadi-Pruneyre N, Letoffe S, Delepierre M, Wandersman C, Briand C, Lecroisey A. Biochemistry. 2003;42:10627–33. doi: 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- 197.Frawley ER, Kranz RG. Proc Natl Acad Sci U S A. 2009;106:10201–6. doi: 10.1073/pnas.0903132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJ. Nucleic Acids Res. 2008;36:D245–9. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hrkal Z, Vodrazka Z, Kalousek I. Eur J Biochem. 1974;43:73–8. doi: 10.1111/j.1432-1033.1974.tb03386.x. [DOI] [PubMed] [Google Scholar]
- 200.Hargrove MS, Barrick D, Olson JS. Biochemistry. 1996;35:11293–9. doi: 10.1021/bi960371l. [DOI] [PubMed] [Google Scholar]
- 201.Delanghe JR, Langlois MR. Clin Chim Acta. 2001;312:13–23. doi: 10.1016/s0009-8981(01)00586-1. [DOI] [PubMed] [Google Scholar]
- 202.Fletcher J, Huehns ER. Nature. 1968;218:1211–4. doi: 10.1038/2181211a0. [DOI] [PubMed] [Google Scholar]
- 203.Holden SA, Steinberg HN, Matzinger EA, Monette FC. Exp Hematol. 1983;11:953–60. [PubMed] [Google Scholar]
- 204.Monette FC, Holden SA. Blood. 1982;60:527–30. [PubMed] [Google Scholar]
- 205.Monette FC, Holden SA, Sheehy MJ, Matzinger EA. Exp Hematol. 1984;12:782–7. [PubMed] [Google Scholar]
- 206.Ibrahim NG, Lutton JD, Levere RD. Br J Haematol. 1982;50:17–28. doi: 10.1111/j.1365-2141.1982.tb01886.x. [DOI] [PubMed] [Google Scholar]
- 207.Fujita H, Sassa S. Br J Haematol. 1989;73:557–60. doi: 10.1111/j.1365-2141.1989.tb00297.x. [DOI] [PubMed] [Google Scholar]
- 208.Chiabrando D, Tolosano E. Adv Hematol 2010. 2010:790632. doi: 10.1155/2010/790632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ito E, Konno Y, Toki T, Terui K. Int J Hematol. 2010;92:413–8. doi: 10.1007/s12185-010-0693-7. [DOI] [PubMed] [Google Scholar]
- 210.Flygare J, Aspesi A, Bailey JC, Miyake K, Caffrey JM, Karlsson S, Ellis SR. Blood. 2007;109:980–6. doi: 10.1182/blood-2006-07-038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, Meerpohl J, Karlsson S, Liu JM, Leblanc T, Paley C, Kang EM, Leder EJ, Atsidaftos E, Shimamura A, Bessler M, Glader B, Lipton JM. Br J Haematol. 2008;142:859–76. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, Aminoff D, Montreuil J. Biochimie. 1998;80:173–95. doi: 10.1016/s0300-9084(98)80024-2. [DOI] [PubMed] [Google Scholar]
- 213.Soe-Lin S, Apte SS, Andriopoulos B, Jr, Andrews MC, Schranzhofer M, Kahawita T, Garcia-Santos D, Ponka P. Proc Natl Acad Sci U S A. 2009;106:5960–5. doi: 10.1073/pnas.0900808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Beaumont C, Canonne-Hergaux F. Transfus Clin Biol. 2005;12:123–30. doi: 10.1016/j.tracli.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 215.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Proc Natl Acad Sci U S A. 2005;102:1324–8. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Moura E, Noordermeer MA, Verhoeven N, Verheul AF, Marx JJ. Blood. 1998;92:2511–9. [PubMed] [Google Scholar]
- 217.Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Antioxid Redox Signal. 2010;12:305–20. doi: 10.1089/ars.2009.2787. [DOI] [PubMed] [Google Scholar]
- 218.Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Blood. 1968;32:811–5. [PubMed] [Google Scholar]
- 219.Tolosano E, Hirsch E, Patrucco E, Camaschella C, Navone R, Silengo L, Altruda F. Blood. 1999;94:3906–14. [PubMed] [Google Scholar]
- 220.Zunszain PA, Ghuman J, Komatsu T, Tsuchida E, Curry S. BMC Struct Biol. 2003;3:6. doi: 10.1186/1472-6807-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sinden RE, Billingsley PF. Trends Parasitol. 2001;17:209–12. doi: 10.1016/s1471-4922(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 222.Lingelbach K, Kirk K, Rogerson S, Langhorne J, Carucci DJ, Waters A. Mol Microbiol. 2004;54:575–87. doi: 10.1111/j.1365-2958.2004.04362.x. [DOI] [PubMed] [Google Scholar]
- 223.Sinden RE. Cell Microbiol. 2002;4:713–24. doi: 10.1046/j.1462-5822.2002.00229.x. [DOI] [PubMed] [Google Scholar]
- 224.Jeong J, Khan AA, Lamm M, Kanzok SM, Quigley JG. Blood. 2010;116:2a. [ http://ash.confex.com/ash/2010/webprogram/Paper30786.html]
