

Abstract
Background
The success of many cell-based therapies is highly dependent on the accurate delivery, dosing and trafficking of the cellular therapeutic. In vivo magnetic resonance (MR) cell tracking provides a means to non-invasively and longitudinally evaluate these parameters for cellular therapy.
Objective
To provide an overview of MR cell tracking and how cellular therapeutics might be improved by utilizing this technology.
Methods
We focused on the technologies utilized for stem cell and immunotherapies in preclinical models of disease.
Results/conclusion
New technologies in MR cell tracking will soon take the field beyond preclinical studies and begin to show benefits in clinical trials of novel experimental cell-based therapies.
Keywords: cell migration, cell tracking, contrast agent, immunotherapy, magnetic resonance imaging, stem cells
1. Introduction
The efficacy of cell-based therapies has, until recently, largely remained a black box. The mechanisms underlying cell migration and the roles the cells themselves play in therapy have not been fully elucidated. An improvement of our knowledge of cell-based therapies will be of benefit to a broad range of fields in medicine. The goal of in vivo cell tracking is to provide insight into the underlying biological processes present in new cell-based therapies. Ideally, in vivo cell tracking will allow us to not only monitor the movement of cells between and within tissues, but also provide us with molecular information depicting cell function and viability within those tissues [1,2]. In this fashion, molecular imaging has expanded the possibilities currently available to evaluate cell-based therapeutics and with the use of different imaging modalities and contrast agents, molecular imaging will continue to push the envelope for non-invasive cellular readouts.
Magnetic resonance (MR) imaging has a long history of application in clinical diagnosis and is rapidly developing as a method for non-invasive imaging of cells during animal development and in laboratory models of disease [3,4]. Several other imaging modalities are available for non-invasive imaging of cell migration including ultrasound (US), optical imaging (OI), computerized tomography (CT), positron emission tomography (PET), and single photon emission computer tomography (SPECT). Limitations of OI-based cell tracking techniques include limited depth of penetration, limited quantification and poor spatial resolution due to photon scatter [5,6]. In comparison, US, CT, PET, SPECT, and MR imaging allow tracking of cell position at any tissue depth at the expense of some detail and sensitivity.
MRI measures the response of the proton that forms the 1H nucleus in water, fat and other biomolecules to applied radio waves in a strong homogeneous magnetic field. Observation of this signal affords MRI a detailed peek into tissues without having to use potentially harmful ionizing radiation, used in imaging modalities such as CT, PET, and SPECT. Besides the detection of regional differences in water abundance in various tissues, MRI can be used to detect local inhomogeneities in proton relaxation produced by paramagnetic or superpara-magnetic particles as well as changes in proton relaxation due to surrounding chemicals and proteins. Sensitivity to such contrast mechanisms allows MRI to respond specifically to contrast-labeled cells, tissues and body fluids. In addition to identifying transplanted cells in their anatomical context, MRI has the unique advantage that it can provide information about the surrounding milieu (i.e., edema, inflammation and lesion size).
In this article, we will discuss the various exogenous contrast agents that are being used for cell labeling and tracking using MRI as well as some of the new reporter genes being used for the same purpose. We will then elaborate upon labeling techniques, cell administration and those MR imaging methods that are being used for in vivo cell tracking, including T2 and T2* weighted images (see next paragraph) as well as chemical exchange saturation transfer (CEST). The limitations of MR imaging for cell tracking will be presented alongside each of the feasible applications in the field of molecular imaging and potential clinical applications.
2. MR contrast agents
Endogenous contrast in MR imaging originates from local variations in tissue water concentration and the chemically bound state of protons. Two relaxation time constants, T1 and T2, are utilized to generate intrinsic tissue contrast. T1 characterizes the relaxation of the nuclear spin to its longitudinal equilibrium following a radio frequency pulse whereas T2 measures the loss of coherency among adjacent nuclear spins. Both of these time constants are affected by the local microenvironment, which may include magnetic inhomogeneities leading to a new time constant T2*. Current contrast agents target one or more of these time constants in an attempt to distinguish transplanted cells from background tissue.
An ideal contrast agent for magnetic resonance imaging would include the following features: biocompatibility, a tolerable toxicity profile, high sensitivity, stable contrast, low background signal with clear enhancement of contrast to noise, it should not dilute with cell division nor transfer to other cells and should allow long term cell tracking [7]. Currently there are no available contrast agents that meet all of these requirements.
2.1 Superparamagnetic agents
2.1.1 Exogenous label
Iron oxide nanoparticles have been used extensively in cell imaging and cell tracking studies due to their strong contrast effect [8–12]. Superparamagnetic iron oxide (SPIO) nanoparticles (50 – 200 nm diameter), ultra small (USPIO) variants (~ 35nm diameter) as well as micron (MPIO) variants act locally to reduce T2 and T2* relaxation via the induction of strong field inhomogeneities. When imaged with a T2 or T2* sensitive pulse sequence, these contrast agents produce a hypointense or negative (black) signal [8]. These iron oxide particles have been coated with varying substrates such as dextran, carboxydextran and polystyrene to ensure stability and solubility in biological media and also to minimize effects on cell function upon cellular uptake.
Currently, only SPIO particles are clinically approved and have been used in the majority of cell tracking studies to date; however, MPIO particles are the most sensitive contrast agent commercially available due to their high iron content. Unlike SPIOs and USPIOs, which are dextran- or carboxydextran-coated, MPIO particles are not biodegradable due to their polystyrene coating and the effects of long-term retention of MPIOs are not know. These concerns have limited the use of MPIOs in their application to animal studies allowing the smaller dextran particles to be used in the bulk of animal studies for cell tracking.
2.1.2 Endogenous label
The labeling of cells with exogenous iron oxides is quite efficient, but is susceptible to a couple of pitfalls. If the cells of interest are not terminally differentiated, they will continue to divide once labeled and will inevitably dilute the contrast agent. Our group has even shown that asymmetric cell division may occur where the contrast agent is not divided evenly amongst daughter cells (Figure 1) [13]. Another major concern with exogenous MR contrast agents is the inability to monitor only the cells which have remained viable after transfer. Once a cell has been labeled with an iron oxide agent, the cell will continue to produce contrast whether it is viable or not. Both of these limitations have led investigators to pursue more reliable methods to track viable and proliferating cells using iron oxides.
Figure 1. High-resolution ex vivo MR imaging of neonate shiverer mouse brain following intracerebroventricular transplantation of superparamagnetic ion oxide-labeled immortalized neural stem cells.

The mismatch between conventional histology and loss of MR detectability following cell proliferation is apparent. (A) Two weeks after grafting, Feridex-labeled C17.2 cells migrated vast distances toward the outer cortical layers of the cerebrum and olfactory bulb as revealed by anti-β-gal staining. This is in sharp contrast with the MR imaging pattern (B), which shows hypointense cells centered in and around the ventricles, but not the cortical layers. The merged histology/MR image (C), in which β-gal+ cells are red, and MRI hypointense cells yellow, further illustrates this mismatch. Scale bar = 1 mm.
Reproduced, with permission, from Walczak et al. [13].
Reporter genes, which involve a gene transfer labeling technique that allows propagation within the cells as they multiply, have been the direction some investigators have turned to in order to address the concerns present with exogenously added iron. Specifically, investigators have turned their attention to iron proteins such as ferritin, transferrin (Tf) and the transferrin receptor (TfR). Each of these proteins plays a role in either transporting iron into the cell or storing iron once inside the cell. Overexpression of the transferrin receptor was first utilized as an MRI reporter gene in 1996 by Koretsky and colleagues in an attempt to increase the amount of iron transported into the cell [14]. Other groups have used overexpression of the ferritin protein in order to sequester iron inside the cell in a manner which would require the cell to upregulate its iron transport naturally [15–17]. Tyrosinase, an enzyme primarily responsible for regulating the synthesis of the protein melanin, has even been used as an MRI reporter gene as melanin has an unusually high affinity and binding capacity for paramagnetic metals such as iron and manganese [18–20]. Most recently, proteins naturally occurring in magnetotactic bacteria have been used to create magnetic iron oxide nanoparticles within cells [21].
