

Abstract
Radiation leaves a fairly characteristic footprint in biological materials, but this is rapidly all but obliterated by the canonical biological responses to the radiation damage. The innate immune recognition systems that sense “danger” through direct radiation damage and through associated collateral damage set in motion a chain of events that, in a tissue compromised by radiation, often unwittingly result in oscillating waves of molecular and cellular responses as tissues attempt to heal. Understanding “nature’s whispers” that inform on these processes will lead to novel forms of intervention targeted more precisely towards modifying them in an appropriate and timely fashion so as to improve the healing process and prevent or mitigate the development of acute and late effects of normal tissue radiation damage, whether it be accidental, as a result of a terrorist incident, or of therapeutic treatment of cancer. Here we attempt to discuss some of the non-free radical scavenging mechanisms that modify radiation responses and comment on where we see them within a conceptual framework of an evolving radiation-induced lesion.
Keywords: TBI, cytokines, RDS, inflammation, NF-κB
“Biological research workers should listen for “nature’s whispers” and stop telling her what to do.”
Peyton Rous
INTRODUCTION
The military use of atomic power during World War II and the subsequent development of the nuclear industry spurred efforts to find agents for the prophylaxis, mitigation, and treatment of radiation injury; efforts that have been reintensified recently by an increased threat of terrorist use of radiation sources. In the 1960s and 1970s considerable research effort went into the discovery of thiol agents that would protect against radiation, working from the knowledge that the cytotoxic effects of ionizing radiation were elicited in large part via production of reactive oxygen species (ROS) such as superoxide and hydroxyl radicals. Many free radical scavengers were studied, none more than WR2721, which became the drug Amifostine. In fact, Amifostine is a prodrug that is converted to the active free aminothiol WR1065 in vivo by alkaline phosphatase, which helps to decrease thiol toxicity, but does not eliminate it [1]. Radio-protection by thiols is thought to reduce initial radiation-induced DNA damage [2], which means that they have to be present at the time of irradiation. This limits their usefulness, but Amifostine has been used to prevent xerostomia in head and neck cancer radiation therapy (RT) [3].
Another important finding from the 1960s was that endotoxin could enhance survival of mice receiving total body irradiation (TBI) [4]. This was of obvious relevance to the leakage of bacterial products across an irradiated gut, but seemed to have limited potential in terms of a useful product for modifying radiation effects given its toxicity. However, the advent of recombinant materials brought a reassessment of this work and the finding that the cytokines interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α) were most likely responsible for the endotoxin-mediated radioprotection [5] spawned numerous studies of other cytokines. This research forged a link between the immune system and radiation-induced normal tissue damage that allows radiation effects to be placed within an appropriate biological context. TNF-α and IL-1 were most active if given 18 hrs before TBI, although significant mitigation was achieved with a single dose of IL-1 administered within 3 h after a lethal dose (LD95/30) of TBI [6]. In contrast, granulocyte colony-stimulating factor (G-CSF), stem cell factor, and granulocyte macrophage colony-stimulating factor (GM-CSF) are most effective if given post-exposure as they accelerated recovery of the hematopoietic system (reviewed in [2, 7]). Human G-CSF, GM-CSF, pegylated G-CSF, and IL-11 are currently approved by FDA for treating acute myelosuppression (reviewed in [8]).
The revolution in biology that has happened since these seminal findings were made has brought considerable understanding of the importance of molecular pathways activated by DNA damage to cellular responses leading to DNA repair, cell cycle arrest and/or cell death - giving a molecular explanation for these classic hallmarks of radiation exposure. The conceptual message is that DNA damage is not the only factor that determines life and death of the irradiated cell, but how the damage is sensed and dealt with is of critical importance.
It soon became clear that genes encoding these DNA damage response pathways were prime candidates for cancer-causing mutations and transfer of such genes often altered intrinsic cellular radiosensitivity, not because the cells were transformed, but because processes relating to cell proliferation and cell death/survival were affected. Since modifications of the internal molecular signaling network could affect cellular responses to radiation, it was not surprising that various cytokines and cytokine gene transfers had similar effects [9] and that compounds that activate or block these same pathways could be used to modify radiation responses, something that has been exploited clinically with the use of anti-EGFR to improve the outcome of radiation therapy in head and neck cancer patients [10], although care must be taken in the extrapolation from responses of cancer cells to normal ones. It is possible to conclude that cells make integrated responses to radiation damage coordinated through DNA repair, cell cycle, and cell death pathways
In the same vein, in recent years there has been considerable progress in our understanding of the coordinated tissue damage response to radiation and the mechanisms by which external “danger” is sensed by the body [11]. The conceptual basis stems largely from studies of innate immunity to pathogens showing that canonical wound healing pathways are activated. In a tissue where cells may be seriously compromised by a large amount of latent radiation-induced DNA damage, the canonical pathways may be disrupted leading to waves of molecular responses with time, similar to a chronic inflammatory lesion, with damage persisting and recurring due to failed efforts at healing [11]. Within this framework, the balance of pro-oxidant/anti-oxidant molecules, pro-inflammatory/anti-inflammatory cytokines, and growth promoting and inhibitory factors, along with involvement of mobilized bone marrow-derived precursor cells affords ample opportunity for intervention aimed at mitigating or treating radiation-damaged tissues, with recognition that the status of the network of molecular processes in a cell and of the cellular interactions within and between tissues has genetic and organ-specific components.
In this review we will discuss classes of agents other than the direct free radical scavenging thiols that modify radiation responses and will comment on how they might fit a conceptual framework of DNA, cell, and tissue responses to radiation damage. We will argue that the best approach to modifying radiation responses may be to promote natural host cytoprotective mechanisms and that by listening to “nature’s whispers”. With this, we may better address the dearth of agents with robust, prolonged efficacy, broad specificity, and minimal toxicity that could protect a large population in the event of a radiological emergency, or that could increase the radiotherapeutic benefit of cancer treatment.
DNA REPAIR AND CHROMATIN MODIFICATION
We will not deal with the “classic” radioprotective mechanisms of thiols here, but it is worth pointing out that there are many agents that affect DNA repair that are not free radical scavengers, and that many thiols may also radioprotect by other mechanisms. One factor affecting DNA repair is chromatin structure. The extent of chromatin condensation has long been known to influence DNA repair and cellular radiosensitivity [12, 13]. Direct visualization of chromatin domains showed that genetically inactive condensed heterochromatin is much less susceptible to radiation-induced DSBs than transcriptionally active, open chromatin [14], presumably because free radicals are less able to penetrate condensed structures. On the other hand, DNA DSB repair was more efficient in euchromatin, presumably in large part because the multiple components of the repair machinery have easier access. The mechanisms linking DNA repair to chromatin remodeling are clearly complex, but there are examples of radioprotection that can be attributed to these mechanisms.
After DNA DSB formation, a prominent change at the chromatin level is the increase in histone H2A.X phosphorrylation (γH2A.X), which elicits recruitment of many effecttor proteins to the site, where the complex interplay of docking and activation events occurs that is essential for DNA repair. One of the binding partners is the tumor suppressor histone acetylase transferase (HAT) Tip60, which binds to DSBs in particular in heterochromatin [15–18], which have a greater requirement for ATM in their repair and which is a slower process than in euchromatin. The finding that Tip60 is activated by histone H3 trimethylated on lysine 9 (H3K9me3), and targeted there by the Mre11–Rad50–Nbs1 (MRN) repair complex, suggests that methylation status of histones will be important in this repair process [19]. Tip60 causes chromatin relaxation by acetylating histone H4, which allows more efficient loading of repair factors and more efficient DSB repair [20]. Acetylation may also be a requirement for dephosphorylation of critical proteins such as -H2AX, which is a required step for efficient recovery from DNA damage checkpoint arrest [21]. Tip60 can also acetylate ATM (ataxia telangiectasia mutated) kinase, which plays so important a role in the DNA repair and damage response that its mutated form represents the poster child for a radiation sensitivity syndrome. In this way, Tip60 can link chromatin remodeling to DNA DSB repair and through ATM to downstream responses like cell cycle arrest and apoptosis. Furthermore, Tip60 activation can repress STAT3 activation with anti-inflammatory and immunosuppressive consequences [22, 23]. Obviously, while allowing access of repair factors, chromatin relaxation prior to, as opposed to after, radiation exposure could also allow better access of radiation-induced free radicals, increasing the extent of DNA damage [24, 25] and the timing of interventions aimed at modifying chromatin structure must be considered if DNA repair is to be enhanced.
Indeed, exclusion of free radicals by chromatin compacttion is a mechanism proposed to account for the radioprotective effects of the DNA minor groove-binding ligand Hoechst 33342 on DNA and intact cell nuclei [26, 27], as well as on cells from cytogenetic damage and death, and on mice from lethality after TBI [28, 29]. Radioprotection by DNA-compaction was also reported for oligolysine-treated DNA [30] and spermine treatment of plasmid or viral DNA and viral minichromosomes, but not native chromatin[31, 32]. RH-3 (an herbal preparation of Hippophae rhamnoides) also causes chromatin compaction [33] and protects mice when given 30 min before lethal TBI [34, 35]. Like many of these agents, RH3 has in fact multiple effects, including acting on death pathways, the immune system, and stem cells [36] that may also affect outcome.
In contrast, tetracycline and ciprofloxacin have been recently shown to activate Tip60 HAT in vitro, presumably by intercalating in DNA, and to aid radiation-induced DNA repair if given before or 1 hr after cell irradiation [37]. These antibiotics also modified cell viability if given after, as well as before, radiation, and tetracycline was an effective mitigator of lethal TBI damage in mice even if given 24 hrs after exposure. This suggests that examination of the effects of agents on post-transcriptional histone modifications may be a useful way to profile the pathways by which they may work. A similar intercalating mechanism was proposed for the radioprotective action of the anti-malaria drug chloroquine, which, unlike the antibiotics, activated ATM [38], suggesting the involvement of different pathways. This profiling may also explain why some DNA intercalating agents, such Hoechst 33342, are mutagenic while others that enhance DNA repair are not. Obviously, not all chromatin remodeling agents intercalate, for example TLK1B, a spliced mRNA variant of the TLK1 kinase, protects cells from the genotoxic effects of radiation [39–41] by promoting chromatin remodeling concurrent with improved repair of DNA damage. Further evidence for the importance of chromatin structure in radiation responses is the finding that histone deacetylase inhibitors may prevent lethality of mice if given 24 hrs before or 1 hr after TBI [42], although they also frequently radiosensitize tumor cells. Clearly, as the role of chromatin structure in the DNA repair and damage response becomes better understood and the tools for its analysis improve, there will be an increase in the number of agents that will use this as a target for intervention in radiation responses.
ANTIOXIDANT PATHWAYS
Cells have powerful innate anti-oxidant defenses. Obviously, these have not evolved to combat radiation-induced ROS but rather the damaging effects of ROS generated in large part through the mitochondrial respiratory chain and oxygen metabolizing enzymes, such as NADPH oxidases, myeloperoxidases, cyclooxygenase and lipoxygenase, and hypoxanthine/xanthine that are notably activated during inflammatory responses. ROS directly produced by radiation-induced ionization events are quantitatively relatively minor in comparison, although the deposition of energy within 2 nm of the DNA and the efficiency of free radicals so produced to cause DNA DSB gives an importance to radiation-induced ROS that transcends their meager quantity. However, the ROS generated through ionization events following irradiation are greatly supplemented by the effects of radiation in causing leakage of ROS from mitochondrial membranes and in generating inflammation. In this way, radiation effects overlap considerably with innate immune mechanisms that cause cell and tissue damage during host responses to other “danger” signals. Importantly, ROS and reactive nitrogen species (RNS) act as second messengers, regulating numerous cellular processes by directly altering protein structures with redox-sensitive cysteines resulting in activation of EGFR, PDGR, and other kinases [43, 44], inactivation of phosphatases such as Cdc25, stimulating NF-κB, JNK, and AP-1 pathways, and affecting ion channel functions [45]. The full spectrum of redox-sensitive proteins is still unknown, but many critical signaling molecules can be categorized in this way and, as a result, altering the redox balance in a cell can have profound effects on radiosensitivity. It is not possible to cover all the anti-oxidant systems here but some examples of the genre will be discussed.
