

Abstract
Preparations of de novo acyclic 2-amido-dienes and 3-amido-trienes through 1,3-hydrogen shifts from allenamides are described. These 1,3-hydrogen shifts could be achieved thermally or they could be promoted by the use of Brønsted acids. Under either condition, these processes are highly regioselective in favour of the α-position, and highly stereoselective in favour of the E-configuration. In addition, 6π-electron electrocyclic ring-closure could be carried out with 3-amido-trienes to afford cyclic 2-amido-dienes, and such electrocyclic ring-closure could be rendered in tandem with the 1,3-hydrogen shift.
Keywords: allenamides; 2-amido-dienes; 3-amido-trienes; electrocyclic ring-closure; 1,3-hydrogen shift; isomerization
Introduction
While allene isomerization to afford conjugated dienes is a well-known and thermodynamically favourable process, it is not trivial kinetically. A concerted allene isomerization leading to a diene involves a 1,3-hydrogen shift, which constitutes a four-electron (2π + 2σ) process that needs an antarafacial approach to fulfil the anti-Hückel (or Möbius) transition state based upon the Woodward–Hoffman rules [1]. Although there is no experimental precedent in an actual allylic system, it is relatively more feasible for an allenic system due to the presence of orthogonally oriented p-orbitals of the sp-hybridized central allenic carbon (Scheme 1), allowing a formal phase change required for an anti-Hückel transition state (in blue, for references on a possible radical pathway, see [2]), and a six-electron (2π + 2σ + 2π) process when considering the possible involvement of the second set of allenic π-electrons. Nevertheless, the calculated ∆Eact value remains high at 77.7 kcal·mol−1 [2]. Whether concerted or not, most thermal isomerizations of allenes require severe reaction conditions (for general reviews on allenes see [3], for some examples of thermal isomerization of exocyclic allenes to dienes via radical intermediates see [4–12]), whereby controlling E/Z ratios of the resulting diene remains a difficult problem. On the other hand, a stepwise isomerization of allenes via acid-, base-, or metal-mediated conditions seem to be more practical, but known examples have issues in controlling stereo- and regioselectivity [3] (for some examples see [13–20]). Therefore, solving these problems can be highly significant.
Scheme 1.

1,3-Hydrogen shifts of allenes.
Because of the popularity of dienes as one of the most utilized organic building blocks, a number of stereoselective preparations are known. The major question here is how viable is it to access conjugated dienes from structurally more challenging allenes through a kinetically difficult and stereochemically undistinguished isomerization. It might not seem like a logical approach; however, our justification is that since there are few well established routes for preparing amido-dienes, our allenamide isomerization strategy (for reviews on allenamide chemistry see [21–23], for reports in 2009, 2010 and 2011 see [24–43], for earlier studies on allenamides see [44–46]) can open the door to construct synthetically useful amido-dienes (for a review on the synthesis of enamides see [47], for reviews on the chemistry of dienamides see [48–50], for reviews on the chemistry of 2-amino or 2-amido-dienes see [51–52]). Problems with the two primary approaches to access amido-dienes [47] are that acid-mediated condensations suffer from functional group tolerances, and metal-mediated coupling methods (for reviews on Cu-mediated C–N and C–O bond formations see [53–55], for some examples see [56–58]) suffer from limited access as well as the instability of halo-dienes (Scheme 2).
Scheme 2.

Synthesizing amido-dienes from allenamides.
In contrast, multi-substituted allenamides can be concisely prepared through α-alkylations of a parent allenamide [47,59] (for the synthesis of parent allenamides see [60]) or amidative cross-couplings of allenyl halides [61–62]. Therefore, our allenamide isomerization strategy has a much greater synthetic potential in constructing amido-dienes. While the chemistry of 1-amido-dienes has been explored in some detail (see Scheme 3 for success in preparing 1-amido-dienes via allenamide isomerizations) [63–64] (for examples see [65–72]), herein, we report details of an efficient entry to synthetically rare 2-amido-dienes [73–77] via a regio- and stereoselective 1,3-hydrogen shift of allenamides.
Scheme 3.

Synthesis of 1-amido-dienes from allenamides.
Results and Discussion
As part of our initial screening efforts, both the thermal and acidic conditions were investigated as shown in Table 1. Allenamide 1 smoothly underwent isomerization via a 1,3-hydrogen shift when heated at 115 °C in CH3CN (sealed tube) to give the desired 2-amido-diene product 2 in 78% isolated yield with a 16:1 E/Z selectivity (Table 1, entry 1). There appears to be some solvent effect on the E/Z selectivity with more polar solvents providing the best ratio (Table 1, entries 2–4). In addition to thermal conditions, we screened several Brønsted acids at room temperature in order to investigate a milder condition. While PTSA resulted in poor E/Z ratio (Table 1, entry 5), a range of Brønsted acids were quite effective in affording the desired 2-amido-diene 2 (Supporting Information File 1 and Supporting Information File 2) with excellent E/Z selectivity [Table 1, entries 6–9].
Table 1.
