

Abstract
This is the first comprehensive HX-MS study of a “robust” 2-Cys peroxiredoxin (Prx), namely Salmonella typhimurium AhpC (StAhpC). Prx proteins control intracellular peroxide levels and are abundant antioxidant proteins in eukaryotes, archaea and bacteria. Crystal structural analyses and structure/activity studies of several bacterial and mammalian 2-Cys Prxs have revealed that the activity of 2-Cys Prxs is regulated by redox-dependent oligmerization and a sensitivity of the active site cysteine residue to overoxidation. The propensity to overoxidation is linked to the conformational flexibility of the peroxidatic active site loop. The HX-MS results emphasize the modulation of the conformational motility of the active site loop by disulfide formation. To obtain information on the conformational impact of decamer formation on the active site loop motility, mutants with Thr77 substituted by Ile, a decamer-disrupting mutation or by Val, a decamer-stabilizing mutation, were studied. For the isoleucine mutant, enhanced mobility was observed for regions encompassing the α4 helix located in the dimer-dimer interface and regions surrounding the peroxidatic loop. In contrast, the T77V mutation resulted in an increase in conformational stability in most regions of the protein except for the active site loop and the region encompassing the resolving cysteine.
Keywords: mass spectrometry, hydrogen exchange, deuterium, conformation, folding, peroxiredoxins
1. Introduction
Peroxiredoxins (Prxs, EC 1.11.1.15) are ubiquitous thiol peroxidases found in archaeal, bacterial and eukaryotic cells. Prxs are highly abundant antioxidant enzymes. In bacteria, removal of H2O2 is predominately achieved by the enzyme alkyl hydroperoxide reductase C22 (AhpC) [1–3]. Because of its peroxide-reducing activities, AhpC helps to protect pathogenic bacteria from the host immune response, and, therefore, AhpC is a possible target for the development of new antibiotics for combating infectious diseases [3]. AhpC belongs to the typical 2-Cys class of Prxs. This means that they contain two redox active Cys residues involved in catalysis and they form an intersubunit disulfide in the oxidized state. Whereas, 1-Cys Prxs only contain one Cys residue which yields a cysteine sulfenic acid (Cys-SOH) upon oxidation [3]. More recently, the dual role of eukaryotic 2-Cys Prxs as antioxidant proteins and key participants in oxidative stress signaling has been recognized [4].
Details of the chemistry and structural changes that accompany catalysis for various Prxs have been recently reviewed [4–6]. All 2-Cys Prxs have a conserved cysteine residue, the peroxidatic cysteine residue (CP or Cys-SPH) to reduce various peroxide substrates (ROOH) to the respective alkyl alcohols (ROH). During this first catalytic step the Cp thiol group will be oxidized to the sulfenic acid (Cys-SPOH) (Figure 1A). All known peroxiredoxins have in close proximity to the active site cysteine CP a conserved arginine residue which likely contributes to the low pKa (between 5 to 6) observed for the peroxidatic thiol by stabilizing its thiolate form. A second free thiol, the resolving SRH, is necessary to complete the catalytic step [4, 5, 7]. In typical 2-Cys Prxs, the resolving cysteine residue resides on the second subunit of the dimer and an intersubunit disulfide bond is formed. The catalytic cycle is completed when the disulfide bond is reduced, which requires a reductase (e.g. AhpF) and reducing equivalents, e.g. NAD(P)H [8].
Figure 1.