- 225.Sinden RE, Alavi Y, Raine JD. Insect Biochem Mol Biol. 2004;34:625–9. doi: 10.1016/j.ibmb.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 226.Molina-Cruz A, DeJong RJ, Charles B, Gupta L, Kumar S, Jaramillo-Gutierrez G, Barillas-Mury C. J Biol Chem. 2008;283:3217–23. doi: 10.1074/jbc.M705873200. [DOI] [PubMed] [Google Scholar]
- 227.Vega-Rodriguez J, Franke-Fayard B, Dinglasan RR, Janse CJ, Pastrana-Mena R, Waters AP, Coppens I, Rodriguez-Orengo JF, Jacobs-Lorena M, Serrano AE. PLoS Pathog. 2009;5:e1000302. doi: 10.1371/journal.ppat.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Leimanis ML, Georges E. Biochem Biophys Res Commun. 2007;354:345–50. doi: 10.1016/j.bbrc.2006.12.219. [DOI] [PubMed] [Google Scholar]
- 229.Krishnamurthy P, Schuetz JD. Annu Rev Pharmacol Toxicol. 2006;46:381–410. doi: 10.1146/annurev.pharmtox.46.120604.141238. [DOI] [PubMed] [Google Scholar]
- 230.Krishnamurthy P, Schuetz JD. Biometals. 2005;18:349–58. doi: 10.1007/s10534-005-3709-7. [DOI] [PubMed] [Google Scholar]
- 231.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. Nat Med. 2001;7:1028–34. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 232.Zhou S, Zong Y, Ney PA, Nair G, Stewart CF, Sorrentino BP. Blood. 2005;105:2571–6. doi: 10.1182/blood-2004-04-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jonker JW, Buitelaar M, Wagenaar E, Van Der Valk MA, Scheffer GL, Scheper RJ, Plosch T, Kuipers F, Elferink RP, Rosing H, Beijnen JH, Schinkel AH. Proc Natl Acad Sci U S A. 2002;99:15649–54. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Proc Natl Acad Sci U S A. 2002;99:12339–44. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Jonker JW, Musters S, Vlaming ML, Plosch T, Gooijert KE, Hillebrand MJ, Rosing H, Beijnen JH, Verkade HJ, Schinkel AH. Am J Physiol Cell Physiol. 2007;292:C2204–12. doi: 10.1152/ajpcell.00359.2006. [DOI] [PubMed] [Google Scholar]
- 236.Desuzinges-Mandon E, Arnaud O, Martinez L, Huche F, Di Pietro A, Falson P. J Biol Chem. 2010 [Google Scholar]
- 237.Lucas ML, Seidel NE, Porada CD, Quigley JG, Anderson SM, Malech HL, Abkowitz JL, Zanjani ED, Bodine DM. Blood. 2005;106:51–8. doi: 10.1182/blood-2004-11-4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M. Proc Natl Acad Sci U S A. 2009;106:10338–42. doi: 10.1073/pnas.0901249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhao R, Unal ES, Shin DS, Goldman ID. Biochemistry. 2010;49:2925–31. doi: 10.1021/bi9021439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rouault TA. Cell. 2005;122:649–51. doi: 10.1016/j.cell.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 241.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Cell. 2006;127:917–28. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 242.Wolf G. Nutr Rev. 2007;65:554–7. doi: 10.1301/nr.2007.dec.554-557. [DOI] [PubMed] [Google Scholar]
- 243.Wang X, Leiendecker-Foster C, Acton RT, Barton JC, McLaren CE, McLaren GD, Gordeuk VR, Eckfeldt JH. Blood Cells Mol Dis. 2009;42:150–4. doi: 10.1016/j.bcmd.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Laftah AH, Latunde-Dada GO, Fakih S, Hider RC, Simpson RJ, McKie AT. Br J Nutr. 2009;101:1150–6. doi: 10.1017/S0007114508066762. [DOI] [PubMed] [Google Scholar]
- 245.Yuasa H, Inoue K, Hayashi Y. J Pharm Sci. 2009;98:1608–16. doi: 10.1002/jps.21515. [DOI] [PubMed] [Google Scholar]
- 246.Schaer CA, Vallelian F, Imhof A, Schoedon G, Schaer DJ. J Leukoc Biol. 2008;83:325–33. doi: 10.1189/jlb.0407226. [DOI] [PubMed] [Google Scholar]
- 247.Rao AU, Carta LK, Lesuisse E, Hamza I. Proc Natl Acad Sci U S A. 2005;102:4270–5. doi: 10.1073/pnas.0500877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Severance S, Rajagopal A, Rao AU, Cerqueira GC, Mitreva M, El-Sayed NM, Krause M, Hamza I. PLoS Genet. 2010;6:e1001044. doi: 10.1371/journal.pgen.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Jakubowski H, Perla-Kajan J, Finnell RH, Cabrera RM, Wang H, Gupta S, Kruger WD, Kraus JP, Shih DM. FASEB J. 2009;23:1721–7. doi: 10.1096/fj.08-127548. [DOI] [PMC free article] [PubMed] [Google Scholar]