Using a metalloprotein-based reporter gene for MRI cell tracking is not without its own limitations. Each of these methods takes a considerable amount of time before a detectable amount of iron has accumulated within each cell [15,16,20]. Additionally, one cannot take iron accumulation as a foregone conclusion since all of these methods rely upon sufficient endogenous iron being present for the accumulation of iron to occur. Due to the body’s tight regulation over free iron stores, there may be insufficient endogenous iron present to create the desired MR contrast.
2.2 Paramagnetic agents
2.2.1 Exogenous label
Paramagnetic metals with unpaired electrons, such as manganese and gadolinium, primarily reduce the T1 relaxation time, consequently producing a hyperintense or positive contrast effect. This brightening of the signal is in stark contrast to superparamagnetic T2 contrast agents, which reduce the signal detected by MRI. However, the paramagnetic metals are naturally toxic to cells and have only been used in their chelated form (gadolinium diethylenetriamine pentaacetic acid (Gd-DPTA) or gadolinium tetraazacyclododecanetertraacetic acid (Gd-DOTA)) and mainly for intravenous use with subsequent rapid renal clearance. Some groups have tried to label cells using Gd-DPTA complexed with albumin protein in order to create a particle with reduced cellular toxicity [22] while others have linked dextran to gadolinium [23] to achieve the same purpose. Unfortunately, since paramagnetic agents interact with protons in exactly the same way as neighboring protons, they are much weaker contrast agents than iron oxides on a molar basis. This interaction is also sensitive to the agents’ access to bulk water protons, as internalized contrast agents will exhibit reduced relaxivity effects in comparison with extracellular agents. In any case, the unknown intracellular biocompatibility coupled with the relatively low T1 contrast effects have led to only a limited number of groups attempting to use Gd as a contrast agent for cellular MRI [24–31].
2.3 Fluorine
2.3.1 Exogenous label
The use of Flourine (19F) as a contrast agent differs greatly from that of iron oxide agents. Iron oxides are imaged indirectly through their influence on the surrounding proton-rich environment whereas 19F is its own distinct element and can be imaged irrespective of the proton image. This distinction, along with the fact that 19F atoms are naturally found within the body in only trace amounts, allows the detection of exogenously added fluorine with the absence of any background signal [32]. Cell labeling with this contrast agent is akin to imaging modalities such as PET, SPECT and bioluminescent imaging in the sense that it is a ‘hot spot’ image which can later be overlain on a familiar anatomical reference such as a simultaneously acquired 1H image [33,34]. In addition, the direct detection of 19F allows the signal to be quantified based upon spectroscopic methods and has the potential to accurately determine the number of trafficking cells [35,36]. Unlike the dextran-coated iron oxides, which become biologically available, most exogenously added fluorine compounds are biologically inert and in many cases eventually exit by exhalation in the form of a gas.
Although this method does remove a great deal of the ambiguity currently present when using iron oxides, as negative contrast can be seen for many reasons other than the presence of the contrast agent, the use of fluorine contrast agents is still limited due to the sensitivity of the imaging technique. The relative sensitivity of 19F is nearly that of 1H but the overwhelming presence of 1H throughout the body makes for a much larger pool to draw signal from. The ‘hot spot’ nature of this technique requires signal to only be obtained from the cells themselves.
2.4 Chemical exchange saturation transfer
2.4.1 Endogenous label
Reporter genes were previously mentioned in the section on iron oxides, and, in some cases, inducible reporter genes were created for these iron oxides. In theory, controlling the induction of protein synthesis would be synonymous with turning a light switch on or off as far as MRI contrast was concerned. In practice, however, the light switch had considerable delay as the accumulation or removal of the protein product is not instantaneous. Our group, as well as others, felt there was a need to be able to turn on or off contrast instantaneously and to achieve this goal turned to chemical exchange saturation transfer (CEST). This technique relies upon chemical shifts generated by macromolecular protons and the exchange of these protons with bulk water protons. The contrast produced using CEST agents can be turned on by applying a saturation radio frequency pulse at the frequency of the macromolecular proton. These saturated protons will then exchange with the bulk water protons creating a reduction in the signal of bulk water. Without this saturation pulse, the image obtained will be void of contrast and the bulk water signal will appear as if the CEST contrast agent is not present.
Our lab described the first case of using a reporter gene to produce CEST contrast in adoptively transferred cells. Gilad and coworkers designed a reporter gene that encoded a lysine rich-protein (LRP) (Figure 2) [37]. The decrease in signal amplitude found during CEST imaging is directly related to how quickly the protons found on the macromolecular contrast agent can exchange with the protons found within the surrounding bulk water. LRP was chosen as an endogenous CEST contrast agent due to the rapid exchange of protons present on the amide groups of poly-L-lysine polypeptides with that of bulk water [38]. In addition, the amide protons provided a significant chemical shift which allowed for targeted saturation of these protons without degrading the bulk water signal directly. This frequency dependence of CEST may prove to be its largest advantage as multiple ‘colors’ could possibly be created by mixing multiple spectrally resolved reporter proteins and saturating each in turn. Our lab has produced spectrally distinct proteins for this very purpose (Figure 3) [39] and a recent study has shown the ability of MRI to produce multiple ‘colors’ using customized spectral shifts created by microfabricated magnetizable elements (Figure 4) [40].
Figure 2. Development of an MR reporter gene using chemical exchange saturation transfer (CEST) imaging.

In vivo anatomical image of mouse brain injected with lysine-rich protein (LRP)-expressing glioma cells in the left hemisphere and control tumor cells in the right hemisphere. (B) CEST signal intensity–difference map overlaid on the anatomical image distinguishes LRP-expressing and control tumor xenografts. Note that, in these initial studies, a proper adjustment of the field homogeneity was only achieved inside the brain, leading to some artifacts at the brain edges.
Reproduced, with permission, from Gilad et al. [37].
Figure 3. Multi-color images of CEST peptides using three different saturation frequencies.

The phantom shown consists of 1 mm tubes inserted into a 5 mm NMR tube, filled with 2.5 mg/ml peptides and buffer as outlined in (A) PBS = phosphate buffered saline; PLR = poly-L-arginine, PLK = poly-L-lysine, PLT = poly-L-threonine. (A) Proton density image. (B) magnetization transfer ratio asymmetry (MTRasym) (± 3.69 p.p.m.) image. (C) MTRasym (± 1.8 p.p.m.) image. (D) MTRasym (± 0.8 p.p.m.) image. (E) Merged image from the three label channels. (F) NH label from the difference between images in (B) and (D) after normalizing the maximum signal in the image. (G) NH2 channel from (C) after normalizing the maximum signal in the image. (H) OH channel from the difference between normalized images (C) and (D).
Reproduced, with permission, from McMahon et al. [39].
Figure 4. Micro-engineered magnetic particles for multi-color MR imaging.

The particle frequency was varied by changing the thickness of electroplated nickel layers that formed the magnetizable disk pairs. (A) As with normal superparamagnetic ion oxide detection, magnetic dephasing due to the particles’ external fields enables the spatial imaging shown in the gradient-echo MRI. However, comparison between (A) and the chemical shift images (B) shows that the additional spectral information both differentiates between particle types and improves particle localization. The particles are shown schematically (not to scale) in (C). With particle spectra [(D), to the right of the corresponding chemical shift images in (B)] shifted well clear of the water proton line, different planes in the chemical shift imaging map isolate different particle types for unambiguous colour-coding with minimal background interference (B, bottom panel).
Reproduced, with permission from Zabow et al. [40].
Although CEST contrast has the ability to produce multiple ‘colors’ for simultaneous labeling of multiple cell types and has the ability to be both turned on or off, it still suffers from a common weakness found throughout many MRI cell tracking applications: sensitivity. A large amount of protein is still currently needed in order to create the CEST contrast, which currently is achieved by injection of large numbers of cells. In addition, the toxicity of each macromolecule must be evaluated for each cellular application.