1,3-Hydrogen shift of allenamides.
 | ||||||
| entry | solvent | acid [10 mol %] | temp [°C] | time [h] | yield [%]a,b | E:Zc |
| 1 | CH3CN | — | 115 | 16 | 91 (78) | 16:1 |
| 2 | THF | — | 115 | 16 | 51 | 9:1 |
| 3 | ClCH2CH2Cl | — | 115 | 16 | 79 | 7:1 |
| 4 | toluene | — | 150 | 16 | 55 | 4:1 |
| 5 | CH2Cl2 | p-toluenesulfonic acid (PTSA) | 25 | 1 | 66 | 2:1 |
| 6 | CH2Cl2 | p-NO2-ArCO2H | 25 | 16 | 81 | 15:1 |
| 7 | CH2Cl2 | PhCO2H | 25 | 16 | 85 (55) | 18:1 |
| 8 | CH2Cl2 | pyridinium p-toluenesulfonate (PPTS) | 25 | 16 | 77 | 15:1 |
| 9 | CH2Cl2 | camphorsulfonic acid (CSA) | 25 | 10 min | 95 (74) | 18:1 |
aNMR yields. bIsolated yields are shown in brackets. cRatios were determined by 1H NMR.
After having established the 1,3-hydrogen shift under thermal and protic conditions, a diverse array of 2-amido-dienes was prepared as summarized in Table 2. Some notable features are: (1) a variety of novel chiral 2-amido-dienes 8–10 were obtained from chiral allenamides 5–7 in synthetically useful yields and with high E/Z ratios (≥95:5) under both thermal or acidic conditions (Table 2, entries 2–12); (2) unsubstituted 2-amido-dienes 8d and 9c could also be prepared in good yields (see R = H in Table 2, entries 7 and 10); (3) even allenamide containing an acyclic carbamate such as 11 underwent an efficient 1,3-hydrogen shift; and (4) the X-ray structure (Supporting Information File 3) of a single crystal of 2-amido-diene 10b was successfully obtained to assign unambiguously the E-configuration (Figure 1).
Table 2.
Synthesis of 2-amido-dienes.
| entry | allenamides | conditions (time)a | amido-dienes | yield [%]b,c | ||
| 1 | 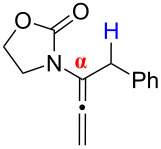 |
3 | 115 °C (16 h) | 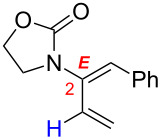 |
4 | 71 |
| 2 | 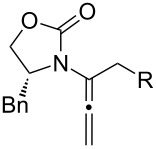 |
5a: R = n-Pr | 115 °C (6 h) | 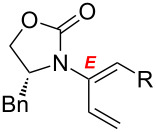 |
8a | 77 |
| 3 | 5a: R = n-Pr | CSA (4 h)d | 8a | 87 | ||
| 4 | 5b: R = Ph | 115 °C (16 h) | 8b | 74 | ||
| 5 | 5b: R = Ph | CSA (2 h) | 8b | 83 | ||
| 6 | 5a: R = 2Nape | 115 °C (16 h)f | 8c | 73 | ||
| 7 | 5a: R = H | 115 °C (16 h) | 8d | 69 | ||
| 8 | 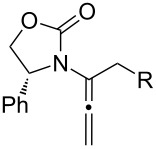 |
6a: R = n-Pr | CSA (10 min) | 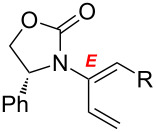 |
9a | 82 |
| 9 | 6b: R = Ph | CSA (10 min) | 9b | 76 | ||
| 10 | 6c: R = H | 115 °C (16 h) | 9c | 69 | ||
| 11 | 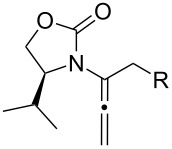 |
7a: R = n-Pr | 115 °C (16 h) | 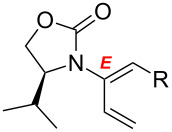 |
10a | 62 |
| 12 | 7b: R = Ph | 115 °C (16 h) | 10b | 82 | ||
| 13 | 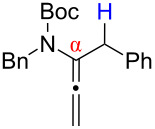 |
11 | 135 °C (16 h) | 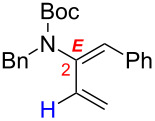 |
12 | 45 |
| 14 | 11 | CSAg (2 h) | 12 | 61 | ||
aUnless otherwise indicated, CH3CN was the solvent for thermal conditions and CH2Cl2 was the solvent when using 10 mol % of CSA at rt. For all reactions, concn = 0.10 M. bAll are isolated yields. cAll 1,3-H shifts were highly E-selective [≥95:5] except for entry 1 in which the E:Z ratio is 6:1 for 4. Ratios were determined by 1H NMR. dTemp started at –78 °C. eThe group 2Nap stands for 2-naphthyl. fClCH2CH2Cl was used. g4Å MS was used.
Figure 1.

X-ray Structure of 10b.
Encouraged by this highly stereoselective isomerization, we turned our attention to the possibility of constructing synthetically much more challenging 3-amido-trienes from allenamides through 1,3-hydrogen shifts. As shown in Table 3, to our satisfaction, a wide variety of 3-amido-trienes could be readily accessed from corresponding α-allylated allenamides. When using a catalytic amount of CSA, both achiral and chiral 3-amido-trienes were obtained in high yields with exclusive E-selectivity, including structurally intriguing examples such as 24–28 (Table 3, entries 9–15). Moreover, a protected alcohol or amine in the allenamide did not impede the isomerization process (Table 3, entries 10–14), leading to more functionalized trienes.
Table 3.