NF-κB and AP-1 are redox-sensitive transcription factors that play critical roles in integrating ROS signaling with cellular response to infectious agents and innate and adaptive immunity. Exposure of mice to TBI induces NF-κB activation that in the spleen, lymph nodes, bone marrow and the intestinal epithelial (GI) cells consist mainly of NF-κB p50/RelA heterodimers, [46, 47]. Genetic mouse models indicate the physiological importance of NF-κB as a survival mechanism in the radioprotection of GI tissue. P50−/− mice have elevated levels of apoptosis in intestinal epithetlial cells, decreased survival of the small intestinal crypts, and increased GI radiosensitivity [48]. Conditional GI cell-specific IkappaB-kinase beta (IKKβ) knockout mice also display a significant increase in radiation-induced epithelial cell apoptosis, indicating that the IKKβ dependency of NF-κB activation, which has been attributed to increased expression and activation of p53 and decreased expression of anti-apoptotic Bcl-2 family proteins[49]. The anti-oxidant caffeic acid phenethyl ester (CAPE) inhibits NF-κB activation [50] and reduces the radiation-induced inflamematory responses in the lung, including IL-1, IL-6, TNF-α, and TGF-β, as well as radiation-induced interstitial pneumonitis. Other free radical scavenging agents such as disulfide metabolites of amifostine (WR-33278), N-acetyl-cysteine, mesna, captopril, and dithiothreitol (DTT) also activate NF-κB although only WR-1065 and WR-33278 displayed immediate activation of NF-κB suggesting direct action on redox-sensitive components [51, 52].
MnSOD (SOD2) is a component of the mitochondrial antioxidant defense system that converts superoxide to hydrogen peroxide, which is in turn reduced by glutathione (GSH) to water and alcohols. It is in the front line of inducible antioxidant defenses being generated through NF-κB-dependent pathways in response to changes in mitochondrial permeability and ROS release. Since ionizing radiation can induce NF-κB, it is no surprise it can induce MnSOD [53]. Although the MnSOD gene promoter contains binding motifs for a number of transcription factors other than NF-κB, it is this that naturally links it to the pro-oxidant tissue destructive activities of pro-inflammatory cytokines such as TNF-α [54] and IL-1 [53]. This is an important mechanism for the radioprotective effects of these cytokines [55, 56] and of other agents. Even thiols such as amifostine [57, 58] and N-acetyl cysteine [59] can induce delayed cytoprotection independent of their direct free radical scavenging abilities through this mechanism. Also, overexpression of heat shock protein HSP25 (HSPB1) induces MnSOD through activating NF-κB [60] and/or Bcl2 expression [61]. Clearly, MnSOD induction could be a common end pathway for many radioprotective agents that activate NF-κB, however it is not the only possible final effector pro-survival mechanism for this pathway and many other downstream molecules could increase radiation resistance, such as bcl-xl, iNOS, and A20, perhaps acting in unison.
Perhaps the most direct evidence that MnSOD can radioprotect cells comes from overexpression of the MnSOD gene in normal cells and tissues. Intratracheal injection of MnSOD plasmid prior to thoracic irradiation of C57B6/6J mice protected the lung and esophagus from damage [62, 63], overexpression in the small intestine protected against radiation enteritis [64], and a single subcutaneous injection of adeno-associated virus expressing MnSOD significantly mitigated acute skin injury following a single dose of 30–35 Gy [65]. Recently, a new mini-circle MnSOD plasmid with improved delivery characteristics was found to be as efficacious as full-length MnSOD plasmid in protecting mice against lethality after TBI [66]. Interestingly, MnSOD-deficient mice, which have a shortened life span and display a multifaceted phenotype [67], showed less radiation-induced neurogenesis defects than wild type mice [68], indicating that compensatory mechanisms might come into play. Radioprotection may not be the outcome for MnSOD gene transfer into tumors, which often have low basal MnSOD levels [53] and even MnSOD gene silencing. Indeed, certain tumors may be radiosensitized by MnSOD overexpression [69] through overproduction of hydrogen peroxide [70] and failure of its scavengers pyruvate or 2-deoxyglucose [71] to remove the high levels. Where tested, the presence of the mitochondrial leader sequence for expressed MnSOD seemed to be essential for radioprotection [72] confirming that MnSOD is normally used largely to control mitochondrial ROS.
As a result of these studies, numerous synthetic SOD mimics with low molecular weight and good membrane permeability have been developed as antioxidant therapies, and some seem to protect against radiation exposure. For example, MnTE-2-PyP5+, a MnPorphyrin, is an effective radioprotector that scavenges ROS/RNS and reduces DNA oxidative damage [73, 74]. Administration of MnTE-2-PyP5+ reduces pulmonary injury and blocks activation of TGF-β and HIF-α in Fischer rats when given daily for 2 weeks after 28 Gy hemithorax irradiation [75]. MnTnHex-2-PyP5+, an analogue of MnTE-2-PyP5+ that has more lipophilic properties, also ameliorated lung damage, although it was not as effective at reducing expression of TGF-β and other key molecules. On the other hand, MnTnHex-2-PyP5+ was more effective than MnTE-2-PyP5+ in protecting against radiation-induced apoptosis and DNA damage in human lymphoblastoid cells [76]. EUK-189, a MnSalen compound, also possesses SOD-like activity and protects from and mitigates against radiation injury. A subcutaneous injection at − 24, − 1, +1, or +6 h relative to lethal TBI enhanced 30 d survival with a dose reduction factor (DRF) of 1.15 [77].
Although antioxidant molecules often display a degree of specialization that may be reflected in the location in which they are expressed, they do not appear to operate in isolation but rather as a coordinated system. MnSOD induction appears to be an early response to ROS production. Haton [78] found that irradiation induced MnSOD and thioredoxin 2 expression in the gut by 6 hours after abdominal exposure, but by 4 days the response had waned and a second level of induced antioxidant genes were expressed including glutathione peroxidases and metallothioneins. Pardo found that overexpression of MnSOD stabilized the antioxidant pool including glutathione and total thiols leading to production of hemeoxygenase-1, glutamate-cysteine ligase, and NF-E2-related factor-2 (Nrf2)[79].
Nrf2 is a transcription factor that binds to the anti-oxidant response element (ARE). Under basal conditions, association of Nrf2 with the redox-sensitive CUL3 adaptor protein Keap1 [80] causes rapid Nrf2 ubiquitylation and degradation. Under oxidative stress Nrf2 is stabilized and acts as a master regulator of multiple cytoprotective pathways such as manganese-superoxide dismutase (MnSOD), glutathione transferases, heme oxygenase-1 (HO-1), and NAD(P) H: quinone oxidoreductase-1 (NQO-1) [81, 82]. Inhibition of 26S proteasome function, which is very sensitive to oxidative stress and leads to increased ROS production perhaps through failure of constitutively produced Bax-beta to be degraded [83], seems intimately linked to Nrf2 levels, which in turn transcriptionally activates genes of certain proteasome subunits [84].
In spite of its obvious relevance, the role of the Nrf2-ARE system in radiation responses is not clear. Nrf2 knockout cells and mice are more sensitive to radiation lethality suggesting that the basal level of anti-oxidants it controls are critical in moderating radiation responses (McDonald, submitted). Irradiation does induce Nrf2 expression, as might be expected, but only after a considerable delay (McDonald, submitted), which is in keeping with the observation of Haton that glutathione peroxidases were produced only 4 days after gut irradiation [78]. This may be in keeping with the need for a level of pro-oxidant/pro-inflammatory response to proceed before the redox balance is restored and a reflection of the involvement of the system in combating prolonged oxidative stress and promoting tissue regeneration. It may also be a reflection of the fact that many Nrf2 products are expressed at high levels and depletion is needed prior to induction. The radioprotective effect of Podophyllum hexandrum (REC-2000) involves up-regulation of the Nrf2 downstream effector HO-1 and is via hemopoietic system stimulation [85]. HO-1 seems to be required for hematopoiesis since bone marrow cells from mice lacking one allele of HO-1 had dramatically compromised ability to rescue lethally irradiated wild-type recipients and HO-1 deficiency disrupted the response of stem cells and progenitors to acute stress [86].
The metallothioneins (MTs) also seem to be induced fairly late after irradiation [78]. These are a group of intracellular metal-binding proteins with high cysteine content that can detoxify heavy metals and protect cells or tissues from oxidative damage by scavenging hydroxyl and superoxide radicals. Heavy metals, glucocorticoids, interferon and other cytokines, free radicals, and certain antitumor drugs will induce MTs, as well as ionizing radiation [87]. Thus, pre-treatment of mice with Zn, Cd, or Mn induce MT synthesis in the liver prior to IR and decrease mortality [88–90]. Bismuth nitrate treatment protected irradiated mice from bone marrow, but not gut, death and increased the number of endogenous spleen colonies [91]. However, overexpression of mouse MT in Chinese hamster ovary cells did not alter cell survival following irradiation [92] and MT pre-induction by cadmium chloride in mice liver did not protect against the lethal effects of combined radiation injury, such as 7 Gy TBI plus thermal burn [93]. Mice deficient in MTs show some effect on radiation sensitivity of bone marrow-derived cells at low doses [94], but the exact role of this system in radiation protection is still not fully clear and, as with all anti-oxidants, it may vary with the cell type, intracellular location, and level of expression.
CELL DEATH
Many modulators of normal tissue radiation responses affect cell death either directly or indirectly. The NF-κB pathway that is so intimately linked to redox control, often promotes cell survival while the AP-1 transcription factor, which is also redox-sensitive, seems more pre-occupied with cell death, although this varies with the cell and tissue type and with genetics [95]. The same is true for the prime sensor of radiation-induced DNA damage, p53, with which NF-κB appears at times to have a mutually antagonistic relationship [49, 96]. Tissues containing cell populations with a high proliferative capacity tend to have strong evidence of rapid p53-mediated apoptosis following even moderate doses of radiation, as cells do not explore the possibility of repair (reviewed in [97]). This may be because cell death is a recurring event in such tissues and of generally little consequence, as in lymphocytes of the hematopoietic system, hair follicles [98], oligodendroblasts of spinal cord, and progenitor cells for epithelial surfaces. On the contrary, normal fibroblasts respond to irradiation predominantly by irreversible p53-dependent growth arrest[99] which is in keeping with their role in tissue replacement. There have been a lot of efforts to suppress p53 activity via pharmacological means in order to reduce p53-dependent radiation-induced rapid apoptosis in normal tissues. Komarov identified a small molecule pifithrin α (PFTα), which was able to temporarily and reversibly suppress p53 via inhibiting p53-mediated transactivation of target genes [96, 100]. Consistently, p53-deficient mice survived doses of radiation that cause lethal hematopoietic syndrome in wild-type animals. However, p53 deficiency also resulted in sensitization of mice to higher doses of radiation, causing lethal GI syndrome [101]. Continuous cell proliferation in p53-deficient epithelium crypts without growth arrest correlated with accelerated death of damaged cells followed by rapid destruction of villi and accelerated lethality. This was also observed in p21-deficient mice indicating that p53/p21-mediated growth arrest plays a protective role in the epithelium of small intestine after high radiation doses.
P53 can induce apoptosis by transcriptional target gene regulation or transcription-independent signaling [102, 103]. Strom et al. took an approach to target non-transcriptional activity of p53; selective inhibition of the mitochondrial branch of the p53 pathway [104]. P53 translocates to the mitochondria where it can directly interact with Bcl2 family members, as can its downstream colleague PUMA, resulting in permeabilization of the outer mitochondrial membrane. They identified a small molecule pifithrin-μ (PFTμ) that inhibits p53 binding to mitochondria reducing its affinity to anti-apoptotic proteins Bcl-xL and Bcl-2 without any effect on p53-dependent transcriptional activation. PFTμ has a high specificity for p53 and rescued primary mouse thymocytes from p53-mediated apoptosis caused by irradiation and protected mice from lethal TBI. Furthermore, suppressing PUMA by antisense oligonucleotides provided significant radioprotection of intestinal progenitor cells reducing the GI syndrome [105].
Modulation of radiation-induced apoptosis has been implicated as a mechanism of action for a number of radioprotectors. A dietary supplement consisting of L-selenomethionine, vitamin C, vitamin E succinate, α-lipoic acid and N-acetyl cysteine significantly increased the 30 day survival of mice when given prior to, or after, lethal TBI [106]. This was associated with increased Bcl2 and decreased Bax, caspase 9 and TGF-β1 mRNA expression in the bone marrow after irradiation. Similar molecular changes accompanied the protective effect of dietary flaxseed against pulmonary fibrosis, inflammation, and oxidative lung damage [107]. Clearly, it is logical that modifying cell death pathways would affect radiation responses and there are numerous examples to support this view, however care must be taken not to compromise the effects of such death pathways in tissue homeostasis and regeneration.