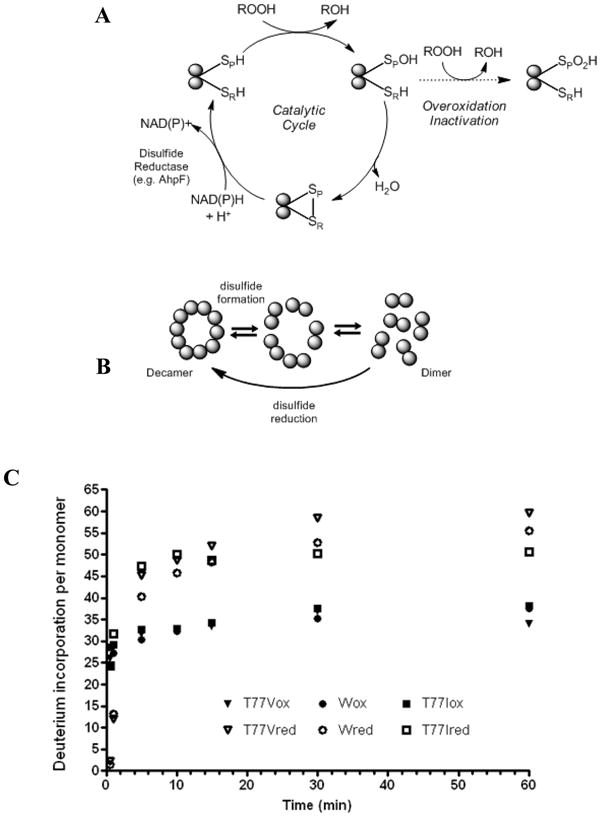
Structure/activity characteristics of StAhpC and HX-MS analysis. (A) Catalytic cycle of the typical 2-Cys peroxiredoxin, StAhpC. Cp, peroxidatic cysteine residue (Cys-46); CR, resolving cysteine residue (Cys-165). (B) Redox-dependent oligomerization of StAhpC. (3) Global deuterium incorporation profiles of wild-type StAphC, and the two single amino acid mutants, T77V and T77I, in their oxidized and reduced forms. Enhanced conformational mobility in StAhpC, T77V and T77I upon reduction of the intersubunit disulfide bridge.
In order to allow for formation of the intersubunit disulfide bond, the active site loop and the C-terminal region encompassing the CR residue must locally unfold [4]. In “sensitive” Prxs, whereby sensitivity refers to the tendency of the protein to become overoxidized, these unfolding events are unfavorable, causing a kinetic arrest which in turn makes the Cys-SP vulnerable to overoxidation by a second molecule of peroxide with formation of a sulfinic acid derivative (Cys-SPO2H) [5]. In contrast, “robust” Prxs, such as Salmonella typhimurium AhpC (StAhpC) and some other bacterial Prxs, seem to be resistant to overoxidation, even at relatively high (mM) concentration of H2O2. In addition, some of the 2-Cys Prxs undergo redox-dependent oligomerization. For example, StAhpC undergoes a decamer to dimer transition upon oxidation (Figure 1B) [1, 5, 9].
Here, we describe conformational studies of StAhpC and two mutants in which Thr-77, located in the decamer-building A-type interface, was replaced by valine, a decamer-stabilizing mutation, and isoleucine, a decamer-disrupting mutation [6]. Hydrogen/deuterium (H/D) exchange monitored by mass spectrometry (HX-MS) was used to study the impact of the intersubunit disulfide linkage on the conformational dynamics of StAhpC,) and to investigate how dimer-dimer interactions affect the conformational properties of the peroxidatic active-site loop in StAhpC.
2. Experimental
2.1. Materials
Deuterium oxide (99.999 atom % D) was purchased from Aldrich Chemical Co. Immobilized pepsin and tris(2-carboxy-ethylphosphine) hydrochloride (TCEP-HCl) were from Thermo/Pierce (Rockford, IL). Biochemical grade potassium phosphate monobasic (KH2PO4) and potassium phosphate dibasic anhydrous (K2HPO4) were obtained from Sigma-Aldrich.
The wild-type protein, and both mutants, T77V and T77I, of StAhpC were prepared and purified in Dr. Poole’s laboratory (Wake Forest University) according to published protocols [10] and stored at −80°C. Reduced proteins were obtained by adding 0.5 μL 0.01 M TCEP-HCl (in 25 mM phosphate buffer, pH 7) to 0.5 μL protein stock solution (10 mg/mL in 25 mM potassium phosphate buffer containing 1mM EDTA, pH 7). Reduction was performed for 30 min at room temperature.
Deuterated phosphate buffer was prepared by lyophilizing potassium phosphate (0.01 M, pH 7) and reconstituting the residue in deuterium oxide. Immobilized pepsin was resuspended in 0.1 M potassium phosphate buffer (pH 2.5) containing 0.01 M TCEP-HCl [11]. The volumetric ratio of pepsin beads to phosphate buffer was 1:2. TCEP-HCl was added during digestion to reduce the disulfide bond in the oxidized proteins which enabled the observation of the peptides encompassing the catalytically active Cys-46 and Cys-165 residue by LC-MS/MS analysis. The immobilized pepsin slurry was stored in ice bath before use.