Synthesis of 3-amido-trienes.a
| entry | α-allylated allenamides | 3-amido-trienes | yield [%]b | ||
| 1 | 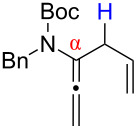 |
13 | 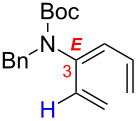 |
14 | 86 |
| 2 |  |
15a | 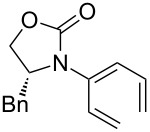 |
22 | 79 |
| 3 | 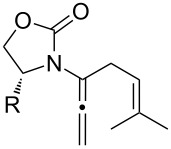 |
16a: R = Bn | 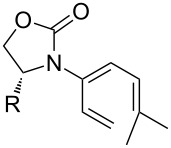 |
23a | 89 |
| 4 | 16b: R = Ph | 23b | 89 | ||
| 5 | 16c: R = iPr | 23c | 91 | ||
| 6 | 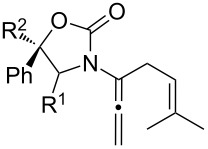 |
16d: R1 = (R)-Me, R2 = H | 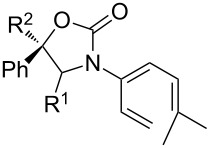 |
23d | 74 |
| 7 | 16e: R1 = (S)-Ph, R2 = H | 23e | 89 | ||
| 8 | 16f: R1 = (R)-Ph, R2 = Ph | 23f | 86 | ||
| 9 | 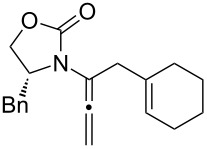 |
17 | 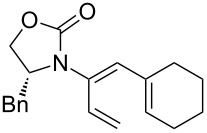 |
24 | 95 |
| 10 | 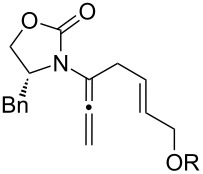 |
18a: R = TBDPS | 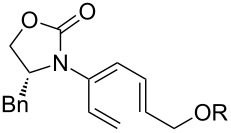 |
25a | 84 |
| 11 | 18b: R = allyl | 25b | 75 | ||
| 12 | 18c: R = cinnamyl | 25c | 54 | ||
| 13 | 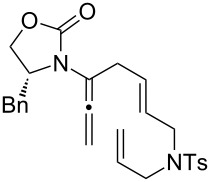 |
19 | 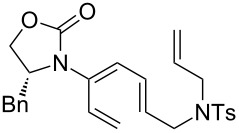 |
26 | 62 |
| 14 | 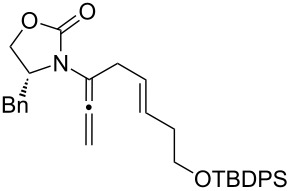 |
20 | 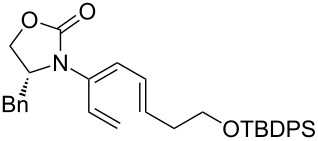 |
27 | 72 |
| 15 | 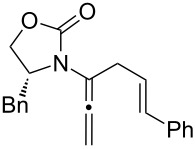 |
21 | 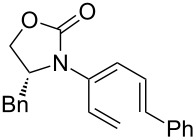 |
28 | 72 |
aAll reactions were run in CH2Cl2 [concn = 0.10 M] with 10 mol % of CSA for 10 min at rt. bAll were isolated yields.
To continue elevating the level of complexity, we examined allenamides with both α- and γ-substitutions and hoped to observe regioselectivity during the 1,3-hydrogen shift. Consequently, as shown in Table 4, isomerizations of tetra-substituted allenamides were examined. When heating α- and γ-substituted allenamides 29a and 30 in CH3CN at 115 °C in a sealed tube, 1,3-hydrogen shift took place exclusively from the α-position affording highly substituted (E)-2-amido-dienes 33a and 34 in 71% and 79% yields, respectively (Table 4, entries 1 and 3). The E-geometry in 33a and 34 was assigned by NOE (Supporting Information File 2).
Table 4.
A regioselective 1,3-hydrogen shift.a
| entry | allenamides | conditions (time) | amido-alkenes | yield [%]b,c | ||
| 1 | 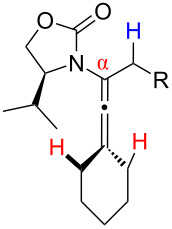 |
29a: R = Ph | 115 °C (16 h) | 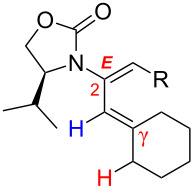 |
33a | 71 |
| 2 | 29b: R = H | —d | 33b | 90 | ||
| 3 | 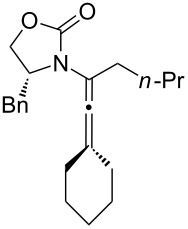 |
30 | 115 °C (16 h) | 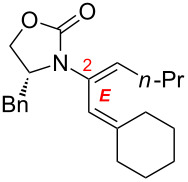 |
34 | 79 |
| 4 | 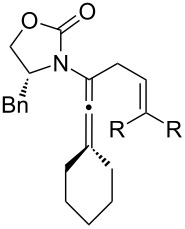 |
31a: R = H | CSA (10 min) |  |
35a | 68 |
| 5 | 31b: R = Me | CSA (10 min) | 35b | 80 | ||
| 6 | 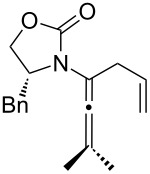 |
32 | CSA (10 min) | 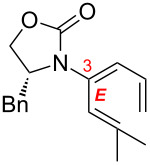 |
36 | 84 |
aUnless otherwise noted, CH3CN was the solvent for thermal conditions and CH2Cl2 was the solvent when using 10 mol % of CSA at rt. For all reactions, concn = 0.10 M. bAll were isolated yields. cAll amido-di- and trienes were exclusively E-selective [≥95:5]. dSee text for this isomerization.