3. Labeling techniques
3.1 Endogenous label
Cells can be forced to produce foreign proteins or overexpress endogenous proteins by placing the protein encoding genes within the control of the promoter of other genes. This process can be used to enable the tracking of cells expressing particular proteins or if placed following a constitutively active promoter, can be used to track all cells that have received the transgene. In vitro labeling is commonly achieved through transduction with the use of vectors (e.g., retrovirus, lentivirus or adenovirus) that encode the given reporter gene [41].
3.2 Exogenous label
Although there are many cell types which naturally sample their surroundings and are capable of phagocytosing nanoparticle contrast agents, there are still many cells which lack this natural property with regards to their surroundings [42]. For these cells, simple incubation with a contrast agent is not sufficient to effectively label them and they require alternate means for contrast uptake [43]. Early attempts at increasing contrast agent uptake by these naturally non-phagocytic cells varied from lectin coupling, to viral capsid incorporation, and even antibody-mediated uptake [44–47]. Other groups aimed at coating contrast agents with polycations in order to facilitate particle binding to the anionic cell membrane and subsequent engulfment [48,49]. Commercially available transfection agents such as lipo-fectamine, poly-L-lysine, and protamine sulfate have been used for this purpose and have now become widely adapted methods for non-specific intracellular labeling or iron oxides [50–52].
Our lab has introduced a technique termed ‘magneto-electroporation’ (MEP) where electroporation of cells in the presence of Feridex is utilized to increase the natural uptake of iron by nonphagocytic cells [31,53,54]. Electroporation induces reversible electromechanical permeability changes in cell membranes which allows for products to pass through the membrane and into the cytosol. We have shown that MEP is much more efficient than incubation with SPIO alone in labeling non-phagocytic cells, such as stem cells, with iron oxides (Figure 5) [53]. The efficiency is similar to that which can be can be obtained using transfection agents only the results are instantaneous and do not require the use of agents not currently clinically approved as cellular agents. One drawback of this method is that cell death can be significant if the electroporation parameters are not carefully optimized for each cell type.
Figure 5. Superparamagnetic ion oxide (SPIO)-labeling of cells using magnetoelectroporation.

C17.2 mouse neural stem cells (A – D) and rat mesenchymal stem cells (rMSC) (E – G) were incubated with Feridex (2 mg Fe/ml) with (A, C, E, G) and without (B, D, F) electroporation (EP). Only MEP-treated cells show significant Feridex uptake as assessed by DAB-enhanced Prussian blue stain (A) and anti-dextran immunofluorescence (C, E). The higher magnification in (G) demonstrates Feridex-containing clusters with a measured diameter of 830 ± 350 nm.
Reproduced, with permission, from Walczak et al. [53].
Before any in vivo application, variation in uptake behavior must be thoroughly evaluated for the given cell labeling technique as well as the stability of the chosen contrast agent. These two variables will play a large role in the time period one can expect to monitor cell trafficking as well as the accuracy of the images obtained. After any cell labeling procedure where exogenous agents were used, thorough washing of the cells must be completed in order to reduce false signals obtained after delivery of cells. Finally, it should be recognized that for cells that are monitored for several days, it is necessary to verify contrast agent retention and dilution following cell division [13,55,56].
4. Applications
MRI provides a dynamic and non-invasive tool to peer inside the inner-workings of cellular therapies. The ability to provide anatomical as well as functional information is key to illuminating the black box which is the current state of cellular therapy. Whether it is a stem cell therapy or an immunotherapy, MRI is perfectly suited to follow the pertinent cells at high resolutions and continuously.
4.1 Immune cells
In order to generate an effective immune response, immunotherapies require the concerted activity of many components of the immune system. Effector T cells such as CD4+ and CD8+ T cells as well as natural killer cells (NK cells) and dendritic cells (DCs) are all needed to create a sufficient and sustainable immune response. In the case of active immunotherapy, the goal is to generate an immune response after the delivery of antigenic DNA, RNA, peptides or cells bearing antigenic peptides. Conversely, passive immunotherapy utilizes ex vivo expanded T cells, which are then re-introduced into the patient in order to have their therapeutic effect.
In each case, the fate of the effector cells is incredibly important to the outcome of the therapy. Our current histological ‘snap shots’ in time do not adequately reflect the true dynamics of the immune response and in many cases, cells that are isolated from patients do not function in vitro as they did in vivo. This disconnect in cellular function demonstrates the need to develop methods for assessing the location and function of T cells and other immune cells in vivo. Additionally, serial tracking is not possible with histological or ex vivo studies and in order to obtain different time points for analysis, many animals or biopsies are needed. With the advent of in vivo cellular imaging, repeated assessment of specific immune cells has become a possibility.
To date, macrophages and other naturally phagocytic cell types such as Kupffer cells in the liver have been the primary focus of in vivo magnetic resonance imaging. However, these cell types have been studied following intravenous injection of contrast material and are not typically monitored following cellular delivery. Dendritic cells, on the other hand, are the body’s other highly phagocytic cell type and have been used in recent studies involving in vivo cellular MR imaging. Cellular therapies using DCs represent the active arm of immunotherapies and a promising approach for tumor immunotherapies [57]. In order to be effective, however, DCs must migrate throughout the lymphatic system in order to present antigens to T cells located within secondary lymph tissues. This migration represents an intermediate biological endpoint which could be used to improve immunological potency of vaccines. In addition, the route of administration, whether it be intradermal, subcutaneous or intranodal is still a parameter ripe for optimization.
The first study aimed at magnetically labeling DCs for in vivo MRI utilized receptor-mediated endocytosis of antibody-conjugated SPIO particles [58]. Targeting of the DC surface marker CD11c was shown to increase SPIO uptake by as much as 50 times compared with naked SPIO. As it was the first study to label DC’s for in vivo tracking, the authors took time to convincingly show that SPIO uptake did not significantly change cell viability and immunological function. This study provided proof that it was possible to monitor labeled DCs in vivo for several days following an intramuscular injection into murine quadriceps. Baumjohann et al. took this study one step further and attempted to not only monitor the presence of DCs but to actually visualize the migration of SPIO labeled DCs into draining lymph nodes of mice [59]. Mature SPIO labeled DCs were injected subcutaneously into the hind footpads of mice and their accumulation in the draining popliteal lymph nodes was monitored in vivo by mutli-slice multi-echo images obtained at 4.7T. Histological evaluation of the dissected lymph nodes displayed a strong correlation between iron containing DCs and the signal reduction pattern found by MRI. Additionally, when lymph node homing deficient mice, CCR7−/−, were used as the DC source for adoptive transfer, no hypointense signal could be found within T cell areas of the lymph nodes, indicating a DC specific MR signal. This important study provided credence for MRI’s ability to aid in the design of DC vaccines.
The use of MRI for cell tracking of SPIO labeled DCs has even made its way to the clinic for use in monitoring of DC vaccines for immunotherapy [60]. De Vries et al., showed that the added spatial resolution of MRI could be used to evaluate the efficacy of ultrasound-guided DC injections in patients (Figure 6). SPIO-labeled DCs as well as 111 In labeled DCs, which had been matured and loaded with tumor-derived antigens ex vivo, were injected intranodally in eight stage-III melanoma patients. MR imaging was found to be more sensitive than gamma scintigraphic imaging when detecting lymph nodes containing labeled DCs. Additionally, MRI revealed that half the patients (four out of eight) had been mis-injected with the cellular therapy revealing a serious defect in the route of cellular delivery in these patients. In this study and its follow up study [61], the authors showed that MRI is ideally suited to track DC therapeutics in vivo.
Figure 6. In vivo MRI of magnetically labeled dendritic cells used as cancer vaccines in melanoma patients.