2.2 HX-MS analysis at the protein level
Protein stock solutions (10 mg/mL) were equilibrated at room temperature for 30 min prior to use. The protein was labeled by diluting 0.5 μl stock solution in 10 μL of 0.01 M deuterated phosphate buffer (pH 7) at various periods of time (30 s, 1, 5, 10, 15, 30, 60 min). The exchange-in reaction was arrested with ice-cold quenching buffer (0.1 M potassium phosphate containing 0.01 M TCEP-HCl, pH 2.5). Quenched samples were flash-frozen in liquid nitrogen until the analysis was performed.
A nanoAcquity UPLC system (Waters, Milford, MA) coupled with a LCT ESI-TOF mass spectrometer (Waters, Milford, MA) was used for LC-MS analysis. Frozen samples were thawed at room temperature, placed in the autosampler and injected. A 6-min time period was needed for thawing the sample and for making the injection. After injection, the samples were trapped on an Acquity UPLC BEH C18 (5 μm) trap immersed in an ice bath (around 0°C) and desalted with 15 μL of 97% of solvent A (acetonitrile/H2O/formic acid, 5/94.9/0.1) and 3% of solvent B (acetonitrile/H2O/formic acid, 94.9/5/0.1) for 1 min. By using a switching valve, the trapped proteins were diverted onto an Acquity UPLC BEH C18 column (1.7 μm particle size, 100 μm × 100 mm) immersed in a ice bath maintained at approximately 0 °C. Proteins were eluted with a gradient from 15% B to 70% B in 3 min and 70% B to 80% B over the next 3 min. The flow rate was 0.47 μL/min. The LC-T time-of-flight (TOF) mass spectrometer was equipped with an electrospray source operated at 3 kV. Mass spectra were acquired over an m/z-range of 400–2000. Cesium iodide was used to calibrate the instrument. Deuterium incorporation at each exchange time point was determined by subtracting the mass of the undeuterated protein from the mass of the protein at each exchange point. The same sets of experiments were performed for the oxidized and reduced forms of wild type protein, StAhpC T77V and StAhpC T77I.
2.3. Peptic proteolysis and mass spectral analysis of deuterium-labeled peptides
The exchange reaction was performed as described above for similar time points. The resulting deuterated samples were quenched and digested with ice-cold digestion buffer and pepsin, 0.1 M potassium phosphate buffer pH 2.5, 0.01M TCEP-HCl) for 1 min. It was thoroughly mixed and then centrifuged to separate the immobilized pepsin from the solution to stop further digestion. GELoader tips were used to pipette the supernatant into the UPLC vials to avoid the immobilized pepsin from entering the vial. Sample vials were flash frozen in liquid nitrogen to avoid the back exchange until the analysis was performed [12, 13]. The peptide solutions were injected into the nanoAcquity UPLC for further analysis as described above. All the parameters and conditions were maintained constant for the UPLC separation of peptides. The resulting peptides were directed into the LC-T ESI mass spectrometer under the similar conditions to those described above except that the mass spectra were acquired over the m/z range of 400–1500. Deuterium levels in peptides were calculated using the Excel-based program HX Express [14]. No correction for back-exchange was performed [15]. Relative deuterium levels (in %) were calculated by dividing the number of deuterons incorporated by the number of backbone amide hydrogens present in each peptide. Three independent experiments were performed for each peptide-level experiment. The same sets of experiments were performed for the oxidized and reduced forms of wild-type (wt) StAhpC, and the T77V and T77I mutants.
2.4. Sequence analysis and assignment of peptic peptides
To obtain sequence assignments for the peptic peptides, an undeuterated peptide sample was analyzed by LC-MS/MS using a Waters/Micromass QTof Ultimate Global mass spectrometer equipped with a lockmass sprayer and coupled to a nanoAcquity UPLC. The chromatographic setup was the same as described above except that for peptide elution the following gradient program was used: 1) 5% B to 35% B in 60 min, 2) 35% B to 85% B by 65 min, and 3) maintain 85% until 72 min. Mass spectra were obtained over a m/z-range of 400–2000. Peptic peptides were identified based on mass measurement and fragment ion information. For the assignment of tandem mass spectral data, Mascot software (Matrix Science) was used. Complete sequence coverage was routinely obtained for the wild-type StAhpC protein and both mutants after disulfide reduction.