ADENOSINERGIC PATHWAYS
Purine nucleosides, like adenosine, have been shown to enhance DNA repair in irradiated leucocytes and repeatedly to radioprotect [108–113] to mitigate lethality in mice receiving TBI [114–116]. The Chinese herbal medicine Cordyceps sinesis that is an effective radioprotector and accelerates bone marrow recovery when given after TBI [117, 118] was recently found to be 3′ adenosine [119]. Mice treated with adenosine and its analog inosine following 12 Gy TBI had increased survival [116], and drugs that elevate extracellular adenosine are known to accelerate myelorecovery of mice when given starting even 3 days after WBI and mobilizing hematopoietic progenitor cells [115, 120–122].
Adenosine is one of nature’s tissue protectants and the production of extracellular adenosine is a primordial homeostatic “danger” signaling mechanism. Its production is enhanced by hypoxia and anti-oxidant stimuli to generate an anti-inflammatory and immunosuppressive milieu. Nucleotides such as ATP that are released in sites of damage are catabolized extracellularly by ectonucleotidases to produce adenosine, which has long been known to play a critical and non-redundant role in the protection of normal tissues from collateral damage during inflammation [123–125]. The exact mechanism of tissue protection is unknown, but classic cytoprotective mechanisms that are involved probably include the production of hypoxia and of the cytokine interferon-alpha (IFN-α). Activating of ectonucleotidases leads to production of adenosine, the anti-inflammatory cytokines interleukin-10 (IL-10) and TGF-β [119, 126], and an anti-oxidant milieu that promotes hematopoiesis, angiogenesis, tissue regeneration, and wound healing [127, 128]. Importantly, IFN-α has recently been shown to activate hematopoietic stem cells [129] and may provide a link between adenosinergic stimuli and hematopoiesis. In addition to controlling immune function, anti-inflammatory adenosinergic pathways also aid enteric function following gut damage [130]. Extracellular adenosine regulates many physiologic functions through four known G-protein coupled receptors (GPCR): A1, A2a, A2b, and A3 that appear to have distinct functions. The available data suggest that agonists of A1 receptors inhibit adenylate cyclase to exert pro-inflammatory effects that attract macrophages to the site of damage but inhibit the bone marrow recovery process, whereas A2 and A3 receptor agonists are anti-inflammatory, act on lymphocytes (largely A2R), and stimulate bone marrow regeneration and progenitor mobilization (largely A3R) [120, 131]. In the future, these drugs will help elucidate the role of the adenosinergic pathways in the tissue response to damage and may offer new reagents for modifying such responses.
TOLL LIKE RECEPTOR (TLR) ACTIVATION AND CYTOKINES
TLRs recognize molecular patterns associated with microbial “non-self” (pathogen-associated molecular patterns, or PAMPs) and “damaged self” (damaged-associated molecular pattern molecules, or DAMPs) to initiate acute inflammation and to combat the “danger” inherent in pathogen invasion or pathological cell death, as is caused by irradiation. To do so effectively, they must be closely linked with the adenosinergic system and again INF-α and NF-κB pathways are critical players. There are currently about 12 members of the TLR subfamily in mice, 10 in humans [132], plus nucleotide-binding oligomerization domain (NOD)-like and retinoic acid-inducible gene (RIG)-like receptors [133], and C-type lectins [134], all of which can recognize PAMPS and/or DAMPS. The ligands for some TLRs have yet to be identified, but TLR4/MD2 dimers are particularly important in the response to PAMPS like lipopolysaccharride (LPS), whereas TLR2 can form heterodimers with TLR1 or TLR6 to respond to lipopeptide components of Gram-positive and -negative bacteria, while TLR5 recognizes bacterial flagellins [135]. In contrast to these cell surface dimers, TLR3, TLR7, and TLR9 are intracellular receptors that sense mainly RNA and DNA, as do NOD- and RIG-like receptors (reviewed in [136]).
DAMPs include the high mobility group box-1 (HMGB1) proteins that bind within the minor DNA groove and are released by damaged cells, including following irradiation. They share with LPS the ability to activate TLR4/MD2 but may also activate TLR2 [137]. Other DAMPs include heat shock proteins, degradation products of extracellular matrix (surfactant protein A, fibronectin extra domain A, and hyaluronan fragments), and other damage-associated proteins, such as beta defensin, uric acid, S100, minimally modified low-density lipoprotein (LDL), and possibly ROS-damaged proteins [138]. In spite of their complexity, all TLRs essentially signal through the adapter proteins, MyD88 and/or TRIF [139], to activate primarily the transcription factors NF-κB, AP-1, and interferon regulatory factor (IRF) 3 or 7.
Radioprotection conferred by LPS pretreatment [4] is presumed to operate through TLR4 and similar receptors to activate NF-κB to give pro-inflammatory cytokines that generate radioprotective, anti-oxidant responses, such as MnSOD, as described above. However, there are other stimuli and pathways associated with these “danger” signaling systems that give different outcomes. Gudkov’s group attempted to suppress radiation-induced apoptosis in the hematopoietic and gut systems by flagellin-induced NF-κB activation. They developed CBLB502, a polypeptide derived from Salmonella flagellin that binds to TLR5 and activates NF-κB without triggering systemic inflammation [140]. Systemic treatment of mice with purified flagellin protected mice if given between 2 h before to 4 h after exposure to lethal WBI and required TLR5 and the TLR signaling adaptor MyD88 [141]. Flagellin-elicited radioprotection was, in part, mediated via effects on bone marrow but without a pronounced rescue of radiation-induced anemia or leukopenia. Again, the TLR system appears to offer some hope of modifying radiation-induced tissue damage responses.
While these innate immune systems provide critical tools for modifying radiation responses, it is still not clear how best to use them in a given situation. The complexity of the responses can be illustrated by IL-1, which is much studied and clearly an excellent radioprotector if given 18 hrs before TBI, although toxic in humans. It acts to generate anti-oxidant defenses such as MnSOD, but also metallothionein, scavenging acute-phase proteins, and GSH, as well as production of hematopoietic growth factors [142–144]. The IL-1 receptor is critical in radioprotection suggesting a role for stromal cell activation [145] and since anti-IL-1 type 1 receptor antibody sensitizes mice to TBI, IL-1 is thought to have intrinsic radioprotective activity [146]. In spite of this effect on hematopoiesis and modest GI radioprotective activity [147] that can be attributed to decreased jejunal progenitor cell apoptosis [148], it has little activity if given after whole body TBI. In general, TNF-α has radioprotective activities that seem to overlap with those of IL-1, as might be expected [6, 142, 149, 150].
In contrast to IL-1 and TNF-α, stem cell factor (SCF), which stimulates primitive multipotential hematopoietic stem cells [151] and more rapid hematopoietic recovery when given after TBI [152], did not induce hematopoietic CSFs, IL-6 or MnSOD [153] and had radioprotective effects against GI death if given after TBI [154]. SCF significantly reduced radiation-induced apoptosis in murine cell lines transfected with the SCF receptor, c-kit [155]. The zinc-finger transcription factor Slug was reported to be a target of c-kit mediated radioprotection [156] since Slug could complement the hematopoietic failure c-kit-deficient mice receiviung TBI, while SCF/c-kit could not return the complement in Slug-deficient radiosensitive animals. Mono-or dual therapy with GM-CSF and/or IL-3, or the human GM-CSF/IL-3 fusion protein (PIXY321), has been shown to decrease the respective periods of neutropenia and thrombocytopenia in sublethally irradiated rhesus monkeys. In mice, the extent of radioprotection seemed to vary with the strains and conditions [142, 157–160], but clearly accelerating bone marrow recovery is an important part of enhancing recovery from TBI. Tissues may however vary in their responses to cytokine intervention. Thus, the immune cytokine IL-12 radioprotected mice when given 18 to 24 h prior to TBI, but sensitized B6D2F1 (12 Gy) and C3H/HeJ (9 Gy) to GI damage increasing circulating TNF and IL-6 levels to LPS through an INF mechanism [153, 161, 162].
A critical aspect of understanding cytokine involvement is realization that these systems work in a coordinated manner and that the balance between pro-inflammatory, pro-oxidant cytokines and their anti-oxidant, anti-inflammatory counterparts affects the hematopoietic system. When given before radiation exposure, the anti-oxidant/pro-oxidant balance seems critical to outcome, while after exposure the ability to generate cellular regeneration becomes more relevant.
CELL PROLIFERATION AND THE HEALING PHASE
One would hope that following radiation exposure the final phase of healing is reached, which involves cell proliferation, angiogenesis, fibrogenesis, and re-epithelialization that aim to limit damage and repair tissue, and that these processes also would offer targets for therapeutic intervention. The triggers for this phase include free radicals [45], ligands for ARs and TLRs [163], and such growth factors as fibroblast (FGF), epithelial (EGF), vascular endothelial (VEGF), and platelet derived (PDGF), acting on epithelial cells, endothelial cells, and fibroblasts. While such responses are generally beneficial, in irradiated tissues the stimulus to proliferate can unmask latent DNA damage and cause a vicious cycle of further cell death or carcinogenesis. Some years ago we proposed a unifying concept: that factors promotive of cell growth are cytoprotective and increase cellular radioresistance [164, 165].
As an example of what appears to be a common paradigm, EGFR is thought to confer tumor resistance to radiation through the activation of survival and cell proliferation pathways, and radiation-induced rapid EGFR activation to confer protection through the activation of the PI3K/AKT pathway [166–168], although how this may differ from ligand-induced EGFR activation is unknown [169–171]. Recent studies identified more a direct role for EGFR in repair of radiation-induced DNA damage. Radiation induces the ligand-independent translocation of the EGFR into the nucleoplasm in a free radical dependent process [172] where it can interact with the catalytic subunit of DNA-dependent protein kinase and the regulatory subunit Ku70 to promote nonhomologous end-joining DNA repair [173]. A few radioprotectors including Bowman-Birk proteinase inhibitor and O-phosphotyrosine have been reported to assist DNA repair via EGFR phosphorylation and nuclear transport [174–176]. It is important to note that induced cytoprotective responses have an inherent risk of producing persisting genomic instability that might drive carcinogenesis since they involve cells that might otherwise have been eliminated by cell death.
Another important family contains the fibroblast growth factors (FGFs) that participate in the control of growth and differentiation of endodermal and mesodermal structures during embryonal development, induce angiogenesis, and facilitate wound healing. Various members have been shown to protect against acute and late radiation damage of normal tissues. Acidic (aFGF, FGF-1) and basic (bFGF, FGF-2) myeloprotect mice from TBI, with FGF2 showing more effect and giving more rapid recovery of hematopoietic cells and peripheral white and red cells, and platelets [177]. FGF1 and FGF2 are radioprotective for small bowel when given 24 h before or 1 h after irradiation [178]. FGFs also promote bone growth and may prevent radiation-induced pneumonitis [2, 179]. Part of the radioprotective effect of FGF2 may be in preventing endothelial cell apoptosis [180], which may how it protected mice from radiation-induced crypt damage and the GI syndrome [181]. FGF1:FGF2 chimeric protein (FGFC) which presents universal FGF receptor specificity, better stability and biological activity than FGF1 or FGF2 has been developed for clinical applications. It stimulated keratinocyte proliferation more effectively than FGF2 alone and enhanced the survival of small intestine crypts [182]. FGF-4 is a radiation-inducible, heparin-binding growth factor whose expression by adenoviral delivery protects mice from bone marrow and GI damage after TBI [183, 184], inhibiting radiation-induced apoptosis of the crypt cells. Prophylactic administration of FGF-20 prior to lethal TBI significantly increased mouse survival, perhaps by activating Nrf2, MnSOD, and the ERK/Akt pathway [185]. FGF7 and FGF10, or keratinocyte growth factors (KGF) 1 and 2, are more epithelial cell-specific than are other FGFs. KGFs could prevent radiation toxicity to the abdomen or pelvis [186] and lung [187], and other epithelial tissues. Palifermin (KGF-1) reduced mucositis in a phase III trial with non-Hodgkin’s lymphoma patients undergoing bone marrow transplantation consisting of 12 Gy TBI [188]. It also showed improved the severity of mucositis and less salivary toxicity among head and neck cancer patients receiving hyperfractionated radiation [189].
