

Abstract
New functional monomers bearing a methacrylate, a bisphosphonate function and, for most, an internal carboxylate group, were prepared for incorporation into copolymers with adhesive or anticorrosive properties. Methanolysis of some trimethylsilyl bisphosphonate esters not only deprotects the desired bisphosphonate function but also regioselectively cleaves the alkyl ester function without affecting the methacrylate ester.
Keywords: bifunctional monomer, phosphonic acid, regioselective ester cleavage
Introduction
The potential applications for polymer products containing phosphorus are numerous; dental adhesives, ion-exchange resins and adhesion promotors are just three of the more common applications [1–7]. Compounds containing phosphorous are excellent promotors with respect to adhesion, and thus anti-corrosion. Commercial anti-corrosion polymer compounds are generally formed from Sipomer® or Phosmer® monomers, which are phosphate-type (meth)acrylates, and can be readily polymerized by emulsion or solution methods [8–9]. Polymers with some phosphonate functionality have long been established as excellent adhesives and anti-corrosion compounds [10–17], however, there has been very little investigation into the use of phosphonate-type methacrylates for the same purpose [8–9]. In the domain of polymer-based materials exhibiting specific properties, bifunctional monomers bearing a methacrylate function and a bisphosphonate function are recognized as useful building blocks for dental materials [11–12,18–19]. Such materials require a high hydrolytical stability that originates in the hydrolytical stability of the monomers. With this requirement in mind, we have investigated the synthesis of bisphosphonates and their deprotection to the corresponding acids.
Results and Discussion
Synthesis of bisphosphonate methacrylate monomers
We have designed new bifunctional monomers 1a–7a bearing a methacrylate and an amino(bismethylene)bisphosphonate (Scheme 1) linked by an aliphatic or an aromatic spacer [20–21].
Scheme 1.

Novel bisphosphonate methacrylate monomers.
To the best of our knowledge, only a single acrylate containing monomer 8 has been previously synthesized and tested, after copolymerization and incorporation, in a desensitizing solution for lithography [22]. More recently we investigated a similar compound 1a for its adhesive or anticorrosive or flame-retardant properties [20–21]. The synthesis of bisphosphonate monomers 1c–7c is described in Scheme 2.
Scheme 2.

Synthesis of novel bisphosphonate methacrylate monomers 1c–7c, by use of the following reagents and conditions: (a) paraformaldehyde, (MeO)2P-OH, THF, reflux, 96%; (b) methacryloyl chloride, NEt3, CHCl3, 68%; (c) paraformaldehyde, (MeO)2P-OH, THF, reflux (n = 5, 92%; n = 10, 95%); (d) BH3-THF, CH2Cl2 (n = 5, 87%; n = 10, 85%); (e) methacryloyl chloride, NEt3, CHCl3 (n = 5, 75%; n = 10, 77%; (f) HEMA (22), DCCI, DMAP, CHCl3 (n = 5, 74%; n = 10, 73%); (g) paraformaldehyde, (MeO)2P-OH, THF, reflux, 92%; (h) B2H6; (i) methacryloyl chloride, NEt3, CHCl3, 62%; (j) HEMA (22), DCCI, DMAP, CHCl3, 65%.
Thus bisphosphonate 1c was simply obtained from 2-aminoethanol (9) by a two-step process involving first Kabachnik–Fields conditions [23–24] to introduce the bisphosphonate moiety followed by esterification of compound 10 with methacryloyl chloride. The synthesis of the next aliphatic target molecules 2c–4c and 3c–5c started from 6-aminohexanoic acid (11) and 11-aminoundecanoic acid (12), respectively. The three component coupling of 11, respectively 12, with paraformaldehyde and dimethyl phosphite furnished bisphosphonates 13 and 14 in excellent yields. These latter compounds were then reduced regioselectively by diborane [25] to the corresponding alcohols 15 and 16, respectively. Their subsequent esterification in the presence of methacryloyl chloride gave the target molecules 2c and 3c. Alternatively, compounds 13 and 14 were esterified with (hydroxyethyl)methacrylate (HEMA, 22) to give the monomers 4c and 5c. The two aromatic targets 6c and 7c were prepared from p-(aminomethyl)benzoic acid (17) which was converted into the bisphosphonate 18 in 92% yield under Kabachnik–Fields conditions. This common intermediate 18 was either reduced by diborane to the alcohol 19 followed by esterification by methacryloyl chloride giving access to compound 6c, or esterified directly with HEMA (22) to furnish the bisphosphonate 7c.