3. Results and Discussion
3.1. Global HX-MS analysis of wild-type StAhpC and Thr77 mutant proteins
To study the impact of the intersubunit disulfide bond on the conformational compactness of the typical 2-Cys Prx, StAhpC, we performed a deuterium exchange-in analysis of StAhpC in the absence and presence of the disulfide reducing agent, TCEP. The time course study of the intact protein, having an intersubunit disulfide involving Cys-46 at one subunit and Cys-165 on the other subunit, showed a mass increase from 41,242.6 +/− 0.3 Da to 41,317.6 +/− 0.4 Da, which corresponds to an uptake of approximately 75 deuteriums per disulfide-linked dimer (for the 60-min time point). With other words, this represents a deuterium incorporation level of ~21 % (based on 178 backbone amide hydrogens per monomer). In contrast, reduction of the intersubunit disulfide bonds resulted in an increase of overall deuterium incorporation levels. For the reduced protein, a deuterium exchange level to ~32 % (of 178 backbone NHs) at the 60-min was determined (Figure 1C). Deuterium incorporation profiles for the two Thr77 mutants were also obtained. In accord to the wild-type StAhpC protein, the global HX-MS analyses showed that deuterium incorporation levels for the oxidized mutants were consistently lower than the deuterium levels observed for the reduced mutant proteins (Figure 1C). Removal of the intersubunit disulfide linkage resulted in an approximately 30 % increase in deuterium uptake for the T77V as well as for the T77I mutant compared to the respective disulfide-containing proteins (Figure 1C).
3.2. Exchange-in characteristics of StAhpC at medium spatial resolution derived by combining peptic proteolysis and HX-MS analysis
The H/D exchange-in profiles revealed overall trends in conformational stability for the disulfide-containing proteins and the thiol-containing forms. In order to obtain information on the conformational dynamics with medium spatial resolution, we added to the exchange-in labeling experiments peptic proteolysis under conditions that maintain the deuterium labeling information. Over 70 peptic peptides were identified of which sixteen peptides were chosen that cover the entire protein sequence for all three proteins investigated. In order to allow the comparison of deuterium uptake at the peptide level, the relative percentage of exchange-in was calculated for each peptide by dividing the number of deuterium incorporated in a particular peptide by the number of backbone amide hydrogens present in the peptide.
Figure 2 summarizes the time-dependent H/D exchange-in data for 16 peptides arranged according to their sequence for the intact StAhpC protein. The bar graph presentation allows to roughly group the peptides into three exchange categories (compiled in Supplemental Material Table S1): low-exchanging peptides, that show little deuterium incorporation over the exchange-in period tested; 2) medium-exchanging peptides, i.e. peptides that exhibit a consistent deuterium uptake over the 60-min time frame; and 3) high-exchanging peptides, that rapidly reach their high exchange-in plateaus. Peptides that belong to the “low-exchanging” category are encompassing the following partial sequences, 68–87, 96–110, 111–122, 123–132 and 148–156. Peptides of the “medium-exchanging” group are part of the N-terminal region of StAhpC and include the peptides 1–20, 21–35, 36–43 and the peptides 61–67 and 88–95. Peptides that were grouped into the “high-exchanging” category included peptides 43–50, 51–60, 133–147, 156–176, 177–182 and 181–186.
Figure 2.

Summary of deuterium incorporation data observed for peptic peptides derived from StAhpC. (A) H/D exchange profile; (B) H/D exchange map, and (C) deuterium exchange-in data overlaid on the X-ray structure of StAhpC (1YEP). For (C), the color coding for the deuterium incorporation levels is simplified to 5 categories ranging from blue to red. Blue regions indicate regions with lowest deuterium incorporation and the highest level of protection against exchange-in. Red regions indicate regions exhibiting the highest levels of deuterium incorporation and hence mark regions with the highest exchange rates and lowest level of protection against deuterium exchange-in.