Fibrosis is a common outcome of tissue irradiation that is considered to be an attempt by the body to replace damaged tissue with ground substance. A major profibrotic cytokine is transforming growth factor (TGF)-β, which regulates many aspects of wound repair including inflammation, chemotaxis, and deposition of extracellular matrix [190] with synthesis of collagen I, III and IV, repression of degrading enzymes, such as MMP-1, and up-regulation of integrin surface receptors, such as α2β1, leading to myofibroblast differentiation and constrictive fibrosis [191]. TGF-β1 levels, and the most important signaling receptor TGF-β R2, are increased in irradiated mouse skin [192, 193] and the rat ileal muscularis layer even on day 1 after irradiation [47]. Signaling leads to increased nucleoplasmatic shuttling of active Smad2/3 and induction of TGF-β1 target genes in fibrotic healing, which is mainly due to decrease in cytoplasmatic levels of the inhibitory Smad7 in irradiated tissue [194]. At the same time, the anti-proliferative and anti-inflammatory action of TGF-β on primitive hematopoietic progenitors sensitizes mice to radiation-induced hematopoietic lethality [162]. A direct role of TGF-β1 in radiation fibrosis was reported using Smad3 knockout (S3KO) mice that had decreased wound widths and enhanced epithelialization that correlated with increased neutrophil migration [195, 196]. S3KO fibroblasts had increased phosphorylation of ERK-MAPK, p53 and H2A.X, and decreased profibrotic gene expression after irradiation [197]. In the lung, anti-TGF-β antibody decreased radiation-induced lung injury [198] and this system has become a popular target for modification of radiation-induced lung injury.
Finally, blockers of the renin-angiotensin system (RAS) have been very successful in ameliorating late radiation tissue injury. The decapeptide angiotensin (Ang) I produced by the RAS is biologically inactive and cleaved to the active AngII by the angiotensin-converting enzyme (ACE) in blood and endothelial cells. AngII, a potent vasoconstrictor, functions by binding to the type 1 (AT1) or type 2 (AT2) receptor. However, recent studies found more complex pathways for the formation/metabolism of Ang peptides and their mediators (reviewed in [199]). Treatments with the RAS blockers are well tolerated and effective against radiation-induced injury to kidney, lung and brain.
In kidney, Moulder and others established that captopril, first generation thiol-containing ACE inhibitor could prevent and treat experimental radiation nephropathy [200–203]. Further studies revealed that a non-thiol ACE inhibitor enalapril as well as AT1R and AT2R antagonists reduced the severity of nephropathy, whole other anti-hypertensive drugs had no effect [204–208]. AT1 blockers such as L-158,809 are superior to ACE inhibitors for prevention/mitigation, while they are equally effective in treatment [209, 210]. Blocking AT2R augments the effect of AT1R antagonists on mitigation of nephropathy while it has no effect on treatment, suggesting potentially different mechanism [211].
In lung, Captopril administered to rats after lung irradiation ameliorated pulmonary endothelial dysfunction and fibrosis [212, 213]. Non-thiol ACEIs and L-158809 were equally effective, as was the AT1R blocker, affirming the role of AngII in modulation of lung fibrosis [214, 215]. In rat brain, chronic administration of ramipril (an ACE inhibitor) started 2 weeks after a 30 Gy dose reduced optic neuropathy [216] and administration of L-158,809 in drinking water prior to 40 Gy fractioned TBI and continued for the duration of experiment prevented cognitive impairment in rats at 6 and 12 months [217].
The mechanisms by which RAS blockers act to mitigate radiation-induced late tissue injuries are not known. However, it has been suggested that they exert their activity by modulating chronic oxidative stress and inflammation [199, 218]. AngII plays a key role in regulating blood pressure and fluid homeostasis under physiological conditions and has mitogenic, pro-inflammatory, and pro-fibrotic properties. Lowering blood pressure does not however seem to be important for modification of radiation-induced late effects [218]. Its effects in upregulating TGF-β and MMP-2, and in altering activity of the fibrinolytic system [219–222], or in generating ROS through NADPH oxidase [223] or aldosterone signaling, which can lead to EGFR transactivation and MAPK activity [224, 225] remain as possible mechanisms.
COMMENTARY
The most obvious conclusion to be drawn from this incomplete review is that numerous agents can modify radiation damage by numerous processes. Indeed, perhaps all radiation response modifiers affect multiple aspects of biology that are of possible relevance to the outcome. Nowhere is the level of integration more striking than when a single agent is found to affect DNA damage and repair, redox regulation, multiple cell signaling pathways, and tissue regeneration. Yet, radiation biologists have long been familiar with the integration of DNA damage and repair with cell cycle arrest and cell death that speak to common coordinated responses and should not be surprised by the spatial and temporal integration of the molecular and cellular networks that contribute to tissue homeostasis.
In thinking of mechanism of action of agents, it is perhaps more useful to consider the procession of events that are initiated by radiation damage. While different types of radiation may leave slightly different footprints in biological material, these are rapidly obliterated by the biological responses to damage that conform to canonical pathways of wound healing that in a heavily radiation damaged tissue results in wave-like oscillations in molecular markers of events that unfold over what may be a considerable time period (Fig. 1). Little is known about the control mechanisms that operate to dampen these oscillations and damaging sequelae of inflammation but they appear to be under genetic and tissue-specific control. It follows that DNA damage with repair or misrepair, cellular damage with death or survival, and tissue damage with regeneration or replacement are not unique to radiation, except in that the tissue may be compromised by latent radiation damage that is expressed only when cells are stimulated to divide and respond with mitotic death or sensecence.
Fig. 1.
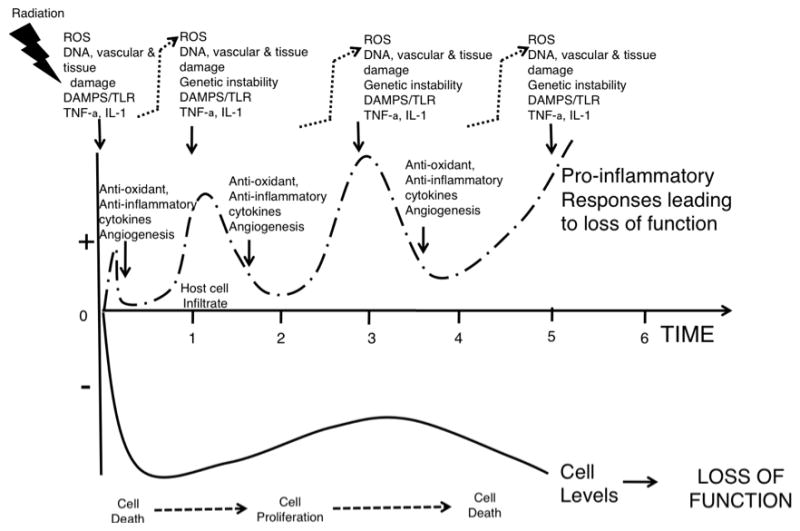
Cartoon showing waves of molecular and cellular pro-oxidant/anti-oxidant and pro-inflammatory/anti-inflammatory events leading to cell death, proliferation, and more cell death with functional loss of homeostasis in a tissue after irradiation.
In an evolutionary sense, these mechanisms have arisen to allow multi-cellular, multi-tissue structures with considerable specialization of organ function. Damage to these structures whether from pathogen or non-pathogen sources is sensed by innate immune recognition mechanisms that respond accordingly. This integrated scenario is much more complex than simply direct radiation-induced cell kill resulting in tissue damage. Host defenses are activated involving pro-inflammatory cytokines and cells that cause vascular leak and initially amplify the damage through production of high levels of ROS. The exact role of these free radicals in cell killing due to radiation is uncertain but cell death receptors and associated molecules activated following radiation generally enhance cell death and are reasonable targets for intervention, as are other molecules responsible for this collateral damage and the cell death process itself. Little is known of the importance of chromatin structure in this scenario, but radiation-induced cell kill is clearly not a random process and this is one factor that can affect it. One can imagine this system as being critical with a day or so of exposure and its manipulation could be of considerable benefit.
Damage sensors, such as the adenosinergic and toll-like receptor systems, play critical roles in sensing the extent of damage and limiting it. The development of cytoprotective hypoxia in concert with activation of cells surrounding the lesion by cytokines and growth factors shifts their molecular balance towards resisting cell death and microbial invasion and protective pathways are induced to limit damage through transcription factors such as NF-κB and Nrf2 that increase anti-oxidant levels. It follows that LPS and the pro-inflammatory cytokines like TNF-α and IL-1 that generate these conditions, if delivered before radiation exposure will be protective. The timing of agents that generate level 1 induced anti-oxidants may differ somewhat from those acting more on level 2 systems, but there may be some crosstalk. The pro-inflammatory, pro-oxidant nature of the initial destructive response to LPS and similar agents is likely to counter any beneficial effects if they are given after irradiation. Since the nature of the adenosinergic and toll-like receptors that are activated can dictate the nature of the subsequent cytokines that are produced, this is another balancing act that critically determines outcome and that needs further investigation. Importantly, after TBI these systems may be bombarded by PAMPS from microorganisms liberated through a radiation-damaged intestine. The nature of the microbiota and the extent to which TLRs expressed on epithelial cells generate innate local immunity are likely to be critical factors.
Importantly, this scenario must change into one where there is angiogenesis and cell proliferation leading to a regenerative response. Proliferative regeneration that follows irradiation has been the subject of numerous normal tissue studies but little is known of the checks and balances that control it. Inherent in this phase are the factors that decide between tissue regeneration and tissue replacement, such as fibrosis. Ongoing inflammation would seem to be a negative factor in this process and the rapid transition to an anti-inflammatory, anti-oxidant milieu that also stimulates angiogenesis would seem of most benefit. However, while it is easy to use words “anti-inflammatory milieu” multiple factors contribute to each situation making a unique blend that is likely to require further more precise definition before fully understanding its consequences. What is known is that there is a time lag before normal tissues start to regenerate following irradiation that can be days, weeks, or months and this is of obvious relevance to interventions aimed at amplifying or prolonging this phase, or at removing negative influences. There is remarkably little research been conducted on how best to achieve these goals, in spite of recognition that recovery of the stem/progenitor cell populations must be critical for fully functional tissue repair.
Finally, the most promising aspect of these scenarios is that the need to restore homeostasis at many levels within multiple tissue systems, suggests multiple distinct targets for mitigation efforts that may be initiated days, weeks, or even months and years after exposure so as to rebalance deranged tissues. DNA repair processes, redox action, cytokines, ARs, TLRs, and stem/progenitor cells obviously serve as potential targets. In addition to tissue-specific targets, the fact that the innate immune system plays a key role not only in sensing “danger” but also in integrating repair efforts and in directing mesenchymal and epithelial cells to restore tissue function offers a wealth of new possibilities for intervention. Understanding how to direct this molecular and cellular orchestra will lead to more informed discovery of novel modifiers of radiation responses and their more extensive use.
Acknowledgments
Grant support: University of California at Los Angeles Center for Biological Radioprotectors grant U19 AI067769/NIAID.