Study of bisphosphonate methacrylate monomer deprotection
As already mentioned in the introduction, the resulting polymers from bisphosphonate methacrylate monomers can be involved in many applications such as dental adhesives, ion-exchange resins and adhesion promotors. However, these polymers must be in the acidic form, i.e., with phosphonic acid groups, to function efficiently [26].
The intermediate bisphosphonates were then subjected to a two-step deprotection process to restore the phosphonic acids by using first trimethylsilyl bromide followed by a methanolysis step [27]. The first step is known to transform alkyl phosphonates into the corresponding trimethylsilyl phosphonates which are then cleaved to the phosphonic acids under hydrolytic conditions [28]. Phosphonates 1c–7c were treated with trimethylsilyl bromide for 16 h at room temperature to give the trimethylsilyl esters 1b–7b which were isolated in quantitative yields (Scheme 3).
Scheme 3.

Schematic procedure for phosphonate methacrylate monomer deprotection.
1H NMR analysis of compounds 1b–7b showed the absence of deprotected products 1a–7a which could have arisen from possible traces of residual HBr. The next challenge was to cleave the trimethylsilyl phosphonates selectively, without affecting alkyl carboxylates, under controlled conditions of both temperature and solvent [23] since alkyl esters including acrylate or methacrylate esters are sensitive to hydrolytic conditions [29]. According to McKenna's recent results, the use of methanol instead of water should achieve the selective deprotection of these trimethylsilyl phosphonates [30]. Our results are summarized in Table 1.
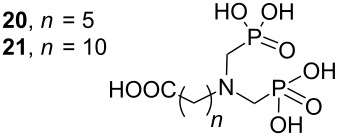
Table 1.
Deprotection of phosphonates 1b–7b obtained with the following conditions: 1) TMSBr, CHCl3, RT, 16 h; 2) MeOH, RT, 2 h.
 | |||
| Entry | Reactant | Product | Yield (%)a |
| 1 | 1b | 1a | 95 |
| 2 | 2b | 2a | 97 |
| 3 | 3b | 3a | 94 |
| 4 | 4b | 4a/9/24 | 97 (30/35/35)b |
| 5 | 5b | 5a/9/25 | 98 (30/35/35)b |
| 6 | 6b | 6a | 98 |
| 7 | 7b | 7a | 99 |
aCrude yield after solvent evaporation. bRelative proportions as determined by 1H NMR.
The crude silyl esters 1b–7b were dissolved in methanol and stirred for 2 h at ambient temperature. Concentration of the reaction mixture furnished phosphonic acids 1a–3a and 6a, 7a in good yields (Table 1, entries 1 and 2) while methanolysis of compounds 4b and 5b resulted in a mixture of the desired phosphonic acids 4a and 5a, HEMA (22) and the phosphonic acids 20 and 21, respectively (entries 4 and 5). In both the latter cases careful 1H NMR examination of the reaction mixture from the methanolysis step revealed that the phosphonic acid 4a, respectively 5a, was the sole compound until evaporation of the solvent. These results showed that the internal carboxylic ester was only cleaved after concentration of the reaction mixture probably due to the higher acidity of the medium and are in agreement with the previously described deprotections using water as solvent [31].
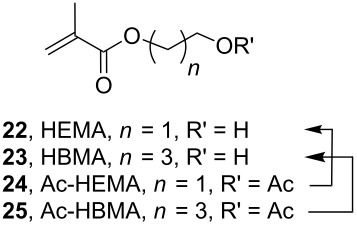
We prepared two model compounds 24 and 25 derived from acetylation of HEMA (22) and (hydroxybutyl)methacrylic acid (HBMA, 23), respectively to study the deprotection of these esters in the methanol under increasing concentrations of hydroxymethylphosphonic acid as a model phosphonic acid. Our results are summarized in Table 2.
Table 2.