Intriguingly, allenamide 29b underwent a 1,3-hydrogen shift at room temperature when simply in contact with silica gel during the purification stage; but again, only the 1,3-hydrogen shift was favoured proceeding from the α-position to give (E)-2-amido-diene 33b (Table 4, entry 2). In addition, highly substituted 3-amido-trienes 35a, 35b, and 36 were regioselectively synthesized in overall high yields using the CSA-catalyzed conditions (Table 4, entries 4–6). Not only are the products from this regioselective isomerization structurally unique, but also mechanistically intriguing.
One of the probable explanations for the significantly lowered thermal activation barrier of 1,3-hydrogen-shifts of allenamides is that the nitrogen atom can serve to stabilize the biradical intermediate [2,4–12] (for another leading reference on related radical intermediates see [78]) which are presumed to be electron deficient. Based on the model in Figure 2 (left side), stabilization of the biradical intermediate is direct when isomerizations proceed from the α-position, whereas the isomerization from the γ-position is “vinylogous”, or remotely stabilized through the olefin. Therefore, thermal isomerizations at the α-position should be faster than at the γ-position.
Figure 2.

Proposed mechanistic models.
While under thermal conditions, a biradical intermediate is at play [2,4,24], under acidic conditions, the isomerization clearly proceeds through an N-acyl iminium intermediate via protonation of the allenamide (Figure 2, center). Consequently, a similar argument could be used to rationalize the regioselective 1,3-hydrogen shift when acid was used. It is noteworthy that this charged transition state could also be adopted for the thermal isomerization. While still being a neutral transition state, the nitrogen atom could facilitate a polarized transition state through increasing negative charge density at the β-carbon. This action would lead to an N-acyl iminium ion-like character with the migrating hydrogen being proton-like with the α-position being favoured. This polarized transition state should also have a lower thermal activation barrier for the 1,3-hydrogen shift than the neutral one.
Lastly, a non-radical proton-transfer like mechanism could also be at play under conditions using protic solvents or owing to the presence of trace of amount of water (Figure 2, right). These last two models also reveal some insight into the E-selectivity given the pro-E configured transition state (TS) (see the R1 group). Along the same line, if the reaction proceeds through a radical pathway, the observed E-selectivity in the thermal 1,3-hydrogen shift should be favoured because the pro-Z transition state experiences a greater allylic strain compared to the pro-E transition state (Scheme 4). A thermodynamically driven equilibration from (Z)- to (E)-enamide post-isomerization is a real possibility that cannot be ruled out, and the observed solvent effect on the (E/Z)-selectivity would particularly support this possible notion.
Scheme 4.

A favored pro-E TS.
An interesting discovery was made during this work. As shown in Scheme 5, when subjected to CSA catalyzed isomerization for protected allyl alcohol-substituted allenamides, the reactions with 37a and 37b did not stop at the intermediate 38, but an unexpected 1,7-H-shift (for some examples of an antarafacial 1,7-H shift see [79–82]) took place at room temperature to afford 5-amido-trienes 39a and 39b stereoselectively in good yields. Furthermore, when heating the protected homo-allyl alcohol-substituted allenamide 40, after the 1,3-hydrogen shift an unprecedented double 1,7-H-shift through intermediate 41 and 42 took place to afford the 6-amido-triene 43 in 45% yield. It is noteworthy that amido-triene 41 could be isolated in 65% yield when using 10 mol % CSA.
Scheme 5.

Unexpected competing 1,7-hydrogen shifts.
The synthesis of 3-amido-trienes from α-isomerization of allenamides allowed us to explore an important pericyclic process for yet another amido-diene synthesis. As shown in Scheme 6, isomerizations of α-allylated allenamides 15a and 13 under acidic conditions can afford 3-amido-trienes 22 and 14 in excellent yields. Given the E-selectivity of this isomerization, these 3-amido-trienes are perfectly suited for thermal 6π-electron electrocyclic ring-closure (for reviews on pericyclic ring-closures see [83–84], for reviews on ring-closure in natural product synthesis see [85–86], for recent examples of 6π-electron electrocyclic ring-closure see [87–93], for examples on accelerated ring-closures of 1,3,5-hexatrienes see [94–99]) to access cyclic 2-amido-dienes that are quite rare (for examples see [100–102]). Chiral amido-triene 22 underwent electrocyclization efficiently to give chiral cyclic 2-amido-diene 44a in 84% yield. Although obtained in only 35% yield, the achiral cyclic 2-amido-diene 45 could also be prepared.
Scheme 6.

Applications in pericyclic ring-closure.