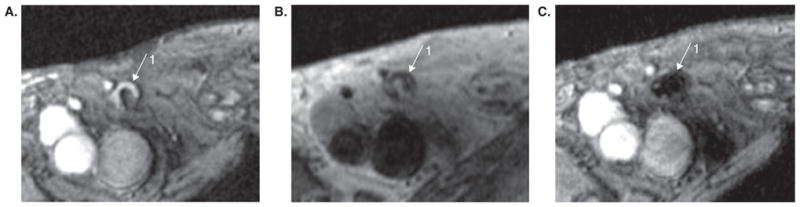
(A) Gradient echo MR image before vaccination showing inguinal lymph node as a hyperintense signal area (1). (B) Spin echo MR image showing same lymph node after vaccination. (C) Gradient echo (technique more sensitive to superparamagnetic ion oxide) MR image after vaccination in same position as (B) showing a marked decrease in signal intensity from lymph node 1.
Reproduced, with permission, from de Vries et al. [60].
Although iron oxides have proven to be quite a successful contrast agent, groups have attempted to achieve similar results for DC monitoring using alternative agents. As mentioned in the contrast section above, fluorine has the potential for being an incredibly useful contrast agent due to its lack of endogenous signal. Groups have taken advantage of this unique characteristic and created fluorinated nanoparticles capable of producing a signal in a 19F image. These particles have been used to track DCs as well as T cells after ex vivo labeling [32,36].
Active immunotherapies in the form of DC vaccines will no doubt play a large role in the future of cancer immunotherapies; however, a current push for cancer therapy has been in the form of passive therapy. The ability of MR to provide detailed serial tracking of T cells in vivo will undoubtedly have a significant effect on our understanding of the in vivo immune response following adoptive transfer of a variety of different T cells. The distribution, persistence, and proliferation of these T cells at target sites could provide critical details pertinent to improving passive immunotherapies.
Tumor antigen-specific T cells, in particular, have been used for adoptive cellular therapies for treatment of a variety of malignancies [62–64]. The cells should specifically home to the sites of tumors in patients and laboratory animals, proliferate, and eventually eradicate the tumor. This ideal response is not often found in clinical situations and the ability to monitor these lymphocytes in vivo with sufficiently high spatial and temporal resolutions may aide in properly evaluating these treatments [65]. There have been attempts to use nuclear and optical imaging in order to follow the distribution of T cells over time in vivo; however, these modalities do not provide the necessary spatial resolution and tissue penetration needed to properly evaluate these cellular therapeutics [66,67].
This is where MRI has stepped in to provide a non-invasive method of tracking T cells with high resolution. Although T cells are not naturally phagocytic, groups have been able to load these cells with SPIO using a variety of transfection techniques which include the use of transfection agents, conjugation with uptake-facilitating molecules, electroporation (magnetoelectroporation) and formation of complexes with poly-L-lysine [47,53,68,69]. HIV-derived Tat peptides have been linked to cross-linked iron oxide (CLIO) particles in order to achieve the most efficient intracellular labeling of these cells [49,70–72]. Of these groups, the work by Kircher et al. has gone the furthest to display the utility of MRI cell tracking in T cell immunotherapies. This group originally developed a novel quantitative high resolution imaging approach to follow the recruitment of antigen-specific CD8+ T cells to target tumors and then followed up this study with another describing the possible mechanisms of tumor immune escape [70,72]. Although these studies have provided a great deal of information about the whereabouts of T cells following adoptive transfer, the problem of contrast agent dilution looms over this field of study. In order for T cell therapies to be effective, the cells need to proliferate to a great extent. With each cell division, there is a dilution of contrast, which will limit our ability to track these cells indefinitely and with a high degree of accuracy.
4.2 Stem cells
Arguably, the most important application for MRI cell tracking may be for monitoring of new stem-cell-based therapies. The pattern of stem cell migration is largely unknown, yet they maintain a significant potential for therapy in many clinical conditions. With a variety of interventional protocols available for stem cell delivery, MRI cell tracking could provide invaluable information as to the optimal delivery method for each specific disease treatment.
Stem cells have been used in cardiovascular disease to treat ischemic injury leading to improved myocardial function, yet the mechanisms remain unknown [7]. Along this same vein, the promise of this therapy has been hindered by limited clinical efficacy in randomized clinical trials [73–75]. Clearly, these therapies have not been optimized so the recent ability to directly label stem cells with MR contrast agents provides a possible mechanism to monitor and improve upon these therapies. The rapid beating of the heart provides a substantial challenge to MR imaging in this context due to the sensitivity of this imaging modality to motion artifacts. As a result, both respiratory and cardiac gating is required to generate good quality images [76]. Despite these technical challenges, groups have attempted to visualize the incorporation of stem cells into cardiac tissue following the (MR-guided) injection of SPIO-labeled mesenchymal stem cells (MSC) [77–80]. These studies have shown that it is possible to use MRI for stem cell tracking in the context of cardiovascular diseases and future clinical trials should benefit greatly from this new ability to precisely inject and track the biological fate of these cells.
The potential of MSC therapy is not restricted to cardiovascular disease as reports have also extensively described their potential to treat neurological diseases. Using ischemic and stroke models of CNS injury, several groups have shown that MSCs may be capable of releasing cytokines and growth factors which aide in the survival of cells at the site of the lesion [81–83]. While the mechanisms responsible for this beneficial affect are largely unknown, it is thought that these factors are released in a paracrine fashion and the cells must migrate to the site of the lesion in order to have their effect. MRI cell tracking studies have shown that MSCs do have the ability to migrate extensively to infarcted areas whether they be on the ipsilateral or contralateral side of the intracranial injection [84].
Neural stem cells have also been shown to migrate and, most importantly, integrate within the CNS; therefore, providing a potential role for treatment of CNS tumors, Parkinson’s disease, and multiple sclerosis (MS) [85–91]. When grown in culture, neural stem cells form neurospheres, which are capable of giving rise to many neural cell types. Our lab has developed methods of labeling these spheres for in vivo MR tracking [48,51]. This method has allowed the labeling of mouse and human neural stem cells and MR detection in mice for up to four weeks following intracranial injection [56,92,93]. Groups have shown that these neurospheres will migrate in response to inflammatory cues present in experimental autoimmune encephalomyelitis (EAE, a model of MS) following intracerebral injection, thus providing a key role for MR cell tracking to identify the optimal time for administering cellular therapy as well the delivery route and cell numbers [94,95].
Most recently MRI cell tracking has been utilized to follow the transplantation of pancreatic islet cells [96–101]. In our lab, we have taken advantage of alginate microcapsules to immunoisolate the islet cells while also providing a membrane permeable to insulin and metabolites. These ‘magnetocapsules’ can be followed by magnetic resonance fluoroscopy and allow for real-time MR-guided injection and monitoring of cell engraftment (Figure 7). Upon rupture, the contrast produced by a given capsule is altered, potentially allowing for distinction of viable capsules from non-viable. This technology should allow for advancements in islet cell transplantations and could also lead to multimodality capsules where one could draw upon the strengths of many imaging modalities to provide a complete picture of cell fate.
Figure 7. MR-guided injection of magnetocapsules in swine.

(A) Conventional magnetic resonance angiography/venography of the mesenteric venous system was performed with Gadolinium- diethylene-triamine-pentaacetic acid before any punctures. White arrow, active needle; black arrow, portal vein. The needle is seen in the inferior vena cava in the proper orientation for porto-caval puncture. (B,C) In vivo MRI of magnetocapsules before (B) and 5 min after (C) intraportal infusion of magnetocapsules in a pig. Magnetocapsules can be seen distributed throughout the liver as hypointense signal voids created by superparamagnetic ion oxide particles. (D,E) MRI follow up at three weeks shows the persistence of magnetocapsules as intact signal voids. (F) Magneto-encapsulated human islets retain functionality in vivo, as assessed by a sustained increase in human C-peptide in plasma.