Deprotection of esters 24 and 25a.
 | |||
| Entry | Acid amountb | Yield of 9c | Yield of 10c |
| 1 | 2 | 30 | 20 |
| 2 | 4 | 50 | 40 |
| 3 | 6 | 62 | 54 |
| 4 | 8 | 70 | 65 |
| 5 | 10 | 75 | 70 |
aStarting esters were stirred in MeOH at RT for 4 h before concentration of the solvent. bMolar percentage of hydroxymethylphosphonic acid. cIsolated yield after evaporation and chromatographic purification.
We observed that: i) Both ester functions were stable until evaporation of the mixture, ii) the sole acetyl group was cleaved by concentrating the reaction mixture leading to increasing proportions of HEMA (22), respectively HBMA (23) as the molar percentage of hydroxymethylphosphonic acid increases. These last results are in agreement with the observed cleavage of compounds 4a and 5a and show the weak influence of the chain length between the methacrylate and acetate groups. It is worth mentioning that the phosphonic acid 7a is stable in the two-step deprotection process (Table 1, entry 7) emphasizing the greater stability of conjugated carboxylic esters over unconjugated ones.
Other examples of similar phosphonate deprotection by TMSBr involved the presence of a tertiary amine but the authors did not mention any cleavage of the carboxylic ester to prove the role of the base used during the selective deprotection of the phosphonic ester into its acid [32]. We finally deprotected the trimethylsilyl phosphonates 4b and 5b with methanol in the presence of aqueous ammonia (reaction time 1 h) to obtain the target phosphonic acids 4a and 5a in quantitative yields as their ammonium salts.
Conclusion
In conclusion we were able to prepare new bifunctional monomers bearing a methacrylate function and a bisphosphonic acid function. We confirmed that the unconjugated alkyl ester function involved in these monomers was cleaved selectively in the presence of conjugated esters by the released phosphonic acid. The use of methanol instead of water during this final deprotection step was essential to preserve the more stable methacrylate and benzoate esters.
References
- 1.Quittmann U, Lecamp L, El Khatib W, Youssef B, Bunel C. Macromol Chem Phys. 2001;202:628–635. doi: 10.1002/1521-3935(20010301)202:5<628::AID-MACP628>3.0.CO;2-T. [DOI] [Google Scholar]
- 2.Hwang K-Y, Chen H-H, Tu A-P, inventors. Phosphorus-containing resins and fire-resistant epoxy resin compositions containing the same. USA 2003073781. 2003 (Chang Chun Plastics Co., Ltd., Taiwan).
- 3.Wan I Y, Keifer L A, McGrath J E, Kashiwagi T. Polym Prepr. 1995;36:491–492. [Google Scholar]
- 4.Zhang Y, Tebby J C, Wheeler J W. Eur Pol J. 1999;35:209–214. doi: 10.1016/S0014-3057(98)00119-0. [DOI] [Google Scholar]
- 5.Horrocks A R, Zhang S. Polymer. 2001;42:8025–8033. doi: 10.1016/S0032-3861(01)00321-4. [DOI] [Google Scholar]
- 6.Fesman G, Lin R Y, Rehder R A, inventors. Flame-retardant mixture for polyurethane materials. EP0138204A1. 1985 Apr 24;
- 7.Ebdon J R, Price D, Hunt B J, Joseph P, Gao F, Milnes G J, Cunliffe L K. Polym Degrad Stab. 2000;69:267–277. doi: 10.1016/S0141-3910(00)00066-5. [DOI] [Google Scholar]
- 8.Zakikhani M, Davis J, inventors. Polymeric adhesive and flame-retardant compositions. EP0765889A1. 1997 Apr 2;
- 9.Okamoto T, Mori H, Matsuda H, inventors. Adhesive compositions. US 4,433,124. 1984 Feb 21;