Finally, this overall process was rendered in tandem under thermal conditions to directly prepare cyclic 2-amido-dienes 44a–c from allenamides 15a–c, respectively, in good yields (Scheme 7). Notably these 6π-electron pericyclic ring-closures took place at relatively low temperature (135 °C), thereby implying an accelerated process of electrocyclization. This feature is consistently observed in related ring-closures of 1,3,5-hexatrienes with an electron-donating substituent at the C3 position of the triene [94–99] (for theoretical studies on substituent effects on electrocyclic ring-closures of 1,3,5-hexatrienes see [97,103–105]. It is also noteworthy that while acyclic 2-amido-dienes and 3-amido-trienes are synthetically challenging to make, cyclic amido-dienes are almost inaccessible synthetically [100–102].
Scheme 7.

Cyclic 2-amido-diene synthesis.
Conclusion
Herein, we have accomplished the preparation of de novo acyclic 2-amido-dienes and 3-amido-trienes through 1,3-hydrogen shifts from allenamides. These 1,3-hydrogen shifts could be achieved under thermal conditions or they could be promoted with Brønsted acids. Under either condition, these processes are highly regioselective in favour of the α-position, and highly stereoselective in favour of the E-configuration. Additionally, 6π-electron electrocyclic ring-closure could be carried out from 3-amido-trienes to afford cyclic 2-amido-dienes, and such electrocyclic ring-closure could be rendered in tandem with the 1,3-hydrogen shift, thereby constituting a facile construction of synthetically rare cyclic 2-amido-dienes.
Supporting Information
Supporting Information features detailed information on synthesis, purification and characterization data of all substances given in this article, proton and selected carbon NMR spectra, and X-ray data of compound 10b.
Experimental section.
Proton and Carbon NMR spectra, and NOE data.
X-Ray structural analysis and information for compound 10b.
Acknowledgments
Authors thank NIH (GM066055) for financial support and Dr. Victor Young (University of Minnesota) for X-ray structural analysis.
This article is part of the Thematic Series "Allene chemistry".
Contributor Information
Ryuji Hayashi, Email: rhayashi@wisc.edu.
John B Feltenberger, Email: jfelten@chem.wisc.edu.
Andrew G Lohse, Email: lohse@wisc.edu.
Mary C Walton, Email: mcwalton@wisc.edu.
Richard P Hsung, Email: rhsung@wisc.edu.
References
- 1.Woodward R B, Hoffmann R. Angew Chem, Int Ed Engl. 1969;8:781. doi: 10.1002/anie.196907811. [DOI] [Google Scholar]
- 2.Jensen F. J Am Chem Soc. 1995;117:7487. doi: 10.1021/ja00133a021. [DOI] [Google Scholar]
- 3.Krause N, Hashmi A S K, editors. Modern allene chemistry. Weinheim, Germany: Wiley-VCH; 2004. [Google Scholar]
- 4.Crandall J K, Paulson D R. J Am Chem Soc. 1966;88:4302. doi: 10.1021/ja00970a063. [DOI] [Google Scholar]
- 5.Paulson D R, Crandall J K, Bunnell C A. J Org Chem. 1970;35:3708. doi: 10.1021/jo00836a027. [DOI] [Google Scholar]
- 6.Bloch R, Perchec P L, Conia J-M. Angew Chem, Int Ed Engl. 1970;9:798. doi: 10.1002/anie.197007981. [DOI] [Google Scholar]
- 7.Jones M, Hendrick M E, Hardie J A. J Org Chem. 1971;36:3061. doi: 10.1021/jo00819a042. [DOI] [Google Scholar]
- 8.Patrick T B, Haynie E C, Probst W J. Tetrahedron Lett. 1971;27:423. doi: 10.1016/S0040-4039(01)96457-3. [DOI] [Google Scholar]
- 9.Lenk W, Hopf H. Tetrahedron Lett. 1982;23:4073. doi: 10.1016/S0040-4039(00)88350-1. [DOI] [Google Scholar]
- 10.Hopf H, Gottschild D, Lenk W. Isr J Chem. 1985;26:79. [Google Scholar]
- 11.Lehrich F, Hopf H. Tetrahedron Lett. 1987;28:2697. doi: 10.1016/S0040-4039(00)96184-7. [DOI] [Google Scholar]
- 12.Meier H, Schmitt M. Tetrahedron Lett. 1989;30:5873. doi: 10.1016/S0040-4039(01)93493-8. [DOI] [Google Scholar]
- 13.Tsuboi S, Masuda T, Takeda A. J Org Chem. 1982;47:4478. doi: 10.1021/jo00144a015. [DOI] [Google Scholar]
- 14.Peng W, Zhu S. Tetrahedron. 2003;59:4641. doi: 10.1016/S0040-4020(03)00663-X. [DOI] [Google Scholar]