“High-exchanging” regions in StAhpC
It is evident from Figure 2A and Table S1 that the active site loop region, which is covered by peptide 43–50, and the C-terminal region represented by the peptides 156–176, 177–182, and 181–186 of the wild-type StAhpC, have higher deuterium incorporation levels compared to the rest of the protein. These “high-exchanging” peptides showed ~35% deuterium incorporation within the first 30 s and little subsequent increase in deuterium uptake for the remaining time points. High deuterium incorporation levels imply that these regions are less protected (or more flexible) compared to the rest of the protein. The high exchange-in rate and associated high flexibility of the C-terminal region is in concurrence with the absence of this region starting at residue 166 in the crystal structure of StAphC (pdb 1YEP).
Local unfolding of the peroxidatic active site loop of StAhpC
The active site region, peptide 43–50, exhibited the highest exchange-in rate in the oxidized StAhpC protein (Figure 2B). Deuterium uptake reached ~40 % within the first 30 s followed by a slow increase at the 5 and 10-min time points reaching thereafter the exchange-in plateau. The active site loop is locally unfolded in the oxidized StAhpC in order to accommodate the conformational constraints imposed by the disulfide linkage between Cys-46 and Cys-165 [1, 2, 4]. The observed HX-MS data gives testimony of this intrinsic unfolding paradigm for StAphC proteins on which their catalytic function is based.
“Medium-exchanging” regions in StAhpC
For the regions encompassing the peptides 1–20, 36–43, 88–95 a steady increase in the exchange was observed over time. The peptide region of 88–95 exhibited an initial deuterium incorporation of 20% after 30 s and deuterium incorporation levels gradually increased to 35% after 60 min.
Exchange-protected, “low-exchanging” regions in StAhpC
A few regions of StAhpC represented by the peptides 68–87, 111–122, 148–156 were well protected from the exchange and showed very low deuterium incorporation levels (around 10%) and no further increase in deuterium uptake with increase in time. The large peptide 68–87, which represents the dimer-to-dimer interfacial region and encompasses the α4 helix and half of 310 helix, showed only a minimal uptake of < 2 deuteriums (~10 % out of a total of 19 possible backbone amide hydrogen) over the time course tested (Figure 2). The low exchange-in rate observed for this partial sequence indicates a substantial protection against exchange-in due to strong hydrogen-bonding and solvent exclusion occurring at the dimer-to-dimer, A-interface of StAhpC protein oligomers. The slow exchanging properties of the partial sequences 96–110, representing the α5-helix, and 110–122, covering the β1' and β2' strands and part of the β6 strand, agree well with their secondary structural properties and the proximity to the A-interface. Another exchange-in protected region in StAhpC is represented by the peptide 148–156. This peptide encompassed residues of the α6 helix and protection against exchange-in is gained by interactions with helices α2, α3 and α6 of the other subunit of the StAhpC homodimer.
3.3. Effect of disulfide bond reduction on the conformational mobility of the active site loop
The disulfide bond involving Cys-46 and Cys-165 in StAhpC was reduced with TCEP and then the deuterium exchange-in experiments at various time points were carried out the same way as for the intact protein. Figure 3 reports the differences in deuterium incorporation observed as a result of reducing the disulfide bond in StAhpC. Most peptic peptide regions of the reduced StAhpC protein exhibited higher deuterium incorporation levels compared to the disulfide-containing protein. However, certain regions showed higher deuterium uptake in oxidized protein than in the reduced form. This was particularly obvious for the regions covered by the peptides 36–43 and 43–50, encompassing the active site loop helix region, and the peptides 156–176, 177–182 and 181–186, representing the C-terminal region which includes the “resolving” cysteine at position 165. The lower deuterium uptakes observed for these peptides derived from the reduced protein allowed us to conclude that the active site loop region in the reduced protein and part of the C-terminal tail region is more protected against exchange-in in absence of the disulfide bond. The adoption of a“fully folded (FF)” state in absence of the disulfide bond has been repeatedly described in structural studies of typical 2-Cys Prxs [3, 4]. The “fully folded” conformation was first seen in the X-ray structure of the StAhpC C46S mutant, which cannot form a disulfide bond [2]. Formation of the disulfide bond demands local unfolding of the active site region around Cys-46 and parts of the C-terminal region encompassing the resolving Cys-165 residue. The increased conformational flexibility in the “locally unfolded” state is well reflected in the higher deuterium uptake observed for the peptides 36–43, 43–50 and 156–186 derived from the reduced StAhpC protein.
Figure 3.