- 10.Herbst W, Ludwig H, Roehlitz F, Vilcsek H, inventors. Method and material for the application of adhering coatings on iron and steel surfaces. DE1187100B. 1965 Feb 11;
- 11.Moszner N, Zeuner F, Fischer U K, Rheinberger V. Macromol Chem Phys. 1999;200:1062–1067. doi: 10.1002/(SICI)1521-3935(19990501)200:5<1062::AID-MACP1062>3.0.CO;2-#. [DOI] [Google Scholar]
- 12.Moszner N, Salz U, Zimmermann J. Dent Mater. 2005;21:895–910. doi: 10.1016/j.dental.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Salz U, Zimmermann J, Zeuner F, Moszner N. Polym Prepr. 2004;45:325–326. [Google Scholar]
- 14.Salz U, Zimmermann J, Zeuner F, Moszner N. J Adhes Dent. 2005;7:107–116. doi: 10.3290/j.jad.a10282. [DOI] [PubMed] [Google Scholar]
- 15.Zeuner F, Moszner N, Völkel T, Vogel K, Rheinberger V. Phosphorus, Sulfur Silicon Relat Elem. 1999;144–146:133–136. doi: 10.1080/10426509908546200. [DOI] [Google Scholar]
- 16.Zeuner F, Moszner N, Drache M, Rheinberger V. Phosphorus, Sulfur Silicon Relat Elem. 2002;177:2263. doi: 10.1080/10426500213431. [DOI] [Google Scholar]
- 17.Senhaji O, Robin J J, Achchoubi M, Boutevin B. Macromol Chem Phys. 2004;205:1039–1050. doi: 10.1002/macp.200400011. [DOI] [Google Scholar]
- 18.Adusei G, Deb S, Nicholson J W, Mou L, Singh G. J Appl Polym Sci. 2003;88:565–569. doi: 10.1002/app.11437. [DOI] [Google Scholar]
- 19.Mou L, Singh G, Nicholson J W. Chem Commun. 2000:345–346. doi: 10.1039/A909877A. [DOI] [Google Scholar]
- 20.Chougrani K, Boutevin B, David G, Boutevin G. Eur Pol J. 2008;44:1771–1781. doi: 10.1016/j.eurpolymj.2008.03.009. [DOI] [Google Scholar]
- 21.Chougrani K, Boutevin B, David G, Seabrook S, Loubat C. J Polym Sci, Part A: Polym Chem. 2008;46:7972–7984. doi: 10.1002/pola.23097. [DOI] [Google Scholar]
- 22.Kasai S, Itakura R, Kato E, inventors. Desensitizing solution for lithography. US 5,965,660. 1999 Oct 12;
- 23.Kabachnik M I, Medved T Y. Dokl Akad Nauk SSSR. 1952;83:689–692. [Google Scholar]
- 24.Medved T Y, Kabachnik M I. Dokl Akad Nauk SSSR. 1952;84:717–720. [Google Scholar]
- 25.Loewe R S, Ambroise A, Muthukumaran K, Padmaja K, Lysenko A B, Mathur G, Li Q, Bocian D F, Misra V, Lindsey J S. J Org Chem. 2004;69:1453–1460. doi: 10.1021/jo034946d. [DOI] [PubMed] [Google Scholar]
- 26.El Asri Z, Chougrani K, Negrell-Guirao C, David G, Boutevin B, Loubat C. J Polym Sci, Part A: Polym Chem. 2008;46:4794–4803. doi: 10.1002/pola.22813. [DOI] [Google Scholar]
- 27.McKenna C E, Higa M T, Cheung N H, McKenna M-C. Tetrahedron Lett. 1977;18:155–158. doi: 10.1016/S0040-4039(01)92575-4. [DOI] [Google Scholar]
- 28.Grison C, Coutrot P, Comoy C, Balas L, Joliez S, Lavecchia G, Oliger P, Penverne B, Serre V, Hervé G. Eur J Med Chem. 2004;39:333–344. doi: 10.1016/j.ejmech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Moszner N, Zeuner F, Rheinberger V. Macromol Symp. 2001;175:133–140. doi: 10.1002/1521-3900(200110)175:1<133::AID-MASY133>3.0.CO;2-8. [DOI] [Google Scholar]
- 30.Marma M S, Khawli L A, Harutunian V, Kashemirov B A, McKenna C E. J Fluorine Chem. 2005;126:1467–1475. doi: 10.1016/j.jfluchem.2005.04.002. [DOI] [Google Scholar]
- 31.Harris W R, Brook C E, Spilling C D, Elleppan S, Wang P, Xin M, Van Wyk J. J Inorg Biochem. 2004;98:1824–1836. doi: 10.1016/j.jinorgbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Engel R. Org React. 1988;36:175–248. [Google Scholar]