Reproduced, with permission, from Barnett et al. [97]
5. Conclusion
With the recent burst in adoptive cell therapies, including tumor vaccines and stem cell therapies, there is an ever-increasing need for surrogate markers of efficacy. Given the complexity of the biology surrounding these cell therapeutics, simple flow cytometric or ex vivo analyses will no longer suffice for monitoring the relevant biomarkers. Novel in vivo systems will need to be developed that are capable of tracking the cells of interest and displaying their functionality. MR cell tracking provides an exciting tool to do just that. The discipline has proven its utility for a variety of different cellular therapies such as immuno-therapies and stem cell transplantation. The field currently has a variety of different contrast agents and pulse sequences, which have been proven effective, yet new mechanisms of developing contrast are still in the pipeline. Preclinical as well as early clinical studies have been performed which demonstrate the feasibility and safety of MR cell tracking. These past studies along with emerging technologies will soon take the field beyond preclinical status and begin to show benefits in clinical trials of novel therapies.
6. Expert opinion
Cell tracking has found wide applications in the preclinical setting. Nuclear imaging techniques, such as SPECT and PET, provide an incredibly sensitive and often quantitative approach to tracking cells; however, these modalities suffer from poor spatial resolution and the lack of an inherent anatomical reference. The physical limitations intrinsic to the radioactive decay being imaged inhibit the visualization of single cells using these modalities. On the other hand, reporter genes have been successfully created and utilized in nuclear imaging, providing a means to serially track cells capable of multiple divisions. Although reporter genes are in the discovery process for MR, this exclusion from the MR repertoire provides a serious restriction to the possible cell types capable of being monitored.
Optical imaging using bioluminescence and fluorescence is another option for in vivo cell tracking; however, scattering and absorption of light prevents these studies from progressing much beyond the point of preclinical studies. Optical imaging is limited to a penetration depth on the order of a few centimeters. The sensitivity of this modality can approach a few thousand cells and the acquisition time is quite fast, obtaining images within seconds in most cases. The imaging time along with the low cost associated with performing optical imaging has made this technique the modality of choice for many small animal preclinical studies. Additionally, multiple colors and the possibility for limited dilution effects make this imaging modality particularly attractive.
The promise of multiple colors for MR contrast is one of the most exciting emerging areas in the field of MR cell tracking. Many cellular therapies would benefit from the ability to monitor not just one but multiple cell types. In the case of tumor immunotherapy, it would be beneficial to be able to label antigen-specific T cells and antigen presenting dendritic cells with different colors and monitor interaction of these two cell types in vivo. Additionally, watching the communication between regulatory T cells and cytotoxic T cells could teach us a great deal about how these cells walk the fine line between immune tolerance and activation. The ability to perform these experiments is nearly in reach with the advent of CEST reporter genes and newly microfabricated magnetizable elements [39,40]. The frequency dependence of MR imaging should allow for many new contrast agents or reporter proteins to come along and utilize frequencies not naturally being utilized by water.
The ability to distinguish multiple cell types extends beyond just different colors as one will need sufficient sensitivity in order to tease single cell populations from one another. True ‘single cell’ tracking with MRI has only recently become a reality with the advent of the 1 μm Bang particles. Prior to development of these large paramagnetic particles, the field was limited to viewing large aggregates of cells and many SPIO particles in order to produce enough contrast to create a hypointense signal in a T2 image. The sensitivity of MRI has always been one of its major hindrances and must be improved in order for all the benefits of this modality to be realized. In the same vein, sensitivity increases should not be gained at the loss of cellular resolution. When using superparamagnetic particles, the surrounding area is affected by the magnetic field produced by the contrast agent. As the particles get larger, so does the affected surrounding area. As our current scanners do not achieve an in plane resolution smaller than one cell, the contrast produced needs to be on the order of nearly 100 μ3 in order to black out a single voxel. These voxels obviously cover more than a single cell, but ideally our contrast agent would stop right at the spatial resolution of our scanners and not produce blooming effects beyond these voxel limitations.
Another major advancement could be made in the field of positive contrast. Contrast agents relying on signal loss require a bit of a priori knowledge in order to locate the cells of interest due to signal dropout present in other nonspecific areas. Positive contrast may be achieved through new positive contrast pulse sequences which rely upon current contrast agents being utilized in a new fashion or new contrast agents entirely. The recent advent of fluorine imaging has proven to be an effective positive contrast agent providing no endogenous background; however, the sensitivity of this technique has hindered its broad use. New pulse sequences such as gradient acquisition for superparamagnetic particles/susceptibility (GRASP), inversion recovery on-resonance water suppression (IRON), fast low angle positive contrast with steady state free precession (FLAPS), as well as others have attempted to create positive contrast by selective excitation and refocusing of the off resonant frequencies produced by a spin echo acquisition [76,102,103].
Another well-documented drawback for the use of MRI for cell tracking is the inability to distinguish live cells from dead. As long as a superparamagnetic contrast agent is in the field of view, it will produce contrast. This means as dead cells deposit their label non-specifically or as resident macrophages carry the contrast agent distantly, erroneous signals will be produced. In order to combat this limitation, groups have begun to combine imaging modalities in order to validate the signals produced by MRI. For example, if the cells of interest are labeled with an optical reporter gene as well as with an iron oxide contrast agent, the dual modality imaging can provide high-resolution MR images as well as validate the viability of the cells being imaged. This method will also eliminate erroneous signal being produced by non-target cells. Beyond simple confirmation, using multiple modalities may provide a means for quantifying cellular engraftment and trafficking, a current capability using PET but not MR. Non-invasive quantification should provide the means to determine the number of cells needed to generate the desired biological effect and monitor the kinetics with which the biological process proceeds. In conclusion, advances in MR technology and novel contrast agents have had a significant effect upon in vivo cell tracking studies and will continue to provide insight into cell distribution, engraftment and survival. Future studies will rely upon larger magnets producing faster scan times at higher resolutions, contrast agents which will not persist after cell death and are not susceptible to cell division, and pulse sequences and post processing techniques which will allow for single cell quantification. Exploiting all of these advances will certainly contribute to the next generation of cell tracking techniques and should allow the discipline to become a mainstay of clinical evaluation of new cellular therapeutics.