Comparative HX-MS study of the oxidized and reduced forms of StAhpC. (A) Differential HX-MS profile, WTred-WTox, comparing relative changes in deuterium incorporation levels between the reduced and oxidized protein. Positive bars indicate regions that showed higher deuterium incorporation levels in the reduced form. (B) Deuterium exchange-in data is overlaid on the X-ray structure of StAhpC (1YEP). Red regions show higher deuterium incorporation levels in the reduced form relative to the oxidized form. Blue regions represent regions that show lower deuterium uptake, i.e. are more protected against exchange-in, in the reduced form relative to the oxidized protein.
H/D exchange profiles were also obtained for both mutants, T77I and T77V, in their oxidized and reduced forms (Figure 4 and Supplemental Materials Figures S2 and S3 (oxidized forms) and S5 and S6 (reduced forms)). As observed for the wild-type protein, reduction of the intersubunit disulfide bond resulted in increased conformational mobility in most parts of both mutant proteins except for the partial sequence encompassing the peroxidatic Cys-46 residue and the C-terminal region beyond Cys-165 which is absent in either crystal structure (1YFO, Ile mutant, and 1YF1, Val mutant). Thus, both mutants obey the conceptual model in which the active site loop mobility represents a locally unfolded region necessary for disulfide formation and this region becomes more compact upon disulfide reduction [4].
Figure 4.

Deuterium incorporation data at the peptide level is overlaid on the respective crystal structures of StAhpC and the two mutant T77V and T77I in presence (ox) and absence (red) of the intersubunit disulfide bridge. The same color coding scheme as in Figure 2C is used. Deuterium incorporation levels increase from blue to red. Blue regions show the lowest levels of deuterium incorporation. Red regions exhibit the highest deuterium incorporation levels. The following X-ray structures were used: 1YEP, wt StAhpC; 1YF1, StAhpC T77V; 1YFO, StAhpC T77I.
3.4. Conformational consequences of mutating Thr77, a critical residue in the decamer building, dimer-dimer interface of StAhpC
The structure/activity relationship of 2-Cys Prxs is governed by their redox-dependent partitioning between dimeric and decameric states. Previous mutation studies involving Thr-77, located at the dimer-to-dimer decamer-building interface, showed that decamer-disrupting mutations, e.g. the replacement of Thr-77 with the larger Ile residue, destabilizes the decamer-building interface leading to decamer dissociation and reduced catalytic efficiency. In contrast, mutation of Thr-77 with Val had a stabilizing effect on the dimer-dimer interface and decamer formation and increased catalytic activity [1, 3, 6]. We therefore set out to look closer at the changes in conformational dynamics caused by the mutations of Thr-77 by H/D exchange.
HX-MS analysis of StAhpC T77I
To observe the altered dynamics at various sites in the protein upon mutation of T77, HX-MS experiments were carried out for the T77I mutant. Peptides were identified for the T77I mutant that provided complete sequence coverage. When the peptides were mapped onto the sequence, approximately 54% of the residues in the T77I mutant showed higher deuterium incorporation suggesting that the T77I mutation overall negatively impacted the conformational integrity of the wild-type protein (Figure 5). Differential analysis of the deuterium uptake reveals that the peptides covering the partial sequence 61–87 encompassing parts of α3, β4, α4 and parts of the 310 helix were less protected against exchange-in and consequently destabilized in the T77I mutant (compared to the wild-type protein). In addition, increased deuterium uptake relative to the wild-type protein was observed in the sequence 96–122 which partly forms the interfacial region and covers the α5 helix (peptide 96–110), the short β1' and β2' strands (peptide 111–122) and subsequently leads into the β7 strands. Increased exchange-in levels were also observed for the peroxidative active site loop covered by peptides 21–35, 36–43 and 43–50. Regions that showed less protection against exchange-in in the Ile mutant compared to the wild-type protein are indicated in red in Figure 5B. In contrast, regions that were found to be more protected against exchange-in in the T77I mutant were located in the region 51–60 and the C-terminal partial sequence 156–186 covered by the peptides 156–176, 177–182, 181–186. These regions are indicated in blue in Figure 5B.
Figure 5.