References
- 1.Seed TM. Radiation protectants: current status and future prospects. Health Phys. 2005;89(5):531–45. doi: 10.1097/01.hp.0000175153.19745.25. [DOI] [PubMed] [Google Scholar]
- 2.Murray D, McBride WH. Radioprotective agents. In: Kroschwitz I, Howe-Grant M, editors. Kirk-Othmer Encyclopedia of Chemical Technology and Encyclopedia of Environmental Analysis and Remediation. John Wiley & Sons; New York: 1996. pp. 963–1006. [Google Scholar]
- 3.Brizel DM. Pharmacologic approaches to radiation protection. J Clin Oncol. 2007;25(26):4084–9. doi: 10.1200/JCO.2007.11.5816. [DOI] [PubMed] [Google Scholar]
- 4.Ainsworth EJ. Bacterial endotoxins and the radiation syndrome. Annu Rep Div Biol Med Res Argonne Natl Lab. 1960;ANL-6264:158–61. [PubMed] [Google Scholar]
- 5.Neta R, Oppenheim JJ. Radioprotection with cytokines--learning from nature to cope with radiation damage. Cancer Cells. 1991;3(10):391–6. [PubMed] [Google Scholar]
- 6.Neta R, Oppenheim JJ, Douches S. Interdependence of the radioprotective effects of human recombinant interleukin 1b, tumor necrosis factora, granulocyte colony stimulating factor, and murine recombinant granulocyte-macrophage colony-stimulating factor. J Immunol. 1988;140:108–11. [PubMed] [Google Scholar]
- 7.Weiss W. Towards a coherent conceptual framework for emergency preparedness/response and rehabilitation - the application of the new ICRP recommendations given in ICRP 103. J Environ Radioact. 2009;100(12):1002–4. doi: 10.1016/j.jenvrad.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Singh VK, Shafran RL, Inal CE, Jackson WE, 3rd, Whitnall MH. Effects of whole-body gamma irradiation and 5-androstenediol administration on serum G-CSF. Immunopharmacol Immunotoxicol. 2005;27(4):521–34. doi: 10.1080/08923970500416707. [DOI] [PubMed] [Google Scholar]
- 9.McBride WH, Dougherty GJ. Radiotherapy for genes that cause cancer. Nat Med. 1995;1(11):1215–7. doi: 10.1038/nm1195-1215. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21–8. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 11.McBride WH, Chiang CS, Olson JL, et al. A sense of danger from radiation. Radiat Res. 2004;162(1):1–19. doi: 10.1667/rr3196. [DOI] [PubMed] [Google Scholar]
- 12.Chapman JD, Stobbe CC, Gales T, et al. Condensed chromatin and cell inactivation by single-hit kinetics. Radiat Res. 1999;151(4):433–41. [PubMed] [Google Scholar]
- 13.Biade S, Stobbe CC, Boyd JT, Chapman JD. Chemical agents that promote chromatin compaction radiosensitize tumour cells. Int J Radiat Biol. 2001;77(10):1033–42. doi: 10.1080/09553000110066068. [DOI] [PubMed] [Google Scholar]
- 14.Falk M, Lukasova E, Kozubek S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim Biophys Acta. 2008;1783(12):2398–414. doi: 10.1016/j.bbamcr.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Ikura T, Ogryzko VV, Grigoriev M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102(4):463–73. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 16.Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16(9):433–42. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19(6):321–9. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 18.Bird AW, Yu DY, Pray-Grant MG, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419(6905):411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11(11):1376–82. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8(1):91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 21.Kusch T, Florens L, Macdonald WH, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306(5704):2084–7. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Chin YE, Weisiger E, et al. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182(10):5899–903. doi: 10.4049/jimmunol.0804388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao H, Chung J, Kao HY, Yang YC. Tip60 is a co-repressor for STAT3. J Biol Chem. 2003;278(13):11197–204. doi: 10.1074/jbc.M210816200. [DOI] [PubMed] [Google Scholar]
- 24.Loontiens FG, Regenfuss P, Zechel A, Dumortier L, Clegg RM. Binding characteristics of Hoechst 33258 with calf thymus DNA, poly[d(A-T)], and d(CCGGAATTCCGG): multiple stoichiometries and determination of tight binding with a wide spectrum of site affinities. Biochemistry. 1990;29(38):9029–39. doi: 10.1021/bi00490a021. [DOI] [PubMed] [Google Scholar]
- 25.Martin RF, Anderson RF. Pulse radiolysis studies indicate that electron transfer is involved in radioprotection by Hoechst 33342 and methylproamine. Int J Radiat Oncol Biol Phys. 1998;42(4):827–31. doi: 10.1016/s0360-3016(98)00316-2. [DOI] [PubMed] [Google Scholar]
- 26.Denison L, Haigh A, D’Cunha G, Martin RF. DNA ligands as radioprotectors: molecular studies with Hoechst 33342 and Hoechst 33258. Int J Radiat Biol. 1992;61(4):561. doi: 10.1080/09553009214551341. [DOI] [PubMed] [Google Scholar]
- 27.Lyubimova NV, Coultas PG, Yuen K, Martin RF. In vivo radioprotection of mouse brain endothelial cells by Hoechst 33342. Br J Radiol. 2001;74(877):77–82. doi: 10.1259/bjr.74.877.740077. [DOI] [PubMed] [Google Scholar]
- 28.Singh SP, Jayanth VR, Chandna S, et al. Radioprotective effects of DNA ligands Hoechst-33342 and 33258 in whole body irradiated mice. Indian J Exp Biol. 1998;36(4):375–84. [PubMed] [Google Scholar]
- 29.Tawar U, Bansal S, Shrimal S, Singh M, Tandon V. Nuclear condensation and free radical scavenging: a dual mechanism of bisbenzimidazoles to modulate radiation damage to DNA. Mol Cell Biochem. 2007;305(1–2):221–33. doi: 10.1007/s11010-007-9546-y. [DOI] [PubMed] [Google Scholar]
- 30.Newton GL, Ly A, Tran NQ, Ward JF, Milligan JR. Radioprotection of plasmid DNA by oligolysines. Int J Radiat Biol. 2004;80(9):643–51. doi: 10.1080/09553000400005510. [DOI] [PubMed] [Google Scholar]
- 31.Chiu S, Oleinick NL. Radioprotection against the formation of DNA double-strand breaks in cellular DNA but not native cellular chromatin by the polyamine spermine. Radiat Res. 1997;148(2):188–92. [PubMed] [Google Scholar]
- 32.Chiu S, Oleinick NL. Radioprotection of cellular chromatin by the polyamines spermine and putrescine: preferential action against formation of DNA-protein crosslinks. Radiat Res. 1998;149(6):543–9. [PubMed] [Google Scholar]
- 33.Kumar IP, Namita S, Goel HC. Modulation of chromatin organization by RH-3, a preparation of Hippophae rhamnoides, a possible role in radioprotection. Mol Cell Biochem. 2002;238(1–2):1–9. doi: 10.1023/a:1019905211392. [DOI] [PubMed] [Google Scholar]
- 34.Goel HC, Prasad J, Singh S, Sagar RK, Kumar IP, Sinha AK. Radioprotection by a herbal preparation of Hippophae rhamnoides, RH-3, against whole body lethal irradiation in mice. Phytomedicine. 2002;9(1):15–25. doi: 10.1078/0944-7113-00077. [DOI] [PubMed] [Google Scholar]
- 35.Goel HC, Kumar IP, Samanta N, Rana SV. Induction of DNA-protein cross-links by Hippophae rhamnoides: implications in radioprotection and cytotoxicity. Mol Cell Biochem. 2003;245(1–2):57–67. doi: 10.1023/a:1022809625826. [DOI] [PubMed] [Google Scholar]
- 36.Goel HC, Prasad J, Singh S, et al. Radioprotective potential of an herbal extract of Tinospora cordifolia. J Radiat Res (Tokyo) 2004;45(1):61–8. doi: 10.1269/jrr.45.61. [DOI] [PubMed] [Google Scholar]
- 37.Kim K, Pollard JM, Norris AJ, et al. High-throughput screening identifies two classes of antibiotics as radioprotectors: tetracyclines and fluoroquinolones. Clin Cancer Res. 2009;15(23):7238–45. doi: 10.1158/1078-0432.CCR-09-1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kastan MB. DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol Cancer Res. 2008;6(4):517–24. doi: 10.1158/1541-7786.MCR-08-0020. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, DeFatta R, Anthony C, Sunavala G, De Benedetti A. A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene. 2001;20(6):726–38. doi: 10.1038/sj.onc.1204147. [DOI] [PubMed] [Google Scholar]
- 40.Sen SP, De Benedetti A. TLK1B promotes repair of UV-damaged DNA through chromatin remodeling by Asf1. BMC Mol Biol. 2006;7:37. doi: 10.1186/1471-2199-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sunavala-Dossabhoy G, De Benedetti A. Tousled homolog, TLK1, binds and phosphorylates Rad9; TLK1 acts as a molecular chaperone in DNA repair. DNA Repair (Amst) 2009;8(1):87–102. doi: 10.1016/j.dnarep.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Brown SL, Kolozsvary A, Liu J, Ryu S, Kim JH. Histone deacetylase inhibitors protect against and mitigate the lethality of total-body irradiation in mice. Radiat Res. 2008;169(4):474–8. doi: 10.1667/RR1245.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae YS, Kang SW, Seo MS, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272(1):217–21. [PubMed] [Google Scholar]
- 44.Joo CK, Kim HS, Park JY, Seomun Y, Son MJ, Kim JT. Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-beta involves multiple signaling pathways. Oncogene. 2008;27(5):614–28. doi: 10.1038/sj.onc.1210649. [DOI] [PubMed] [Google Scholar]
- 45.Sarsour E, Kumar MG, Chaudhuri L, Kalen AL, Goswami P. Redox Control of the Cell Cycle in Health and Disease. Antioxid Redox Signal. 2009;11(12):2985–3011. doi: 10.1089/ars.2009.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rithidech KN, Tungjai M, Arbab E, Simon SR. Activation of NF-kappaB in bone marrow cells of BALB/cJ mice following exposure in vivo to low doses of (137)Cs gamma-rays. Radiat Environ Biophys. 2005;44(2):139–43. doi: 10.1007/s00411-005-0004-5. [DOI] [PubMed] [Google Scholar]
- 47.Linard C, Ropenga A, Vozenin-Brotons MC, Chapel A, Mathe D. Abdominal irradiation increases inflammatory cytokine expression and activates NF-kappaB in rat ileal muscularis layer. Am J Physiol Gastrointest Liver Physiol. 2003;285(3):G556–65. doi: 10.1152/ajpgi.00094.2003. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Meng A, Lang H, et al. Activation of nuclear factor kappaB In vivo selectively protects the murine small intestine against ionizing radiation-induced damage. Cancer Res. 2004;64(17):6240–6. doi: 10.1158/0008-5472.CAN-04-0591. [DOI] [PubMed] [Google Scholar]
- 49.Egan LJ, Eckmann L, Greten FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA. 2004;101(8):2452–7. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YJ, Liao HF, Tsai TH, Wang SY, Shiao MS. Caffeic acid phenethyl ester preferentially sensitizes CT26 colorectal adenocarcinoma to ionizing radiation without affecting bone marrow radioresponse. Int J Radiat Oncol Biol Phys. 2005;63(4):1252–61. doi: 10.1016/j.ijrobp.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 51.Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFkappaB-mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162(5):536–46. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 52.Murley JS, Kataoka Y, Hallahan DE, Roberts JC, Grdina DJ. Activation of NFkappaB and MnSOD gene expression by free radical scavengers in human microvascular endothelial cells. Free Radic Biol Med. 2001;30(12):1426–39. doi: 10.1016/s0891-5849(01)00554-8. [DOI] [PubMed] [Google Scholar]
- 53.Eastgate J, Moreb J, Nick HS, Suzuki K, Taniguchi N, Zucali JR. A role for manganese superoxide dismutase in radioprotection of hematopoietic stem cells by interleukin-1. Blood. 1993;81(3):639–46. [PubMed] [Google Scholar]
- 54.Wong GHW, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–4. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- 55.Janssen YM, Van Houten B, Borm PJ, Mossman BT. Cell and tissue responses to oxidative damage. Lab Invest. 1993;69(3):261–74. [PubMed] [Google Scholar]
- 56.Wong GH. Protective roles of cytokines against radiation: induction of mitochondrial MnSOD. Biochim Biophys Acta. 1995;1271(1):205–9. doi: 10.1016/0925-4439(95)00029-4. [DOI] [PubMed] [Google Scholar]
- 57.Murley JS, Kataoka Y, Baker KL, Diamond AM, Morgan WF, Grdina DJ. Manganese superoxide dismutase (SOD2)-mediated delayed radioprotection induced by the free thiol form of amifostine and tumor necrosis factor alpha. Radiat Res. 2007;167(4):465–74. doi: 10.1667/RR0758.1. [DOI] [PubMed] [Google Scholar]