Bibliography
- 1.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17(5):545–80. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 2.Massoud TF, Gambhir SS. Integrating noninvasive molecular imaging into molecular medicine: an evolving paradigm. Trends Mol Med. 2007;13(5):183–91. doi: 10.1016/j.molmed.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Bulte JW, Kraitchman DL. Monitoring cell therapy using iron oxide MR contrast agents. Curr Pharm Biotechnol. 2004;5(6):567–84. doi: 10.2174/1389201043376526. [DOI] [PubMed] [Google Scholar]
- 4.Modo M, Hoehn M, Bulte JW. Cellular MR imaging. Mol Imaging. 2005;4(3):143–64. doi: 10.1162/15353500200505145. [DOI] [PubMed] [Google Scholar]
- 5.Ntziachristos V, Bremer C, Weissleder R. Fluorescence imaging with near-infrared light: new technological advances that enable in vivo molecular imaging. Eur Radiol. 2003;13(1):195–208. doi: 10.1007/s00330-002-1524-x. [DOI] [PubMed] [Google Scholar]
- 6.Rice BW, Cable MD, Nelson MB. In vivo imaging of light-emitting probes. J Biomed Opt. 2001;6(4):432–40. doi: 10.1117/1.1413210. [DOI] [PubMed] [Google Scholar]
- 7.Frangioni JV, Hajjar RJ. In vivo tracking of stem cells for clinical trials in cardiovascular disease. Circulation. 2004;110(21):3378–83. doi: 10.1161/01.CIR.0000149840.46523.FC. [DOI] [PubMed] [Google Scholar]
- 8.Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17(7):484–99. doi: 10.1002/nbm.924. [DOI] [PubMed] [Google Scholar]
- 9.Weissleder R, Elizondo G, Wittenberg J, et al. Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology. 1990;175(2):489–93. doi: 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro EM, Sharer K, Skrtic S, Koretsky AP. In vivo detection of single cells by MRI. Magn Reson Med. 2006;55(2):242–9. doi: 10.1002/mrm.20718. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro EM, Skrtic S, Sharer K, et al. MRI detection of single particles for cellular imaging. Proc Natl Acad Sci USA. 2004;101(30):10901–6. doi: 10.1073/pnas.0403918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walczak P, Zhang J, Gilad AA, et al. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke. 2008;39(5):1569–74. doi: 10.1161/STROKEAHA.107.502047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walczak P, Kedziorek DA, Gilad AA, et al. Applicability and limitations of MR tracking of neural stem cells with asymmetric cell division and rapid turnover: the case of the shiverer dysmyelinated mouse brain. Magn Reson Med. 2007;58(2):261–9. doi: 10.1002/mrm.21280. [DOI] [PubMed] [Google Scholar]
- 14.Koretsky ALYJ, Schorle H, Jaenish R. Genetic control of MRI contrast by expression of the transferrin receptor. Proceedings of the International Society of Magnetic Resonance Medicine; 1996. p. 69. [Google Scholar]
- 15.Cohen B, Dafni H, Meir G, et al. Ferritin as an endogenous MRI reporter for noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia. 2005;7(2):109–17. doi: 10.1593/neo.04436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen B, Ziv K, Plaks V, et al. MRI detection of transcriptional regulation of gene expression in transgenic mice. Nat Med. 2007;13(4):498–503. doi: 10.1038/nm1497. [DOI] [PubMed] [Google Scholar]
- 17.Genove G, DeMarco U, Xu H, et al. A new transgene reporter for in vivo magnetic resonance imaging. Nat Med. 2005;11(4):450–4. doi: 10.1038/nm1208. [DOI] [PubMed] [Google Scholar]
- 18.Enochs WS, Petherick P, Bogdanova A, et al. Paramagnetic metal scavenging by melanin: MR imaging. Radiology. 1997;204(2):417–23. doi: 10.1148/radiology.204.2.9240529. [DOI] [PubMed] [Google Scholar]
- 19.Weissleder R, Simonova M, Bogdanova A, et al. MR imaging and scintigraphy of gene expression through melanin induction. Radiology. 1997;204(2):425–9. doi: 10.1148/radiology.204.2.9240530. [DOI] [PubMed] [Google Scholar]
- 20.Alfke H, Stoppler H, Nocken F, et al. In vitro MR imaging of regulated gene expression. Radiology. 2003;228(2):488–92. doi: 10.1148/radiol.2282012006. [DOI] [PubMed] [Google Scholar]
- 21.Zurkiya O, Chan AW, Hu X. MagA is sufficient for producing magnetic nanoparticles in mammalian cells, making it an MRI reporter. Magn Reson Med. 2008;59(6):1225–31. doi: 10.1002/mrm.21606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granot D, Kunz-Schughart LA, Neeman M. Labeling fibroblasts with biotin-BSA-GdDTPA-FAM for tracking of tumor-associated stroma by fluorescence and MR imaging. Magn Reson Med. 2005;54(4):789–97. doi: 10.1002/mrm.20628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobs RE, Fraser SE. Magnetic resonance microscopy of embryonic cell lineages and movements. Science. 1994;263(5147):681–4. doi: 10.1126/science.7508143. [DOI] [PubMed] [Google Scholar]
- 24.Modo M, Cash D, Mellodew K, et al. Tracking transplanted stem cell migration using bifunctional, contrast agent-enhanced, magnetic resonance imaging. Neuroimage. 2002;17(2):803–11. [PubMed] [Google Scholar]
- 25.Anderson SA, Lee KK, Frank JA. Gadolinium–fullerenol as a paramagnetic contrast agent for cellular imaging. Invest Radiol. 2006;41(3):332–8. doi: 10.1097/01.rli.0000192420.94038.9e. [DOI] [PubMed] [Google Scholar]
- 26.Biancone L, Crich SG, Cantaluppi V, et al. Magnetic resonance imaging of gadolinium-labeled pancreatic islets for experimental transplantation. NMR Biomed. 2007;20(1):40–8. doi: 10.1002/nbm.1088. [DOI] [PubMed] [Google Scholar]
- 27.Crich SG, Biancone L, Cantaluppi V, et al. Improved route for the visualization of stem cells labeled with a Gd-/Eu-chelate as dual (MRI and fluorescence) agent. Magn Reson Med. 2004;51(5):938–44. doi: 10.1002/mrm.20072. [DOI] [PubMed] [Google Scholar]
- 28.Daldrup-Link HE, Rudelius M, Metz S, et al. Cell tracking with gadophrin-2: a bifunctional contrast agent for MR imaging, optical imaging, and fluorescence microscopy. Eur J Nucl Med Mol Imaging. 2004;31(9):1312–21. doi: 10.1007/s00259-004-1484-2. [DOI] [PubMed] [Google Scholar]
- 29.Giesel FL, Stroick M, Griebe M, et al. Gadofluorine m uptake in stem cells as a new magnetic resonance imaging tracking method: an in vitro and in vivo study. Invest Radiol. 2006;41(12):868–73. doi: 10.1097/01.rli.0000246147.44835.4c. [DOI] [PubMed] [Google Scholar]
- 30.Vuu K, Xie J, McDonald MA, et al. Gadolinium–rhodamine nanoparticles for cell labeling and tracking via magnetic resonance and optical imaging. Bioconjug Chem. 2005;16(4):995–9. doi: 10.1021/bc050085z. [DOI] [PubMed] [Google Scholar]
- 31.Gilad AA, Walczak P, McMahon MT, et al. MR tracking of transplanted cells with “positive contrast” using manganese oxide nanoparticles. Magn Reson Med. 2008;60(1):1–7. doi: 10.1002/mrm.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahrens ET, Flores R, Xu H, Morel PA. In vivo imaging platform for tracking immunotherapeutic cells. Nat Biotechnol. 2005;23(8):983–7. doi: 10.1038/nbt1121. [DOI] [PubMed] [Google Scholar]
- 33.Bulte JW. Hot spot MRI emerges from the background. Nat Biotechnol. 2005;23(8):945–6. doi: 10.1038/nbt0805-945. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz-Cabello J, Walczak P, Kedziorek DA, et al. In vivo “hot spot” MR imaging of neural stem cells using fluorinated nanoparticles. Magn Reson Med. 2008;60:1506–11. doi: 10.1002/mrm.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Partlow KC, Chen J, Brant JA, et al. 19F magnetic resonance imaging for stem/progenitor cell tracking with multiple unique perfluorocarbon nanobeacons. FASEB J. 2007;21(8):1647–54. doi: 10.1096/fj.06-6505com. [DOI] [PubMed] [Google Scholar]