- 15.Al-Masum M, Yamamoto Y. J Am Chem Soc. 1998;120:3809. doi: 10.1021/ja974223+. [DOI] [Google Scholar]
- 16.Bibas H, Koch R, Wentrup C. J Org Chem. 1998;63:2619. doi: 10.1021/jo972137m. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs T L, Johnson R N. J Am Chem Soc. 1960;82:6397. doi: 10.1021/ja01509a050. [DOI] [Google Scholar]
- 18.Buzas A K, Istrate F M, Gagosz F. Org Lett. 2007;9:985. doi: 10.1021/ol063031t. [DOI] [PubMed] [Google Scholar]
- 19.Trost B M, Kazmaier U. J Am Chem Soc. 1992;114:7933. doi: 10.1021/ja00046a062. [DOI] [Google Scholar]
- 20.Guo C, Lu X. J Chem Soc, Perkin Trans 1. 1993:1921. doi: 10.1039/P19930001921. [DOI] [Google Scholar]
- 21.Wei L-L, Xiong H, Hsung R P. Acc Chem Res. 2003;36:773. doi: 10.1021/ar030029i. [DOI] [PubMed] [Google Scholar]
- 22.Standen P E, Kimber M C. Curr Opin Drug Discovery Dev. 2010;13:645. [PubMed] [Google Scholar]
- 23.Deagostino A, Prandi C, Tabasso S, Venturello P. Molecules. 2010;15:2667. doi: 10.3390/molecules15042667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Yin G, Hong D, Lu P, Wang Y. Org Lett. 2011;13:1024. doi: 10.1021/ol103074d. [DOI] [PubMed] [Google Scholar]
- 25.Yin G, Zhu Y, Zhang L, Lu P, Wang Y. Org Lett. 2011;13:940. doi: 10.1021/ol102992n. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi R, Walton M C, Hsung R P, Schwab J H, Yu X. Org Lett. 2010;12:5768. doi: 10.1021/ol102693e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lohse A G, Krenske E H, Antoline J E, Houk K N, Hsung R P. Org Lett. 2010;12:5506. doi: 10.1021/ol1023745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beccalli E M, Bernasconi A, Borsini E, Broggini G, Rigamonti M, Zecchi G. J Org Chem. 2010;75:6923. doi: 10.1021/jo101501u. [DOI] [PubMed] [Google Scholar]
- 29.Hill A W, Elsegood M R J, Kimber M C. J Org Chem. 2010;75:5406. doi: 10.1021/jo101035n. [DOI] [PubMed] [Google Scholar]
- 30.Persson A K Å, Bäckvall J-E. Angew Chem, Int Ed. 2010;49:4624. doi: 10.1002/anie.201000726. [DOI] [PubMed] [Google Scholar]
- 31.Krenske E H, Houk K N, Lohse A G, Antoline J E, Hsung R P. Chem Sci. 2010;1:387. doi: 10.1039/c0sc00280a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danowitz A M, Taylor C E, Shrikian T M, Mapp A K. Org Lett. 2010;12:2574. doi: 10.1021/ol1007845. [DOI] [PubMed] [Google Scholar]
- 33.Zbieg J R, McInturff E L, Krische M J. Org Lett. 2010;12:2514. doi: 10.1021/ol1007235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordier P, Aubert C, Malacria M, Gandon V, Lacôte E. Chem–Eur J. 2010;16:9973. doi: 10.1002/chem.201000914. [DOI] [PubMed] [Google Scholar]
- 35.Kimber M C. Org Lett. 2010;12:1128. doi: 10.1021/ol1001494. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto K, Horino Y, Kuroda S. Heterocycles. 2010;80:187. doi: 10.3987/COM-09-S(S)54. [DOI] [Google Scholar]
- 37.Persson A K Å, Johnston E V, Bäckvall J-E. Org Lett. 2009;11:3814. doi: 10.1021/ol901294j. [DOI] [PubMed] [Google Scholar]
- 38.Skucas E, Zbieg J R, Krische M J. J Am Chem Soc. 2009;131:5054. doi: 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong A, Emmerson D P G. Org Lett. 2009;11:1547. doi: 10.1021/ol900146s. [DOI] [PubMed] [Google Scholar]
- 40.Beccalli E M, Broggini G, Clerici F, Galli S, Kammerer C, Rigamonti M, Sottocornola S. Org Lett. 2009;11:1563. doi: 10.1021/ol900171g. [DOI] [PubMed] [Google Scholar]
- 41.Broggini G, Galli S, Rigamonti M, Sottocornola S, Zecchi G. Tetrahedron Lett. 2009;50:1447. doi: 10.1016/j.tetlet.2009.01.074. [DOI] [Google Scholar]
- 42.Lohse A G, Hsung R P. Org Lett. 2009;11:3430. doi: 10.1021/ol901283m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu T, Hayashi R, Hsung R P, DeKorver K A, Lohse A G, Song Z, Tang Y. Org Biomol Chem. 2009;7:3331. doi: 10.1039/b908205k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Overman L E, Clizbe L A, Freerks R L, Marlowe C K. J Am Chem Soc. 1981;103:2807. doi: 10.1021/ja00400a053. [DOI] [Google Scholar]