- 58.Grdina DJ, Murley JS, Kataoka Y, et al. Amifostine induces antioxidant enzymatic activities in normal tissues and a transplantable tumor that can affect radiation response. Int J Radiat Oncol Biol Phys. 2009;73(3):886–96. doi: 10.1016/j.ijrobp.2008.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagata K, Iwasaki Y, Yamada T, et al. Overexpression of manganese superoxide dismutase by N-acetylcysteine in hyperoxic lung injury. Respir Med. 2007;101(4):800–7. doi: 10.1016/j.rmed.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 60.Yi MJ, Park SH, Cho HN, et al. Heat-shock protein 25 (Hspb1) regulates manganese superoxide dismutase through activation of Nfkb (NF-kappaB) Radiat Res. 2002;158(5):641–9. doi: 10.1667/0033-7587(2002)158[0641:hsphrm]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 61.Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000;6(8):324–9. doi: 10.1016/s1357-4310(00)01756-1. [DOI] [PubMed] [Google Scholar]
- 62.Carpenter M, Epperly MW, Agarwal A, et al. Inhalation delivery of manganese superoxide dismutase-plasmid/liposomes protects the murine lung from irradiation damage. Gene Ther. 2005;12(8):685–93. doi: 10.1038/sj.gt.3302468. [DOI] [PubMed] [Google Scholar]
- 63.Epperly MW, Defilippi S, Sikora C, Gretton J, Greenberger JS. Radioprotection of lung and esophagus by overexpression of the human manganese superoxide dismutase transgene. Mil Med. 2002;167(2 Suppl):71–3. [PubMed] [Google Scholar]
- 64.Epperly MW, Guo HL, Jefferson M, et al. Cell phenotype specific kinetics of expression of intratracheally injected manganese superoxide dismutase-plasmid/liposomes (MnSOD-PL) during lung radioprotective gene therapy. Gene Ther. 2003;10(2):163–71. doi: 10.1038/sj.gt.3301852. [DOI] [PubMed] [Google Scholar]
- 65.Yan S, Brown SL, Kolozsvary A, Freytag SO, Lu M, Kim JH. Mitigation of radiation-induced skin injury by AAV2-mediated MnSOD gene therapy. J Gene Med. 2008;10(9):1012–8. doi: 10.1002/jgm.1226. [DOI] [PubMed] [Google Scholar]
- 66.Zhang M, Qian J, Xing X, et al. Inhibition of the tumor necrosis factor-alpha pathway is radioprotective for the lung. Clin Cancer Res. 2008;14(6):1868–76. doi: 10.1158/1078-0432.CCR-07-1894. [DOI] [PubMed] [Google Scholar]
- 67.Huang LE, Willmore WG, Gu J, Goldberg MA, Bunn HF. Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J Biol Chem. 1999;274(13):9038–44. doi: 10.1074/jbc.274.13.9038. [DOI] [PubMed] [Google Scholar]
- 68.Fishman K, Baure J, Zou Y, et al. Radiation-induced reductions in neurogenesis are ameliorated in mice deficient in CuZnSOD or MnSOD. Free Radic Biol Med. 2009;47(10):1459–67. doi: 10.1016/j.freeradbiomed.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Urano M, Kuroda M, Reynolds R, Oberley TD, St Clair DK. Expression of manganese superoxide dismutase reduces tumor control radiation dose: gene-radiotherapy. Cancer Res. 1995;55(12):2490–3. [PubMed] [Google Scholar]
- 70.Sun W, Kalen AL, Smith BJ, Cullen JJ, Oberley LW. Enhancing the antitumor activity of adriamycin and ionizing radiation. Cancer Res. 2009;69(10):4294–300. doi: 10.1158/0008-5472.CAN-09-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thongphasuk J, Oberley LW, Oberley TD. Induction of superoxide dismutase and cytotoxicity by manganese in human breast cancer cells. Arch Biochem Biophys. 1999;365(2):317–27. doi: 10.1006/abbi.1999.1179. [DOI] [PubMed] [Google Scholar]
- 72.Greenberger JS, Epperly MW. Antioxidant gene therapeutic approaches to normal tissue radioprotection and tumor radiosensitization. In vivo. 2007;21(2):141–6. [PubMed] [Google Scholar]
- 73.Mitchell JA, Sheng H, Forstermann U, Murad F. Characterization of nitric oxide synthases in non-adrenergic non-cholinergic nerve containing tissue from the rat anococcygeus muscle. Br J Pharmacol. 1991;104(2):289–91. doi: 10.1111/j.1476-5381.1991.tb12422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vujaskovic Z, Batinic-Haberle I, Rabbani ZN, et al. A small molecular weight catalytic metalloporphyrin antioxidant with superoxide dismutase (SOD) mimetic properties protects lungs from radiation-induced injury. Free Radic Biol Med. 2002;33(6):857–63. doi: 10.1016/s0891-5849(02)00980-2. [DOI] [PubMed] [Google Scholar]
- 75.Gauter-Fleckenstein B, Fleckenstein K, Owzar K, Jiang C, Batinic-Haberle I, Vujaskovic Z. Comparison of two Mn porphyrin-based mimics of superoxide dismutase in pulmonary radioprotection. Free Radic Biol Med. 2008;44(6):982–9. doi: 10.1016/j.freeradbiomed.2007.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pollard JM, Reboucas JS, Durazo A, et al. Radioprotective effects of manganese-containing superoxide dismutase mimics on ataxiatelangiectasia cells. Free Radic Biol Med. 2009;47(3):250–60. doi: 10.1016/j.freeradbiomed.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Srinivasan V, Doctrow S, Singh VK, Whitnall MH. Evaluation of EUK-189, a synthetic superoxide dismutase/catalase mimetic as a radiation counter measure. Immunopharmacol Immunotoxicol. 2008;30(2):271–90. doi: 10.1080/08923970801925331. [DOI] [PubMed] [Google Scholar]
- 78.Haton C, Francois A, Vandamme M, Wysocki J, Griffiths NM, Benderitter M. Imbalance of the antioxidant network of mouse small intestinal mucosa after radiation exposure. Radiat Res. 2007;167(4):445–53. doi: 10.1667/RR0581.1. [DOI] [PubMed] [Google Scholar]
- 79.Pardo M, Tirosh O. Protective signalling effect of manganese superoxide dismutase in hypoxia-reoxygenation of hepatocytes. Free Radic Res. 2009;43(12):1225–39. doi: 10.3109/10715760903271256. [DOI] [PubMed] [Google Scholar]
- 80.Sekhar KR, Rachakonda G, Freeman ML. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol Appl Pharmacol. 2010;244(1):21–6. doi: 10.1016/j.taap.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Banning A, Brigelius-Flohe R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid Redox Signal. 2005;7(7–8):889–99. doi: 10.1089/ars.2005.7.889. [DOI] [PubMed] [Google Scholar]
- 82.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278(14):12029–38. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 83.Fu NY, Sukumaran SK, Kerk SY, Yu VC. Baxbeta: a constitutively active human Bax isoform that is under tight regulatory control by the proteasomal degradation mechanism. Mol Cell. 2009;33(1):15–29. doi: 10.1016/j.molcel.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 84.Kraft DC, Deocaris CC, Wadhwa R, Rattan SI. Preincubation with the proteasome inhibitor MG-132 enhances proteasome activity via the Nrf2 transcription factor in aging human skin fibroblasts. Ann N Y Acad Sci. 2006;1067:420–4. doi: 10.1196/annals.1354.060. [DOI] [PubMed] [Google Scholar]
- 85.Rajesh A, Sagar R, Singh S, et al. Cytoprotective effect of Podophyllum hexandrum against gamma radiation is mediated via hemopoietic system stimulation and up-regulation of heme-oxygenase-1 and the prosurvival multidomain protein Bcl-2. Integr Cancer Ther. 2007;6(1):54–65. doi: 10.1177/1534735406298303. [DOI] [PubMed] [Google Scholar]
- 86.Cao C, Lu S, Kivlin R, et al. AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes. J Biol Chem. 2008;283(43):28897–908. doi: 10.1074/jbc.M804144200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Achanta P, Thompson KJ, Fuss M, Martinez JL., Jr Gene expression changes in the rodent hippocampus following whole brain irradiation. Neurosci Lett. 2007;418(2):143–8. doi: 10.1016/j.neulet.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 88.Matsubara J, Tajima Y, Karasawa M. Metallothionein induction as a potent means of radiation protection in mice. Radiat Res. 1987;111(2):267–75. [PubMed] [Google Scholar]
- 89.Matsubara J, Tajima Y, Karasawa M. Promotion of radioresistance by metallothionein induction prior to irradiation. Environ Res. 1987;43(1):66–74. doi: 10.1016/s0013-9351(87)80058-0. [DOI] [PubMed] [Google Scholar]
- 90.Ogata H, Izumo Y. Mortality reduction in mice administered a single abundant dose of zinc, manganese or magnesium after irradiation by gamma-rays at sublethal doses. Radioisotopes. 1990;39(12):573–6. doi: 10.3769/radioisotopes.39.12_573. [DOI] [PubMed] [Google Scholar]
- 91.Kagimoto O, Naganuma A, Imura N, Toge T, Niwa O, Yokoro K. Effect of the administration of bismuth nitrate on radiogenic thymoma induction in mice. J Radiat Res (Tokyo) 1991;32(4):417–28. doi: 10.1269/jrr.32.417. [DOI] [PubMed] [Google Scholar]
- 92.Koropatnick J, Pearson J. Altered cisplatin and cadmium resistance and cell survival in Chinese hamster ovary cells expressing mouse metallothionein. Mol Pharmacol. 1993;44(1):44–50. [PubMed] [Google Scholar]
- 93.Budagov RS, Ul’ianova LP, Smoryzanova OA, Rott GM. Increase in the level of metallothioneins in mouse liver after administration of cadmium chloride does not protect from combined radiation-thermal injury. Radiats Biol Radioecol. 2001;41(6):671–6. [PubMed] [Google Scholar]
- 94.Shibuya K, Suzuki JS, Kito H, Naganuma A, Tohyama C, Satoh M. Protective role of metallothionein in bone marrow injury caused by X-irradiation. J Toxicol Sci. 2008;33(4):479–84. doi: 10.2131/jts.33.479. [DOI] [PubMed] [Google Scholar]
- 95.Lorimore SA, Coates PJ, Wright EG. Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene. 2003;22(45):7058–69. doi: 10.1038/sj.onc.1207044. [DOI] [PubMed] [Google Scholar]
- 96.Gurova KV, Hill JE, Guo C, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci USA. 2005;102(48):17448–53. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3(2):117–29. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 98.Botchkarev VA, Komarova EA, Siebenhaar F, et al. p53 Involvement in the control of murine hair follicle regression. Am J Pathol. 2001;158(6):1913–9. doi: 10.1016/S0002-9440(10)64659-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burger A, Loffler H, Bamberg M, Rodemann HP. Molecular and cellular basis of radiation fibrosis. Int J Radiat Biol. 1998;73(4):401–8. doi: 10.1080/095530098142239. [DOI] [PubMed] [Google Scholar]
- 100.Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 101.Komarova EA, Kondratov RV, Wang K, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23(19):3265–71. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- 102.Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 103.Chipuk JE, Green DR. Cytoplasmic p53: bax and forward. Cell Cycle. 2004;3(4):429–31. [PubMed] [Google Scholar]
- 104.Strom E, Sathe S, Komarov PG, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2(9):474–9. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- 105.Qiu W, Carson-Walter EB, Liu H, et al. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell. 2008;2(6):576–83. doi: 10.1016/j.stem.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wambi C, Sanzari J, Wan XS, et al. Dietary antioxidants protect hematopoietic cells and improve animal survival after total-body irradiation. Radiat Res. 2008;169(4):384–96. doi: 10.1667/RR1204.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee JH, Lee B, Lee HS, et al. Lactobacillus suntoryeus inhibits pro-inflammatory cytokine expression and TLR-4-linked NF-kappaB activation in experimental colitis. Int J Colorectal Dis. 2009;24(2):231–7. doi: 10.1007/s00384-008-0618-6. [DOI] [PubMed] [Google Scholar]
- 108.Grant GA, Barlow JA, Leach KE. Modification of survival of gamma irradiated mice by adenosine nucleotides. Strahlentherapie. 1976;152(3):285–91. [PubMed] [Google Scholar]
- 109.Kozyreva EV, Eliseenko NN, Iashkin PN, Tikhomirova MV. ADP as one of the possible regulators of recovery processes in the cell and body following irradiation. Radiobiologiia. 1977;17(5):733–8. [PubMed] [Google Scholar]
- 110.Langendorff H, Langendorff M. Chemical radiation protection and the c AMP mechanism. Int J Radiat Biol Relat Stud Phys Chem Med. 1971;19(5):493–5. doi: 10.1080/09553007114550631. [DOI] [PubMed] [Google Scholar]
- 111.Tikhomirova MV, Iashkin PN. Comparative radioprotective activity of adenylates in short-term and prolonged gamma-irradiation. Radiobiologiia. 1983;23(1):100–4. [PubMed] [Google Scholar]
- 112.Tikhomirova MV, Rogozkim VD. Action of ADP in irradiation at a high and low dosage rate. Radiobiologiia. 1979;19(2):241–5. [PubMed] [Google Scholar]
- 113.Gudkov SV, Gudkova OY, Chernikov AV, Bruskov VI. Protection of mice against X-ray injuries by the post-irradiation administration of guanosine and inosine. Int J Radiat Biol. 2009;85(2):116–25. doi: 10.1080/09553000802641144. [DOI] [PubMed] [Google Scholar]