Comparative HX-MS study of StAhpC and the T77I mutant, a decamer disruption mutant. (A) Differential HX-MS profile chart, WT Ox - T77I Ox. In this plot, positive bars indicate regions exhibiting higher deuterium levels in the wild-type protein compared to the isoleucine mutant. Negative bars indicate regions that showed higher deuterium incorporation in the mutant protein relative to the wild-type protein. Regions that show more deuterium increase and thus less protection in the T77I mutant are depicted in red in Figure 5B. Bars indicate average differences in deuterium uptake from three independent measurements. (B) Deuterium exchange-in data is overlaid on the X-ray structure of StAhpC (1YEP). Red regions mark regions that show higher deuterium uptake and hence less protection against exchange-in indicating increased conformational mobility in the T77I mutant compared to the wild-type StAhpC protein. The Thr residue in position 77 is depicted in CPK style.
The replacement of Thr-77 with Ile disturbs and destabilizes not only the decamer-building interface but also allosterically destabilized the loop that encompasses the peroxidatic active site. The observed allosteric changes in the conformational dynamics caused by mutating Thr77 to Ile in AhpC supports our working hypothesis that destabilization of the active site loop associated with increased flexibility will remove conformational restraints allowing facile disulfide formation and confer protection against overoxidation [6]. Earlier reported sedimentation velocity studies showed that the T77I mutant is predominantly present as dimer in solution [1]. The results of the H/D exchange-in studies support well a structural model in which destabilization of the decamer-building interface by the T77I mutation shifts the oligmerization equilibrium towards the dimeric form.
HX-MS analysis of StAhpC T77V
When the deuterium incorporation data of the T77V mutant was compared to the data obtained for wild-type StAhpC (Figure 6), it became apparent that about 75% of the protein backbone of T77V protein exhibited more protection against deuterium exchange-in than the wild-type AhpC protein. Less than 10% of the protein backbone exhibited higher deuterium incorporation in the T77V mutant. For the remaining regions (about 15% of the residues) similar levels of deuterium incorporation were observed for both the wild-type protein and the T77V mutant. Differential analysis of the H/D exchange-in profiles of T77V and wild-type protein revealed that the N-terminal parts of the protein backbone (1–43), regions in proximity to the A-interface (51–67 and 88–110) and a large section encompassing the residues 111 to 176 gained conformational stability upon replacement of Thr-77 by valine. These regions are indicated in blue in Figure 6B. For the interfacial peptides, 68–87 (α4 helix) and 111–122 (β1’-β2’ loop and part of β6), no or little change in exchange-in dynamics was observed. In contrast, the active site loop peptide (43–50) and the C-terminal region (177–182) displayed higher deuterium incorporation in the T77V mutant compared to the wild-type AhpC protein. Regions with reduced levels of protection against exchange-in are indicated in red in the Figure 6B. The exchange-in behavior observed for the T77V mutant allowed concluding that the introduction of the valine residue at the dimer-dimer interface resulted in gain of conformational stabilization in the majority of the protein except for active site loop encompassing Cys-46 and the C-terminal partial sequence.
Figure 6.

Comparative HX-MS study of StAhpC and the T77V mutant, a decamer promoting mutant. HX-MS data demonstrates increased conformational rigidity for the T77V mutant relative to the wild-type protein. (A) Differential HX-MS profile chart, WT Ox - T77V Ox. In this plot, positive bars indicate regions that exhibit higher deuterium incorporation levels in the wild-type protein relative to the valine mutant. Whereas, negative bars indicate regions that showed increased deuterium incorporation in the mutant protein and hence indicate regions that are less protected against exchange-in in the T77V mutant. These regions are indicated in red in Figure 6B. Bars indicate average differences in deuterium uptake from three independent measurements. (B) Deuterium exchange-in data is overlaid on the X-ray structure of StAhpC (1YEP). Red regions mark regions that show elevated deuterium uptake and less protection against exchange-in as a consequence of the T77V mutation relative to the wild-type StAhpC protein. The Thr residue in position 77 is depicted in CPK style.