- 45.Farmer M L, Billups W E, Greenlee R B, Kurtz A N. J Org Chem. 1966;31:2885. doi: 10.1021/jo01347a035. [DOI] [Google Scholar]
- 46.Kinderman S S, van Maarseveen J H, Schoemaker H E, Hiemstra H, Rutjes F P J T. Org Lett. 2001;3:2045. doi: 10.1021/ol016013e. [DOI] [PubMed] [Google Scholar]
- 47.Tracey M R, Hsung R P, Antoline J, Kurtz K C M, Shen L, Slafer B W, Zhang Y. Product Class 4: N-Arylalkanamides, Ynamides, Enamides, Dienamides, and Allenamides. In: Weinreb S M, editor. Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Stuttgart: Thieme; 2005. pp. 387–476. [Google Scholar]
- 48.Overman L E. Acc Chem Res. 1980;13:218. doi: 10.1021/ar50151a005. [DOI] [Google Scholar]
- 49.Petrzilka M, Grayson J I. Synthesis. 1981:753. doi: 10.1055/s-1981-29592. [DOI] [Google Scholar]
- 50.Campbell A L, Lenz G R. Synthesis. 1987:421. doi: 10.1055/s-1987-27972. [DOI] [Google Scholar]
- 51.Krohn K. Angew Chem, Int Ed Engl. 1993;32:1582. doi: 10.1002/anie.199315821. [DOI] [Google Scholar]
- 52.Enders D, Meyer O. Liebigs Ann. 1996:1023. doi: 10.1002/jlac.199619960702. [DOI] [Google Scholar]
- 53.Lindley J. Tetrahedron. 1984;40:1433. doi: 10.1016/S0040-4020(01)91791-0. [DOI] [Google Scholar]
- 54.Ley S V, Thomas A W. Angew Chem, Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 55.Dehli J R, Legros J, Bolm C. Chem Commun. 2005:973. doi: 10.1039/b415954c. [DOI] [PubMed] [Google Scholar]
- 56.Klapper A, Huang X, Buchwald S L. J Am Chem Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 57.Han C, Shen R, Su S, Porco J A., Jr Org Lett. 2004;6:27. doi: 10.1021/ol0360041. [DOI] [PubMed] [Google Scholar]
- 58.Shen R, Lin C T, Bowman E J, Bowman B J, Porco J A., Jr J Am Chem Soc. 2003;125:7889. doi: 10.1021/ja0352350. [DOI] [PubMed] [Google Scholar]
- 59.Xiong H, Hsung R P, Wei L-L, Berry C R, Mulder J A, Stockwell B. Org Lett. 2000;2:2869. doi: 10.1021/ol000181+. [DOI] [PubMed] [Google Scholar]
- 60.Wei L-L, Mulder J A, Xiong H, Zificsak C A, Douglas C J, Hsung R P. Tetrahedron. 2001;57:459. doi: 10.1016/S0040-4020(00)01014-0. [DOI] [Google Scholar]
- 61.Trost B M, Stiles D T. Org Lett. 2005;7:2117. doi: 10.1021/ol050395x. [DOI] [PubMed] [Google Scholar]
- 62.Shen L, Hsung R P, Zhang Y, Antoline J E, Zhang X. Org Lett. 2005;7:3081. doi: 10.1021/ol051094q. [DOI] [PubMed] [Google Scholar]
- 63.Hayashi R, Hsung R P, Feltenberger J B, Lohse A G. Org Lett. 2009;11:2125. doi: 10.1021/ol900647s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayashi R, Feltenberger J B, Hsung R P. Org Lett. 2010;12:1152. doi: 10.1021/ol902821w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Terada A, Murata K. Bull Chem Soc Jpn. 1967;40:1644. doi: 10.1246/bcsj.40.1644. [DOI] [Google Scholar]
- 66.Overman L E, Clizbe L A. J Am Chem Soc. 1976;98:2352. doi: 10.1021/ja00424a068. [DOI] [Google Scholar]
- 67.Oppolzer W, Bieber L, Francotte E. Tetrahedron Lett. 1979;20:4537. doi: 10.1016/S0040-4039(01)86643-0. [DOI] [Google Scholar]
- 68.Smith A B, III, Wexler B A, Tu C-Y, Konopelski J P. J Am Chem Soc. 1985;107:1308. doi: 10.1021/ja00291a034. [DOI] [Google Scholar]
- 69.Schlessinger R H, Pettus T R R, Springer J P, Hoogsteen K. J Org Chem. 1994;59:3246. doi: 10.1021/jo00091a002. [DOI] [Google Scholar]
- 70.Kozmin S A, Rawal V H. J Am Chem Soc. 1997;119:7165. doi: 10.1021/ja971272d. [DOI] [Google Scholar]
- 71.Huang Y, Iwama T, Rawal V H. J Am Chem Soc. 2002;124:5950. doi: 10.1021/ja026088t. [DOI] [PubMed] [Google Scholar]
- 72.Robiette R, Cheboub-Benchaba K, Peeters D, Marchand-Brynaert J. J Org Chem. 2003;68:9809. doi: 10.1021/jo0302049. [DOI] [PubMed] [Google Scholar]
- 73.Ha J D, Kang C H, Belmore K A, Cha J K. J Org Chem. 1998;63:3810. doi: 10.1021/jo980593k. [DOI] [Google Scholar]
- 74.González-Romero C, Bernal P, Jiménez F, Cruz M C, Fuentes-Benites A, Benavides A, Bautista R, Tamariz J. Pure Appl Chem. 2007;79:181. [Google Scholar]
- 75.Movassaghi M, Hunt D K, Tjandra M. J Am Chem Soc. 2006;128:8126. doi: 10.1021/ja0626180. [DOI] [PubMed] [Google Scholar]