- 114.Hennig UG, Wang Q, Gee NH, von Borstel RC. Protection and repair of gamma-radiation-induced lesions in mice with DNA or deoxyribonucleoside treatments. Mutat Res. 1996;350(1):247–54. doi: 10.1016/0027-5107(95)00109-3. [DOI] [PubMed] [Google Scholar]
- 115.Hofer M, Pospisil M, Znojil V, et al. Drugs elevating extracellular adenosine promote regeneration of haematopoietic progenitor cells in severely myelosuppressed mice: their comparison and joint effects with the granulocyte colony-stimulating factor. Eur J Haematol. 2002;68(1):4–11. doi: 10.1034/j.1600-0609.2002.00564.x. [DOI] [PubMed] [Google Scholar]
- 116.Hou B, Xu ZW, Yang CW, Gao Y, Zhao SF, Zhang CG. Protective effects of inosine on mice subjected to lethal total-body ionizing irradiation. J Radiat Res (Tokyo) 2007;48(1):57–62. doi: 10.1269/jrr.06067. [DOI] [PubMed] [Google Scholar]
- 117.Liu WC, Wang SC, Tsai ML, et al. Protection against Radiation-Induced Bone Marrow and Intestinal Injuries by Cordyceps sinensis, a Chinese Herbal Medicine. Radiat Res. 2006;166(6):900–7. doi: 10.1667/RR0670.1. [DOI] [PubMed] [Google Scholar]
- 118.Liu WC, Chuang WL, Tsai ML, Hong JH, McBride WH, Chiang CS. Cordyceps sinensis health supplement enhances recovery from taxol-induced leukopenia. Exp Biol Med (Maywood) 2008;233(4):447–55. doi: 10.3181/0708-RM-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou X, Luo L, Dressel W, et al. Cordycepin is an immunoregulatory active ingredient of Cordyceps sinensis. Am J Chin Med. 2008;36(5):967–80. doi: 10.1142/S0192415X08006387. [DOI] [PubMed] [Google Scholar]
- 120.Hofer M, Pospisil M, Znojil V, Hola J, Streitova D, Vacek A. Homeostatic action of adenosine A3 and A1 receptor agonists on proliferation of hematopoietic precursor cells. Exp Biol Med (Maywood) 2008;233(7):897–900. doi: 10.3181/0802-RM-43. [DOI] [PubMed] [Google Scholar]
- 121.Hofer M, Pospisil M, Znojil V, Hola J, Vacek A, Streitova D. Adenosine A(3) receptor agonist acts as a homeostatic regulator of bone marrow hematopoiesis. Biomed Pharmacother. 2007;61(6):356–9. doi: 10.1016/j.biopha.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 122.Hofer M, Vacek A, Pospisil M, Hola J, Streitova D, Znojil V. Activation of adenosine A(3) receptors potentiates stimulatory effects of IL-3, SCF, and GM-CSF on mouse granulocyte-macrophage hematopoietic progenitor cells. Physiol Res. 2009;58(2):247–52. doi: 10.33549/physiolres.931454. [DOI] [PubMed] [Google Scholar]
- 123.Ohta A, Madasu M, Kini R, Subramanian M, Goel N, Sitkovsky M. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. 2009;183(9):5487–93. doi: 10.4049/jimmunol.0901247. [DOI] [PubMed] [Google Scholar]
- 124.Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A. Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol. 2008;153 (Suppl 1):S457–64. doi: 10.1038/bjp.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008 Oct 1;14(19):5947–52. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]
- 126.Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180(6):4246–55. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- 127.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181(12):8633–41. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Su Y, Huang X, Raskovalova T, et al. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother. 2008;57(11):1611–23. doi: 10.1007/s00262-008-0494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 130.Antonioli L, Fornai M, Colucci R, et al. Regulation of enteric functions by adenosine: pathophysiological and pharmacological implications. Pharmacol Ther. 2008;120(3):233–53. doi: 10.1016/j.pharmthera.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 131.Ohta A, Sitkovsky M. The adenosinergic immunomodulatory drugs. Curr Opin Pharmacol. 2009;9(4):501–6. doi: 10.1016/j.coph.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–8. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 133.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442(7098):39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 134.Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7(12):1258–65. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 135.Feuillet V, Medjane S, Mondor I, et al. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci USA. 2006;103(33):12487–92. doi: 10.1073/pnas.0605200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kluwe J, Mencin A, Schwabe RF. Toll-like receptors, wound healing, and carcinogenesis. J Mol Med. 2009;87(2):125–38. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 138.Lotze MT, Zeh HJ, Rubartelli A, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 139.Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- 140.Burdelya LG, Krivokrysenko VI, Tallant TC, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008;320(5873):226–30. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vijay-Kumar M, Aitken JD, Sanders CJ, et al. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol. 2008;180(12):8280–5. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 142.Neta R. Role of cytokines in radioprotection. Pharmacol Ther. 1988;39(1–3):261–6. doi: 10.1016/0163-7258(88)90070-8. [DOI] [PubMed] [Google Scholar]
- 143.Zucali JR, Suresh A, Tung F, Eastgate J, Nick H, Moreb J. Cytokine protection of hematopoietic stem cells. Prog Clin Biol Res. 1994;389(207):207–16. [PubMed] [Google Scholar]
- 144.Schwartz GN, Patchen ML, Neta R, MacVittie TJ. Radioprotection of mice with interleukin-1: relationship to the number of erythroid and granulocyte-macrophage colony-forming cells. Radiat Res. 1990;121:220–6. [PubMed] [Google Scholar]
- 145.Dubois CM, Neta R, Keller JR, Jacobsen SE, Oppenheim JJ, Ruscetti F. Hematopoietic growth factors and glucocorticoids synergize to mimic the effects of IL-1 on granulocyte differentiation and IL-1 receptor induction on bone marrow cells in vivo. Exp Hematol. 1993;21(2):303–10. [PubMed] [Google Scholar]
- 146.Neta R, Perlstein R, Vogel SN, Ledney GD, Abrams J. Role of interleukin 6 (IL-6) in protection from lethal irradiation and in endocrine responses to IL-1 and tumor necrosis factor. J Exp Med. 1992;175(3):689–94. doi: 10.1084/jem.175.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu SG, Miyamoto T. Radioprotection of the intestinal crypts of mice by recombinant human interleukin-1 alpha. J Immunol. 1990;145(2):489–98. [PubMed] [Google Scholar]
- 148.Johnke RM, Smith ES, Cariveau MJ, et al. Radioprotection of murine gastrointestinal epithelium by interleukin-1alpha involves down-regulation of the apoptotic response. Anticancer Res. 2008;28(6A):3601–7. [PubMed] [Google Scholar]
- 149.Slordal L, Muench MO, Warren DJ, Moore MA. Radioprotection by murine and human tumor-necrosis factor: dose-dependent effects on hematopoiesis in the mouse. Eur J Haematol. 1989;43(5):428–34. doi: 10.1111/j.1600-0609.1989.tb00331.x. [DOI] [PubMed] [Google Scholar]
- 150.Dalmau SR, Freitas CS, Tabak DG. Interleukin-1 and tumor necrosis factor-alpha as radio- and chemoprotectors of bone marrow. Bone Marrow Transplant. 1993;12(6):551–63. [PubMed] [Google Scholar]
- 151.Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood. 1992;80(12):3044–50. [PubMed] [Google Scholar]
- 152.Zsebo KM, Smith KA, Hartley CA, et al. Radioprotection of mice by recombinant rat stem cell factor. Proc Natl Acad Sci USA. 1992;89(20):9464–8. doi: 10.1073/pnas.89.20.9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Neta R, Oppenheim JJ, Wang JM, Snapper CM, Moorman MA, Dubois CM. Synergy of IL-1 and stem cell factor in radioprotection of mice is associated with IL-1 up-regulation of mRNA and protein expression for c-kit on bone marrow cells. J Immunol. 1994;153(4):1536–43. [PubMed] [Google Scholar]
- 154.Leigh BR, Khan W, Hancock SL, Knox SJ. Stem cell factor enhances the survival of murine intestinal stem cells after photon irradiation. Radiat Res. 1995;142(1):12–5. [PubMed] [Google Scholar]
- 155.Maddens S, Charruyer A, Plo I, et al. Kit signaling inhibits the sphingomyelin-ceramide pathway through PLC gamma 1: implication in stem cell factor radioprotective effect. Blood. 2002;100(4):1294–301. [PubMed] [Google Scholar]
- 156.Perez-Losada J, Sanchez-Martin M, Perez-Caro M, Perez-Mancera PA, Sanchez-Garcia I. The radioresistance biological function of the SCF/kit signaling pathway is mediated by the zinc-finger transcription factor Slug. Oncogene. 2003;22(27):4205–11. doi: 10.1038/sj.onc.1206467. [DOI] [PubMed] [Google Scholar]
- 157.Neta R, Douches S, Oppenheim JJ. Interleukin 1 is a radioprotector. J Immunol. 1986;136:2483–5. [PubMed] [Google Scholar]
- 158.Uckun FM, Souza L, Waddick KG, Wick M, Song CW. In vivo radioprotective effects of recombinant human granulocyte colony-stimulating factor in lethally irradiated mice. Blood. 1990;75(3):638–45. [PubMed] [Google Scholar]
- 159.Neta R. Radioprotection and therapy of radiation injury with cytokines. Int J Immunopharmacol. 1990;12(5):491–6. [PubMed] [Google Scholar]
- 160.Patchen ML, Fischer R, MacVittie TJ, Seiler FR, Williams DE. Mast cell growth factor (C-kit ligand) in combination with granulocyte-macrophage colony-stimulating factor and interleukin-3: in vivo hemopoietic effects in irradiated mice compared to in vitro effects. Biotherapy. 1993;7(1):13–26. doi: 10.1007/BF01878150. [DOI] [PubMed] [Google Scholar]
- 161.Neta R, Stiefel SM, Ali N. In lethally irradiated mice interleukin-12 protects bone marrow but sensitizes intestinal tract to damage from ionizing radiation. Ann N Y Acad Sci. 1995;762:274–80. doi: 10.1111/j.1749-6632.1995.tb32332.x. discussion 80–1. [DOI] [PubMed] [Google Scholar]
- 162.Neta R. Modulation with cytokines of radiation injury: suggested mechanisms of action. Environ Health Perspect. 1997;1056(Suppl)(3):1463–5. doi: 10.1289/ehp.97105s61463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102(1):99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McBride WH. Cytokine cascades in late normal tissue radiation responses. Int J Radiat Oncol Biol Phys. 1995;33(1):233–4. doi: 10.1016/0360-3016(95)02019-8. [DOI] [PubMed] [Google Scholar]
- 165.Chiang CS, Sawyers CL, McBride WH. Oncogene Expression and Cellular Radiation Resistance: A Modulatory Role for c-myc. Mol Diagn. 1998;3(1):21–7. doi: 10.154/MODI00300021. [DOI] [PubMed] [Google Scholar]
- 166.Contessa JN, Hampton J, Lammering G, et al. Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene. 2002;21(25):4032–41. doi: 10.1038/sj.onc.1205500. [DOI] [PubMed] [Google Scholar]
- 167.Dent P, Reardon DB, Park JS, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol Biol Cell. 1999;10(8):2493–506. doi: 10.1091/mbc.10.8.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhan M, Han ZC. Phosphatidylinositide 3-kinase/AKT in radiation responses. Histol Histopathol. 2004;19(3):915–23. doi: 10.14670/HH-19.915. [DOI] [PubMed] [Google Scholar]
- 169.Schmidt-Ullrich RK, Mikkelsen RB, Dent P, et al. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15(10):1191–7. doi: 10.1038/sj.onc.1201275. [DOI] [PubMed] [Google Scholar]
- 170.Dent P, Yacoub A, Contessa J, et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res. 2003;159(3):283–300. doi: 10.1667/0033-7587(2003)159[0283:sariao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 171.Withers HR, Taylor JMG, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncologica. 1988;27:131–45. doi: 10.3109/02841868809090333. [DOI] [PubMed] [Google Scholar]
- 172.Dittmann K, Mayer C, Fehrenbacher B, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280(35):31182–9. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 173.Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–27. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 174.Dittmann K, Mayer C, Wanner G, Kehlbach R, Rodemann HP. The radioprotector O-phosphotyrosine stimulates DNA-repair via epidermal growth factor receptor- and DNA-dependent kinase phosphorylation. Radiother Oncol. 2007;84(3):328–34. doi: 10.1016/j.radonc.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 175.Dittmann KH, Mayer C, Ohneseit PA, et al. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int J Radiat Oncol Biol Phys. 2008;70(1):203–12. doi: 10.1016/j.ijrobp.2007.08.065. [DOI] [PubMed] [Google Scholar]