- 36.Srinivas M, Morel PA, Ernst LA, et al. Fluorine-19 MRI for visualization and quantification of cell migration in a diabetes model. Magn Reson Med. 2007;58(4):725–34. doi: 10.1002/mrm.21352. [DOI] [PubMed] [Google Scholar]
- 37.Gilad AA, McMahon MT, Walczak P, et al. Artificial reporter gene providing MRI contrast based on proton exchange. Nat Biotechnol. 2007;25(2):217–9. doi: 10.1038/nbt1277. [DOI] [PubMed] [Google Scholar]
- 38.Goffeney N, Bulte JW, Duyn J, et al. Sensitive NMR detection of cationic-polymer-based gene delivery systems using saturation transfer via proton exchange. J Am Chem Soc. 2001;123(35):8628–9. doi: 10.1021/ja0158455. [DOI] [PubMed] [Google Scholar]
- 39.McMahon MT, Gilad AA, Deliso MA, et al. New “multicolor” polypeptide diamagnetic chemical exchange saturation transfer (DIACEST) contrast agents for MRI. Magn Reson Med. 2008;60(4):803–12. doi: 10.1002/mrm.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zabow G, Dodd S, Moreland J, Koretsky A. Micro-engineered local field control for high-sensitivity multispectral MRI. Nature. 2008;453(7198):1058–63. doi: 10.1038/nature07048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilad AA, Winnard PT, Jr, van Zijl PC, Bulte JW. Developing MR reporter genes: promises and pitfalls. NMR Biomed. 2007;20(3):275–90. doi: 10.1002/nbm.1134. [DOI] [PubMed] [Google Scholar]
- 42.Weissleder R, Cheng HC, Bogdanova A, Bogdanov A. Magnetically labeled cells can be detected by MR imaging. J Magn Reson Imaging. 1997;7(1):258–63. doi: 10.1002/jmri.1880070140. [DOI] [PubMed] [Google Scholar]
- 43.Bulte JW, Arbab AS, Douglas T, Frank JA. Preparation of magnetically labeled cells for cell tracking by magnetic resonance imaging. Methods Enzymol. 2004;386:275–99. doi: 10.1016/S0076-6879(04)86013-0. [DOI] [PubMed] [Google Scholar]
- 44.Bulte JW, Hoekstra Y, Kamman RL, et al. Specific MR imaging of human lymphocytes by monoclonal antibody-guided dextran-magnetite particles. Magn Reson Med. 1992;25(1):148–57. doi: 10.1002/mrm.1910250115. [DOI] [PubMed] [Google Scholar]
- 45.Hawrylak N, Ghosh P, Broadus J, et al. Nuclear magnetic resonance (NMR) imaging of iron oxide-labeled neural transplants. Exp Neurol. 1993;121(2):181–92. doi: 10.1006/exnr.1993.1085. [DOI] [PubMed] [Google Scholar]
- 46.Norman AB, Thomas SR, Pratt RG, et al. Magnetic resonance imaging of neural transplants in rat brain using a superparamagnetic contrast agent. Brain Res. 1992;594(2):279–83. doi: 10.1016/0006-8993(92)91135-2. [DOI] [PubMed] [Google Scholar]
- 47.Bulte JW, Laughlin PG, Jordan EK, et al. Tagging of T cells with superparamagnetic iron oxide: uptake kinetics and relaxometry. Acad Radiol. 1996;3(Suppl 2):S301–3. doi: 10.1016/s1076-6332(96)80564-2. [DOI] [PubMed] [Google Scholar]
- 48.Bulte JW, Douglas T, Witwer B, et al. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat Biotechnol. 2001;19(12):1141–7. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- 49.Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem. 1999;10(2):186–91. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 50.Arbab AS, Yocum GT, Kalish H, et al. Efficient magnetic cell labeling with protamine sulfate complexed to ferumoxides for cellular MRI. Blood. 2004;104(4):1217–23. doi: 10.1182/blood-2004-02-0655. [DOI] [PubMed] [Google Scholar]
- 51.Frank JA, Miller BR, Arbab AS, et al. Clinically applicable labeling of mammalian and stem cells by combining superparamagnetic iron oxides and transfection agents. Radiology. 2003;228(2):480–7. doi: 10.1148/radiol.2281020638. [DOI] [PubMed] [Google Scholar]
- 52.Hoehn M, Kustermann E, Blunk J, et al. Monitoring of implanted stem cell migration in vivo: a highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc Natl Acad Sci USA. 2002;99(25):16267–72. doi: 10.1073/pnas.242435499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walczak P, Kedziorek DA, Gilad AA, et al. Instant MR labeling of stem cells using magnetoelectroporation. Magn Reson Med. 2005;54(4):769–74. doi: 10.1002/mrm.20701. [DOI] [PubMed] [Google Scholar]
- 54.Walczak P, Ruiz-Cabello J, Kedziorek DA, et al. Magnetoelectroporation: improved labeling of neural stem cells and leukocytes for cellular magnetic resonance imaging using a single FDA-approved agent. Nanomedicine. 2006;2(2):89–94. doi: 10.1016/j.nano.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Magnitsky S, Watson DJ, Walton RM, et al. In vivo and ex vivo MRI detection of localized and disseminated neural stem cell grafts in the mouse brain. Neuroimage. 2005;26(3):744–54. doi: 10.1016/j.neuroimage.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 56.Neri M, Maderna C, Cavazzin C, et al. Efficient in vitro labeling of human neural precursor cells with superparamagnetic iron oxide particles: relevance for in vivo cell tracking. Stem Cells. 2008;26(2):505–16. doi: 10.1634/stemcells.2007-0251. [DOI] [PubMed] [Google Scholar]
- 57.Aarntzen EH, Figdor CG, Adema GJ, et al. Dendritic cell vaccination and immune monitoring. Cancer Immunol Immunother. 2008;57(10):1559–68. doi: 10.1007/s00262-008-0553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahrens ET, Feili-Hariri M, Xu H, et al. Receptor-mediated endocytosis of iron-oxide particles provides efficient labeling of dendritic cells for in vivo MR imaging. Magn Reson Med. 2003;49(6):1006–13. doi: 10.1002/mrm.10465. [DOI] [PubMed] [Google Scholar]
- 59.Baumjohann D, Hess A, Budinsky L, et al. In vivo magnetic resonance imaging of dendritic cell migration into the draining lymph nodes of mice. Eur J Immunol. 2006;36(9):2544–55. doi: 10.1002/eji.200535742. [DOI] [PubMed] [Google Scholar]
- 60.de Vries IJ, Lesterhuis WJ, Barentsz JO, et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol. 2005;23(11):1407–13. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 61.Verdijk P, Scheenen TW, Lesterhuis WJ, et al. Sensitivity of magnetic resonance imaging of dendritic cells for in vivo tracking of cellular cancer vaccines. Int J Cancer. 2007;120(5):978–84. doi: 10.1002/ijc.22385. [DOI] [PubMed] [Google Scholar]
- 62.Hanson HL, Donermeyer DL, Ikeda H, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13(2):265–76. doi: 10.1016/s1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 63.Wang RF, Rosenberg SA. Human tumor antigens for cancer vaccine development. Immunol Rev. 1999;170:85–100. doi: 10.1111/j.1600-065x.1999.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 64.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99(25):16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yee C, Riddell SR, Greenberg PD. In vivo tracking of tumor-specific T cells. Curr Opin Immunol. 2001;13(2):141–6. doi: 10.1016/s0952-7915(00)00196-5. [DOI] [PubMed] [Google Scholar]
- 66.Adonai N, Nguyen KN, Walsh J, et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis (N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci USA. 2002;99(5):3030–5. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hardy J, Edinger M, Bachmann MH, et al. Bioluminescence imaging of lymphocyte trafficking in vivo. Exp Hematol. 2001;29(12):1353–60. doi: 10.1016/s0301-472x(01)00756-1. [DOI] [PubMed] [Google Scholar]
- 68.Arbab AS, Rad AM, Iskander AS, et al. Magnetically-labeled sensitized splenocytes to identify glioma by MRI: a preliminary study. Magn Reson Med. 2007;58(3):519–26. doi: 10.1002/mrm.21343. [DOI] [PubMed] [Google Scholar]
- 69.Anderson SA, Shukaliak-Quandt J, Jordan EK, et al. Magnetic resonance imaging of labeled T-cells in a mouse model of multiple sclerosis. Ann Neurol. 2004;55(5):654–9. doi: 10.1002/ana.20066. [DOI] [PubMed] [Google Scholar]
- 70.Kircher MF, Allport JR, Graves EE, et al. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003;63(20):6838–46. [PubMed] [Google Scholar]