4. Conclusion
We report the first HX-MS analyses for a “robust” 2-Cys Prxs, namely of the bacterial peroxiredoxin from Salmonella typhimurium, StAhpC. The current studies provide proof of principle that HX-MS is a suitable approach for studying the modulation of the conformational properties of Prxs by redox state and oligomerization. A combination of H/D exchange labeling experiments, peptic proteolysis under conditions that retain the deuterium labeling information and LC-ESI-MS was used to study how the conformational flexibility of the active site loop is governed by the redox state and associated competence of forming decameric assemblies. The reported H/D exchange studies emphasize the adaption of a locally unfolded active site loop in the disulfide-linked dimeric protein. Whereas, increased conformational rigidity in the active site loop region was observed upon reduction of the intersubunit disulfide bond. Thus, our HX-MS results provide support to a structure/activity concept in which the conformational flexibility of the active site loop determines the sensitivity of the peroxidative cysteine to overoxidation [2–4].
In addition, two single amino acid substitution mutants, StAhpC T77V and T77I in their oxidized and reduced forms were studied. Previous ultracentrifugation studies indicated that the T77V is a decamer promoting mutation, whereas T77V is a decamer-disruptive mutation [1]. The HX-MS studies revealed that the Thr77 mutation located at the A-interface impacts not only the dimer-dimer interactions but also the active site loop motility. Compared to the wild-type protein, enhanced motility was observed for the T77V mutation. There is no other technique that will allow delineate the impact of a single mutation on the allosteric interactions within a protein assembly of this size. Future work will focus on defining differences in the conformational properties of “robust” Prxs proteins, such as the bacterial AhpCs, compared to “sensitive” Prxs, such as the mammalian peroxiredoxins, e.g. the human PrxII protein [16].
Research Highlights.
First HX-MS studies of the robust peroxiredoxin, StAhpC, and two mutants in which the Thr-77 was substituted by isoleucine, a decamer-disruptive mutation, or valine, a decamer-promoting mutation.
Global HX-MS studies indicate that disulfide reduction causes a reduction in overall conformational stability.
HX-MS at the peptide level demonstrate enhanced conformational mobility in the peroxidatic active site of loop as a consequence of disulfide formation.
HX-MS studies reveal allosteric interaction between the mutations in the dimer-dimer interface and the active site loop.
Supplementary Material
Acknowledgments
This study was supported by a grant from the National Institutes of Health to L.B.P. with a subcontract to P.A.K. and C.S.M. (R01 GM050389). The OSU/EHSC mass spectrometry facility and services core is supported in part by NIH/NIEHS grant P30 ES000210.
Abbreviations
- AhpC
Alkyl hydroperoxide reductase C-22 (peroxidase component)
- AhpF
alkyl hydroperoxide reductase F component (flavoprotein reductase)
- CP
peroxidatic cysteine (Cys-46 of Salmonella typhimurium AhpC)
- CR
resolving cysteine (Cys-165 of Salmonella typhimurium AhpC)
- StAhpC
Salmonella typhimurium AhpC
- wt
wild type
Footnotes
Supplementary Data: Table S1 summarizes observed exchange behaviors of peptic peptides derived from StAhpC and two single amino acid mutants, T77V and T77I, in their oxidized (ox) and reduced (red) forms. Figures S1 to S6 show deuterium incorporation charts and H/D exchange maps for oxidized and reduced forms of StAhpC, T77I and T77V. This material is available free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 2.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 3.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 4.Hall A, Karplus PA, Poole LB. Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. Febs J. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci U S A. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 8.Jonsson TJ, Ellis HR, Poole LB. Cysteine reactivity and thiol-disulfide interchange pathways in AhpF and AhpC of the bacterial alkyl hydroperoxide reductase system. Biochemistry. 2007;46:5709–5721. doi: 10.1021/bi7001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barranco-Medina S, Lazaro JJ, Dietz KJ. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009;583:1809–1816. doi: 10.1016/j.febslet.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 10.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 11.Yan X, Zhang H, Watson J, Schimerlik MI, Deinzer ML. Hydrogen/deuterium exchange and mass spectrometric analysis of a protein containing multiple disulfide bonds: Solution structure of recombinant macrophage colony stimulating factor-beta (rhM-CSFbeta) Protein Sci. 2002;11:2113–2124. doi: 10.1110/ps.0204402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2009;81:2644–2651. doi: 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barranco-Medina S, Kakorin S, Lazaro JJ, Dietz KJ. Thermodynamics of the dimer-decamer transition of reduced human and plant 2-cys peroxiredoxin. Biochemistry. 2008;47:7196–7204. doi: 10.1021/bi8002956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.