- 176.Wanner G, Mayer C, Kehlbach R, Rodemann HP, Dittmann K. Activation of protein kinase Cepsilon stimulates DNA-repair via epidermal growth factor receptor nuclear accumulation. Radiother Oncol. 2008;86(3):383–90. doi: 10.1016/j.radonc.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 177.Ding I, Wu T, Matsubara H, et al. Acidic fibroblast growth factor (FGF1) increases survival and haematopoietic recovery in total body irradiated C3H/HeNCr mice. Cytokine. 1997;9(1):59–65. doi: 10.1006/cyto.1996.0136. [DOI] [PubMed] [Google Scholar]
- 178.Okunieff P, Mester M, Wang J, et al. In vivo radioprotective effects of angiogenic growth factors on the small bowel of C3H mice. Radiat Res. 1998;150(2):204–11. [PubMed] [Google Scholar]
- 179.Okunieff P, Wu T, Huang K, Ding I. Differential radioprotection of three mouse strains by basic or acidic fibroblast growth factor. Br J Cancer Suppl. 1996;27:S105–8. [PMC free article] [PubMed] [Google Scholar]
- 180.Kolesnick RN, Haimovitz-Friedman A, Fuks Z. The sphingomyelin signal transduction pathway mediates apoptosis for tumor necrosis factor, Fas, and ionizing radiation. Biochem Cell Biol. 1994;72(11–12):471–4. doi: 10.1139/o94-063. [DOI] [PubMed] [Google Scholar]
- 181.Paris F, Fuks Z, Kang A, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293(5528):293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 182.Motomura K, Hagiwara A, Komi-Kuramochi A, et al. An FGF1: FGF2 chimeric growth factor exhibits universal FGF receptor specificity, enhanced stability and augmented activity useful for epithelial proliferation and radioprotection. Biochim Biophys Acta. 2008;1780(12):1432–40. doi: 10.1016/j.bbagen.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 183.Takahama Y, Ochiya T, Tanooka H, et al. Adenovirus-mediated transfer of HST-1/FGF-4 gene protects mice from lethal irradiation. Oncogene. 1999;18(43):5943–7. doi: 10.1038/sj.onc.1203171. [DOI] [PubMed] [Google Scholar]
- 184.Konishi H, Kuroda S, Tanaka M, et al. Molecular cloning and characterization of a new member of the RAC protein kinase family: association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem Biophys Res Commun. 1995;216(2):526–34. doi: 10.1006/bbrc.1995.2654. [DOI] [PubMed] [Google Scholar]
- 185.Maclachlan T, Narayanan B, Gerlach VL, et al. Human fibroblast growth factor 20 (FGF-20; CG53135–05): a novel cytoprotectant with radioprotective potential. Int J Radiat Biol. 2005;81(8):567–79. doi: 10.1080/09553000500211091. [DOI] [PubMed] [Google Scholar]
- 186.Okunieff P, Hammond E, Grignon D, et al. Radiation Therapy Oncology Group. Research Plan 2002–2006. Translational Research Program. Int J Radiat Oncol Biol Phys. 2001;51(3 Suppl 2):75–87. [PubMed] [Google Scholar]
- 187.Ma J, Doig AJ, Patel N, Cui NJ, Terry NH. Modulating effect of keratinocyte growth factor on cellular kinetics of murine lung(I) Ai Zheng. 2003;22(11):1127–34. [PubMed] [Google Scholar]
- 188.Spielberger R, Stiff P, Bensinger W, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med. 2004;351(25):2590–8. doi: 10.1056/NEJMoa040125. [DOI] [PubMed] [Google Scholar]
- 189.Brizel DM, Herman TS, Goffinet D, et al. A phase I/II trial of escalating doses of recombinant human keratinocyte growth factor (rHuKGF) in head & neck cancer (HNC) patients receiving radiotherapy (RT) with concurrent chemotherapy (CCT) Int J Radiat Oncol Biol Phys. 2001;51(3):40. [Google Scholar]
- 190.Pohlers D, Brenmoehl J, Loffler I, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792(8):746–56. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 191.Arora PD, McCulloch CA. The deletion of transforming growth factor-beta-induced myofibroblasts depends on growth conditions and actin organization. Am J Pathol. 1999;155(6):2087–99. doi: 10.1016/s0002-9440(10)65527-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Randall K, Coggle JE. Expression of transforming growth factor-beta 1 in mouse skin during the acute phase of radiation damage. Int J Radiat Biol. 1995;68(3):301–9. doi: 10.1080/09553009514551231. [DOI] [PubMed] [Google Scholar]
- 193.Randall K, Coggle JE. Long-term expression of transforming growth factor TGF beta 1 in mouse skin after localized beta-irradiation. Int J Radiat Biol. 1996;70(3):351–60. doi: 10.1080/095530096145085. [DOI] [PubMed] [Google Scholar]
- 194.Schultze-Mosgau S, Wehrhan F, Rodel F, et al. Anti-TGFbeta1 antibody for modulation of expression of endogenous transforming growth factor beta 1 to prevent fibrosis after plastic surgery in rats. Br J Oral Maxillofac Surg. 2004;42(2):112–9. doi: 10.1016/S0266-4356(03)00257-2. [DOI] [PubMed] [Google Scholar]
- 195.Flanders KC, Ho BM, Arany PR, et al. Absence of Smad3 induces neutrophil migration after cutaneous irradiation: possible contribution to subsequent radioprotection. Am J Pathol. 2008;173(1):68–76. doi: 10.2353/ajpath.2008.070937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Flanders KC, Major CD, Arabshahi A, et al. Interference with transforming growth factor-beta/Smad3 signaling results in accelerated healing of wounds in previously irradiated skin. Am J Pathol. 2003;163(6):2247–57. doi: 10.1016/s0002-9440(10)63582-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Arany PR, Flanders KC, DeGraff W, Cook J, Mitchell JB, Roberts AB. Absence of Smad3 confers radioprotection through modulation of ERK-MAPK in primary dermal fibroblasts. J Dermatol Sci. 2007;48(1):35–42. doi: 10.1016/j.jdermsci.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Anscher MS, Thrasher B, Rabbani Z, Teicher B, Vujaskovic Z. Antitransforming growth factor-beta antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation. Int J Radiat Oncol Biol Phys. 2006;65(3):876–81. doi: 10.1016/j.ijrobp.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 199.Robbins ME, Diz DI. Pathogenic role of the renin-angiotensin system in modulating radiation-induced late effects. Int J Radiat Oncol Biol Phys. 2006;64(1):6–12. doi: 10.1016/j.ijrobp.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 200.Cohen EP, Fish BL, Moulder JE. Treatment of radiation nephropathy with captopril. Radiat Res. 1992;132:346–50. [PubMed] [Google Scholar]
- 201.Geraci JP, Sun MC, Mariano MS. Amelioration of radiation nephropathy in rats by postirradiation treatment with dexamethasone and/or captopril. Radiat Res. 1995;143:58–68. [PubMed] [Google Scholar]
- 202.Moulder JE, Cohen EP, Fish BL, Hill P. Prophylaxis of bone marrow transplant nephropathy with captopril, an inhibitor of angiotensin-converting enzyme. Radiat Res. 1993;136(3):404–7. [PubMed] [Google Scholar]
- 203.Moulder JE, Fish BL, Cohen EP. Treatment of radiation nephropathy with ACE inhibitors. Int J Radiat Oncol Biol Phys. 1993;27(1):93–9. doi: 10.1016/0360-3016(93)90425-u. [DOI] [PubMed] [Google Scholar]
- 204.Molteni A, Moulder JE, Cohen EP, et al. Prevention of radiation-induced nephropathy and fibrosis in a model of bone marrow transplant by an angiotensin II receptor blocker. Exp Biol Med (Maywood) 2001;226(11):1016–23. doi: 10.1177/153537020122601108. [DOI] [PubMed] [Google Scholar]
- 205.Moulder JE, Robbins ME, Cohen EP, Hopewell JW, Ward WF. Pharmacologic modification of radiation-induced late normal tissue injury. Cancer Treat Res. 1998;93:129–51. doi: 10.1007/978-1-4615-5769-2_6. [DOI] [PubMed] [Google Scholar]
- 206.Moulder JE, Fish BL, Cohen EP. Treatment of radiation nephropathy with ACE inhibitors and AII type-1 and type-2 receptor antagonists. Curr Pharm Des. 2007;13(13):1317–25. doi: 10.2174/138161207780618821. [DOI] [PubMed] [Google Scholar]
- 207.Coleman CN, Stone HB, Moulder JE, Pellmar TC. Medicine. Modulation of radiation injury. Science. 2004;304(5671):693–4. doi: 10.1126/science.1095956. [DOI] [PubMed] [Google Scholar]
- 208.Stone HB, Moulder JE, Coleman CN, et al. Models for evaluating agents intended for the prophylaxis, mitigation and treatment of radiation injuries. Report of an NCI Workshop, December 3–4, 2003. Radiat Res. 2004;162(6):711–28. doi: 10.1667/rr3276. [DOI] [PubMed] [Google Scholar]
- 209.Moulder JE, Fish BL, Cohen EP. ACE Inhibitors and AII Receptor Antagonists in the Treatment and Prevention of Bone Marrow Transplant Nephropathy. Curr Pharm Des. 2003;9(9):737–49. doi: 10.2174/1381612033455422. [DOI] [PubMed] [Google Scholar]
- 210.Moulder JE, Fish BL, Cohen EP, Bonsib SM. Angiotensin II receptor antagonists in the prevention of radiation nephropathy. Radiat Res. 1996;146(1):106–10. [PubMed] [Google Scholar]
- 211.Moulder JE, Cohen EP. Future strategies for mitigation and treatment of chronic radiation-induced normal tissue injury. Semin Radiat Oncol. 2007;17(2):141–8. doi: 10.1016/j.semradonc.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 212.Ward WF, Lin PJ, Wong PS, Behnia R, Jalali N. Radiation pneumonitis in rats and its modification by the angiotensin-converting enzyme inhibitor captopril evaluated by high-resolution computed tomography. Radiat Res. 1993;135(1):81–7. [PubMed] [Google Scholar]
- 213.Ward WF, Molteni A, Ts’ao CH, Hinz JM. Captopril reduces collagen and mast cell accumulation in irradiated rat lung. Int J Radiat Oncol Biol Phys. 1990;19(6):1405–9. doi: 10.1016/0360-3016(90)90351-j. [DOI] [PubMed] [Google Scholar]
- 214.Molteni A, Moulder JE, Cohen EF, et al. Control of radiation-induced pneumopathy and lung fibrosis by angiotensin-converting enzyme inhibitors and an angiotensin II type 1 receptor blocker. Int J Radiat Biol. 2000;76(4):523–32. doi: 10.1080/095530000138538. [DOI] [PubMed] [Google Scholar]
- 215.Ward WF, Molteni A, Ts’ao CH, Kim YT, Hinz JM. Radiation pneumotoxicity in rats: modification by inhibitors of angiotensin converting enzyme. Int J Radiat Oncol Biol Phys. 1992;22(3):623–5. doi: 10.1016/0360-3016(92)90890-t. [DOI] [PubMed] [Google Scholar]
- 216.Kim JH, Brown SL, Kolozsvary A, et al. Modification of radiation injury by ramipril, inhibitor of angiotensin-converting enzyme, on optic neuropathy in the rat. Radiat Res. 2004;161(2):137–42. doi: 10.1667/rr3124. [DOI] [PubMed] [Google Scholar]
- 217.Robbins ME, Payne V, Tommasi E, et al. The AT1 receptor antagonist, L-158, 809, prevents or ameliorates fractionated whole-brain irradiation-induced cognitive impairment. Int J Radiat Oncol Biol Phys. 2009;73(2):499–505. doi: 10.1016/j.ijrobp.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhao W, Robbins ME. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr Med Chem. 2009;16(2):130–43. doi: 10.2174/092986709787002790. [DOI] [PubMed] [Google Scholar]
- 219.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension. 1998;31(1 Pt 2):181–8. doi: 10.1161/01.hyp.31.1.181. [DOI] [PubMed] [Google Scholar]
- 220.Peters H, Border WA, Noble NA. Targeting TGF-beta overexpression in renal disease: maximizing the antifibrotic action of angiotensin II blockade. Kidney Int. 1998;54(5):1570–80. doi: 10.1046/j.1523-1755.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- 221.Gomez-Garre D, Ruiz-Ortega M, Ortego M, et al. Effects and interactions of endothelin-1 and angiotensin II on matrix protein expression and synthesis and mesangial cell growth. Hypertension. 1996;27(4):885–92. doi: 10.1161/01.hyp.27.4.885. [DOI] [PubMed] [Google Scholar]
- 222.Kagami S, Kuhara T, Okada K, Kuroda Y, Border WA, Noble NA. Dual effects of angiotensin II on the plasminogen/plasmin system in rat mesangial cells. Kidney Int. 1997;51(3):664–71. doi: 10.1038/ki.1997.96. [DOI] [PubMed] [Google Scholar]
- 223.Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4(6):899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 224.Komazaki T, Nagai H, Emi M, et al. Hypermethylation-associated inactivation of the SOCS-1 gene, a JAK/STAT inhibitor, in human pancreatic cancers. Jpn J Clin Oncol. 2004;34(4):191–4. doi: 10.1093/jjco/hyh035. [DOI] [PubMed] [Google Scholar]
- 225.Xiao F, Puddefoot JR, Barker S, Vinson GP. Mechanism for aldosterone potentiation of angiotensin II-stimulated rat arterial smooth muscle cell proliferation. Hypertension. 2004;44(3):340–5. doi: 10.1161/01.HYP.0000140771.21243.ed. [DOI] [PubMed] [Google Scholar]