- 71.Lewin M, Carlesso N, Tung CH, et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18(4):410–4. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 72.Vianello F, Papeta N, Chen T, et al. Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-specific T cell chemorepulsion and escape from immune control. J Immunol. 2006;176(5):2902–14. doi: 10.4049/jimmunol.176.5.2902. [DOI] [PubMed] [Google Scholar]
- 73.Assmus B, Honold J, Schachinger V, et al. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006;355(12):1222–32. doi: 10.1056/NEJMoa051779. [DOI] [PubMed] [Google Scholar]
- 74.Schachinger V, Erbs S, Elsasser A, et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355(12):1210–21. doi: 10.1056/NEJMoa060186. [DOI] [PubMed] [Google Scholar]
- 75.Schachinger V, Erbs S, Elsasser A, et al. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: final 1-year results of the REPAIR-AMI trial. Eur Heart J. 2006;27(23):2775–83. doi: 10.1093/eurheartj/ehl388. [DOI] [PubMed] [Google Scholar]
- 76.Mani V, Adler E, Briley-Saebo KC, et al. Serial in vivo positive contrast MRI of iron oxide-labeled embryonic stem cell-derived cardiac precursor cells in a mouse model of myocardial infarction. Magn Reson Med. 2008;60(1):73–81. doi: 10.1002/mrm.21642. [DOI] [PubMed] [Google Scholar]
- 77.Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci USA. 2005;102(32):11474–9. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amsalem Y, Mardor Y, Feinberg MS, et al. Iron-oxide labeling and outcome of transplanted mesenchymal stem cells in the infarcted myocardium. Circulation. 2007;116(11 Suppl):I38–45. doi: 10.1161/CIRCULATIONAHA.106.680231. [DOI] [PubMed] [Google Scholar]
- 79.Kraitchman DL, Heldman AW, Atalar E, et al. In vivo magnetic resonance imaging of mesenchymal stem cells in myocardial infarction. Circulation. 2003;107(18):2290–3. doi: 10.1161/01.CIR.0000070931.62772.4E. [DOI] [PubMed] [Google Scholar]
- 80.Karmarkar PV, Kraitchman DL, Izbudak I, et al. MR-trackable intramyocardial injection catheter. Magn Reson Med. 2004;51(6):1163–72. doi: 10.1002/mrm.20086. [DOI] [PubMed] [Google Scholar]
- 81.Jendelova P, Herynek V, DeCroos J, et al. Imaging the fate of implanted bone marrow stromal cells labeled with superparamagnetic nanoparticles. Magn Reson Med. 2003;50(4):767–76. doi: 10.1002/mrm.10585. [DOI] [PubMed] [Google Scholar]
- 82.Jendelova P, Herynek V, Urdzikova L, et al. Magnetic resonance tracking of transplanted bone marrow and embryonic stem cells labeled by iron oxide nanoparticles in rat brain and spinal cord. J Neurosci Res. 2004;76(2):232–43. doi: 10.1002/jnr.20041. [DOI] [PubMed] [Google Scholar]
- 83.Li Y, Chen J, Wang L, et al. Treatment of stroke in rat with intracarotid administration of marrow stromal cells. Neurology. 2001;56(12):1666–72. doi: 10.1212/wnl.56.12.1666. [DOI] [PubMed] [Google Scholar]
- 84.Kim D, Chun BG, Kim YK, et al. In vivo tracking of human mesenchymal stem cells in experimental stroke. Cell Transplant. 2007;16(10):1007–12. doi: 10.3727/000000007783472381. [DOI] [PubMed] [Google Scholar]
- 85.Shah K, Bureau E, Kim DE, et al. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. 2005;57(1):34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- 86.Wu X, Hu J, Zhou L, et al. In vivo tracking of superparamagnetic iron oxide nanoparticle-labeled mesenchymal stem cell tropism to malignant gliomas using magnetic resonance imaging. Laboratory investigation. J Neurosurg. 2008;108(2):320–9. doi: 10.3171/JNS/2008/108/2/0320. [DOI] [PubMed] [Google Scholar]
- 87.Einstein O, Fainstein N, Vaknin I, et al. Neural precursors attenuate autoimmune encephalomyelitis by peripheral immunosuppression. Ann Neurol. 2007;61(3):209–18. doi: 10.1002/ana.21033. [DOI] [PubMed] [Google Scholar]
- 88.Einstein O, Grigoriadis N, Mizrachi-Kol R, et al. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol. 2006;198(2):275–84. doi: 10.1016/j.expneurol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 89.Einstein O, Karussis D, Grigoriadis N, et al. Intraventricular transplantation of neural precursor cell spheres attenuates acute experimental allergic encephalomyelitis. Mol Cell Neurosci. 2003;24(4):1074–82. doi: 10.1016/j.mcn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 90.Pluchino S, Zanotti L, Rossi B, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436(7048):266–71. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 91.Aharonowiz M, Einstein O, Fainstein N, et al. Neuroprotective effect of transplanted human embryonic stem cell-derived neural precursors in an animal model of multiple sclerosis. PLoS One. 2008;3(9):e3145. doi: 10.1371/journal.pone.0003145. Published online September 5, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ben-Hur T, van Heeswijk RB, Einstein O, et al. Serial in vivo MR tracking of magnetically labeled neural spheres transplanted in chronic EAE mice. Magn Reson Med. 2007;57(1):164–71. doi: 10.1002/mrm.21116. [DOI] [PubMed] [Google Scholar]
- 93.Guzman R, Uchida N, Bliss TM, et al. Long-term monitoring of transplanted human neural stem cells in developmental and pathological contexts with MRI. Proc Natl Acad Sci USA. 2007;104(24):10211–6. doi: 10.1073/pnas.0608519104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bulte JWM, Ben-Hur T, Miller BR, et al. MR microscopy of magnetically labeled neurospheres transplanted into the Lewis EAE rat brain. Magn Reson Med. 2003;50(1):201–5. doi: 10.1002/mrm.10511. [DOI] [PubMed] [Google Scholar]
- 95.Politi LS, Bacigaluppi M, Brambilla E, et al. Magnetic-resonance-based tracking and quantification of intravenously injected neural stem cell accumulation in the brains of mice with experimental multiple sclerosis. Stem Cells. 2007;25(10):2583–92. doi: 10.1634/stemcells.2007-0037. [DOI] [PubMed] [Google Scholar]
- 96.Jirak D, Kriz J, Herynek V, et al. MRI of transplanted pancreatic islets. Magn Reson Med. 2004;52(6):1228–33. doi: 10.1002/mrm.20282. [DOI] [PubMed] [Google Scholar]
- 97.Barnett BP, Arepally A, Karmarkar PV, et al. Magnetic resonance-guided, real-time targeted delivery and imaging of magnetocapsules immunoprotecting pancreatic islet cells. Nat Med. 2007;13(8):986–91. doi: 10.1038/nm1581. [DOI] [PubMed] [Google Scholar]
- 98.Evgenov NV, Medarova Z, Dai G, et al. In vivo imaging of islet transplantation. Nat Med. 2006;12(1):144–8. doi: 10.1038/nm1316. [DOI] [PubMed] [Google Scholar]
- 99.Medarova Z, Evgenov NV, Dai G, et al. In vivo multimodal imaging of transplanted pancreatic islets. Nat Protoc. 2006;1(1):429–35. doi: 10.1038/nprot.2006.63. [DOI] [PubMed] [Google Scholar]
- 100.Toso C, Vallee JP, Morel P, et al. Clinical magnetic resonance imaging of pancreatic islet grafts after iron nanoparticle labeling. Am J Transplant. 2008;8(3):701–6. doi: 10.1111/j.1600-6143.2007.02120.x. [DOI] [PubMed] [Google Scholar]
- 101.Tai JH, Foster P, Rosales A, et al. Imaging islets labeled with magnetic nanoparticles at 1.5 Tesla. Diabetes. 2006;55(11):2931–8. doi: 10.2337/db06-0393. [DOI] [PubMed] [Google Scholar]
- 102.Stuber M, Gilson WD, Schar M, et al. Positive contrast visualization of iron oxide-labeled stem cells using inversion-recovery with ON-resonant water suppression (IRON) Magn Reson Med. 2007;58(5):1072–7. doi: 10.1002/mrm.21399. [DOI] [PubMed] [Google Scholar]
- 103.Koktzoglou I, Li D, Dharmakumar R. Dephased FLAPS for improved visualization of susceptibility-shifted passive devices for real-time interventional MRI. Phys Med Biol. 2007;52(13):N277–86. doi: 10.1088/0031-9155/52/13/N01. [DOI] [PubMed] [Google Scholar]