- 76.Enders D, Meyer O, Raabe G. Synthesis. 1992:1242. doi: 10.1055/s-1992-26349. [DOI] [Google Scholar]
- 77.Barluenga J, Canteli R-M, Flórez J, García-Granda S, Gutiérrez-Rodríguez A, Martín E. J Am Chem Soc. 1998;120:2514. doi: 10.1021/ja972588o. [DOI] [Google Scholar]
- 78.Siebert M, Osbourn J M, Brummond K M, Tantillo D J. J Am Chem Soc. 2010;132:11952. doi: 10.1021/ja102848z. [DOI] [PubMed] [Google Scholar]
- 79.Kerr D J, Willis A C, Flynn B L. Org Lett. 2004;6:457. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]
- 80.Mousavipour S H, Fernández-Ramos A, Meana-Pañeda R, Martínez-Núñez E, Vázquez S A, Ríos M A. J Phys Chem A. 2007;111:719. doi: 10.1021/jp0665269. [DOI] [PubMed] [Google Scholar]
- 81.Gu Z, Ma S. Chem–Eur J. 2008;14:2453. doi: 10.1002/chem.200701171. [DOI] [PubMed] [Google Scholar]
- 82.Shu X-Z, Ji K-G, Zhao S-C, Zheng Z-J, Chen J, Lu L, Liu X-Y, Liang Y-M. Chem–Eur J. 2008;14:10556. doi: 10.1002/chem.200801591. [DOI] [PubMed] [Google Scholar]
- 83.Marvell E N. Thermal Electrocyclic Reactions. New York: Academic Press; 1980. [Google Scholar]
- 84.Okamura W H, de Lera A R. In: Comprehensive Organic Synthesis. Trost B M, Fleming I, Paquette L A, editors. Vol. 5. New York: Pergamon Press; 1991. pp. 699–750. [DOI] [Google Scholar]
- 85.Pindur U, Schneider G H. Chem Soc Rev. 1994;23:409. doi: 10.1039/cs9942300409. [DOI] [Google Scholar]
- 86.Beaudry C M, Malerich J P, Trauner D. Chem Rev. 2005;105:4757. doi: 10.1021/cr0406110. [DOI] [PubMed] [Google Scholar]
- 87.Bishop L M, Barbarow J E, Bergmen R G, Trauner D. Angew Chem, Int Ed. 2008;47:8100. doi: 10.1002/anie.200803336. [DOI] [PubMed] [Google Scholar]
- 88.Sofiyev V, Navarro G, Trauner D. Org Lett. 2008;10:149. doi: 10.1021/ol702806v. [DOI] [PubMed] [Google Scholar]
- 89.Kan S B J, Anderson E A. Org Lett. 2008;10:2323. doi: 10.1021/ol8007952. [DOI] [PubMed] [Google Scholar]
- 90.Hulot C, Blond G, Suffert J. J Am Chem Soc. 2008;130:5046. doi: 10.1021/ja800691c. [DOI] [PubMed] [Google Scholar]
- 91.Benson C L, West F G. Org Lett. 2007;9:2545. doi: 10.1021/ol070924s. [DOI] [PubMed] [Google Scholar]
- 92.Pouwer R H, Schill H, Williams C M, Bernhardt P V. Eur J Org Chem. 2007:4699. doi: 10.1002/ejoc.200700367. [DOI] [Google Scholar]
- 93.Jung M E, Min S-J. Tetrahedron. 2007;63:3682. doi: 10.1016/j.tet.2007.02.085. [DOI] [Google Scholar]
- 94.Sünnemann H W, Banwell M G, de Meijere A. Eur J Org Chem. 2007:3879. doi: 10.1002/ejoc.200700201. [DOI] [Google Scholar]
- 95.Tessier P E, Nguyen N, Clay M D, Fallis A G. Org Lett. 2005;7:767. doi: 10.1021/ol047602y. [DOI] [PubMed] [Google Scholar]
- 96.Greshock T J, Funk R L. J Am Chem Soc. 2006;128:4946. doi: 10.1021/ja060282o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu T-Q, Fu Y, Liu L, Guo Q-X. J Org Chem. 2006;71:6157. doi: 10.1021/jo060885i. [DOI] [PubMed] [Google Scholar]
- 98.Magomedov N A, Ruggiero P L, Tang Y. J Am Chem Soc. 2004;126:1624. doi: 10.1021/ja0399066. [DOI] [PubMed] [Google Scholar]
- 99.Barluenga J, Merino I, Palacios F. Tetrahedron Lett. 1990;31:6713. doi: 10.1016/S0040-4039(00)97155-7. [DOI] [Google Scholar]
- 100.Martínez R, Jiménez-Vázquez H A, Delgado F, Tamariz J. Tetrahedron. 2003;59:481. doi: 10.1016/S0040-4020(02)01536-3. [DOI] [Google Scholar]
- 101.Wallace D J, Klauber D J, Chen C Y, Volante R P. Org Lett. 2003;5:4749. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]
- 102.Wabnitz T C, Yu J-Q, Spencer J B. Chem–Eur J. 2004;10:484. doi: 10.1002/chem.200305407. [DOI] [PubMed] [Google Scholar]
- 103.Spangler C W, Jondahl T P, Spangler B. J Org Chem. 1973;38:2478. doi: 10.1021/jo00954a013. [DOI] [Google Scholar]
- 104.Guner V A, Houk K N, Davies I A. J Org Chem. 2004;69:8024. doi: 10.1021/jo048540s. [DOI] [PubMed] [Google Scholar]
- 105.Duncan J A, Calkins D E G, Chavarha M. J Am Chem Soc. 2008;130:6740. doi: 10.1021/ja074402j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental section.
Proton and Carbon NMR spectra, and NOE data.
X-Ray structural analysis and information for compound 10b.